

Abstract
Biological water oxidation, performed by a single enzyme, photosystem II, is a central research topic not only in understanding the photosynthetic apparatus but also for the development of water splitting catalysts for technological applications. Great progress has been made in this endeavor following the report of a high-resolution X-ray crystallographic structure in 2011 resolving the cofactor site (Umena et al. in Nature 473:55–60, 2011), a tetra-manganese calcium complex. The electronic properties of the protein-bound water oxidizing Mn4OxCa complex are crucial to understand its catalytic activity. These properties include: its redox state(s) which are tuned by the protein matrix, the distribution of the manganese valence and spin states and the complex interactions that exist between the four manganese ions. In this short review we describe how magnetic resonance techniques, particularly EPR, complemented by quantum chemical calculations, have played an important role in understanding the electronic structure of the cofactor. Together with isotope labeling, these techniques have also been instrumental in deciphering the binding of the two substrate water molecules to the cluster. These results are briefly described in the context of the history of biological water oxidation with special emphasis on recent work using time resolved X-ray diffraction with free electron lasers. It is shown that these data are instrumental for developing a model of the biological water oxidation cycle.
Keywords: Photosystem II, Oxygen-evolving complex, Water binding, Triplet oxygen formation, EPR spectroscopy, Quantum chemical calculations
Introduction
More than three billion years ago, the cyanobacteria evolved a light-driven enzyme that was able to split water into molecular oxygen and hydrogen. Biology couples this to the reduction of carbon dioxide (CO2) to carbohydrates and in so doing stores the energy of the sun in energy-rich compounds with water acting as the electron source. We call this whole process photosynthesis and it represents the central metabolic pathway of the biosphere. While there exists a wide variety of photosynthetic organisms from cyanobacteria to algae and higher plants, the cellular machinery responsible for water splitting is unique and uses the same mechanism across all species.
A byproduct of biological water splitting is dioxygen (O2) (Fig. 1). Its accumulation over the last billion years has led to the formation of the oxygen-rich atmosphere of our planet (Fischer et al. 2016; Hamilton et al. 2016). It has also caused build-up of the ozone layer in the stratosphere that provides an efficient shield against high-energy UV radiation from the sun, thus protecting and sustaining life on earth. The increase in the concentration of O2 in the atmosphere started about two and a half billion years ago (Bekker et al. 2004) and led to the ‘great oxygenation event’ (GOE), a catastrophe for most early life forms on earth which had no defense mechanism to cope with the toxic oxygen, leading to mass extinction. On the other hand, the presence of O2 in the atmosphere fostered the development of higher, aerobic life forms, i.e., organisms that evolved to use oxygen for their metabolism. Thus, without the earlier invention of oxygenic photosynthesis, the development of complex life forms with cellular respiration—from simple vertebrates to human beings—would not have taken place. It is, therefore, not only of scientific but also of cultural value for mankind to understand how water oxidation and oxygen release functions in photosynthesis.
Fig. 1.

Oxygen build-up in the Earth’s atmosphere on a time scale of billions of years (Ga) and some major events in the development of our planet. The red and green curves denote an upper and lower estimate of the oxygen in the atmosphere. Oxygenic photosynthesis started about ≈ 3.5 Ga ago (Planavsky et al. 2014), the release of O2 in the atmosphere ≈ 2.4 Ga ago (Bekker et al. 2004). The present level of O2 is ≈ 21% (Holland 2006)
We benefit from photosynthesis in many ways. It is our only source of food and supplies us with much of our energy in the form of fossilized photosynthetic material—oil, coal and natural gas. Furthermore, it provides us with valuable natural materials like wood, paper, cotton, peat and biomass in general. Plants also contain many important bioactive materials that are used as drugs, in cosmetics, as natural dyes etc.
A basic understanding of the way how Nature stores energy and, in particular, uses sunlight to split water, could open future pathways to harvest and store the sun’s abundant energy, satisfying the energy demands of society in a sustainable fashion i.e., by an “artificial photosynthesis” (acatech et al. 2018; Chabi et al. 2017; Collings and Critchley 2005; Cox and Lubitz 2013; Cox et al. 2015; Faunce et al. 2013; Gratzel 2005; McKone et al. 2016; Messinger et al. 2014; Nocera 2012, 2017; Wydrzynski and Hillier 2012). In the short review below the current knowledge of the principles of photosynthetic water oxidation in photosystem II of oxygenic organisms are described with special emphasis on the recent X-ray crystallographic data and the results obtained on the electronic structure of the water oxidizing cluster and the water binding and processing derived from advanced EPR techniques in combination with quantum chemical calculations. For a more extensive and detailed description of photosynthetic water oxidation in PS II the reader is referred to some recent reviews (Junge 2019; Pantazis 2018; Shen 2015; Vinyard and Brudvig 2017; Yano and Yachandra 2014).
The structure of photosystem II and the oxygen-evolving complex
In oxygenic photosynthesis, sunlight is collected by the light-harvesting complexes (LHCs), specialized chlorophyll (and carotenoid) containing proteins, which show a large variation depending on the type of organism (Croce and van Amerongen 2014). The light energy is funneled to a reaction center (RC) where charge separation across the photosynthetic membrane takes place. Two RCs—photosystem I and II (PS I, PS II)—work in tandem to create the redox potential necessary to drive the coupled reactions (Blankenship 2014). This process provides electrons with sufficiently negative potential needed to reduce NADP+ to NADPH, which can be considered “bound biological hydrogen”. The electrons for this process originate from water oxidation that takes place in PS II. This process, and subsequent reactions, also generate a proton gradient across the photosynthetic membrane, which is used by the enzyme ATPase to produce ATP from ADP (Junge and Nelson 2015). In a different cell compartment, ATP and NADPH are then used to reduce CO2 to carbohydrates in the so-called dark reactions (Calvin–Benson cycle) (Calvin 1962).
Much of our current understanding of the photosynthetic machinery has come from high-resolution crystal structures of its constituents, particular photosystem I and II. The first crystal structure of a bacterial photosynthetic reaction center (BRC) was reported by Deisenhofer, Michel and Huber in the early eighties (Deisenhofer et al. 1985; Michel 1982, 1983)—who subsequently received the Nobel Prize in 1988 for this work. The structure of the simpler BRC provided a model for the functional core of PS I and PS II, in particular the processes of charge separation and progressive radical pair migration. It did not, however, provide any information on the water oxidizing complex (WOC) which is also called the oxygen-evolving complex (OEC), as this component is not found in the BRC that instead uses another electron source.
A crystal structure of a functional oxygen-evolving PS II core complex took almost two decades to be obtained. This was first achieved by the groups of Horst T. Witt and Wolfram Saenger in Berlin who reported the first crystallization of PS II from the cyanobacterium Synechococcus (S.) elongatus (later renamed Thermosynechococcus (T.) elongatus) and obtained a structure with 3.8 Å resolution in 2001 (Zouni et al. 2001). Although this first crystallographic structure provided valuable insight into the arrangement of the protein subunits and the cofactors of PS II it did not allow the positions of the atoms in the OEC, a tetranuclear manganese cluster, to be determined. This is because of radiation damage induced by the intense X-ray beam used for X-ray diffraction (XRD) data collection in modern synchrotrons. X-ray absorption spectroscopy (XAS) experiments with varying beam intensity by Yano and coworkers have shown photoreduction of the oxidized Mn ions, disintegration of the complex and blurring of the electron density associated with the cluster (Yano et al. 2005). Before the advent of a PS II crystal structure with sufficient resolution attempts have been made to obtain models of the tetranuclear Mn cluster from spectroscopic experiments, in particular from XAS spectroscopy (Yachandra et al. 1996; Yano et al. 2006). In 2004 the group of James Barber (Imperial College London) presented an improved structure of PS II from S. elongatus at 3.5 Å resolution (Ferreira et al. 2004). Based on their refined data, including anomalous diffraction to identify the Ca, and also on previous EXAFS data (Robblee et al. 2001) the authors proposed a heterometallic cubane-type structure of the OEC, which contained a Mn3Ca unit along with a more distant dangler Mn. Such a “dangler model” had been proposed earlier by David Britt’s group (UC Davis) based on constraints from EPR and ENDOR experiments and the distances obtained from EXAFS (Peloquin and Britt 2001; Peloquin et al. 2000). Finally, in 2011 the groups of Jian-Ren Shen (Okayama, Japan) and Nobuo Kamija (Osaka, Japan), who had reported a low resolution structure of PS II in 2003 (Kamiya and Shen 2003) published a significantly improved structure from Thermosynechococcus (T.) vulcanus at 1.9 Å resolution, in which the atomic arrangement of the ions in the OEC was finally resolved (see below) (Umena et al. 2011). Subsequent theoretical studies showed that the actual oxidation state of the OEC in this structure was a mixture of overreduced states, not present in the catalytic cycle (Galstyan et al. 2012; Luber et al. 2011). The Shen group also succeeded in obtaining a structure with reduced radiation damage using femtosecond (fs) pulsed X-ray crystallography with a free electron laser (XFEL) (Suga et al. 2015). More recently, time resolved XFEL measurements (serial femtosecond crystallography—SFX) near ambient temperature has emerged as an important tool for the investigation of the various states of the catalytic cycle of the OEC (see below) (Kern et al. 2012, 2013, 2014, 2018; Kupitz et al. 2014; Suga et al. 2015, 2017).
In Fig. 2 the dimeric PS II protein is shown (Suga et al. 2015), located in the thylakoid membrane. Each monomer is made up of 20 protein subunits (in cyanobacteria). A few important ones are indicated in Fig. 2A. In Fig. 2B the cofactor arrangement in the D1/D2 subunits is shown—comprising chlorophylls (PD1, PD2, ChlD1, ChlD2, ChlzD1, ChlzD2), carotenoids (CarD1, CarD2), pheophytins (PheoD1, PheoD2), plastoquinones (QA, QB), non-heme iron (Fe) and also the WOC or OEC (Fig. 2C). This cubane-like cluster is bound in the PS II protein and comprises four Mn ions and one Ca ion linked by five oxygen bridges (six in the last stage of the catalytic cycle). Remarkably, it also binds four water molecules, two at the Ca and two at the dangling Mn4. The cluster is connected to the protein surface via specific channels for the efficient uptake of water, and the release of protons and molecular oxygen (Bondar and Dau 2012; Gabdulkhakov et al. 2009; Ho and Styring 2008; Linke and Ho 2014; Murray and Barber 2007; Retegan and Pantazis 2017; Saito et al. 2012; Umena et al. 2011; Vassiliev et al. 2012). For each PS II monomer about 1300 water molecules have been detected in the crystal structure (Umena et al. 2011); they are mostly found at the outside membrane region but quite a few are located inside of the PS II protein. The water access channels to the Mn cluster are shown together with a few key amino acid ligands in Fig. 2D. The first channel is via the Asp 61 (shaded blue) the second via the Ca (shaded yellow) and a possible third one via the Cl− binding site (green). In all channels several water molecules have been found in the crystal structure (Ferreira et al. 2004; Gabdulkhakov et al. 2009; Ho and Styring 2008; Murray and Barber 2007; Saito et al. 2012; Umena et al. 2011; Vassiliev et al. 2012). Cl− depletion inhibits water oxidation; exchange of Cl− with Br− or I− slows the water oxidation kinetics (Damoder et al. 1986; Ono et al. 1987). The effect of these halogen anions is probably related to charge effects in the protein and proton egress.
Fig. 2.

X-ray crystallographic structure of photosystem II (PS II) from T. vulcanus. A View of the dimeric protein (molecular weight of the dimer ≈ 700 kDa); the two monomers are related to each other by a C2 axis. The most important subunits are indicated, CP43, CP47, in which the core antenna pigments are located; D1 and D2, which bind all pigments of the reaction center and all cofactors of the electron transport chain, and the small subunits PsbO, PsbU and PsbV, which stabilize the water splitting unit. The D1 protein (yellow) holds the active branch cofactors and the Mn cluster, the D2 protein (orange) the cofactors of the second pigment branch. B Pigment arrangement in one PS II monomer; the two branches are related by a pseudo-C2 axis (dotted line). Shown are the primary donor P680 (four chlorophylls: PD1, PD2, ChlD1, ChlD2), the two pheophytins (PheoD1, PheoD2), the two plastoquinones (QA, QB), and the non-heme iron Fe. Additional chlorophylls (Chlz) and carotenoids (Car), as well as two redox-active tyrosines (TyrZ, TyrD) are also indicated. Next to the active branch (D1) and close to TyrZ/P680, the Mn4O5Ca cluster is located. C Structure of the water oxidizing Mn4O5Ca cluster in PS II with four Mn ions (Mn1 to Mn4, purple) and one Ca (yellow), bridged by oxygen ligands (red). Three Mn ions (1 to 3) and the Ca form a distorted cube bridged by oxygen ligands, the fourth Mn (Mn4) is dangling. Mn4 and the Ca carry two water molecules each (W1 to W4, orange). The coordination of the metal ions by amino acid ligands is also shown; for further details see (Suga et al. 2015; Umena et al. 2011). D Water channels leading to the OEC: Three channels have been localized. Note that also the essential chloride ion is shown in this picture (Suga et al. 2015; Umena et al. 2011). A second Cl− is found farther away from the OEC. It has been pointed out that the channels could be multi-functional
Basic function of photosystem II: kinetics and energetics of water oxidation
Light excitation of the reaction center of PS II, a multi-pigment assembly of four chlorophyll-a and two pheophytin-a molecules, leads to primary charge separation on a picosecond timescale. This is a single electron transfer process resulting in a charge-separated radical pair (RP) state comprising the radical cation P680·+ and the radical anion Pheo·− (Cardona et al. 2012). During primary charge separation, the positive charge is initially localized on the accessory chlorophyll ChlD1 and it is stabilized by subsequent transfer on PD1 (see Fig. 2A) (Groot et al. 2005; Holzwarth et al. 2006; Kammel et al. 2003). The primary acceptor Pheo is the pheophytin pigment bound to the D1 protein (Holzwarth et al. 2006). The oxidation potential of P680·+ is estimated to + 1.2 to + 1.3 V, the highest known in biology (Rappaport et al. 2002). The primary RP P680·+Pheo·− is stabilized by subsequent electron/hole transfer steps. This is important as the radical cation can, in principle, oxidize neighboring chlorophylls, and the protein itself. To suppress both side and back reactions, nature has placed secondary donor/acceptors in close proximity to the primary RP. On the donor side of the protein (lumen), it is the redox-active tyrosine residue D1-Tyr161 (known as TyrZ or Yz) next to P680·+ that is able to quickly donate an electron to this species on a nanosecond (20–250 ns) time scale, thereby reducing the radical cation to its initial state P680. The tyrosine radical cation Y·+z is stabilized by losing a proton to the neighboring D1-His190 in a reversible fashion, forming a neutral tyrosine radical (Y·z) (Berthomieu et al. 1998; Chu et al. 1995; Saito et al. 2011). Y·z in turn oxidizes the adjacent Mn cluster in 40–1600 µs dependent on the Si transition (Babcock et al. 1976; Brettel et al. 1984; Dekker et al. 1984a; Haumann et al. 2005a; Karge et al. 1997; Klauss et al. 2012a; Noguchi et al. 2012; Rappaport et al. 1994; Razeghifard and Pace 1997). On the acceptor side of the protein (cytoplasm), the acceptor Pheo·− passes its electron to the bound plastoquinone molecules (to QA and subsequently to QB). Reducing equivalents derived from this process are temporarily stored as reduced plastoquinol (QBH2), a mobile electron carrier, generated after two light absorption/charge separation and protonation events.
Other light-induced processes that occur in PS II are equally dangerous, for which nature has not found a perfect solution to circumvent them. This is the creation of triplet states, e.g., of the chlorophylls in the core antenna via intersystem crossing or in the RC via (triplet) radical pair recombination. While carotenoid molecules are present in PS II to suppress (quench) triplet chlorophyll formation, these states can still react with triplet oxygen (3O2) formed in the water oxidation process yielding singlet oxygen (1O2) (Macpherson et al. 1993). This very aggressive species can then react with the pigments and/or the protein, resulting in damage of the D1 protein subunit of the PSII supercomplex. The production of such reactive oxygen species (ROS) (Vass 2012) limits the lifetime of PS II to only 30 min under normal light conditions. Owing to its remarkable efficiency (the turnover time of the OEC is ≈ 2 ms, that of the whole PS II ≈ 10 ms)(Vinyard and Brudvig 2017), it performs more than 105 reaction cycles before it must be replaced. Fortunately, all organisms performing oxygenic photosynthesis have developed an efficient repair mechanism for the PS II supercomplex by the discrete replacement of the D1 protein (Nixon et al. 2010).
PS II thus acts as a water:plastoquinone oxidoreductase (Wydrzynski and Satoh 2005). The products are molecular (triplet) oxygen (3O2), protons and reduced plastohydroquinone. The light-induced process in PS II is very efficient, with a quantum yield of over 90% and an energy efficiency of about 20%. This value is, however, strongly attenuated when all the subsequent processes are considered, such that the total biomass generated often contains only less than 1% of the original light energy input (Blankenship et al. 2011; Michel 2008).
It was discovered by Pierre Joliot almost half a century ago that PS II releases oxygen after four consecutive light flashes (Joliot et al. 1969) (Fig. 3A). This shows that four light-induced trans-membrane charge separation events are necessary before one O2 molecule is formed and released. This result is in agreement with the fact that the oxidation of two water molecules to produce a single O2 molecule is a four-electron process,
Fig. 3.
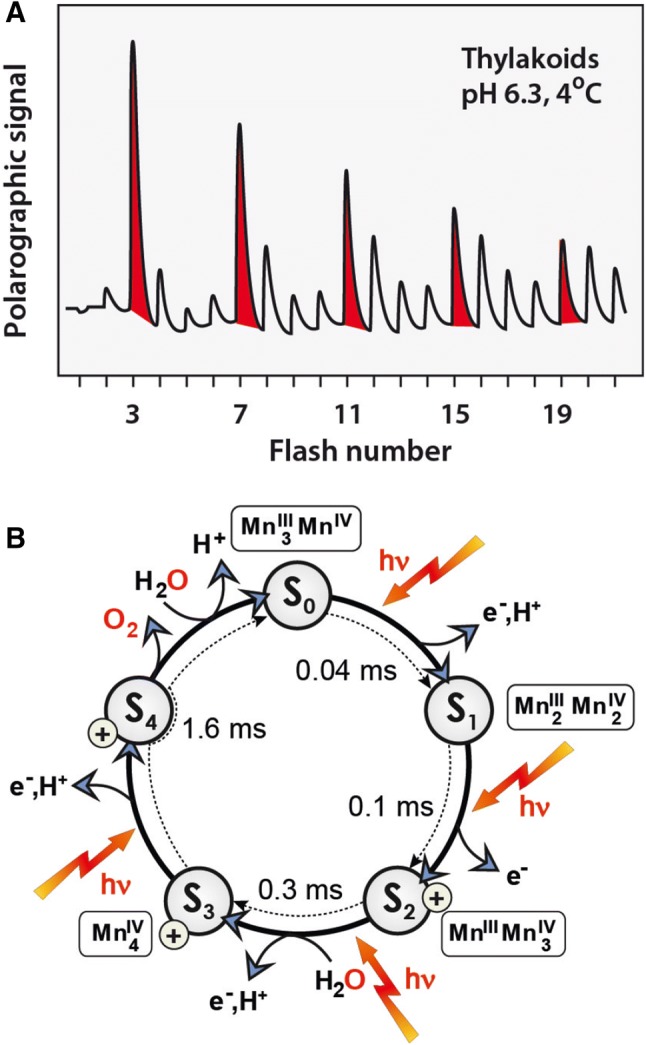
A Release pattern of molecular oxygen (O2) measured polarographically following successive light flashes of spinach thylakoids at 4 °C (Messinger and Renger 2008). Note that O2 release follows a 4-flash pattern (the starting dark stable state is S1). The original experiment was performed by Pierre Joliot as early as 1969 (Joliot et al. 1969). B Water oxidation cycle (Kok cycle) (Kok et al. 1970) detailing the five basic S states (S0 to S4), the light-induced 1e− oxidation steps and the proton release pattern (Dau and Haumann 2008), the uptake of the two substrate waters (Hillier and Wydrzynski 2008) and the Mn oxidation states (Krewald et al. 2015) (vide infra). The reaction times for the single electron oxidation steps are also indicated (Klauss et al. 2012a). Note that here the “S” stands for “state” and not for the electron spin quantum number S
| 1 |
whereas the charge separation across the membrane is a one-electron process. The oxidizing equivalents for the water oxidation reaction are stored transiently by the tetranuclear manganese cluster, an insight that came from Kok et al. (1970). His so-called Kok cycle (S-state cycle) of water oxidation, shown in Fig. 3B, comprises five distinct states, S0 to S4, which differ by the number of oxidizing equivalents transiently stored in the cofactor, as indicated by their subscript. The cycle is driven by four light absorption events (hν), by which electrons are withdrawn from the metal ions and protons are released to avoid the accumulation of positive charges and thus prevent a “coulombic explosion” of the cluster. O2 is released in a concerted reaction in the last reaction step with an equilibrium constant k > 1.0 × 107 (Nilsson et al. 2016). Kinetic measurements showed that the reaction times lie in the micro- to millisecond range (40 µs to 1.6 ms) (Klauss et al. 2012a), see Fig. 3B; the complete cycle turns over in about 2 ms. The kinetics of the proton release, which follow a 1:0:1:2 pattern (or 1:0:1:1:1) have also been determined, see (Dau and Haumann 2007; Klauss et al. 2012a, 2015; Schlodder and Witt 1999; Suzuki et al. 2009). The (alternating) electron and proton release led to an extended catalytic cycle for the water oxidation reaction in PS II with nine states that differ in their net electron and proton count (Klauss et al. 2012a, 2015; Perez-Navarro et al. 2016).
In this sequence, YZ promotes both electron and proton transfer in the catalytic cycle displaying a dual function (Bovi et al. 2013; Klauss et al. 2012b; Perez-Navarro et al. 2016). Between Si and Si+1 states (for i = 0–3) the short-lived SiYZ˙ intermediates exist. Studying these intermediates is critical for understanding the role of YZ˙ in water oxidation and especially in proton removal from the Mn4OxCa cluster (Boussac et al. 2008; Havelius et al. 2010; Nugent et al. 2002; Peloquin et al. 1998; Petrouleas et al. 2005; Retegan et al. 2014; Styring et al. 2012).
There are three interlinked reasons why the Mn4OxCa cluster functions in this way: (i) The energetic cost is significantly lowered by first storing oxidizing equivalents and then performing the four-electron chemistry; (ii) this sequence avoids the formation of reactive oxygen species (ROS) as intermediates resulting from partial oxidation of substrate water, which are highly reactive and can destroy the PS II protein complex; and (iii) proton release during charge accumulation leads to charge neutrality so that about the same oxidizing potential (oxidant P680·+) can be used for all Mn oxidation events. This is illustrated by the scheme in Fig. 4 (Messinger and Renger 2008). Here the Mn ions (and/or their ligands) are oxidized four times by the neighboring tyrosine Y·Z triggered by four charge separation events in PS II before the cluster removes four electrons from two bound water molecules in a concerted reaction leading to O2 release and regeneration of the original starting state of the Mn4OxCa cluster. The Mn cluster thus acts as an interface and storage device between the very fast light reaction (ps time scale) and the slow catalytic reaction (ms time scale) of the 4-electron water oxidation chemistry, bridging a kinetic gap of nine orders of magnitude.
Fig. 4.
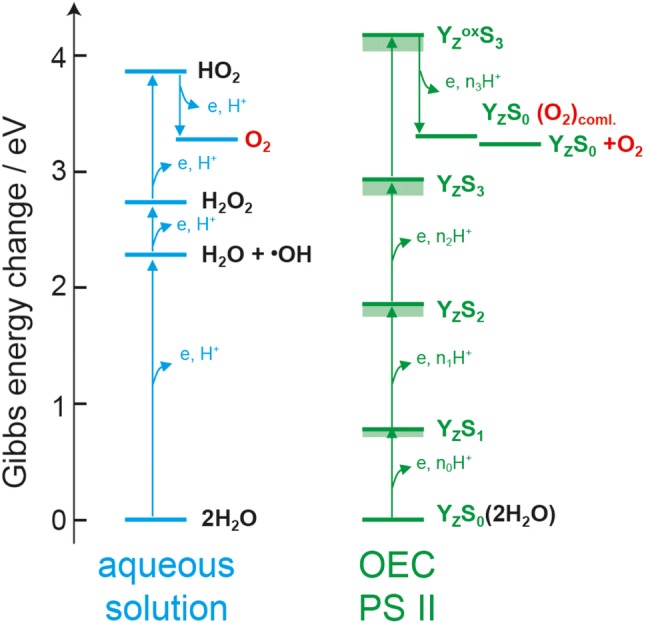
Gibbs energy (in eV) required to oxidize water stepwise in aqueous solution (left) and in the OEC of PS II (right). In particular the removal of the first electron from water (left) requires much energy (> 2 eV) that cannot be provided in a biological system. Thus, in the OEC, water is not oxidized by subsequent single electron removal from substrate water. Instead, it is the Mn cluster that is oxidized by four successive oxidation events; the two attached substrate water molecules release the protons (for charge neutrality), and O2 is released only in the last step after O–O bond formation in a concerted reaction. Thereby, high-energy steps are avoided and the redox process is leveled. Figure adapted from (Messinger and Renger 2008)
The electronic structure of the Mn4OxCa cluster
A detailed understanding of the catalytic process of water oxidation in PS II requires knowledge of the electronic structure, i.e., the distribution of the electrons in the cluster, in all consecutive reaction steps. The oxidation and spin states of the Mn ions, representing the total number and configuration of electrons in the Mn valence orbitals, give a basic description thereof. These together with the magnetic interactions between the spin-bearing Mn ions, depending to a large part on the metal ligands, provide a comprehensive picture of the respective electronic state, which governs the chemical and catalytic properties of each S state. Thus, the spin states provide information about how the structure of the cofactor evolves during the S-state cycle, for a recent review see (Krewald et al. 2016).
An experimental method to investigate the electronic structure of transition metal complexes is electron paramagnetic resonance (EPR) spectroscopy (Goldfarb and Stoll 2018; Schweiger and Jeschke 2001; Weil and Bolton 2007). It exploits a fundamental property of matter, which is that unpaired electrons have an intrinsic angular momentum (spin), which can be excited by microwave radiation in a magnetic field. The unpaired electron spin also interacts with other electron and nuclear spins as well as with local electric field gradients, making it a sensitive reporter of its chemical environment. It is thus analogous to other magnetic spectroscopies such as nuclear magnetic resonance (NMR). Since the Mn ions are open-shell species, i.e., exhibit orbitals with single electron occupancy, whereas most of the electrons of the protein and solvent surrounding are paired, the EPR signals of the Mn4OxCa cluster can be detected selectively. It has also been shown that all S states in the Kok cycle can be trapped (except for the elusive S4 state) and that all exhibit paramagnetism (Haddy 2007).
The first EPR signal observed by Charles Dismukes in 1981 (Dismukes and Siderer 1981) came from the trapped S2 state. Centered at g ≈ 2 it included over 20 resolved lines and was thus called “multiline signal” (see Fig. 5). The structure is due to the coupling of the electron spin with the four 55Mn nuclei (I = 5/2) via the electron nuclear hyperfine interaction. The EPR signal represents an effective low-spin ground state of Seff = ½ (Bencini and Gatteschi 1990). The spin state depends on the oxidation state of the Mn ions, their geometry and in particular on the (bridging) ligands which connect the metal ions. These mediate antiferromagnetic or ferromagnetic exchange interactions between the Mn ions leading to either a low-spin state, minimizing the number of unpaired electrons, or a high-spin state, maximizing the number of unpaired electrons. The studies of the groups of Ono and Kusunoki on oriented PS II membranes and theoretical investigations (Hasegawa et al. 1998, 1999) and subsequent advanced EPR studies from the Britt (UC Davis) (Britt et al. 2000, 2004; Peloquin et al. 2000) and Lubitz (MPI Mülheim) laboratories (Cox et al. 2011; Kulik et al. 2005a, b, 2007; Lohmiller et al. 2012, 2014; Su et al. 2011) further constrained all four 55Mn hyperfine tensors in the S2 state and allowed the spin coupling in the tetranuclear manganese cluster to be interrogated. These data together with results collected on the S0 state, which also resolves a multiline signal (Ahrling et al. 1997; Kulik et al. 2005b, 2007; Lohmiller et al. 2017; Messinger et al. 1997), and density functional theory calculations allowed the local oxidation states and set of coupling pathways to be determined (Ames et al. 2011; Krewald et al. 2013, 2015, 2016; Pantazis et al. 2012, 2009). The oxidation state assignment for the S2 state, which comes from this analysis, is shown in Fig. 5 (top), i.e., Mn4(III, IV, IV, IV). According to the low oxidation scheme S0 includes a MnII (Kolling et al. 2012; Pace et al. 1991; Terrett et al. 2016), but this was excluded by the 55Mn ENDOR experiments and analysis of the S0 state (Kulik et al. 2005b, 2007; Lohmiller et al. 2017). The reader interested in details on how the electronic configuration of a polynuclear Mn cluster, especially the Mn4CaO5, can be probed by EPR spectroscopy and double resonance techniques in relation with calculations of magnetic properties by quantum mechanical methods is referred to the following review (Lohmiller et al. 2013). Additional support for the high oxidation state scheme has come from the detection of 14N (I = 1), 2H (I = 1) and 13C (I = ½) hyperfine couplings of the S2 state using ESEEM and ENDOR techniques performed by the group of R. D. Britt (Britt et al. 1989; Marchiori et al. 2018; Perez-Navarro et al. 2013; Oyala et al. 2015; Stich et al. 2011; Stull et al. 2010); see also (Lohmiller et al. 2017; Perez-Navarro et al. 2013). X-ray spectroscopies also agree with this assignment (Chatterjee et al. 2016; Zaharieva et al. 2016a, b). These studies rule out the alternative lower oxidation state models for the manganese cluster (Kolling et al. 2012; Pace et al. 1991) that are, however, still discussed in the literature (Chen et al. 2018; Terrett et al. 2016).
Fig. 5.
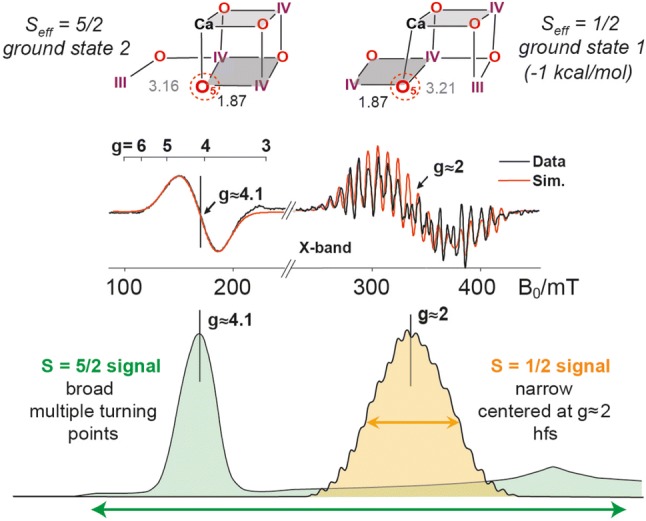
Top: DFT-optimized structures of the Mn4O5Ca cluster in the S2 state (distances given in Ångström). Note that the “closed cubane” (left) and “open cubane” (right) structures have almost the same energy but different positions of the MnIII ion (Mn4 vs. Mn1, respectively) and different total (effective) spin ground states. This explains the two EPR signals observed for this state (shown below as first derivative X-band EPR spectra), with g = 4.1 (Seff = 5/2; no hyperfine structure) and g = 2 (Seff = ½; multiline signal). The bottom trace shows simulations of the two EPR absorption signals.
Figure modified from (Krewald et al. 2016; Pantazis et al. 2012)
In the S2 state, in addition to the low-spin Seff = ½ form showing the characteristic multiline signal around g ≈ 2, the cluster can also be found in a high-spin Seff = 5/2 state under certain conditions. This is evident from an EPR signal around g = 4.1 (see Fig. 5), which has been observed earlier by several research groups (Boussac et al. 1996; Casey and Sauer 1984; Haddy et al. 2004; Zimmermann and Rutherford 1984). Pantazis et al. could show computationally that the two electronic structures are a direct consequence of two different spatial conformations of the manganese cluster, namely a “closed cubane” (Seff = 5/2) and an “open cubane” (Seff = ½) form, which have almost the same energy (Bovi et al. 2013; Isobe et al. 2012; Pantazis et al. 2012). In these two structures one of the oxygen bridges, O5, is occupying different positions. In turn this also led to a change of the MnIII position (Jahn–Teller ion) and thus of the open coordination site of the cluster resulting in a change of the precise electronic properties of the cofactor. EXAFS data also support this model (Chatterjee et al. 2019). This shows that the effective spin Seff is a crucial parameter for describing a particular state of the cluster and assigning it a spatial structure. It is postulated that O5 has not a fixed position but toggles between two structures in the dynamic S2 state, which is important for the water binding and the catalytic mechanism (see below).
Modifications of the Mn4OxCa cluster, such as exchange of Ca2+ with Sr2+ (Boussac and Rutherford 1988; Boussac et al. 2015; Chu et al. 2000; Cox et al. 2011; Koua et al. 2013; Yachandra and Yano 2011), its complete removal (Boussac et al. 1989; Lohmiller et al. 2012; Styring et al. 2003; Vrettos et al. 2001b) and the binding of small molecules (Beck et al. 1986; Perez-Navarro et al. 2013; Oyala et al. 2015; Oyala et al. 2014; Retegan and Pantazis 2016; Su et al. 2011), provided further insight into the conformation and number of ligands of individual Mn ions (coordination geometry) and substrate access in the S2 state. Upon Ca depletion water oxidation functionality is lost, but the S2 state (S2′) is still formed (Boussac et al. 1989, 1990). Interestingly, the EPR and 55Mn ENDOR data of S2′ show that the calcium has no substantial effect on the magnetic parameters of the S2 state; this ion is thus not crucial for maintaining the electronic structure of the tetranuclear Mn cluster (Lohmiller et al. 2012). Instead, it might serve as stage for the delivery of water molecules to the reaction site (Nakamura et al. 2016; Service et al. 2014; Ugur et al. 2016), affect the function of YZ, (Miqyass et al. 2008; Retegan et al. 2014) and introduce some structural flexibility allowing the cofactor to toggle between the different motifs of the open and closed cubane structures (see Fig. 5). The interactions of small molecules which mimic the substrate water (methanol, ammonia) also support this potential role for Ca2+. These molecules associate with the Ca2+ and Asp61 water channels that lead to the Mn4OxCa cofactor (Marchiori et al. 2018; Navarro et al. 2013; Oyala et al. 2014; Retegan and Pantazis 2016; Schraut and Kaupp 2014) (see Fig. 2D). In addition, both of these molecules reduce turnover efficiency; these results implicate that at least one (or possibly both) of these channels are involved in substrate delivery.
The EPR measurements have been extended to the S3 state. These results are of particular importance since this is the last metastable state prior to O–O bond formation and O2 release. The EPR signal of the S3 state has first been reported to originate from a ground state with an integer spin of Seff = 3 by the groups of Petrouleas (Sanakis et al. 2008) and Boussac (Boussac et al. 2009). Recent pulse high field (W-band) EPR experiments by Cox et al. (see Fig. 6A) corroborated this assignment and directly proved that the effective spin state is indeed Seff = 3 by measuring the Rabi oscillations via a spin nutation experiment (Cox et al. 2014). In addition 55Mn ELDOR-detected-NMR (EDNMR) experiments at W-band could successfully be performed showing that in the S3 state all Mn ions are in the MnIV state Mn4 (IV,IV,IV,IV) and octahedrally coordinated. Thus, no ligand oxidation takes place as previously proposed (Kawashima et al. 2018; Messinger et al. 2001). The results also show that a sixth ligand is binding to the open coordination site of the MnIII present in the S2 state when it is oxidized. This ligand is most probably the second substrate water molecule which also loses a proton in the S2 → S3 transition. Data from XAS, XES and FTIR further support these finding (Noguchi 2008; Sakamoto et al. 2017; Zaharieva et al. 2016a, b). Mn oxidation in the S2 to S3 transition has been proposed by Dekker (Dekker et al. 1984a, b) and Ono (Ono et al. 1992) and substantiated by Dau and coworkers (Haumann et al. 2005a, b). It has been included in mechanistic models (Pecoraro et al. 1998; Vrettos et al. 2001a) and has also theoretically been supported by Siegbahn (Siegbahn 2009). Very recently, SFX data have shown additional electron density acquired during the S2 to S3 transition, consistent with the binding of a light atom (e.g., an oxo or hydroxyl ligand) in the manganese cluster (Kern et al. 2018; Suga et al. 2017).
Fig. 6.

A W-band EPR spectrum of the OEC in the S3 state in PS II of T. elongatus (S3–S1 light–dark difference spectrum) characteristic of an Seff = 3 ground state. The red-dashed line shows a simulation with g and fine structure (zero field splitting) parameters D and E/D; the shaded curves show the contributions of the allowed EPR transitions to the simulated powder spectrum, the color code is indicated. As insert a 55Mn EDNMR spectrum is shown obtained at the EPR position indicated by the arrow. The used EDNMR pulse sequence is given above. Figure modified from (Cox et al. 2014). B Structure of the manganese cluster proceeding through the S2 → S3 → S4 states including water Wf binding, proton release and O2 formation as suggested by magnetic spectroscopy
The magnetic resonance experiments described above together with theoretical calculations allowed a reliable characterization of the S0, S2 and S3 states with respect to oxidation and spin states of individual ions and their spin coupling in the tetranuclear Mn cluster summarized by Krewald et al. (2015, 2016). Together with information about binding of the two substrate water molecules described below spatial models of the S states could be obtained that form the basis for developing a catalytic mechanism for the OEC (Fig. 8).
Fig. 8.

Model for the water oxidation cycle in PS II based on spectroscopic and theoretical work (Cox et al. 2015; Krewald et al. 2016) detailing the structures of the Mn cluster in the different S states, the water binding events and the O–O bond formation and O2 release in S4. The boxes show the determined oxidation states of the Mn ions in the respective S state. A color code is used for the assignment of the MnIII (light purple) and MnIV (dark purple) ions when the OEC is passing through the S states. Note that the oxygens of the waters and µ-oxo bridges are given in red except for the proposed substrate oxygens O5 and O6 (green). The S2 state exists in two conformations (closed/open cubanes), see text. S3 may also exist in an open (SA3) and a closed (SB3) cubane form (not shown). For S3 a state S3′ is shown in which manganese oxidation has occurred but the 2nd water has not been introduced. This state is present in native sample in a minority of PS II centers, but it is stabilized upon specific chemical treatment (methanol addition or Ca2+ to Sr2+ exchange) (Chrysina et al. 2019). A switching of the preferred total spin ground state configuration of the cluster is thought to take place in S2 from low to high spin and between S4 and S0 back from high to low spin; this is indicated by the diagonal line dividing the green and blue boxes. This switching of the spin state may be necessary for the formation of triplet dioxygen 3O2 in the final step of the cycle (see also Fig. 10).
Figure changed from (Krewald et al. 2016)
Identification of the substrate water molecules in water oxidation
Knowledge of the binding and dynamics of the substrate water molecules is crucial for formulating the reaction mechanism of photosynthetic water oxidation. Candidates for substrate waters, besides the four H2O/OH− molecules (Fig. 2C) directly attached to the cluster (Ames et al. 2011; Suga et al. 2015; Umena et al. 2011), are oxygen bridges within the cluster as well as possible water molecules binding at a later point in the catalytic cycle. Apart from X-ray diffraction spectroscopic techniques are used to investigate water binding, in particular EPR (Cox et al. 2013b; Fiege et al. 1996; Kawamori et al. 1989; McConnell et al. 2012; Nagashima and Mino 2013; Rapatskiy et al. 2012) and vibrational (infrared) spectroscopy (Debus 2014, 2015; Kim et al. 2018; Kim and Debus 2017; Nakamura et al. 2016; Noguchi 2008; Sakamoto et al. 2017; Suzuki et al. 2008). These are used in conjunction with time-resolved mass spectrometry (MIMS) which detects the uptake of H182O labeled water into the product O2; for a comprehensive review on the identification of possible water substrates by MIMS see (Cox and Messinger 2013). MIMS experiments showed that the two substrate water molecules exchange at different rates in all of the S states (Hillier and Wydrzynski 2000, 2004, 2008). The more slowly exchanging water (Ws) has an exchange rate of the order of seconds (Messinger et al. 1995) while the faster exchanging water (Wf) has an exchange rate of milliseconds (Hillier et al. 1998) (see Fig. 3B). The observation of two rates implies that the two substrates bind at two chemically distinct sites. They also demonstrate that the O–O bond is not formed until the (S4) state is reached. Interestingly, it was recently observed that during the lifetime of the S3YZ˙ state, that lies between S3 and the elusive S4 state while the YZ˙ radical exists, the water exchange is slowing down, which may suggest that the two oxygen species are “arrested” in a bonding formation (Nilsson et al. 2014).
For the detection of substrate water molecules, EPR spectroscopy makes use of interactions with nuclear spins, e.g. 2H (in 2H2O) (Fiege et al. 1996; Kawamori et al. 1989; Nagashima and Mino 2013) or preferentially 17O (nuclear spin I = 5/2) in PS II samples with isotope-labeled H172O as the solvent (Lohmiller et al. 2017; McConnell et al. 2012; Rapatskiy et al. 2012). Methodological and instrumental developments in our laboratory (Cox et al. 2013a, 2017) allowed us to characterize the 17O signals in the S2 and S0 states using electron–electron double resonance (ELDOR)-detected NMR (EDNMR) at high frequency (94 GHz) (Lohmiller et al. 2017; Rapatskiy et al. 2012), which yields significantly better sensitivity compared with 17O ENDOR (McConnell et al. 2012; Rapatskiy et al. 2012). Through spectral simulations, we could identify the µ-oxo bridge O5 to be an exchangeable ligand in the S2 state (Fig. 7A, B) (Lohmiller et al. 2014; Perez-Navarro et al. 2013; Rapatskiy et al. 2012). O5 represents the most probable candidate for the two-fold deprotonated substrate Ws for the following reasons:
-
(i)
It is the only exchangeable 17O nucleus bound both to Mn and Ca, a requirement imposed by MIMS experiments (Cox and Messinger 2013; Hendry and Wydrzynski 2003; Messinger 2004).
-
(ii)
Shortening of an Mn–Mn bond observed by EXAFS has been explained by a deprotonation of an O-bridge and has been assigned to O5 (Robblee et al. 2002; Messinger 2004).
-
(iii)
Similar timescales for the water exchange were found both for Ws in the MIMS, see ref. (Hillier and Wydrzynski 2008), and for O5 in the EPR experiments, i.e., complete exchange in < 10 s (Rapatskiy et al. 2012).
-
(iv)
Orientation dependent EDNMR studies showed that the measured hyperfine coupling can only be assigned to O5 or O4) (Rapatskiy et al. 2012).
-
(v)
Both Ca/Sr exchange experiments and ammonia inhibition studies demonstrate O5 and only O5 to be an exchangeable oxygen bridge (Cox et al. 2011; Perez-Navarro et al. 2013).
Fig. 7.

A Mn cluster with the µ-oxo bridges and bound H2O/OH− molecules in the S2 state; the same color code as in panel (B) is used. B17O EDNMR spectrum at W-band (94 GHz) obtained from PS II core preparations (T. elongatus) in the S2 state. The buffer has been exchanged with H172O (90% enrichment) for 30 min (3 ×) in the dark (S1 state) prior to flash-advancement to the S2 state (black trace). EDNMR spectra are also shown for H162O buffer, containing only 14N signals (dark blue trace, 14N from histidine His332, see Fig. 2C). Three classes of oxygen ligands were assigned: the largest 17O splitting belongs to the exchangeable µ-oxo bridge O5 (green), the smaller ones to the H2O/OH− molecules (orange, purple) indicated in the figure.
For further details see (Rapatskiy et al. 2012)
This is in agreement with earlier low frequency FTIR studies of the OEC that showed that a µ2-oxo or µ3-oxo bridge is exchangeable (Chu et al. 2000). It is remarkable that in the OEC a µ-oxo bridge is readily exchangeable. For structurally similar model compounds an exchange has only been observed on much longer timescales (Rapatskiy et al. 2015; Tagore et al. 2006, 2007). These results further demonstrate the dynamic nature of this O5 ligand in the OEC (see above, Fig. 5).
Recent 17O EDNMR investigations of H172O binding to the S0 state of the OEC show that an OH− is introduced in the initial step of the cycle (the 1st substrate water that has lost a proton) after O2 release in the S4 to S0 transition (Lohmiller et al. 2017). In the S0 and S1 state the O5 is thought to exist in an open cubane-type structure (in S0 with S = ½, and in S1 with S = 0 ground state) as shown in Fig. 8.
As described above, recent spectroscopic results, including high field EPR indicate that a new water molecule (O6) is inserted into the manganese cluster in the S3 state as an OH− at the open coordination site of Mn1 (Cox et al. 2014). Its close proximity to O5 suggests it is the second substrate Wf (Fig. 6B). How it is exactly inserted remains an open question. Recent SFX data (Kern et al. 2018; Suga et al. 2017) indicate that the Glu189, which bridges the Ca2+ ion and Mn1 in the S1 state detaches from the Ca2+ ion upon formation of the S3 state. This removes steric hindrance to direct water binding via the Ca2+ channel, i.e., it is originally one of the Ca2+ waters. Another possibility is that this OH− (O6) is introduced in an indirect fashion by toggling between the open and closed cubane structures and is thus derived from W2, one of the terminal ligands of Mn4 (compare structure S3′ in Fig. 8). Within this sequence the water that binds during the S2 to S3 transition is not one of the substrates of the current cycle, but a substrate of the next cycle (Cox and Messinger 2013). It also implies that O5 (the existing μ-oxo bridge) and O6 (the new water-derived oxygen ligand) may switch identities. There is significant theoretical support for this sequence, which explains how redox tuning of the cofactor can precede water binding (Retegan et al. 2016). This hypothesis also better agrees with MIMS data which shows both substrates are bound to the cofactor (most likely to Mn ions) in all S-states and that the rate of exchange does not dramatically change upon moving from S2 to S3 (Cox and Messinger 2013; Hendry and Wydrzynski 2002, 2004).
The water oxidation cycle in PS II
As a consequence of the above results, it is assumed in the following discussion of the catalytic cycle of the OEC that the O5 bridge derives from one of the substrate waters (Ws). A possible cycle is shown in Fig. 8 (Cox et al. 2015; Krewald et al. 2016). The first water Ws binds as an OH− (O5) in the S0 state, being deprotonated during the subsequent oxidation step to S1. In the S3 state the second substrate water enters the reaction as OH− (O6) to Mn4 (or Mn1) as suggested by the results obtained from EPR and XFEL experiments on the S3 state (Cox et al. 2014; Kern et al. 2018; Suga et al. 2017). A further oxidation and deprotonation step leads to S4. Experimental evidence shows that formation of S4 is triggered by proton release (Haumann et al. 2005a; Klauss et al. 2012a). Current theoretical models suggest that a proton is transferred from W1 (H2O) to Asp61, as in the preceding S2 to S3 transition (Siegbahn 2012). Mutants of PSII lacking Asp61 show a pH dependency of the lag phase during the S3–S0 transition which has been attributed to a deprotonation of the Mn4CaO5 (Bao and Burnap 2015; Dilbeck et al. 2012) in support of this model.
In the current literature, there are several proposed mechanisms that suggest the early onset of O–O bond formation in the S3 state (Corry and O’Malley 2018). This is mainly based on the SFX crystal structure of Suga et al. which modeled a short distance between the O5 and O6 bridges in the S3 state (Suga et al. 2017). We note that the more recent higher resolution SFX structure of the S3 state (Kern et al. 2018), does not exhibit this short O5–O6 distance, and is instead more consistent with the high field EPR structure described above. Furthermore, earlier biophysical measurements categorically rule out O–O bond formation in the S3 state. The MIMS data described above shows that both substrates still rapidly exchange with bulk water in the S3 state (Messinger et al. 1995). It is only upon oxidation and proton release to form the S3Y·z state directly preceding S4 that substrate exchange is ‘arrested’ indicating the onset of O–O bond formation, i.e., formation of a peroxo type intermediate (Nilsson et al. 2014).
How the O–O bond is precisely formed in the S4 state is still unknown, owing to a lack of experimental data. There are two popular chemical mechanisms for this transition that differ with regard to what component of the cofactor is oxidized to form S4: (i) a ligand (one of the substrate waters); or (ii) a metal (one of the Mn ions). In the case of a ligand oxidation event, forming a MnIV-oxyl moiety, O–O bond formation is proposed to proceed by a radical coupling mechanism described below. In the case of a metal oxidation event, forming a MnV-oxo type moiety, O–O bond formation is instead proposed to proceed by nucleophilic attack of the electrophilic oxygen (O5) bound to the MnV by a nearby water, e.g., W3 bound to the Ca2+. Unfortunately the nature of the last oxidation (ligand vs. metal centered) does not resolve this question. XAS data which monitors an oxidation state change of the Mn ions during the S3 to S0 transition appears to rule out a MnV intermediate (Haumann et al. 2005a), favoring instead a ligand (oxygen) oxidation. We note that while the nucleophilic attack mechanism initially envisaged the involvement of a MnV intermediate, it was later suggested to have substantial MnIV oxyl character in the S4 state based on theoretical studies (Vinyard and Brudvig 2017).
Only a nucleophilic attack mechanism has been observed for first row molecular transition metal water oxidation catalysts, for which there is mechanistic data, (Codola et al. 2015) with radical coupling to be thus far only described for second row transition metals, e.g., ruthenium (Cox and Lubitz 2013; Romain et al. 2009). Nucleophilic attack mechanisms have thus been proposed for the OEC by a number of groups (Ferreira et al. 2004; Pecoraro et al. 1998); see (Barber 2017; Vinyard and Brudvig 2017; Vinyard et al. 2015) for recent reviews. In this sequence, a nearby water (e.g., W3) attacks a manganese bound oxygen, most likely O5. Upon re-reduction of the four Mn ion and loss of the product O2, the newly inserted O6 bridge presumably fills the site vacated by O5 leading to rapid recovery of the S0 state.
In contrast the oxo-oxyl radical coupling mechanism involving two Mn bound oxygens, is most compatible with the assignment of the two substrates sites described in the previous section (O5 and O6). Such a mechanism was first proposed ten years ago by Per Siegbahn (Siegbahn 2009). Importantly high field S3 state EPR data has demonstrated that there is the requisite spin alignment of the two putative substrate oxygens and the Mn ions to which they are bound (Mn4-O5-O6-Mn1 = βαβα) on which this mechanism is based.
To illustrate the current situation, in Fig. 9 three different structures are shown preceding the O–O bond formation in S4. They are derived from the S3 open and closed cubane structures, SA3 and SB3, and from S3′ in which a second water has not (yet) been bound (Fig. 8). In panel (A) and (B) the open and closed cubane structures are shown with all Mn(IV) and O5 and O6 in close contact that could react to form dioxygen via an oxyl-oxo coupling mechanism. In panel C) the dangling Mn4 is 5-coordinate; in the shown geometry the formation of a Mn(V) = O species is preferred in which the electrophilic oxygen could be attacked by a nucleophilic water (e.g., bound to Ca) or a neighboring oxo group as pointed out by Pantazis (2018). In this context it should be mentioned that in the recent SFX structure (Kern et al. 2018) only the open cubane structure has been observed for S2 and S3 which led the authors to propose O5 to react with either O6 (here called Ox) bound to Mn1 or with W3 (at Ca) or W2 (at Mn4).
Fig. 9.

Three representative model structures for the final step in the Kok cycle with O5 and O6 bound (see Figs. 8, 10); A Mn(IV)-oxyl formation at Mn1, octahedral coordination, all Mn(IV), open cubane structure; B Mn(IV)-oxyl formation at Mn4, octahedral, closed cubane structure; C Mn(V) = O formation at Mn4, trigonal bipyramidal, closed cubane. The putative reacting oxygens are indicated by dotted red circles; for details see text.
Figure adopted from (Pantazis 2018)
There are also other alternative mechanisms. A very recent example, proposed by the Yamaguchi group, involves an O–O˙ radical intermediate, formed following electron transfer to YZ (Shoji et al. 2018). Similarly, there are proposed mechanisms that do not fall into either of the two categories, such as a recent proposal from the Sun group. In this novel mechanism, the dangler Mn acts as the site of catalysis, as in a nucleophilic attack like mechanism, forming a MnVII-dioxo intermediate following charge rearrangement of the Mn cluster in the S4 state (Zhang and Sun 2018). As above for the nucleophilic attack mechanism, it is unclear if this mechanism is compatible with XAS data (Haumann et al. 2005a). For a more detailed comparison of alternative mechanisms the reader is directed to a recent in-depth review (Pantazis 2018).
Owing to the increasing experimental support for the oxo-oxyl coupling mechanism first proposed by Siegbahn, a brief description of its key steps is given below (Fig. 10). The first stage of the mechanism involves oxidation of O6 to form an oxyl radical, in concert with deprotonation. In the Siegbahn mechanism, the proton associated with O6 in S3 migrates to W1, which is already deprotonated during S3 formation. The generated oxyl radical carries an unpaired β spin electron, because O6 is antiferromagnetically coupled to Mn1. The oxyl radical then attacks the O5 bridge which carries a small excess of α spin. This results in formation of an σ O–O bond between O5 and O6—with the new σ bond consisting of the α spin electron of O5 and the β spin electron of O6 (Fig. 10 [S4 oxyl]). The α spin electron comes from the cleavage of the existing Mn–O bond between O5 and Mn4. The β spin electron of the cleaved Mn–O bond is transferred back to Mn4, reducing its oxidation state to III+. Note that the β electron of the cleaved Mn–O bond has the same spin as the three existing d-electrons of Mn4, and thus this transfer can occur without energy penalty due to spin conversion.
Fig. 10.

Spin alignment in the S3, [S4] and S0 states that fosters low barrier O–O bond formation as described by Siegbahn (Siegbahn 2009). S3 and S0 have been characterized experimentally; [S4] has not been observed. The spin ground state SG is indicated. The numbering of the Mn ions (1–4) is given for the S3 state; the oxidation state of each Mn ion is given in light purple (III) or dark purple (IV); antiferromagnetic interaction between adjacent Mn ions is represented by yellow shading whereas ferromagnetic interaction by green shading. The spin alignment of the Mn ions in each state is given on the top (the order is: Mn4-Mn3-Mn2-Mn1); the spin of the oxyl radical at the [S4 oxyl] state is indicated by the red colored β. The Jahn–Teller (JT) axis of the MnIII of the [S4 peroxo] intermediate is depicted by a dashed purple line. Evidence of the spin alignment of Mn4-O5-O6-Mn1 in the [S4 oxyl] state (βαβα) is derived from experiments on the S3 state (Cox et al. 2014; Krewald et al. 2016). For details of the mechanism see text
Subsequently, a peroxo product (a bound O2 with a single σ bond) is formed bridging Mn3 and Mn1 (Fig. 10 [S4 peroxo]). Mn4 is now five coordinate with the product peroxide sitting along its Jahn–Teller (JT) axis (Li and Siegbahn 2015; Siegbahn 2009). Cleavage of the two remaining Mn–O bonds leads to formation of the final O2 product. Importantly the cleavage of each Mn–O bond results in an α spin electron being transferred to each of the two Mn ions that were attached to the peroxide and a β spin electron being transferred to O5 and O6. As before, this is an energetically favorable situation as the electron gained by each metal is of the same spin (α) as their existing d-electrons. The two new β spin electrons occupy two π* orbitals giving rise to the triplet O2 product. The displacement of O2, via binding of the first substrate water of the next S cycle (coupled to its deprotonation), leads to formation of the observed (metastable) S0 state (Fig. 10, S0). The O2 may detach in a stepwise fashion, during its concerted replacement with OH−, passing through a transient superoxo intermediate (Li and Siegbahn 2015).
New experimental data is needed to probe these key stages in O–O bond formation and release. SFX crystallography (Kern et al. 2014, 2018; Kupitz et al. 2014; Suga et al. 2015, 2017; Young et al. 2016) is poised to address structural changes during the S3 to S0 transition, and may suggest an alternative proton transfer sequence. Specifically, as described above, the Siegbahn mechanism starts with internal proton transfer from O6 to W1. We note, however, that recent SFX measurements indicate that Glu189 detaches from the Ca2+ ion during S-state progression. Glu189 is proximal to Mn1 and could act as an internal base, accepting a proton from O6, similar to old ideas put forward by Gerry Babcock (Hoganson and Babcock 1997). Within this revised scheme W1 does not need to undergo deprotonation—with proton transfer to Asp61 switched off due to a conformational change (gating) (Kern et al. 2018).
Conclusions
In biology, there is only one catalytic site that is able to split water—the OEC—and its structure and function is identical (Su et al. 2011) in cyanobacteria, algae and all higher plants; however, differences in the second coordination sphere between higher plants and cyanobacteria have been reported (Retegan and Pantazis 2017). Many other enzymes have found different ways to perform their task—but not the unique, highly optimized OEC in PS II. The work compiled above shows that this unique metal cluster requires the presence and concerted action of all four manganese ions in the Mn4OxCa complex. The oxidation states of the Mn ions are always III or IV—there exists no MnII in the whole cycle. The possible presence of a MnV in the last step (S4) has, however, not yet been finally clarified. The importance of the total electron spin state of the cluster has been highlighted—and is understandable considering the necessary restriction to form and release triplet dioxygen 3O2. The Ca2+ is probably important for efficient water delivery and the Cl− for maintaining the correct charge balance of the OEC. Water delivery is optimized through highly efficient water channels leading to the Mn cluster. In the favored model the two substrate waters bind to neighboring redox-active Mn ions, and deprotonation of these waters is coupled to the oxidation events of the cluster. Close proximity and proper alignment of the two active oxygens are assured to efficiently form the O–O bond and finally release the 3O2 molecule with high efficiency.
In spite of the great progress in the understanding of the OEC in PS II there are still several challenges to be tackled. One pertinent open question is the structure of the elusive S4 state. A possible stabilization and characterization of this state would finally complete our knowledge of the S states of the Kok cycle. A missing key experiment is further the detection of the 6th oxygen in the manganese cluster by EPR (using 17O labeling) to support the recent crystallographic XFEL (SFX) data on the S3 state (Kern et al. 2018; Suga et al. 2017). Furthermore the determination of the water binding kinetics should be followed for all state transitions by rapid freeze quench (RFQ) techniques combined with advanced EPR techniques. First experiments along these lines have successfully been performed in our laboratory using time resolved RFQ 17O EDNMR measurements (Rapatskiy et al., unpublished data). The exchange of certain amino acids in the vicinity of the OEC could shed light on the fine-tuning of its electronic structure in the S state cycle and would be important for explaining its high turnover frequency. Furthermore, a great challenge for chemists working in the field of water oxidation is the understanding and modeling of the sophisticated mechanism by which PS II repairs photo-damage of its protein, of the pigments and of the manganese cluster.
The properties of the OEC in PS II described above represent valuable design features for bioinspired light-driven molecular catalysts for water oxidation. The following points are considered essential for such catalysts:
The material of the catalyst must be abundant, easily accessible, inexpensive (i.e., no precious metals), non-toxic, sufficiently stable under working conditions and should be scalable.
The required four oxidizing equivalents must be stored in the catalyst to couple the fast 1-electron photochemical reaction with the slow 4-electron chemical water oxidation process.
The binding of the two substrate water molecules should preferentially take place at two well-defined neighboring redox-active metal centers, accompanied by successive deprotonation and activation of the water molecules.
The redox steps of the catalytic metal centers should be of similar magnitude and in the range of 1 eV, and should finally lead to a concerted water oxidation, O2 formation and O2 release, to avoid reactive oxygen intermediates. This also requires a sequential release of the 4 protons.
A functional matrix mimicking the protein (in PS II) is needed for efficient transport of water to the reaction zone, release of dioxygen and for the correct proton management.
In the photochemical act, a light-induced species must be generated with sufficient oxidative power to oxidize water (+ 1.23 eV).
Effective coupling of the charge separation unit with the catalytic center (metal cluster) is necessary to diminish the overpotential (comparable to the function of the tyrosine YZ in PS II).
The catalytic unit should be stable; in case of deterioration/destruction a mechanism of self-repair or healing should be in place to avoid deactivation of the catalytic process.
At present, it is still a great challenge to create a catalyst that fulfills the above criteria and could thus compete with the Mn cluster in PS II. The future will show if at least part of the above points can be fulfilled. Attempts to synthesize water splitting chemical catalysts and devices are too numerous in the literature to be discussed here—but show the great importance of this research field for a future society using (only) renewable, sustainable energy based on sunlight driven processes (Andreiadis et al. 2011; Blakemore et al. 2015; Kanan and Nocera 2008; Kurz 2016; Najafpour et al. 2016; Tran et al. 2012; Zhang et al. 2015; Matheu et al. 2019).
Acknowledgements
Open access funding provided by Max Planck Society. Financial support of this work by the Max Planck Society and MANGAN (03EK3545) funded by the Bundesministeriums für Bildung und Forschung is gratefully acknowledged. N.C. acknowledges the support of the Australian Research Council (FT140100834).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wolfgang Lubitz, Email: wolfgang.lubitz@cec.mpg.de.
Maria Chrysina, Email: maria.chrysina@cec.mpg.de.
Nicholas Cox, Email: nick.cox@anu.edu.au.
References
- acatech—National Academy of Science and Engineering, Leopoldina—German National Academy of Sciences, Union of the German Academies of Sciences and Humanities, editor. Artificial photosynthesis. Munich: National Academy of Science and Engineering; 2018. [Google Scholar]
- Ahrling KA, Peterson S, Styring S. An oscillating manganese electron paramagnetic resonance signal from the S0 state of the oxygen evolving complex in photosystem II. Biochemistry. 1997;36:13148–13152. doi: 10.1021/bi971815w. [DOI] [PubMed] [Google Scholar]
- Ames W, Pantazis DA, Krewald V, Cox N, Messinger J, Lubitz W, Neese F. Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II: protonation states and magnetic interactions. J Am Chem Soc. 2011;133:19743–19757. doi: 10.1021/ja2041805. [DOI] [PubMed] [Google Scholar]
- Andreiadis ES, Chavarot-Kerlidou M, Fontecave M, Artero V. Artificial photosynthesis: from molecular catalysts for light-driven water splitting to photoelectrochemical cells. Photochem Photobiol. 2011;87:946–964. doi: 10.1111/j.1751-1097.2011.00966.x. [DOI] [PubMed] [Google Scholar]
- Babcock GT, Blankenship RE, Sauer K. Reaction-kinetics for positive charge accumulation on water side of chloroplast photosystem 2. FEBS Lett. 1976;61:286–289. doi: 10.1016/0014-5793(76)81058-7. [DOI] [PubMed] [Google Scholar]
- Bao H, Burnap RL. Structural rearrangements preceding dioxygen formation by the water oxidation complex of photosystem II. Proc Natl Acad Sci USA. 2015;112:6139–6147. doi: 10.1073/pnas.1512008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber J. A mechanism for water splitting and oxygen production in photosynthesis. Nat Plants. 2017;3:17041. doi: 10.1038/nplants.2017.41. [DOI] [PubMed] [Google Scholar]
- Beck WF, Depaula JC, Brudvig GW. Ammonia binds to the manganese site of the O2-evolving complex of photosystem-II in the S2 state. J Am Chem Soc. 1986;108:4018–4022. doi: 10.1021/ja00274a027. [DOI] [Google Scholar]
- Bekker A, Holland HD, Wang PL, Rumble D, Stein HJ, Hannah JL, Coetzee LL, Beukes NJ. Dating the rise of atmospheric oxygen. Nature. 2004;427:117–120. doi: 10.1038/nature02260. [DOI] [PubMed] [Google Scholar]
- Bencini A, Gatteschi D. Electron paramagnetic resonance of exchange coupled systems. New York: Springer; 1990. [Google Scholar]
- Berthomieu C, Hienerwadel R, Boussac A, Breton J, Diner BA. Hydrogen bonding of redox-active tyrosine Z of photosystem II probed by FTIR difference spectroscopy. Biochemistry. 1998;37:10547–10554. doi: 10.1021/bi980788m. [DOI] [PubMed] [Google Scholar]
- Blakemore JD, Crabtree RH, Brudvig GW. Molecular catalysts for water oxidation. Chem Rev. 2015;115:12974–13005. doi: 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- Blankenship RE, et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science. 2011;332:805–809. doi: 10.1126/science.1200165. [DOI] [PubMed] [Google Scholar]
- Blankenship RE, editor. Molecular mechanisms of photosynthesis. 2. Chichester: Wiley-Blackwell; 2014. [Google Scholar]
- Bondar AN, Dau H. Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim Biophys Acta. 2012;1817:1177–1190. doi: 10.1016/j.bbabio.2012.03.031. [DOI] [PubMed] [Google Scholar]
- Boussac A, Rutherford AW. Nature of the inhibition of the oxygen-evolving enzyme of photosystem-II induced by NaCl washing and reversed by the addition of Ca2+ or Sr2+ Biochemistry. 1988;27:3476–3483. doi: 10.1021/bi00409a052. [DOI] [Google Scholar]
- Boussac A, Zimmermann JL, Rutherford AW. Epr signals from modified charge accumulation states of the oxygen evolving enzyme in Ca2+ deficient photosystem II. Biochemistry. 1989;28:8984–8989. doi: 10.1021/bi00449a005. [DOI] [PubMed] [Google Scholar]
- Boussac A, Zimmermann JL, Rutherford AW. Further characterization of the modified S2 and S3 Epr signals observed in Ca2+ depleted photosystem-II reconstituted with the 17 and 23 kDa polypeptides. Curr Res Photosynth. 1990;1–4:713–716. doi: 10.1007/978-94-009-0511-5_162. [DOI] [Google Scholar]
- Boussac A, Girerd JJ, Rutherford AW. Conversion of the spin state of the manganese complex in photosystem II induced by near-infrared light. Biochemistry. 1996;35:6984–6989. doi: 10.1021/bi960636w. [DOI] [PubMed] [Google Scholar]
- Boussac A, Sugiura M, Lai TL, Rutherford AW. Low-temperature photochemistry in photosystem II from Thermosynechococcus elongatus induced by visible and near-infrared light. Philos Trans R Soc B. 2008;363:1203–1210. doi: 10.1098/rstb.2007.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussac A, Sugiura M, Rutherford AW, Dorlet P. Complete EPR spectrum of the S3 state of the oxygen-evolving photosystem II. J Am Chem Soc. 2009;131:5050–5051. doi: 10.1021/ja900680t. [DOI] [PubMed] [Google Scholar]
- Boussac A, Rutherford AW, Sugiura M. Electron transfer pathways from the S2 states to the S3 states either after a Ca2+/Sr2+ or a Cl−/I− exchange in photosystem II from Thermosynechococcus elongatus. Biochim Biophys Acta. 2015;1847:576–586. doi: 10.1016/j.bbabio.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Bovi D, Narzi D, Guidoni L. The S2 state of the oxygen evolving complex of photosystem II explored by QM/MM dynamics: Spin surfaces and metastable states suggest a reaction path towards the S3 state. Angew Chem. 2013;52:11744–11749. doi: 10.1002/anie.201306667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettel K, Schlodder E, Witt HT. Nanosecond reduction kinetics of photooxidized chlorophyll-alpha-II (P680) in single flashes as a probe for the electron pathway, H+ release and charge accumulation in the O2 evolving complex. Biochim Biophys Acta. 1984;766:403–415. doi: 10.1016/0005-2728(84)90256-1. [DOI] [Google Scholar]
- Britt RD, Zimmermann JL, Sauer K, Klein MP. Ammonia binds to the catalytic Mn of the oxygen-evolving complex of photosystem-II: evidence by electron-spin echo envelope modulation spectroscopy. J Am Chem Soc. 1989;111:3522–3532. doi: 10.1021/ja00192a006. [DOI] [Google Scholar]
- Britt RD, Peloquin JM, Campbell KA. Pulsed and parallel-polarization EPR characterization of the photosystem II oxygen-evolving complex. Annu Rev Biophys Biomed. 2000;29:463–495. doi: 10.1146/annurev.biophys.29.1.463. [DOI] [PubMed] [Google Scholar]
- Britt RD, et al. Recent pulsed EPR studies of the photosystem II oxygen-evolving complex: implications as to water oxidation mechanisms. Biochim Biophys Acta. 2004;1655:158–171. doi: 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Calvin M. Path of carbon in photosynthesis. Science. 1962;135:879–889. doi: 10.1126/science.135.3507.879. [DOI] [PubMed] [Google Scholar]
- Cardona T, Sedoud A, Cox N, Rutherford AW. Charge separation in photosystem II: a comparative and evolutionary overview. Biochim Biophys Acta. 2012;1817:26–43. doi: 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Casey JL, Sauer K. Electron-paramagnetic-res detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim Biophys Acta. 1984;767:21–28. doi: 10.1016/0005-2728(84)90075-6. [DOI] [Google Scholar]
- Chabi S, Papadantonakis KM, Lewis NS, Freund MS. Membranes for artificial photosynthesis. Energy Environ Sci. 2017;10:1320–1338. doi: 10.1039/C7EE00294G. [DOI] [Google Scholar]
- Chatterjee R, et al. Structural changes correlated with magnetic spin state isomorphism in the S2 state of the Mn4CaO5 cluster in the oxygen-evolving complex of photosystem II. Chem Sci. 2016;7:5236–5248. doi: 10.1039/C6SC00512H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee R, et al. Structural isomers of the S2 state in photosystem II: do they exist at room temperature and are they important for function? Physiol Plant. 2019;166:60–72. doi: 10.1111/ppl.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Dismukes GC, Case DA. Resolving ambiguous protonation and oxidation states in the oxygen evolving complex of photosystem II. J Phys Chem B. 2018;122:8654–8664. doi: 10.1021/acs.jpcb.8b05577. [DOI] [PubMed] [Google Scholar]
- Chrysina M et al (2019) New intermediates in the activation of nature’s water splitting cofactor. Proc Natl Acad Sci USA, Under Revision [DOI] [PMC free article] [PubMed]
- Chu HA, Nguyen AP, Debus RJ. Amino acid residues that influence the binding of manganese or calcium to photosystem-II. The lumenal interhelical domains of the D1 polypeptide. Biochemistry. 1995;34:5839–5858. doi: 10.1021/bi00017a016. [DOI] [PubMed] [Google Scholar]
- Chu HA, Sackett H, Babcock GT. Identification of a Mn-O-Mn cluster vibrational mode of the oxygen-evolving complex in photosystem II by low-frequency FTIR spectroscopy. Biochemistry. 2000;39:14371–14376. doi: 10.1021/bi001751g. [DOI] [PubMed] [Google Scholar]
- Codola Z, Gomez L, Kleespies ST, Que L, Costas M, Lloret-Fillol J. Evidence for an oxygen evolving iron-oxo-cerium intermediate in iron-catalysed water oxidation. Nat Commun. 2015;6:5865. doi: 10.1038/ncomms6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collings AF, Critchley C. Artificial photosynthesis: from basic biology to industrial application. Chichester: Wiley-VCH Weinheim; 2005. [Google Scholar]
- Corry TA, O’Malley PJ. Evidence of O–O bond formation in the final metastable S3 state of nature’s water oxidizing complex implying a novel mechanism of water oxidation. J Phys Chem Lett. 2018;9:6269–6274. doi: 10.1021/acs.jpclett.8b02793. [DOI] [PubMed] [Google Scholar]
- Cox N, Lubitz W. Molecular concepts of water splitting: nature’s approach. In: Schlögl R, editor. Chemical energy storage. Berlin/Boston: De Gruyter GmbH; 2013. pp. 185–224. [Google Scholar]
- Cox N, Messinger J. Reflections on substrate water and dioxygen formation. Biochim Biophys Acta. 2013;1827:1020–1030. doi: 10.1016/j.bbabio.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Cox N, et al. Effect of Ca2+/Sr2+ substitution on the electronic structure of the oxygen-evolving complex of photosystem II: a combined multifrequency EPR, 55Mn-ENDOR, and DFT study of the S2 state. J Am Chem Soc. 2011;133:3635–3648. doi: 10.1021/ja110145v. [DOI] [PubMed] [Google Scholar]
- Cox N, Lubitz W, Savitsky A. W-band ELDOR-detected NMR (EDNMR) spectroscopy as a versatile technique for the characterisation of transition metal–ligand interactions. Mol Phys. 2013;111:2788–2808. doi: 10.1080/00268976.2013.830783. [DOI] [Google Scholar]
- Cox N, Pantazis DA, Neese F, Lubitz W. Biological water oxidation. Acc Chem Res. 2013;46:1588–1596. doi: 10.1021/ar3003249. [DOI] [PubMed] [Google Scholar]
- Cox N, Retegan M, Neese F, Pantazis DA, Boussac A, Lubitz W. Electronic structure of the oxygen-evolving complex in photosystem II prior to O-O bond formation. Science. 2014;345:804–808. doi: 10.1126/science.1254910. [DOI] [PubMed] [Google Scholar]
- Cox N, Pantazis DA, Neese F, Lubitz W. Artificial photosynthesis: understanding water splitting in nature. Interface Focus. 2015;5:20150009. doi: 10.1098/rsfs.2015.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N, Nalepa A, Lubitz W, Savitsky A. ELDOR-detected NMR: a general and robust method for electron-nuclear hyperfine spectroscopy? J Magn Reson. 2017;280:63–78. doi: 10.1016/j.jmr.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Croce R, van Amerongen H. Natural strategies for photosynthetic light harvesting. Nat Chem Biol. 2014;10:492–501. doi: 10.1038/nchembio.1555. [DOI] [PubMed] [Google Scholar]
- Damoder R, Klimov VV, Dismukes GC. The Effect of Cl− depletion and X− reconstitution on the oxygen-evolution rate, the yield of the multiline manganese electron-paramagnetic-res signal and electron-paramagnetic-res signal II in the isolated photosystem II complex. Biochim Biophys Acta. 1986;848:378–391. doi: 10.1016/0005-2728(86)90214-8. [DOI] [PubMed] [Google Scholar]
- Dau H, Haumann M. Eight steps preceding O–O bond formation in oxygenic photo synthesis: a basic reaction cycle of the photosystem II manganese complex. Biochim Biophys Acta. 2007;1767:472–483. doi: 10.1016/j.bbabio.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Dau H, Haumann M. The manganese complex of photosystem II in its reaction cycle—basic framework and possible realization at the atomic level. Coord Chem Rev. 2008;252:273–295. doi: 10.1016/j.ccr.2007.09.001. [DOI] [Google Scholar]
- Debus RJ. Evidence from FTIR difference spectroscopy that D1-Asp61 influences the water reactions of the oxygen-evolving Mn4CaO5 cluster of Photosystem II. Biochemistry. 2014;53:2941–2955. doi: 10.1021/bi500309f. [DOI] [PubMed] [Google Scholar]
- Debus RJ. FTIR studies of metal ligands, networks of hydrogen bonds, and water molecules near the active site Mn4CaO5 cluster in photosystem II. Biochim Biophys Acta. 2015;1847:19–34. doi: 10.1016/j.bbabio.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J, Epp O, Miki K, Huber R, Michel H. Structure of the protein subunits in the photosynthetic reaction center of Rhodopseudomonas viridis at 3 Å resolution. Nature. 1985;318:618–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- Dekker JP, Plijter JJ, Ouwehand L, Vangorkom HJ. Kinetics of manganese redox transitions in the oxygen-evolving apparatus of photosynthesis. Biochim Biophys Acta. 1984;767:176–179. doi: 10.1016/0005-2728(84)90093-8. [DOI] [Google Scholar]
- Dekker JP, Van Gorkom HJ, Wensink J, Ouwehand L. Absorbance difference spectra of the successive redox states of the oxygen-evolving apparatus of photosynthesis. Biochim Biophys Acta. 1984;767:1–9. doi: 10.1016/0005-2728(84)90073-2. [DOI] [Google Scholar]
- Dilbeck PL, Hwang HJ, Zaharieva I, Gerencser L, Dau H, Burnap RL. The D1–D61 N mutation in Synechocystis sp. PCC 6803 allows the observation of pH-sensitive intermediates in the formation and release of O2 from photosystem II. Biochemistry. 2012;51:1079–1091. doi: 10.1021/bi201659f. [DOI] [PubMed] [Google Scholar]
- Dismukes GC, Siderer Y. Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc Natl Acad Sci USA. 1981;78:274–278. doi: 10.1073/pnas.78.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faunce TA, et al. Energy and environment policy case for a global project on artificial photosynthesis. Energy Environ Sci. 2013;6:695–698. doi: 10.1039/c3ee00063j. [DOI] [Google Scholar]
- Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Fiege R, Zweygart W, Bittl R, Adir N, Renger G, Lubitz W. EPR and ENDOR studies of the water oxidizing complex of photosystem II. Photosynth Res. 1996;48:227–237. doi: 10.1007/BF00041013. [DOI] [PubMed] [Google Scholar]
- Fischer WW, Hemp J, Johnson JE. Evolution of oxygenic photosynthesis. Annu Rev Earth Plant Sci. 2016;44:647–683. doi: 10.1146/annurev-earth-060313-054810. [DOI] [Google Scholar]
- Gabdulkhakov A, Guskov A, Broser M, Kern J, Muh F, Saenger W, Zouni A. Probing the accessibility of the Mn4Ca cluster in photosystem II: channels calculation, noble gas derivatization, and cocrystallization with DMSO. Structure. 2009;17:1223–1234. doi: 10.1016/j.str.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Galstyan A, Robertazzi A, Knapp EW. Oxygen-evolving Mn cluster in photosystem II: the protonation pattern and oxidation state in the high-resolution crystal structure. J Am Chem Soc. 2012;134:7442–7449. doi: 10.1021/ja300254n. [DOI] [PubMed] [Google Scholar]
- Goldfarb D, Stoll S, editors. EPR spectroscopy: fundamentals and methods. New York: eMagRes Books and Wiley; 2018. [Google Scholar]
- Gratzel M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg Chem. 2005;44:6841–6851. doi: 10.1021/ic0508371. [DOI] [PubMed] [Google Scholar]
- Groot ML, Pawlowicz NP, van Wilderen LJGW, Breton J, van Stokkum IHM, van Grondelle R. Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc Natl Acad Sci USA. 2005;102:13087–13092. doi: 10.1073/pnas.0503483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddy A. EPR spectroscopy of the manganese cluster of photosystem II. Photosynth Res. 2007;92:357–368. doi: 10.1007/s11120-007-9194-9. [DOI] [PubMed] [Google Scholar]
- Haddy A, Lakshmi KV, Brudvig GW, Frank HA. Q-band EPR of the S2 state of Photosystem II confirms an S = 5/2 origin of the X-band g = 4.1 signal. Biophys J. 2004;87:2885–2896. doi: 10.1529/biophysj.104.040238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TL, Bryant DA, Macalady JL. The role of biology in planetary evolution: cyanobacterial primary production in low-oxygen proterozoic oceans. Environ Microbiol. 2016;18:325–340. doi: 10.1111/1462-2920.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Kusunoki M, Inoue Y, Ono TA. Simulation of S2 state multiline EPR signal in oriented photosystem II membranes: structural implications for the manganese cluster in an oxygen-evolving complex. Biochemistry. 1998;37:9457–9465. doi: 10.1021/bi973057f. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Ono TA, Inoue Y, Kusunoki M. Spin-exchange interactions in the S2 state manganese tetramer in photosynthetic oxygen-evolving complex deduced from g = 2 multiline EPR signal. Chem Phys Lett. 1999;300:9–19. doi: 10.1016/S0009-2614(98)01369-4. [DOI] [Google Scholar]
- Haumann M, Liebisch P, Müller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- Haumann M, et al. Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3, S4 → S0) characterized by X-ray absorption spectroscopy at 20 K and room temperature. Biochemistry. 2005;44:1894–1908. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- Havelius KGV, Sjoholm J, Ho FM, Mamedov F, Styring S. Metalloradical EPR signals from the YZ S-state intermediates in photosystem II. Appl Magn Reson. 2010;37:151–176. doi: 10.1007/s00723-009-0045-z. [DOI] [Google Scholar]
- Hendry G, Wydrzynski T. The two substrate-water molecules are already bound to the oxygen-evolving complex in the S2 state of photosystem II. Biochemistry. 2002;41:13328–13334. doi: 10.1021/bi026246t. [DOI] [PubMed] [Google Scholar]
- Hendry G, Wydrzynski T. O-18 isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule to the oxygen-evolving complex in photosystem II. Biochemistry. 2003;42:6209–6217. doi: 10.1021/bi034279i. [DOI] [PubMed] [Google Scholar]
- Hillier W, Wydrzynski T. The affinities for the two substrate water binding sites in the O2 evolving complex of photosystem II vary independently during S-state turnover. Biochemistry. 2000;39:4399–4405. doi: 10.1021/bi992318d. [DOI] [PubMed] [Google Scholar]
- Hillier W, Wydrzynski T. Substrate water interactions within the photosystem II oxygen evolving complex. Phys Chem Chem Phys. 2004;6:4882–4889. doi: 10.1039/b407269c. [DOI] [Google Scholar]
- Hillier W, Wydrzynski T. O-18 water exchange in photosystem II: Substrate binding and intermediates of the water splitting cycle. Coordin Chem Rev. 2008;252:306–317. doi: 10.1016/j.ccr.2007.09.004. [DOI] [Google Scholar]
- Hillier W, Messinger J, Wydrzynski T. Kinetic determination of the fast exchanging substrate water molecule in the S3 state of photosystem II. Biochemistry. 1998;37:16908–16914. doi: 10.1021/bi980756z. [DOI] [PubMed] [Google Scholar]
- Ho FM, Styring S. Access channels and methanol binding site to the CaMn4 cluster in photosystem II based on solvent accessibility simulations, with implications for substrate water access. Biochim Biophys Acta. 2008;1777:140–153. doi: 10.1016/j.bbabio.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Hoganson CW, Babcock GT. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- Holland HD. The oxygenation of the atmosphere and oceans. Philos Trans R Soc B. 2006;361:903–915. doi: 10.1098/rstb.2006.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzwarth AR, Muller MG, Reus M, Nowaczyk M, Sander J, Rogner M. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: pheophytin is the primary electron acceptor. Proc Natl Acad Sci USA. 2006;103:6895–6900. doi: 10.1073/pnas.0505371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe H, et al. Theoretical illumination of water-inserted structures of the CaMn4O5 cluster in the S2 and S3 states of oxygen-evolving complex of photosystem II: full geometry optimizations by B3LYP hybrid density functional. Dalton Trans. 2012;41:13727–13740. doi: 10.1039/c2dt31420g. [DOI] [PubMed] [Google Scholar]
- Joliot P, Barbieri G, Chabaud R. A new model of photochemical centers in system 2. Photochem Photobiol. 1969;10:309–329. doi: 10.1111/j.1751-1097.1969.tb05696.x. [DOI] [Google Scholar]
- Junge W. Oxygenic photosynthesis: history, status and perspective. Q Rev Biophys. 2019;52:1–17. doi: 10.1017/S0033583518000112. [DOI] [PubMed] [Google Scholar]
- Junge W, Nelson N. ATP synthase. Annu Rev Biochem. 2015;84:631–657. doi: 10.1146/annurev-biochem-060614-034124. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Shen JR. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc Natl Acad Sci USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammel M, Kern J, Lubitz W, Bittl R. Photosystem II single crystals studied by transient EPR: the light-induced triplet state. Biochim Biophys Acta. 2003;1605:47–54. doi: 10.1016/S0005-2728(03)00063-X. [DOI] [PubMed] [Google Scholar]
- Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- Karge M, Irrgang KD, Renger G. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 1997;36:8904–8913. doi: 10.1021/bi962342g. [DOI] [PubMed] [Google Scholar]
- Kawamori A, Inui T, Ono T, Inoue Y. Endor study on the position of hydrogens close to the manganese cluster in S2 state of photosystem II. FEBS Lett. 1989;254:219–224. doi: 10.1016/0014-5793(89)81043-9. [DOI] [Google Scholar]
- Kawashima K, Takaoka T, Kimura H, Saito K, Ishikita H. O2 evolution and recovery of the water-oxidizing enzyme. Nat Commun. 2018;9:1247. doi: 10.1038/s41467-018-03545-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, et al. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc Natl Acad Sci USA. 2012;109:9721–9726. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science. 2013;340:491–495. doi: 10.1126/science.1234273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat Commun. 2014;5:4371. doi: 10.1038/ncomms5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature. 2018;563:421–425. doi: 10.1038/s41586-018-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CJ, Debus RJ. Evidence from FTIR difference spectroscopy that a substrate H2O molecule for O2 formation in photosystem II is provided by the Ca ion of the catalytic Mn4CaO5 cluster. Biochemistry. 2017;56:2558–2570. doi: 10.1021/acs.biochem.6b01278. [DOI] [PubMed] [Google Scholar]
- Kim CJ, Bao H, Burnap RL, Debus RJ. Impact of D1-V185 on the water molecules that facilitate O2 formation by the catalytic Mn4CaO5 cluster in photosystem II. Biochemistry. 2018;57:4299–4311. doi: 10.1021/acs.biochem.8b00630. [DOI] [PubMed] [Google Scholar]
- Klauss A, Haumann M, Dau H. Alternating electron and proton transfer steps in photosynthetic water oxidation. Proc Natl Acad Sci USA. 2012;109:16035–16040. doi: 10.1073/pnas.1206266109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauss A, Sikora T, Suss B, Dau H. Fast structural changes (200–900 ns) may prepare the photosynthetic manganese complex for oxidation by the adjacent tyrosine radical. Biochim Biophys Acta. 2012;1817:1196–1207. doi: 10.1016/j.bbabio.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Klauss A, Haumann M, Dau H. Seven steps of alternating electron and proton transfer in photosystem II water oxidation traced by time-resolved photothermal beam deflection at improved sensitivity. J Phys Chem B. 2015;119:2677–2689. doi: 10.1021/jp509069p. [DOI] [PubMed] [Google Scholar]
- Kok B, Forbush B, Mcgloin M. Cooperation of charges in photosynthetic O2 evolution 1. A linear 4 step mechanism. Photochem Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- Kolling DR, Cox N, Ananyev GM, Pace RJ, Dismukes GC. What are the oxidation states of manganese required to catalyze photosynthetic water oxidation? Biophys J. 2012;103:313–322. doi: 10.1016/j.bpj.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koua FHM, Umena Y, Kawakami K, Shen J-R. Structure of Sr-substituted photosystem II at 2.1 Å resolution and its implications in the mechanism of water oxidation. Proc Natl Acad Sci USA. 2013;110:3889–3894. doi: 10.1073/pnas.1219922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewald V, Neese F, Pantazis DA. On the magnetic and spectroscopic properties of high-valent Mn3CaO4 cubanes as structural units of natural and artificial water-oxidizing catalysts. J Am Chem Soc. 2013;135:5726–5739. doi: 10.1021/ja312552f. [DOI] [PubMed] [Google Scholar]
- Krewald V, et al. Metal oxidation states in biological water splitting. Chem Sci. 2015;6:1676–1695. doi: 10.1039/C4SC03720K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewald V, Retegan M, Neese F, Lubitz W, Pantazis DA, Cox N. Spin state as a marker for the structural evolution of nature’s water splitting catalyst. Inorg Chem. 2016;55:488–501. doi: 10.1021/acs.inorgchem.5b02578. [DOI] [PubMed] [Google Scholar]
- Kulik L, Epel B, Messinger J, Lubitz W. Pulse EPR, 55Mn ENDOR and ELDOR-detected NMR of the S2 state of the oxygen evolving complex in photosystem II. Photosynth Res. 2005;84:347–353. doi: 10.1007/s11120-005-2438-7. [DOI] [PubMed] [Google Scholar]
- Kulik LV, Epel B, Lubitz W, Messinger J. 55Mn pulse ENDOR at 34 GHz of the S0 and S2 states of the oxygen-evolving complex in photosystem II. J Am Chem Soc. 2005;127:2392–2393. doi: 10.1021/ja043012j. [DOI] [PubMed] [Google Scholar]
- Kulik LV, Epel B, Lubitz W, Messinger J. Electronic structure of the Mn4OxCa cluster in the S0 and S2 states of the oxygen-evolving complex of photosystem II based on pulse 55Mn ENDOR and EPR spectroscopy. J Am Chem Soc. 2007;129:13421–13435. doi: 10.1021/ja071487f. [DOI] [PubMed] [Google Scholar]
- Kupitz C, et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature. 2014;513:261–265. doi: 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz P. Biomimetic water-oxidation catalysts: manganese oxides. In: Tüysüz H, Chan CK, editors. Solar energy for fuels. Topics in current chemistry. Cham: Springer; 2016. pp. 49–72. [DOI] [PubMed] [Google Scholar]
- Li X, Siegbahn PEM. Alternative mechanisms for O2 release and O–O bond formation in the oxygen evolving complex of photosystem II. Phys Chem Chem Phys. 2015;17:12168–12174. doi: 10.1039/C5CP00138B. [DOI] [PubMed] [Google Scholar]
- Linke K, Ho FM. Water in Photosystem II: structural, functional and mechanistic considerations. Biochim Biophys Acta. 2014;1837:14–32. doi: 10.1016/j.bbabio.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Lohmiller T, Cox N, Su J-H, Messinger J, Lubitz W. The basic properties of the electronic structure of the oxygen-evolving complex of photosystem II are not perturbed by Ca2+ removal. J Biol Chem. 2012;287:24721–24733. doi: 10.1074/jbc.M112.365288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmiller T, Ames W, Lubitz W, Cox N, Misra SK. EPR Spectroscopy and the electronic structure of the oxygen-evolving complex of photosystem II. Appl Magn Reson. 2013;44:691–720. doi: 10.1007/s00723-012-0437-3. [DOI] [Google Scholar]
- Lohmiller T, et al. Structure, ligands and substrate coordination of the oxygen-evolving complex of photosystem II in the S2 state: a combined EPR and DFT study. Phys Chem Chem Phys. 2014;16:11877–11892. doi: 10.1039/c3cp55017f. [DOI] [PubMed] [Google Scholar]
- Lohmiller T, et al. The first state in the catalytic cycle of the water-oxidizing enzyme: identification of a water-derived μ-hydroxo bridge. J Am Chem Soc. 2017;139:14412–14424. doi: 10.1021/jacs.7b05263. [DOI] [PubMed] [Google Scholar]
- Luber S, et al. S1 state model of the O2 evolving complex of photosystem II. Biochemistry. 2011;50:6308–6311. doi: 10.1021/bi200681q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AN, Telfer A, Barber J, Truscott TG. Direct detection of singlet oxygen from isolated photosystem II reaction centers. Biochim Biophys Acta. 1993;1143:301–309. doi: 10.1016/0005-2728(93)90201-P. [DOI] [Google Scholar]
- Marchiori DA, Oyala PH, Debus RJ, Stich TA, Britt RD. Structural effects of ammonia binding to the Mn4CaO5 cluster of photosystem II. J Phys Chem B. 2018;122:1588–1599. doi: 10.1021/acs.jpcb.7b11101. [DOI] [PubMed] [Google Scholar]
- Matheu R, Garrido-Barros R, Gil-Sepulcre M, Ertem MZ, Sala X, Gimbert-Suriñach C, Llobet A. The development of molecular water oxidation catalysts. Nat Rev Chem. 2019;3:331–341. doi: 10.1038/s41570-019-0096-0. [DOI] [Google Scholar]
- McConnell IL, Grigoryants VM, Scholes CP, Myers WK, Chen P-Y, Whittaker JW, Brudvig GW. EPR-ENDOR characterization of (17O, 1H, 2H) water in manganese catalase and its relevance to the oxygen-evolving complex of photosystem II. J Am Chem Soc. 2012;134:1504–1512. doi: 10.1021/ja203465y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKone JR, Crans DC, Martin C, Turner J, Duggal AR, Gray HB. Translational science for energy and beyond. Inorg Chem. 2016;55:9131–9143. doi: 10.1021/acs.inorgchem.6b01097. [DOI] [PubMed] [Google Scholar]
- Messinger J. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys Chem Chem Phys. 2004;6:4764–4771. doi: 10.1039/b406437b. [DOI] [Google Scholar]
- Messinger J, Renger G. Photosynthetic water splitting. In: Renger G, editor. Primary processes of photosynthesis, Part 2: principles and apparatus. Cambridge, UK: RSC Publishing; 2008. pp. 291–349. [Google Scholar]
- Messinger J, Badger M, Wydrzynski T. Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem-II. Proc Natl Acad Sci USA. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger J, Robblee JH, Yu WO, Sauer K, Yachandra VK, Klein MP. The S0 state of the oxygen-evolving complex in photosystem II is paramagnetic: detection of EPR multiline signal. J Am Chem Soc. 1997;119:11349–11350. doi: 10.1021/ja972696a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger J, et al. Absence of Mn-centered oxidation in the S2 → S3 transition: implications for the mechanism of photosynthetic water oxidation. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger J, Lubitz W, Shen JR (eds) (2014) Special issue: photosynthesis: from natural to artificial. Phys Chem Chem Phys 24 [DOI] [PubMed]
- Michel H. 3-dimensional crystals of a membrane-protein complex—the photosynthetic reaction center from Rhodopseudomonas viridis. J Mol Biol. 1982;158:567–572. doi: 10.1016/0022-2836(82)90216-9. [DOI] [PubMed] [Google Scholar]
- Michel H. Crystallization of membrane proteins. Trends Biochem Sci. 1983;8:56–59. doi: 10.1016/0968-0004(83)90390-0. [DOI] [Google Scholar]
- Michel H. Die natürliche Photosynthese: Ihre Effizienz und die Konsequenzen. In: Gruss P, Schüth F, editors. Die Zukunft der Energie. München: C.H.Beck; 2008. pp. 71–85. [Google Scholar]
- Miqyass M, Marosvolgyi MA, Nagel Z, Yocum CF, van Gorkom HJ. S-state dependence of the calcium requirement and binding characteristics in the oxygen-evolving complex of photosystem II. Biochemistry. 2008;47:7915–7924. doi: 10.1021/bi8006059. [DOI] [PubMed] [Google Scholar]
- Murray JW, Barber J. Structural characteristics of channels and pathways in photosystem II including the identification of an oxygen channel. J Struct Biol. 2007;159:228–237. doi: 10.1016/j.jsb.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Nagashima H, Mino H. Highly resolved proton matrix ENDOR of oriented photosystem II membranes in the S2 state. Biochim Biophys Acta. 2013;1827:1165–1173. doi: 10.1016/j.bbabio.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Najafpour MM, et al. Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem Rev. 2016;116:2886–2936. doi: 10.1021/acs.chemrev.5b00340. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Ota K, Shibuya Y, Noguchi T. Role of a water network around the Mn4CaO5 cluster in photosynthetic water oxidation: a fourier transform infrared spectroscopy and quantum mechanics/molecular mechanics calculation study. Biochemistry. 2016;55:597–607. doi: 10.1021/acs.biochem.5b01120. [DOI] [PubMed] [Google Scholar]
- Nilsson H, Rappaport F, Boussac A, Messinger J. Substrate–water exchange in photosystem II is arrested before dioxygen formation. Nat Commun. 2014;5:4305. doi: 10.1038/ncomms5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson H, Cournac L, Rappaport F, Messinger J, Lavergne J. Estimation of the driving force for dioxygen formation in photosynthesis. Biochim Biophys Acta. 2016;1857:23–33. doi: 10.1016/j.bbabio.2015.09.011. [DOI] [PubMed] [Google Scholar]
- Nixon PJ, Michoux F, Yu JF, Boehm M, Komenda J. Recent advances in understanding the assembly and repair of photosystem II. Ann Bot. 2010;106:1–16. doi: 10.1093/aob/mcq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocera DG. The artificial leaf. Acc Chem Res. 2012;45:767–776. doi: 10.1021/ar2003013. [DOI] [PubMed] [Google Scholar]
- Nocera DG. Solar fuels and solar chemicals industry. Acc Chem Res. 2017;50:616–619. doi: 10.1021/acs.accounts.6b00615. [DOI] [PubMed] [Google Scholar]
- Noguchi T. FTIR detection of water reactions in the oxygen-evolving centre of photosystem II. Philos Trans R Soc B. 2008;363:1189–1195. doi: 10.1098/rstb.2007.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T, Suzuki H, Tsuno M, Sugiura M, Kato C. Time-resolved infrared detection of the proton and protein dynamics during photosynthetic oxygen evolution. Biochemistry. 2012;51:3205–3214. doi: 10.1021/bi300294n. [DOI] [PubMed] [Google Scholar]
- Nugent JHA, Muhiuddin IP, Evans MCW. Electron transfer from the water oxidizing complex at cryogenic temperatures: The S1 to S2 step. Biochemistry. 2002;41:4117–4126. doi: 10.1021/bi011320d. [DOI] [PubMed] [Google Scholar]
- Ono TA, Nakayama H, Gleiter H, Inoue Y, Kawamori A. Modification of the properties of S2 state in photosynthetic O2 evolving center by replacement of chloride with other anions. Arch Biochem Biophys. 1987;256:618–624. doi: 10.1016/0003-9861(87)90619-9. [DOI] [PubMed] [Google Scholar]
- Ono TA, Noguchi T, Inoue Y, Kusunoki M, Matsushita T, Oyanagi H. X-ray detection of the period-four cycling of the manganese cluster in photosynthetic water oxidizing enzyme. Science. 1992;258:1335–1337. doi: 10.1126/science.258.5086.1335. [DOI] [PubMed] [Google Scholar]
- Oyala PH, Stich TA, Stull JA, Yu F, Pecoraro VL, Britt RD. Pulse electron paramagnetic resonance studies of the interaction of methanol with the S2 state of the Mn4O5Ca cluster of photosystem II. Biochemistry. 2014;53:7914–7928. doi: 10.1021/bi501323h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyala PH, Stich TA, Debus RJ, Britt RD. Ammonia binds to the dangler manganese of the photosystem II oxygen-evolving complex. J Am Chem Soc. 2015;137:8829–8837. doi: 10.1021/jacs.5b04768. [DOI] [PubMed] [Google Scholar]
- Pace RJ, Smith P, Bramley R, Stehlik D. Epr saturation and temperature dependence studies on signals from the oxygen-evolving center of photosystem II. Biochim Biophys Acta. 1991;1058:161–170. doi: 10.1016/S0005-2728(05)80233-6. [DOI] [Google Scholar]
- Pantazis DA. Missing pieces in the puzzle of biological water oxidation. ACS Catal. 2018;8:9477–9507. doi: 10.1021/acscatal.8b01928. [DOI] [Google Scholar]
- Pantazis DA, Orio M, Petrenko T, Zein S, Lubitz W, Messinger J, Neese F. Structure of the oxygen-evolving complex of photosystem II: information on the S2 state through quantum chemical calculation of its magnetic properties. Phys Chem Chem Phys. 2009;11:6788–6798. doi: 10.1039/b907038a. [DOI] [PubMed] [Google Scholar]
- Pantazis DA, Ames W, Cox N, Lubitz W, Neese F. Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew Chem Int Ed. 2012;51:9935–9940. doi: 10.1002/anie.201204705. [DOI] [PubMed] [Google Scholar]
- Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh WY, Law NA. A proposal for water oxidation in photosystem II. Pure Appl Chem. 1998;70:925–929. doi: 10.1351/pac199870040925. [DOI] [Google Scholar]
- Peloquin JM, Britt RD. EPR/ENDOR characterization of the physical and electronic structure of the OEC Mn cluster. Biochim Biophys Acta. 2001;1503:96–111. doi: 10.1016/S0005-2728(00)00219-X. [DOI] [PubMed] [Google Scholar]
- Peloquin JM, Campbell KA, Britt RD. 55Mn pulsed ENDOR demonstrates that the photosystem II “split” EPR signal arises from a magnetically-coupled mangano-tyrosyl complex. J Am Chem Soc. 1998;120:6840–6841. doi: 10.1021/ja981196u. [DOI] [Google Scholar]
- Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD. 55Mn ENDOR of the S2 state multiline EPR signal of photosystem II: implications on the structure of the tetranuclear Mn cluster. J Am Chem Soc. 2000;122:10926–10942. doi: 10.1021/ja002104f. [DOI] [Google Scholar]
- Perez-Navarro MP, et al. Ammonia binding to the oxygen-evolving complex of photosystem II identifies the solvent-exchangeable oxygen bridge (mu-oxo) of the manganese tetramer. Proc Natl Acad Sci USA. 2013;110:15561–15566. doi: 10.1073/pnas.1304334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Navarro M, Neese F, Lubitz W, Pantazis DA, Cox N. Recent developments in biological water oxidation. Curr Opin Chem Biol. 2016;31:113–119. doi: 10.1016/j.cbpa.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Petrouleas V, Koulougliotis D, Ioannidis N. Trapping of metalloradical intermediates of the S-states at liquid helium temperatures. Overview of the phenomenology and mechanistic implications. Biochemistry. 2005;44:6723–6728. doi: 10.1021/bi0503201. [DOI] [PubMed] [Google Scholar]
- Planavsky NJ, et al. Evidence for oxygenic photosynthesis half a billion years before the great oxidation event. Nat Geosci. 2014;7:283–286. doi: 10.1038/ngeo2122. [DOI] [Google Scholar]
- Rapatskiy L, et al. Detection of the water-binding sites of the oxygen-evolving complex of photosystem II using W-band 17O electron–electron double resonance-detected NMR spectroscopy. J Am Chem Soc. 2012;134:16619–16634. doi: 10.1021/ja3053267. [DOI] [PubMed] [Google Scholar]
- Rapatskiy L, et al. CCharacterization of oxygen bridged manganese model complexes using multifrequency 17O hyperfine EPR spectroscopies and density functional theory. J Phys Chem B. 2015;119:13904–13921. doi: 10.1021/acs.jpcb.5b04614. [DOI] [PubMed] [Google Scholar]
- Rappaport F, Blancharddesce M, Lavergne J. Kinetics of electron-transfer and electrochromic change during the redox transitions of the photosynthetic oxygen-evolving complex. Biochim Biophys Acta. 1994;1184:178–192. doi: 10.1016/0005-2728(94)90222-4. [DOI] [Google Scholar]
- Rappaport F, Guergova-Kuras M, Nixon PJ, Diner BA, Lavergne J. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- Razeghifard MR, Pace RJ. Electron paramagnetic resonance kinetic studies of the S states in spinach PSII membranes. Biochim Biophys Acta. 1997;1322:141–150. doi: 10.1016/S0005-2728(97)00069-8. [DOI] [Google Scholar]
- Retegan M, Pantazis DA. Interaction of methanol with the oxygen-evolving complex: atomistic models, channel identification, species dependence, and mechanistic implications. Chem Sci. 2016;7:6463–6476. doi: 10.1039/C6SC02340A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retegan M, Pantazis DA. Differences in the active site of water oxidation among photosynthetic organisms. J Am Chem Soc. 2017;139:14340–14343. doi: 10.1021/jacs.7b06351. [DOI] [PubMed] [Google Scholar]
- Retegan M, Cox N, Lubitz W, Neese F, Pantazis DA. The first tyrosyl radical intermediate formed in the S2–S3 transition of photosystem II. Phys Chem Chem Phys. 2014;16:11901–11910. doi: 10.1039/c4cp00696h. [DOI] [PubMed] [Google Scholar]
- Retegan M, Krewald V, Mamedov F, Neese F, Lubitz W, Cox N, Pantazis DA. A five-coordinate Mn(IV) intermediate in biological water oxidation: spectroscopic signature and a pivot mechanism for water binding. Chem Sci. 2016;7:72–84. doi: 10.1039/C5SC03124A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robblee JH, Cinco RM, Yachandra VK. X-ray spectroscopy-based structure of the Mn cluster and mechanism of photosynthetic oxygen evolution. Biochim Biophys Acta. 2001;1503:7–23. doi: 10.1016/S0005-2728(00)00217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robblee JH, et al. The Mn cluster in the S0 state of the oxygen-evolving complex of photosystem II studied by EXAFS spectroscopy: are there three di-mu-oxo-bridged Mn2 moieties in the tetranuclear Mn complex? J Am Chem Soc. 2002;124:7459–7471. doi: 10.1021/ja011621a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romain S, Vigara L, Llobet A. Oxygen-oxygen bond formation pathways promoted by ruthenium complexes. Acc Chem Res. 2009;42:1944–1953. doi: 10.1021/ar900240w. [DOI] [PubMed] [Google Scholar]
- Saito K, Shen JR, Ishida T, Ishikita H. Short hydrogen bond between redox-active tyrosine Yz and D1-His190 in the photosystem II crystal structure. Biochemistry. 2011;50:9836–9844. doi: 10.1021/bi201366j. [DOI] [PubMed] [Google Scholar]
- Saito T, et al. Possible mechanisms of water splitting reaction based on proton and electron release pathways revealed for CaMn4O5 cluster of PSII refined to 1.9 Å X-ray resolution. Int J Quantum Chem. 2012;112:253–276. doi: 10.1002/qua.23218. [DOI] [Google Scholar]
- Sakamoto H, Shimizu T, Nagao R, Noguchi T. Monitoring the reaction process during the S2–S3 transition in photosynthetic water oxidation using time-resolved infrared spectroscopy. J Am Chem Soc. 2017;139:2022–2029. doi: 10.1021/jacs.6b11989. [DOI] [PubMed] [Google Scholar]
- Sanakis Yiannis, Sarrou Josephine, Zahariou Georgia, Petrouleas Vasili. Photosynthesis. Energy from the Sun. Dordrecht: Springer Netherlands; 2008. Q-band Electron Paramagnetic Resonance Studies of the S3 State of the OEC of Photosystem II; pp. 479–482. [Google Scholar]
- Schlodder E, Witt HT. Stoichiometry of proton release from the catalytic center in photosynthetic water oxidation: reexamination by a glass electrode study at pH 5.5–7.2. J Biol Chem. 1999;274:30387–30392. doi: 10.1074/jbc.274.43.30387. [DOI] [PubMed] [Google Scholar]
- Schraut J, Kaupp M. On ammonia binding to the oxygen-evolving complex of photosystem II: a quantum chemical study. Chem Eur J. 2014;20:7300–7308. doi: 10.1002/chem.201304464. [DOI] [PubMed] [Google Scholar]
- Schweiger A, Jeschke G. Principles of pulse electron paramagnetic resonance. Oxford: Oxford University Press; 2001. [Google Scholar]
- Service RJ, Hillier W, Debus RJ. Network of hydrogen bonds near the oxygen-evolving Mn4CaO5 cluster of photosystem II probed with FTIR difference spectroscopy. Biochemistry. 2014;53:1001–1017. doi: 10.1021/bi401450y. [DOI] [PubMed] [Google Scholar]
- Shen J-R. The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu Rev Plant Biol. 2015;66:23–48. doi: 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- Shoji M, Isobe H, Shigeta Y, Nakajima T, Yamaguchi K. Nonadiabatic one-electron transfer mechanism for the O–O bond formation in the oxygen-evolving complex of photosystem II. Chem Phys Lett. 2018;698:138–146. doi: 10.1016/j.cplett.2018.02.056. [DOI] [Google Scholar]
- Siegbahn PEM. Structures and energetics for O2 formation in photosystem II. Acc Chem Res. 2009;42:1871–1880. doi: 10.1021/ar900117k. [DOI] [PubMed] [Google Scholar]
- Siegbahn PEM. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys Chem Chem Phys. 2012;14:4849–4856. doi: 10.1039/c2cp00034b. [DOI] [PubMed] [Google Scholar]
- Stich TA, Yeagle GJ, Service RJ, Debus RJ, Britt RD. Ligation of D1-His332 and D1-Asp170 to the manganese cluster of photosystem II from Synechocystis assessed by multifrequency pulse EPR spectroscopy. Biochemistry. 2011;50:7390–7404. doi: 10.1021/bi2010703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull JA, Stich TA, Service RJ, Debus RJ, Mandal SK, Armstrong WH, Britt RD. 13C ENDOR reveals that the D1 polypeptide C-terminus is directly bound to Mn in the photosystem II oxygen evolving complex. J Am Chem Soc. 2010;132:446–447. doi: 10.1021/ja908688t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styring S, Feyziyev Y, Mamedov F, Hillier W, Babcock GT. pH dependence of the donor side reactions in Ca2+-depleted photosystem II. Biochemistry. 2003;42:6185–6192. doi: 10.1021/bi027035r. [DOI] [PubMed] [Google Scholar]
- Styring S, Sjoholm J, Mamedov F. Two tyrosines that changed the world: interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim Biophys Acta. 2012;1817:76–87. doi: 10.1016/j.bbabio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Su JH, et al. The electronic structures of the S2 states of the oxygen-evolving complexes of photosystem II in plants and cyanobacteria in the presence and absence of methanol. Biochim Biophys Acta. 2011;1807:829–840. doi: 10.1016/j.bbabio.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Suga M, et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature. 2015;517:99–103. doi: 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- Suga M, et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature. 2017;543:131–135. doi: 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sugiura M, Noguchi T. Monitoring water reactions during the S-state cycle of the photosynthetic water-oxidizing center: detection of the DOD bending vibrations by means of Fourier transform infrared spectroscopy. Biochemistry. 2008;47:11024–11030. doi: 10.1021/bi801580e. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sugiura M, Noguchi T. Monitoring proton release during photosynthetic water oxidation in Photosystem II by means of isotope-edited infrared spectroscopy. J Am Chem Soc. 2009;131:7849–7857. doi: 10.1021/ja901696m. [DOI] [PubMed] [Google Scholar]
- Tagore R, Chen HY, Crabtree RH, Brudvig GW. Determination of μ-oxo exchange rates in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J Am Chem Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. [DOI] [PubMed] [Google Scholar]
- Tagore R, Crabtree RH, Brudvig GW. Distinct mechanisms of bridging-oxo exchange in di-μ-O dimanganese complexes with and without water-binding sites: Implications for water binding in the O2-evolving complex of photosystem II. Inorg Chem. 2007;46:2193–2203. doi: 10.1021/ic061968k. [DOI] [PubMed] [Google Scholar]
- Terrett R, Petrie S, Stranger R, Pace RJ. What computational chemistry and magnetic resonance reveal concerning the oxygen evolving centre in photosystem II. J Inorg Biochem. 2016;162:178–189. doi: 10.1016/j.jinorgbio.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Tran PD, Wong LH, Barber J, Loo JSC. Recent advances in hybrid photocatalysts for solar fuel production. Energy Environ Sci. 2012;5:5902–5918. doi: 10.1039/c2ee02849b. [DOI] [Google Scholar]
- Ugur I, Rutherford AW, Kaila VRI. Redox-coupled substrate water reorganization in the active site of Photosystem II-The role of calcium in substrate water delivery. Biochim Biophys Acta. 2016;1857:740–748. doi: 10.1016/j.bbabio.2016.01.015. [DOI] [PubMed] [Google Scholar]
- Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Vass I. Molecular mechanisms of photodamage in the photosystem II complex. Biochim Biophys Acta. 2012;1817:209–217. doi: 10.1016/j.bbabio.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Vassiliev S, Zaraiskaya T, Bruce D. Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim Biophys Acta. 2012;1817:1671–1678. doi: 10.1016/j.bbabio.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Vinyard DJ, Brudvig GW. Progress toward a molecular mechanism of water oxidation in photosystem II. Annu Rev Phys Chem. 2017;68:101–116. doi: 10.1146/annurev-physchem-052516-044820. [DOI] [PubMed] [Google Scholar]
- Vinyard DJ, Khan S, Brudvig GW. Photosynthetic water oxidation: binding and activation of substrate waters for O–O bond formation. Faraday Discuss. 2015;185:37–50. doi: 10.1039/C5FD00087D. [DOI] [PubMed] [Google Scholar]
- Vrettos JS, Limburg J, Brudvig GW. Mechanism of photosynthetic water oxidation: combining biophysical studies of photosystem II with inorganic model chemistry. Biochim Biophys Acta. 2001;1503:229–245. doi: 10.1016/S0005-2728(00)00214-0. [DOI] [PubMed] [Google Scholar]
- Vrettos JS, Stone DA, Brudvig GW. Quantifying the ion selectivity of the Ca2+ site in photosystem II: evidence for direct involvement of Ca2+ in O2 formation. Biochemistry. 2001;40:7937–7945. doi: 10.1021/bi010679z. [DOI] [PubMed] [Google Scholar]
- Weil JA, Bolton JR. Electron paramagnetic resonance: elementary theory and practical applications. Hoboken: Wiley; 2007. [Google Scholar]
- Wydrzynski TJ, Hillier W, editors. Molecular solar fuels. RSC energy and environment series. Cambridge: RSC Pub., Royal Society of Chemistry (Great Britain); 2012. [Google Scholar]
- Wydrzynski T, Satoh K, editors. Photosystem II, the light-driven water: plastoquinone oxidoreductase. Dordrecht: Springer; 2005. [Google Scholar]
- Yachandra VK, Sauer K, Klein MP. Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- Yachandra VK, Yano J. Calcium in the oxygen-evolving complex: structural and mechanistic role determined by X-ray spectroscopy. J Photochem Photobiol B. 2011;104:51–59. doi: 10.1016/j.jphotobiol.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Yachandra V. Mn4Ca cluster in photosynthesis: where and how water is oxidized to dioxygen. Chem Rev. 2014;114:4175–4205. doi: 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: a case study for metalloprotein crystallography. Proc Natl Acad Sci USA. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, et al. Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ID, et al. Structure of photosystem II and substrate binding at room temperature. Nature. 2016;540:453–457. doi: 10.1038/nature20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaharieva I, Chernev P, Berggren G, Anderlund M, Styring S, Dau H, Haumann M. Room-temperature energy-sampling K beta X-ray emission spectroscopy of the Mn4Ca complex of Photosynthesis reveals three manganese-centered oxidation steps and suggests a coordination change prior to O2 formation. Biochemistry. 2016;55:4197–4211. doi: 10.1021/acs.biochem.6b00491. [DOI] [PubMed] [Google Scholar]
- Zaharieva I, Dau H, Haumann M. Sequential and coupled proton and electron transfer events in the S2–S3 transition of photosynthetic water oxidation revealed by time-resolved X-ray absorption spectroscopy. Biochemistry. 2016;55:6996–7004. doi: 10.1021/acs.biochem.6b01078. [DOI] [PubMed] [Google Scholar]
- Zhang B, Sun L. Why nature chose the Mn4CaO5 cluster as water-splitting catalyst in photosystem II: a new hypothesis for the mechanism of O-O bond formation. Dalton Trans. 2018;47:14381–14387. doi: 10.1039/C8DT01931B. [DOI] [PubMed] [Google Scholar]
- Zhang C, Chen C, Dong H, Shen J-R, Dau H, Zhao J. A synthetic Mn4Ca-cluster mimicking the oxygen-evolving center of photosynthesis. Science. 2015;348:690–693. doi: 10.1126/science.aaa6550. [DOI] [PubMed] [Google Scholar]
- Zimmermann JL, Rutherford AW. Electron-paramagnetic-res studies of the oxygen-evolving enzyme of photosystem II. Biochim Biophys Acta. 1984;767:160–167. doi: 10.1016/0005-2728(84)90091-4. [DOI] [Google Scholar]
- Zouni A, Witt HT, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]