

Abstract
Autophagy is a conserved self‐eating process that delivers cytoplasmic material to the lysosome to allow degradation of intracellular components, including soluble, unfolded and aggregated proteins, damaged organelles, and invading microorganisms. Autophagy provides a homeostatic control mechanism and is essential for balancing sources of energy in response to nutrient stress. Autophagic dysfunction or dysregulation has been implicated in several human pathologies, including cancer and neurodegeneration, and its modulation has substantial potential as a therapeutic strategy. Given the relevant clinical and therapeutic implications of autophagy, there is emerging intense interest in the identification of the key factors regulating the components of the autophagic machinery. Various post‐translational modifications, including ubiquitylation, have been implicated in autophagy control. The list of the E3 ubiquitin protein ligases involved in the regulation of several steps of the autophagic process is continuously growing. In this review, we will focus on recent advances in the understanding of the role of the homologous to the E6AP carboxyl terminus‐type E3 ubiquitin ligases in autophagy control.
Keywords: autophagy, HECT E3 ubiquitin ligases, proteasomal degradation, ubiquitylation
Abbreviations
- AMBRA1
activating molecule in BECLIN 1‐regulated autophagy protein 1
- AML
acute myeloid leukemia
- ATG
autophagy‐related proteins
- BH3
Bcl‐2 homology 3 domain
- CDL
cullin‐dependent ligase
- E1
E1 activating enzyme
- E2
E2 conjugating enzyme
- E3
E3 ubiquitin ligase
- ER
endoplasmic reticulum
- HECT
homologous to the E6AP carboxyl terminus
- LC3
microtubule‐associated protein light‐chain 3 protein
- LIR
LC3‐interacting region
- Lm
L. monocytogenes
- MFN2
mitofusin 2
- Mtb
M. tuberculosis
- mTORC1
mammalian target of rapamycin complex 1
- NBR1
neighbor of BRCA1 gene
- NEDD4
neural precursor cell expressed developmentally down‐regulated protein 4
- OPTN
optineurin
- PE
phosphatidylethanolamine
- PI3P
phosphatidylinositol‐3‐phosphate
- PtdIns3K
class‐III phosphatidylinositol 3‐kinase
- RDL
regulator of chromosome condensation 1 (RCC1)‐like domain
- RING
really interesting new gene
- SMURFs
Smad ubiquitin regulatory factors
- SPRY
SPRY/B30.2 domain
- SQSTM 1
sequestosome 1
- TRIM
TRIpartit motif
- UBA
ubiquitin‐associated domain
- ULK1
Unc‐51‐like kinase 1
- UPS
ubiquitin/proteasome system
- UVRAG
UV resistance‐associated gene
- WD40
W‐D repeat domain
- WWE
WWE domain
- WWP1
WW domain‐containing E3 ubiquitin protein ligase 1
1. The ubiquitin/proteasome system
Ubiquitylation is an ATP‐dependent and reversible enzymatic process that involves the covalent attachment of the conserved polypeptide ubiquitin to target proteins and is highly relevant in a number of physiological and pathological processes (El‐Hachem et al., 2018; Ma et al., 2018; Sane et al., 2018). The outcome of the ubiquitylation reaction is the formation of an isopeptide bond between the ε‐amino group of a Lys residue of the substrate and the C‐terminal Gly76 carboxyl group of ubiquitin. Ubiquitin conjugation to proteins occurs through the sequential and coordinate action of E1 activating (E1), E2 conjugating (E2), and E3 ubiquitin ligase (E3) enzymes that activate, transfer, and ligate ubiquitin to substrates (Ciechanover, 2005). The E3s transfer ubiquitin from the E2 to a substrate or directly promote the attachment of ubiquitin to the target molecule. Given that E3s are the final executioners of ubiquitin tagging, they mainly account for substrate specificity and versatility of the ubiquitylation reaction. Depending on the structural properties of their catalytic structural domains as well as to the mechanism of catalysis, the E3s fall into two major classes, really interesting new gene (RING)‐ and homologous to the E6AP carboxyl terminus (HECT)‐type enzymes. RING‐type E3s contain RING‐finger motifs that serve as scaffolds to bring the E2 in proximity of the substrate and facilitate the transfer of ubiquitin (Ozkan et al., 2005). Members of the HECT family of E3s are instead characterized by a highly conserved HECT domain, which directly catalyzes the covalent attachment of ubiquitin to substrate proteins (Rotin and Kumar, 2009).
Proteins can be modified by mono‐ubiquitylation, as a result of the attachment of a single ubiquitin, or by polyubiquitylation through the sequential attachment of ubiquitin molecules on a Lys residue. Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) that can become acceptors of another ubiquitin moiety in subsequent rounds of ubiquitylation, eventually leading to the generation of different types of polyubiquitin chains (Xu et al., 2009).
Mono‐ or polyubiquitylation and the exact linkage chain composition and topology dictate the distinct fate of the substrates. Recognition and proteolysis of protein substrates by the proteasome are mainly, but not solely, associated with K48‐linked ubiquitin chains. Additionally, K29 and K11 polymers mediate proteasomal degradation, more frequently when found in a mixed or branched chain with K48 and K63 linkages. Substrates conjugated to K63‐linked ubiquitin chains as well as mono‐ubiquitylated proteins are preferentially degraded by the autophagy/lysosome system (Kwon and Ciechanover, 2017).
Chains of at least four ubiquitins mark proteins for transportation and recognition by the proteasome, where proteins are degraded to oligopeptides. These are then released into either the cytoplasm or nucleoplasm, where they are digested into amino acids by soluble peptidases. The proteasome is a large multisubunit organelle, consisting of a central 20S cylinder‐shaped multiprotein complex displaying the proteolytic activity and a 19S regulatory particle at either of its ends. Substrate entry is a complex process which is controlled by the 19S particle. The 19S subunit enables the proteasome to drive binding, deubiquitylation, unfolding, and translocation of target proteins to proteolytic sites. After the substrate enters the 20S's central chamber, polypeptides are broken down by chymotrypsin‐, caspase‐ or trypsin‐like proteolytic sites.
The ubiquitin/proteasome system (UPS) is the major intracellular protein degradation pathway in all eukaryotes. It is responsible for turnover of short‐lived proteins and for the disposal of misfolded proteins. However, ubiquitin does not always signal for protein degradation, but it also controls a number of biological processes such as transcription, enzymatic activation, chromatin remodeling, subcellular relocalization, intracellular trafficking, and DNA repair.
2. General features of HECT‐type domain E3s
The HECT E3s (28 known enzymes in human) are endowed with intrinsic enzymatic activity. They employ a catalytic Cys residue located in the HECT domain as an acceptor of ubiquitin from E2s. After loading activated ubiquitin on themselves through the formation of a ubiquitin‐thioester catalytic intermediate, they transfer ubiquitin to a Lys residue in the target protein. HECT E3s recruit substrates through specific protein–protein interaction module within the N‐terminal region, but they can require accessory or adaptor proteins for substrate recognition.
On the basis of the distinct structural features of their N‐terminal protein–protein interaction domains, the HECT E3s have been grouped into three subfamilies (Fig. 1). The C2‐WW‐HECT [also designated Neural precursor cell expressed developmentally down‐regulated protein 4 (NEDD4)‐like] subfamily displays a common general modular architecture consisting of an N‐terminal protein kinase C‐related C2 domain, and two to four central tryptophan–tryptophan (WW) protein interacting modules that precede the HECT domain (Fig. 1). This group includes nine members in human: NEDD4‐1, NEDD4‐2, ITCH, Smad ubiquitin regulatory factors (SMURF)1, SMURF2, WW domain‐containing E3 ubiquitin protein ligase 1 (WWP1), WWP2, NEDL1, and NEDL2 (Huibregtse et al., 1995; reviewed in Rotin and Kumar, 2009). The C2 domain is a Ca+2‐dependent binding domain that mediates the interactions of the C2‐WW‐HECT E3s with phospholipids, inositol phosphate and proteins. As a result, these enzymes are targeted to membrane compartments, including plasma membrane, Golgi apparatus, endosomes, and lysosomes (Angers et al., 2004). The WW domains are responsible for the recruitment of protein substrates and adaptors through the recognition of Pro‐rich motifs (preferentially PPxY, and LPxY), and phosphorylated Ser/Thr‐Pro sites.
Figure 1.
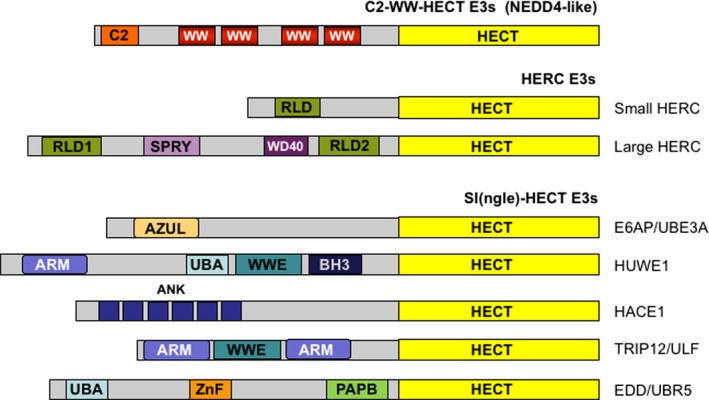
Domain organization of HECT‐type E3 enzymes. The HECT E3s have been assigned to three subgroups according to their N‐terminal protein–protein interaction domains. The C2‐WW‐HECT members are composed of an N‐terminal protein kinase C‐related C2 domain, two‐four central WW domains for substrate recruitment and a C‐terminal HECT domain. HERC E3s have one (small HERC) or more RLDs (large HERC) that precede the HECT domain. Large HERCs contain additional domains, such as SPRY and WD40 domains. The SI(ngle)‐HECT subfamily is characterized by the presence of a HECT domain but lack both WW and RDL domains. These enzymes recruit the substrates through several distinct structural modules including armadillo repeat‐containing domain, amino‐terminal Zn‐finger of Ube3a ligase domain, WWE, BH3 domain, ankyrin repeat‐containing domain, polyadenylate‐binding protein C‐terminal domain, UBA, and zinc finger domain.
The HERC subfamily of E3s is characterized by having one or more regulator of chromosome condensation 1 (RCC1)‐like domains (RLDs) for substrate recruitment (Fig. 1). This subgroup can be further classified into the small and the large HERC family members (six enzymes in human). The small HERC E3s (100–120 kDa) contain a single RLD, while the large HERC E3s (> 500 kDa) carry more than one RLDs and additional domains, including SPRY/B30.2 domain (SPRY) and the W‐D repeat domain (WD40).
The HECT family also includes SI(ngle)‐HECT enzymes that contain neither RLDs nor WW domains and recruit substrates through various number and types of protein interacting modules (Fig. 1).
Different HECTs display distinct chain type specificities. They can indeed generate K6, K11, K27, K29, K33, K48, and K63 linkages (Rotin and Kumar, 2009). The activity of HECT E3s can be regulated through the association of their interaction motifs with regulatory proteins that can facilitate substrate recruitment (adaptor and auxiliary proteins) or instead interfere with substrate binding (negative regulators). In addition, intramolecular interactions (e.g., between the C2 and the HECT domains) force the C2‐WW‐HECT E3s in a catalytically inactive state. More generally, the acquisition of an inactive conformation can be achieved through intermolecular associations, oligomerization, or post‐translational modification events (Chen et al., 2017; Courivaud et al., 2015; Wan et al., 2011).
Recent emerging evidence has unveiled a critical connection between HECT‐type domain E3s and the autophagy machinery. The involvement of HECTs in autophagy regulation will be discussed in detail in the next sections.
3. Autophagy
Macroautophagy, hereafter termed autophagy, is a highly conserved intracellular catabolic pathway in which cytoplasmic constituents are engulfed by double‐membrane vesicles and subsequently delivered to lysosomes for degradation (Cho et al., 2018; Denton et al., 2019; Fu et al., 2018; Galluzzi et al., 2018; Goiran et al., 2018; Kim et al., 2018; Lindqvist et al., 2018). Under homeostatic conditions, basal levels of autophagy are required to remove misfolded or aggregated proteins and damaged organelles, such as mitochondria, endoplasmic reticulum (ER), and peroxisomes. However, autophagic degradation is significantly activated in response to stress conditions to reprogram cell metabolism, meet biosynthetic demands, and allow cell survival. Indeed, autophagy represents an adaptive cellular response to nutrient deprivation, growth factor depletion, infection, oxidative stress hypoxia, and ER stress. In response to these conditions, autophagy preserves the biosynthetic capacity of the cell by supplying amino acids for de novo protein synthesis and maintains ATP levels by providing amino acids and free fatty acids for the Krebs cycle. In particular, starvation triggers nonselective autophagy that nonspecifically uptakes any cytoplasmic material. Selective autophagic pathways are instead engaged to target specific potentially harmful cellular components such as aggregated proteins (aggrephagy), damaged organelles (e.g., mitophagy and ERphagy for mitochondria and ER disposal, respectively), or invading microorganisms (xenophagy) for degradation.
Autophagy consists of several sequential steps: initiation, phagophore nucleation, autophagosome formation, autophagosome–lysosome fusion, and cargo degradation and recycling (Fig. 2). Induction of autophagy begins with nucleation of an isolation membrane, forming a cup‐shaped structure known as the phagophore, which originates from lipid bilayers contributed mainly by the ER, but also by the Golgi apparatus and endosomes (Abada and Elazar, 2014). A portion of cytoplasm, including organelles, is then sequestered by the elongating phagophore to form an autophagosome, a double‐membrane organelle. Then, the outer membrane of the autophagosome fuses with the lysosomal membrane to form an autolysosome, followed by degradation of the engulfed cytosolic material by the acidic lysosomal hydrolases.
Figure 2.
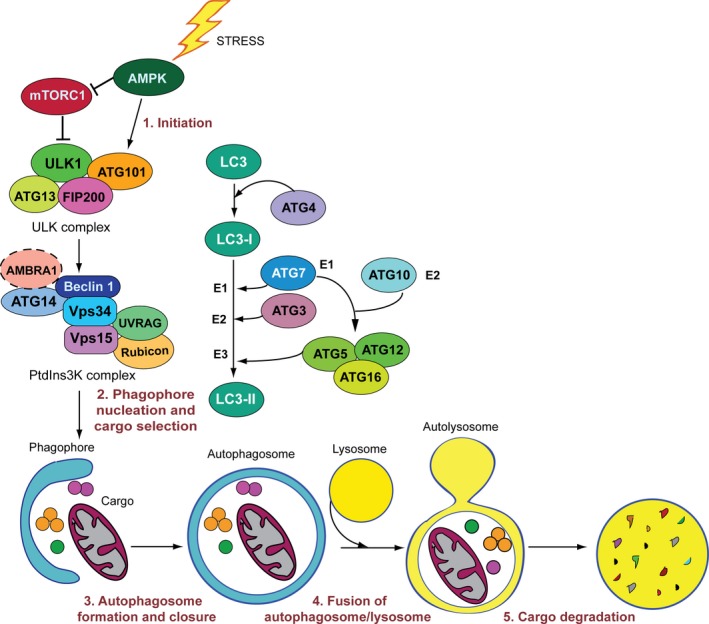
Schematic overview of the autophagy core machinery regulating autophagosome formation. The hierarchy of recruitment, assembly, and activation of ATG proteins during the main phases of the autophagic process are shown. Autophagy is regulated by four ATG protein‐formed complexes, including the ULK1 complex, the PtdIns3K complex, and two ubiquitin‐like protein conjugation systems. The ULK and the PtdIns3K complexes are involved in autophagy initiation and phagophore nucleation, respectively. The ATG4‐processed ATG8s are activated by the E1‐like enzyme ATG7 and then conjugated to membrane‐associated PE, as a result of the coordinated activity of ATG3 and the ATG5‐ATG12‐ATG16L complex, which acts as an E3 ligase. The ATG8 family is essential for biogenesis and elongation of the phagophore and for the recruitment of cargos to the growing phagophore during selective autophagy. A master regulator of the autophagic cascade is mTOR that suppresses PI3P synthesis, indispensable for membrane nucleation, by phosphorylating ULK1, ATG13, and Atg14L.
The core autophagic machinery relies on the autophagy‐related (ATG) proteins, which assemble into four functional complexes that are recruited to autophagy membrane compartments and work in sequential order to deliver the cytosolic cargo to the lysosomes (Klionsky et al., 2011; Yorimitsu and Klionsky, 2005) (Fig. 2). The upstream regulator of autophagy is the mammalian target of rapamycin complex 1 (mTORC1) (Yang and Klionsky, 2010). Under nutrient‐rich conditions, mTORC1 inhibits autophagy at several steps, such as membrane nucleation, autophagosomal maturation, and recycling of lysosomes from autolysosomes (Kim et al., 2011, 2015; Wan et al., 2018). Stress conditions that induce the autophagic process (initiation) inhibit mTORC1, causing consequent activation of the serine–threonine kinase Unc‐51‐like kinase 1 complex (ULK1). Stress‐induced autophagy is positively regulated by AMP‐activated protein kinase, which, under conditions of restricted cellular energy, activates the autophagic pathway by phosphorylating ULK1 and by inhibiting mTORC1 (Fig. 2).
Once activated, the ULK1 complex translocates at discrete location sites on the ER, which have been marked by ATG9, and phosphorylates components of the class‐III phosphatidylinositol 3‐kinase (PtdIns3K) complex (Karanasios et al., 2016), which, as results, regulate phagophore formation. The PtdIns3K complex comprises BECLIN 1, Vps34/PI3K, Vps15, ATG14L, UV resistance‐associated gene (UVRAG), and Rubicon (Fig. 2). Other proteins, such as activating molecule in BECLIN 1‐regulated autophagy protein 1 (AMBRA1), transiently and less stably associate with this core complex in a condition‐specific manner (Nazio et al., 2016). Once phosphorylated, the PtdIns3K complex activates local phosphatidylinositol‐3‐phosphate (PI3P) production at characteristic ER structures called omegasomes. PI3P then recruits the effector proteins, such as DFCP1 and WIPIs, to the omegasome to trigger nucleation of the phagophore. The elongation and expansion of the phagophore membrane are regulated by two ubiquitin‐like conjugation systems (Fig. 2). First, the E1‐like enzyme ATG7 and the E2‐like enzyme ATG10 catalyze the covalent attachment of ATG12 to ATG5 (ATG12~ATG5) (Romanov et al., 2012). Then, ATG16L1 forms an E3‐like complex with ATG12~ATG5 (ATG12~ATG5‐ATG16L1). This complex acts in concert with the E1‐like enzyme ATG7 and the E2‐like enzyme ATG3 to drive the lipidation of the ATG8 family members including microtubule‐associated protein light‐chain 3 (LC3) proteins (Hamasaki et al., 2013). The conjugation process is preceded by the action of the ATG4 protease, which cleaves the ATG8 proteins to expose the C‐terminal Gly for the subsequent lipidation reaction. Conjugation of membrane‐associated phosphatidylethanolamine (PE) converts LC3 from a freely diffuse into a membrane‐anchored form and is indispensable for phagophore expansion and for facilitating cargo recruitment in selective autophagy. The latter generally requires adaptor proteins, such as p62 [also termed sequestosome 1 (SQSTM 1)] and neighbor of BRCA1 gene (NBR1), which possess a ubiquitin‐associated domain (UBA) and a LC3‐interacting region (LIR) facilitating their binding to LC3. They function as receptors for ubiquitin‐modified cargoes interacting with ubiquitin or polyubiquitin chains via the UBA domain, and then, autophagy adaptors deliver ubiquitylated cargoes to the autophagosome via the LIR domain (Pankiv et al., 2007). During mitophagy, a clearance system for removal of damaged mitochondria through the autophagosome–lysosome pathway, recognition of ubiquitin‐modified substrates is mainly mediated by the optineurin (OPTN), NDP52 and AMBRA1 receptors (Di Rita et al., 2018; Heo et al., 2015; Yamano et al., 2016). By acting as bridges between ubiquitin‐tagged mitochondria and LC3, they allow mitochondria delivery to autophagosomes. Similarly, autophagic elimination of intracellular microorganisms requires their targeting with ubiquitin chains, which are then recognized by specific autophagy adaptors, such as p62, NDP52, NBR1, and OPTN, and delivered to autophagosomes (Gomes and Dikic, 2014).
Autophagy controls a variety of physiological and pathophysiological processes including development, aging, neurodegenerative disorders, cancer, and elimination of intracellular pathogens such as viruses, parasites, and bacteria (Levine and Kroemer, 2008).
4. Interplay between ubiquitylation and autophagy
The UPS and autophagy were originally regarded as independent degradative pathways with no or few interactions. It is now evident that ubiquitylation regulates multiple steps in autophagy. In addition, the autophagic pathway is activated to compensate for reduced UPS activity, allowing cells to reduce the load of accumulated proteasome‐specific substrates. A link between ubiquitylation and autophagy is established by sharing common regulators or substrates. The most obvious molecule in common between the two pathways is the ubiquitin itself that acts as a signal to mark target substrates for degradation by the proteasome or to be recognized by the adaptor proteins (e.g., p62) to recruit targets into autophagosomes for selective autophagy (Pankiv et al., 2007). Regulation of autophagy by protein ubiquitylation is also achieved by affecting the activity, the recruitment as well as the turnover of autophagic components. The existence of cross‐talks among different E3s and deubiquitinases alternate the addition of K48‐ubiquitin chains with K63‐linked polyubiquitylation of constituents of the autophagy core machinery. K63‐linked polyubiquitylation and K48‐linked polyubiquitylation compete each other to activate autophagic proteins in response to stress conditions or to degrade them when the stress situation is resolved, respectively. The contribution of RING‐finger E3s to autophagy has been recently reviewed (Antonioli et al., 2017; Cui et al., 2016; Grumati and Dikic, 2018). Overall, they have been primarily implicated in the regulation of upstream components of the autophagic machinery, namely mTORC1, the ULK1 and the PtdIns3K complexes. For instance, mTOR is a substrate of the cullin‐dependent ligase complex (CDL) SCFFBW7 that negatively regulates its protein stability (Mao et al., 2008). Both ULK1 and PtdIns3K complexes are regulated by TRAF6‐mediated ubiquitylation. In particular, K63‐polyubiquitylation promotes stabilization, self‐association, and activation of ULK1 (Nazio et al., 2013). Interestingly, here there is a relevant interaction with the major regulator of cell death: BCL‐2 (Adams and Cory, 2018; Montero and Letai, 2018; Pekarsky et al., 2018; Pentimalli, 2018; Reinhart et al., 2018; Strasser and Vaux, 2018). The attachment of K63 polyubiquitin chains to BECLIN 1 disrupts its inhibitory interaction with BCL‐2, therefore promoting autophagy induction (Shi and Kehrl, 2010). Various members of the TRIpartit motif (TRIM) protein family of E3s have been reported to induce autophagy (reviewed in Antonioli et al., 2017). The TRIMs can either participate in the selective recognition of autophagy cargos by cooperating with scaffold protein (e.g., p62), or directly regulate ATGs. Among these E3s, TRIM50 promotes starvation‐induced autophagy, through binding and K63‐polyubiquitylation of BECLIN 1 that facilitates the assembly of BECLIN 1 with ULK1 and, eventually, promotes BECLIN 1 activation (Fusco et al., 2018). In response to endomembrane damage affecting phagosomes or lysosomes, TRIM16 associates with and catalyzes K63‐linked polyubiquitylation of ULK1 and BECLIN 1, leading to their stabilization (Chauhan et al., 2016). TRIM32 was found to bind ULK1 and AMBRA1 and to stimulate ULK1 phosphorylating activity through AMBRA1‐dependent attachment of K63‐linked polyubiquitin chains (Di Rienzo et al., 2019).
On the other hand, K48‐polyubiquitylation‐mediated degradation of autophagy proteins is indispensable to terminate the autophagy response. The main E3s involved in this step are the CDL complexes. Specifically, in unstressed conditions, CDL4 negatively regulates AMBRA1 protein stability (Antonioli et al., 2014). Upon nutrient deprivation, ULK1‐dependent Cullin‐4 release determines a transient AMBRA1 stabilization, which is then reverted when the stress becomes prolonged to stop autophagy. In addition, the CDL3 complex catalyzes K48‐linked ubiquitylation of ULK1, BECLIN 1, and VPS34, leading to their degradation (Liu et al., 2016).
Among other RING‐finger E3s involved in autophagy inhibition, RNF5 negatively controls the stability of ATG4B (Kuang et al., 2012). This Cys protease cleaves the ATG8 family member precursors to expose the C‐terminal Gly that is subsequently conjugated to PE during autophagosome formation. In the next paragraphs, we will highlight the emerging roles of HECT‐type E3s in autophagy regulation (Table 1).
Table 1.
Protein substrates of the HECT‐type E3s in autophagy regulation
| E3 | Type of autophagy | Target | Type of poly‐ub chains | References |
|---|---|---|---|---|
| SMURF1 |
Xenophagy Starvation‐induced autophagy |
Cargo UVRAG |
K48‐Ub chains K29, K33‐Ub chains |
Orvedahl et al. (2011); Feng et al. (2019) |
| SMURF2 | Selective autophagy | Cargo (Lamin A) | Unknown | Borroni et al. (2018) |
| NEDD4‐1 |
Basal and starvation‐induced autophagy Mitophagy Xenophagy |
BECLIN 1 p62 |
K6, K27‐Ub chains K63‐Ub chains |
Pei et al. (2017); Sun et al. (2017); Lin et al. (2017) |
| NEDD4‐2 | Basal and ER‐induced autophagy | ULK1 | K27, K29 and K63‐Ub chains |
Wang et al. (2016) Nazio et al. (2016) |
| ITCH | Basal autophagy | Unknown | Unknown | Chhangani et al. (2014) |
| WWP1 | Basal autophagy | Unknown | Unknown | Sanarico et al. (2018) |
| HUWE1 |
Starvation‐induced autophagy Mitophagy |
WIPI2 MFN2 |
Unknown |
Wan et al. (2018) Di Rita et al. (2018) |
5. HECT‐type E3s in autophagy regulation
5.1. SMURFs
Smad ubiquitin regulatory factors SMURF1 and SMURF2 are related members of the NEDD4‐like subfamily. Accessibility of SMURFs toward different substrates is mainly controlled at the level of their subcellular localization. They primarily localize in the nucleus, but continuously shuttle between the nucleus and cytoplasm. Nuclear export arises as a result of binding to adaptor molecules that enrich SMURFs predominantly at the plasma membrane (Kavsak et al., 2000). SMURFs also localize to the ER (Guo et al., 2011). They are not required for general autophagy, but are regulators of selective autophagy, including xenophagy and mitophagy (Table 1) (Borroni et al., 2018; Franco et al.,2017; Orvedahl et al., 2011).
SMURF1 is indispensable for xenophagy‐induced host defense. Smurf1 −/− murine embryonic fibroblasts are indeed defective in targeting herpes simplex and Sindbis viruses to autophagosomes, suggesting a potential role for SMURF1 in immune defense by targeting viruses to the autophagic machinery (Orvedahl et al., 2011). In addition, SMURF1 participates to ubiquitin‐dependent autophagic elimination of intracellular bacteria including M. tuberculosis (Mtb) and L. monocytogenes (Lm). Selective autophagy is achieved by recruiting K48‐linked polyubiquitin chains to bacterial‐associated structures, directing them to LC3 and, subsequently, trafficking Mtb to lysosomes (Franco et al., 2017). As a result, SMURF1 restricts bacterial replication in macrophages. Induction of selective autophagy by SMURF1 requires its C2 domain, which is essential for anchoring SMURF1 to membrane phospholipids, specifically to Mtb‐associated structures, and possibly, to the ER. Notably, SMURF1 and the E3 PARKIN work in concert to promote ubiquitin‐dependent selective autophagy of Mtb. The biological relevance of SMURF1‐catalyzed ubiquitylation of Mtb‐associated structures is well exemplified by increased lung bacterial load, pulmonary inflammation, and accelerated mortality that follow the chronic phase of Mtb infection in Smurf1 −/− mice (Franco et al., 2017). Further studies are needed to establish the identity of the substrates targeted for ubiquitylation by SMURF1 and whether it also functions in the autophagic disposal of other intracellular microorganisms.
A recent report by Feng and colleagues has shown that SMURF1 recruits and forms a complex with UVRAG through the PPxY motif (Feng et al., 2019). This association results in K29‐ and K33‐linked UVRAG polyubiquitylation, which becomes more pronounced when cells are glucose deprived. This observation is apparently in contrast with all previous findings implying SMURF1 uniquely in selective autophagy (Franco et al.,2017; Orvedahl et al., 2011). Ubiquitylation of UVRAG by SMURF1 impairs its interaction with Rubicon (Fig. 3), which acts as a negative regulator of autophagosome maturation through binding to the PtdIns3K complex. Rubicon interacts with the Vps34/PI3K catalytic subunit thus inhibiting its PI3K lipid kinase activity (Sun et al., 2011). Interestingly, ubiquitylation of UVRAG impairs PIK3‐Rubicon interaction, enhances the activity of PIK3, and, eventually, promotes autophagosome maturation.
Figure 3.
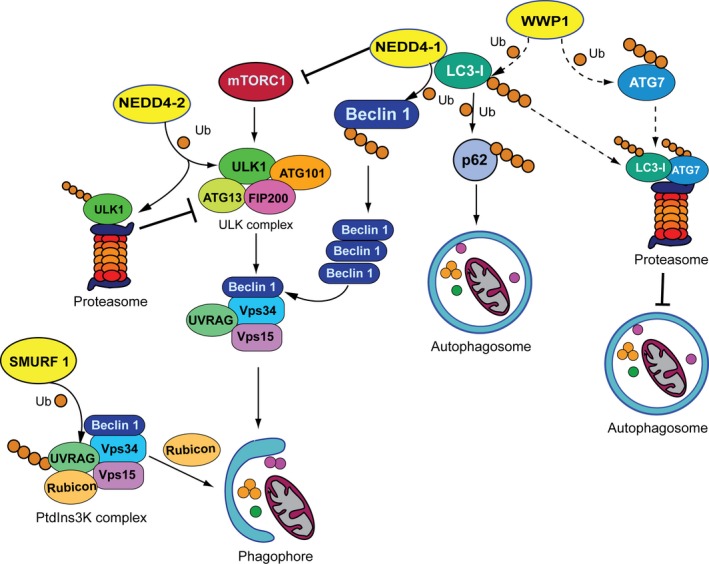
Regulation of the main phases of the autophagic pathway by the C2‐WW‐HECT E3s. Ubiquitin modification of substrates by the C2‐WW‐HECT E3s is mainly nondegradative and affects different steps of the autophagic process: initiation, phagophore nucleation, and autophagosome formation. NEDD4‐1 takes part in the initiation step by negatively modulating the phosphorylation levels of mTOR. In addition, NEDD4‐1 interferes with phagophore formation through K6‐ and K27‐mediated stabilization of BECLIN 1, and with autophagosomal biogenesis by interacting with LC3 and promoting K63‐conjugation of p62. NEDD4‐2 acts as an E3 for ULK1 and regulates its protein stability as an early response to nutrient deprivation. SMURF1 promotes phagophore nucleation by catalyzing K29‐ and K33‐linked UVRAG polyubiquitylation, and subsequent displacement of the inhibitory component Rubicon from the PtdIns3K complex. WWP1 negatively regulates autophagy possibly through the disposal of both LC3 and ATG7.
SMURF2 binds and ubiquitylates Lamin A and its mutant pathogenetic variant progerin (Borroni et al., 2018). Lamin filaments provide the nucleus with mechanical stability. Mutations in the gene encoding Lamin A cause genetic premature aging disorders, including Hutchinson–Gilford progeria syndrome (HGPS). Interestingly, SMURF2‐mediated modification of Lamin A promotes its disposal through the autophagic/lysosomal degradation pathway. As a result, SMURF2 negatively affects cellular levels of Lamin A and progerin in primary human dermal fibroblasts derived from healthy individuals and from patients affected by HGPS, respectively. Regulation of progerin by SMURF2 appears to be biologically relevant since ectopic expression of SMURF2 in progeria fibroblasts is able to reduce the nuclear deformability and improve the nuclear circularity, characteristic mechanical features of progeric cells. The clinical implication of this study is that SMURF2‐mediated clearance of progerin represents a potential therapeutic approach in progeria treatment.
5.2. NEDD4‐1 and NEDD4‐2
Neural precursor cell expressed developmentally down‐regulated protein 4 is the founding member of the C2‐WW‐HECT subfamily. Along with its closely related homolog NEDD4‐2, NEDD4‐1 plays a crucial role in the regulation of electrolyte homeostasis by controlling the surface abundance of the epithelial Na+ channel (Staub et al., 1997). Though some controversial findings have been reported on the contribution of NEDD4‐1 to autophagy regulation, the majority of available experimental evidence seems to support a promoting function in basal and starvation‐induced autophagy as well as mitophagy (Table 1) (Lin et al., 2017; Pei et al., 2017; Sun et al., 2017). In addition, similarly to SMURF1, NEDD4‐1 contributes to killing of intracellular Mtb, Lm, and S. pneumoniae during bacterial infection (Pei et al., 2017; Ogawa et al., 2018). A role in the initiation phase of the autophagic process has been suggested by studies reported by Li and collaborators (Li et al., 2015). Silencing of NEDD4‐1 expression impairs starvation‐ and rapamycin‐induced activation of autophagy (Sun et al., 2017) and increases the phosphorylation levels of mTOR, suggesting a role for NEDD4‐1 in promoting autophagy activation through the down‐regulation of mTORC1 signaling (Fig. 3). In addition, NEDD4‐1 controls autophagosomal biogenesis by interacting with LC3 through a LIR motif located between the C2 and the WW domains (Pei et al., 2017; Sun et al., 2017). Interestingly, inactivation of NEDD4‐1 expression impairs autophagosome building and causes the formation of enormous mitochondria, which points out at a severe defect in mitophagy. Mechanistically, LC3 is not a ubiquitylation substrate for NEDD4‐1. However, it has been proposed that the interaction of LC3 with NEDD4‐1 is relevant to recruit NEDD4‐1 to the phagophore and to activate its ligase activity, which ultimately would be required for p62 ubiquitylation (Sun et al., 2017) (Fig. 3). NEDD4‐1 polyubiquitylates p62 through K63 conjugation and is indispensable for its function in inclusion body autophagy (Lin et al., 2017).
Another component of the autophagic machinery that is ubiquitylated by NEDD4‐1 is BECLIN 1 (Pei et al., 2017). NEDD4‐mediated K6‐ and K27‐linked polyubiquitylation of BECLIN 1 results in protein stabilization of BECLIN 1, which, in turn, potentiates autophagy (Fig. 3).
Nazio et al. (2016) reported that, in response to nutrient deprivation, ULK1 protein levels are initially destabilized through the activity of NEDD4‐2, while they are restored to basal levels in response to prolonged starvation (Fig. 3). This event is thought to provide a negative control mechanism that limits autophagy overactivation and allows cells to survive prolonged starvation conditions. Furthermore, NEDD4‐2, but not NEDD4‐1, is upregulated in response to ER stress (Wang et al., 2016). Perturbations in ER homeostasis cause the accumulation of misfolded proteins and the activation of a signaling network called unfolded protein response. This system ensures the refolding of misfolded polypeptides or their disposal via ubiquitin‐proteasomal degradation and ER stress‐activated autophagy. In particular, in response to ER stress inducers, NEDD4‐2 is upregulated through sXBP1, a transcription factor that regulates the expression of genes necessary to recover ER function. Once induced, NEDD4‐2 modulates ER stress‐induced autophagy, though the mechanism through which this occurs remains to be clarified.
5.3. WWP1
WW domain‐containing E3 ubiquitin protein ligase 1 is a well‐established oncogene that has been implicated in the development of several human cancers (Chen et al., 2007; Lee et al., 2019; Sanarico et al., 2018). Unlike the other members of the HECT family, WWP1 functions as a negative regulator of the autophagic pathway (Sanarico et al., 2018). WWP1 knockdown in acute myeloid leukemia (AML) cells indeed leads to autophagy induction, which is accompanied by increased total levels of LC3 and ATG7 and accumulation of lipidated LC3. On the contrary, other components of the autophagic machinery seem to be unaltered by WWP1 inactivation. Autophagy activation reduces blast cell survival and contributes to delay leukemia progression in AML cancer xenografts. Although whether or not LC3 and ATG7 might be substrates for WWP1 during autophagy activation is still unexplored, this possibility may represent a conceivable explanation for clarifying how WWP1 interferes with the autophagic machinery. This hypothesis would imply that WWP1 favors degradation of proteins that are involved in the elongation and closure of the autophagosomal membranes to prevent the formation of autophagosomes (Fig. 3). Another observation that supports WWP1 as a regulator of the early steps of autophagy is that the enzyme predominantly localizes to cell membranes (Chen et al., 2008), which represent the main nucleation sites for autophagosome formation. Additional studies are required to identify the relevant targets of WWP1 in the context of autophagy regulation.
5.4. ITCH
Using a high‐throughput screening approach, Rossi and collaborators have identified clomipramine, an antidepressant drug, as an ITCH inhibitor compound (Rossi et al., 2014). Clomipramine efficiently blocks ITCH auto‐ubiquitylation, as well as the modification of its protein substrates. Clomipramine displays some general degree of specificity for other HECT E3s, but it does not inhibit RING‐type E3s. Interestingly, this compound interferes with the autophagic flux blocking the degradation of autophagic cargo (Rossi et al., 2009; Rossi et al., 2014). Clomipramine exerts its biological effects at lower micromolar concentrations than those required to inhibit ITCH activity in vitro, suggesting that the interference with the autophagic flux may be ITCH‐independent. Nevertheless, these findings arise the intriguing possibility that ITCH may be involved in autophagy regulation to promote the degradation of the serotonin receptor or its recycling mechanism. Clearly, this is an interesting point for future investigations.
Data reported by the group of Chhangani and colleagues seem to corroborate an implication of ITCH in autophagy regulation (Chhangani et al., 2014). They provided evidence that ITCH recruits both misfolded and aggregated (e.g., polyglutamine‐containing huntingtin aggregates) cytosolic proteins. They also observed the recruitment of other components of the UPS and of the autophagy machinery to ITCH‐positive protein aggregates. As a result, ITCH promotes the clearance of denatured proteins and expanded‐polyglutamine polypeptides and diminishes protein aggregates. Overall, this study identifies ITCH as a crucial component of the cytoplasmic protein quality control pathway, which provides cytoprotection against misfolded protein‐mediated stress.
5.5. HUWE1
HUWE1 (also known as ARF‐BP1, HectH9, URE‐B1, and Mule) is a large E3 (482‐kDa) belonging to the SI(ngle)‐HECT subgroup (Fig. 1). The enzyme contains two N‐terminal ARLDs, an UBA domain, followed by a WWE domain (WWE) and a Bcl‐2 homology 3 (BH3) domain (Fig. 1). HUWE1 catalyzes mono‐ubiquitylation as well as K48‐, K63‐ and K6‐linked ubiquitylation of protein targets. Modification of its substrates crucially regulates cell proliferation, apoptosis, DNA repair, and tumorigenesis (Atsumi et al., 2015; Bernassola et al., 2008; Chen et al., 2005; Zhong et al., 2005).
A report by Wan et al. (2018) has recently highlighted WIPI2 as a novel ubiquitylation substrate for HUWE1 (Fig. 4A). During the initial steps of the autophagic process, the recruitment of effector proteins such as WIPI1 and WIPI2 facilitates LC3 lipidation and the subsequent growth and elongation of the phagophore by recruiting the ATG12‐ATG5‐ATG16L complex to the PI3P‐rich phagophores. Binding of WIPI2 by HUWE1 requires mTORC1‐mediated phosphorylation and results in its ubiquitin‐dependent proteasomal degradation, which eventually keeps in check WIPI2 cellular levels and controls the intensity of basal autophagy (Fig. 4A, left). Following autophagy induction, mTORC1 is inactivated and, hence, WIPI2 adopts a dephosphorylated form, which is unable to interact with HUWE1 and therefore undergoes protein stabilization (Fig. 4A, right). WIPI2 is a WD40‐repeat‐containing protein. Some of the HUWE1‐binding proteins contain WD40 domains and are recruited by HUWE1 through this protein–protein interaction module (Thompson et al., 2014). Therefore, the authors suggest that mTORC1‐induced phosphorylation of WIPI2 may disrupt the interaction with HUWE1 by interfering with binding mediated by the WD40 domain. An interesting speculation arising from the report by Wan et al. is that metabolic adaptation and high mTORC1 activity in cancer cells may have an impact on autophagy regulation, at least in part, through WIPI2 stability control.
Figure 4.
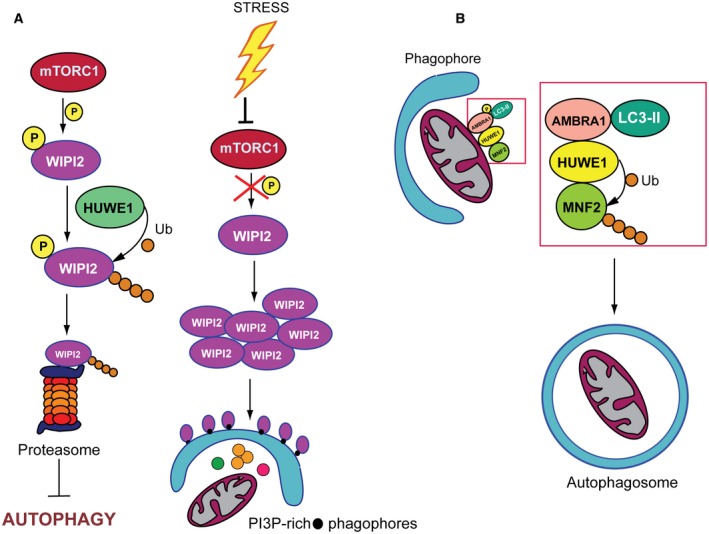
Role of HUWE1 in autophagy regulation. (A) Contribution of HUWE1 to elongation of the phagophore. Under basal conditions, mTORC1‐dependent phosphorylation instructs WIPI2 to interact with HUWE1, resulting in its ubiquitylation and subsequent proteasomal degradation, which, ultimately, prevents autophagy activation. In response to stressors, inhibition of mTORC1 impairs the association of HUWE1 and WIP2. WIP2 then accumulates and recruits the ATG12‐ATG5‐ATG16L complex to the PI3P‐rich phagophores. (B) During mitophagy, AMBRA1 acts as a cofactor for HUWE1 to recruit and ubiquitylates MFN2. Proteasomal degradation of MFN2 facilitates the dissociation of mitochondria from the ER, which is required for mitochondrial degradation.
HUWE1 has been also implicated in mitophagy regulation (Di Rita et al., 2018). During mitophagy, ubiquitylation is a key process that contributes to normal turnover of mitochondrial proteins. PARKIN participates in removing damaged mitochondria by modifying several outer mitochondrial membrane proteins (Heo et al., 2015). A central role in selective mitophagy regulation is exerted by AMBRA1 that binds LC3 through a LIR motif, and, acting as a mitophagy receptor, it eventually controls both PARKIN‐dependent and PARKIN‐independent mitochondrial clearance (Strappazzon et al., 2015). During PARKIN‐independent mitophagy, AMBRA1 functions as a cofactor for HUWE1 to favor recruitment of HUWE1 to mitochondria and its interaction with mitofusin 2 (MFN2) (Fig. 4B). Cooperation of AMBRA1 with HUWE1 leads to MFN2 ubiquitin‐mediated degradation. Disposal of MFN2 then favors dissociation of mitochondria from the ER, which, ultimately, enhances the rate of mitochondrial degradation (Ashrafi and Schwarz, 2013). AMBRA1‐mediated mitophagy is dependent on IKKα‐induced phosphorylation at Ser1014, flanking its LIR motif. This modification increases binding of AMBRA1 to LC3B and thus fosters its mitophagy receptor function. Notably, HUWE1 potentiates AMBRA1‐activated phosphorylation state, thus establishing a functional crosstalk between HUWE1 and AMBRA1 in mitophagy regulation (Di Rita et al., 2018).
6. Clinical implications
Deregulation of HECT E3s plays a relevant role in human diseases including neurological disorders (Zhang et al., 2011), viral infections (Medina et al., 2005), inflammatory diseases (Melino et al., 2008), and cancer (Bernassola et al., 2008). Emerging evidence has highlighted that HECT E3s play relevant roles in cancer development and progression by regulating the degradation or the activity of both oncogenes and tumor suppressors. As a consequence, abnormal expression, dysfunction, and mutations of the HECT enzymes have been associated with cancer development and chemoresistance (Kao et al., 2018; Koganti et al., 2018; Lee et al., 2019; Melino et al., 2008; Sanarico et al., 2018).
Similarly, autophagy has been linked to several pathological conditions (Murrow and Debnath, 2013). Paradoxically, autophagy can serve as a protection mechanism but can also contribute to cell damage. The effect of autophagy on cell survival can be variable depending on the cellular context and the intensity and persistence of the stress condition. Autophagy endows cells with an adaptation defense mechanism against cellular stress. This homeostatic function protects against a wide variety of diseases, including neurodegeneration, myopathy, and diabetes. In contrast, the prosurvival functions of autophagy may be deleterious in other disease settings. Autophagy can indeed favor the survival of cancer cells by providing the required cellular intermediates to satisfy their metabolic demands. On the other hand, excessive or prolonged degradation of cellular components ultimately leads to cell death that may or may not display hallmarks of apoptosis (Høyer‐Hansen et al., 2005). In this respect, autophagy induction may be beneficial across different pathological conditions including cancer.
Hence, cancer properly exemplifies the dual role of autophagy, which indeed provides a tumor suppressive function during cancer initiation, by removing damaged organelles that may generate free radicals. This eventually limits genomic instability. During cancer progression, autophagy instead favors tumor cell survival within the low‐oxygen and nutrient‐deprived environment and during metastatic dormancy (Lu et al., 2008; Smith and Macleod, 2019). In addition, autophagy promotes tumor cell migration and invasion and facilitates the resistance of tumors to radiation and chemotherapy (Kenific et al., 2010; Smith and Macleod, 2019).
It becomes evident that HECT E3s might contribute to pathological conditions, at least in part, through autophagy regulation. Modification of UVRAG by SMURF1 was found to be functionally relevant for autophagy‐dependent degradation of the epidermal growth factor receptor and, as a result, for inhibiting hepatocellular carcinoma cell proliferation and tumor growth (Feng et al., 2019).
Silencing WWP1 expression in AML blasts activates autophagy which, in turn, decreases viability and induces differentiation of leukemic cells (Sanarico et al., 2018). WWP1 overexpression in AML patients indeed causes stabilization of oncoproteins such as PML‐RARα and FLT3/ITD, whose turnover is regulated in an autophagy‐dependent manner. Accumulation of these oncoproteins then accounts for the myeloid maturation block of leukemic cells.
By supporting the high bioenergetic demand of cancer cells, mitophagy sustains tumor growth. HUWE1 may influence cancer metabolism and tumor progression by promoting mitophagy through MFN2 ubiquitylation (Di Rita et al., 2018). The role of HUWE1 in tumorigenesis has been very debated. Both tumor suppressive and oncogenic functions have been reported (Kao et al., 2018). It has been proposed that the dual role of HUWE1 in cancer is influenced by different protein adaptors or post‐translational modifications. In this respect, AMBRA1 acts as a cofactor for HUWE1 to favor mitophagy induction (Di Rita et al., 2018).
7. Concluding remarks
Clearly, HECT‐type E3s play a relevant role in the regulation of several steps of the autophagic pathway. With few exceptions (e.g., WWP1, HUWE1), HECT E3s act as autophagy inducers. Current evidence indicates that HECT E3s are mainly involved in the early steps of the autophagy response. Their activities converge toward the regulation of autophagy initiation, nucleation of the phagophore, autophagosome formation, and cargo recruitment in selective autophagy. Autophagy initiation is regulated through the inhibition of mTORC1 by NEDD4‐1, though the underlying molecular mechanism has not been elucidated yet. Phagophore formation regulation is achieved as a result of UVRAG ubiquitylation by SMURF1 that, ultimately, enhances PIK3 activity, and via NEDD4‐1‐mediated modification and stabilization of BECLIN 1. Cargo selection and autophagosome building are regulated by SMURF1 that catalyzes the addition of polyubiquitin chains to bacterial‐associated structures to direct them to LC3. NEDD4‐1 also promotes these steps through p62 ubiquitylation. An example of negative regulation of autophagy by HECT E3s is provided by HUWE1. This enzyme is involved in the ubiquitin‐mediated degradation of the effector protein WIPI2 that is relevant to restrain the intensity of basal autophagy. Furthermore, WWP1 prevents autophagy possibly by interfering with the formation of autophagosomes, though the substrates mediating its function have not been identified yet.
Further exploring the contribution of HECT‐type E3s in autophagy regulation will implement our understanding of the autophagic process. Few inhibitors of the HECT enzymes have been reported to display cytotoxic activities against cancer cells or to increase the response of tumor cells to conventional therapies (Rossi et al., 2014; Watt et al., 2018; Lee et al., 2019). It would be of clinical interest to assess whether these compounds function, at least in part, by interfering with the autophagic pathway. Future studies aimed to identify and characterize HECT E3 inhibitors may lead to the development of novel therapeutic strategies for the treatment of autophagy‐associated pathologies such as cancer and inflammatory disorders.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
All authors contributed in writing the manuscript, read, edited, and approved the final manuscript.
Acknowledgements
This work was supported by PRIN 2017 to FB, ‘the Medical Research Council (to GM), and Associazione Italiana per la Ricerca contro il Cancro (AIRC) to GM IG#20473 (2018‐2022)’.
References
- Abada A and Elazar Z (2014) Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep 15, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM and Cory S (2018) The BCL‐2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ 25, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers A, Ramjaun AR and McPherson PS (2004) The HECT domain ligase itch ubiquitinates endophilin and localizes to the trans‐Golgi network and endosomal system. J Biol Chem 279, 11471–11479. [DOI] [PubMed] [Google Scholar]
- Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, Marsella C, Piselli P, Gretzmeier C, Dengjel J et al (2014) AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell 31, 734–746. [DOI] [PubMed] [Google Scholar]
- Antonioli M, Di Rienzo M, Piacentini M and Fimia GM (2017) Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci 42, 28–41. [DOI] [PubMed] [Google Scholar]
- Ashrafi G and Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H et al (2015) ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient gammaH2AX foci formation. Cell Rep 13, 2728–2740. [DOI] [PubMed] [Google Scholar]
- Bernassola F, Karin M, Ciechanover A and Melino G (2008) The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14, 10–21. [DOI] [PubMed] [Google Scholar]
- Borroni AP, Emanuelli A, Shah PA, Ilić N, Apel‐Sarid L, Paolini B, Manikoth Ayyathan D, Koganti P, Levy‐Cohen G and Blank M (2018) Smurf2 regulates stability and the autophagic‐lysosomal turnover of lamin A and its disease‐associated form progerin. Aging Cell 17, e12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA et al (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jiang H, Xu W, Li X, Dempsey DR, Zhang X, Devreotes P, Wolberger C, Amzel LM, Gabelli SB et al (2017) A tunable brake for HECT ubiquitin ligases. Mol Cell 66, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J and Gu W (2005) ARF‐BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhou Z, Liu R, Li Y, Azmi PB and Seth AK (2008) The WW domain containing E3 ubiquitin protein ligase 1 upregulates ErbB2 and EGFR through RING finger protein 11. Oncogene 27, 6845–6855. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhou Z, Ross JS, Zhou W and Dong JT (2007) The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer 121, 80–87. [DOI] [PubMed] [Google Scholar]
- Chhangani D, Upadhyay A, Amanullah A, Joshi V and Mishra A (2014) Ubiquitin ligase ITCH recruitment suppresses the aggregation and cellular toxicity of cytoplasmic misfolded proteins. Sci Rep 4, 5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YR, Lee JH, Kim JH, Lee SY, Yoo S, Jung MK, Kim SJ, Yoo HJ, Pack CG, Rho JK et al (2018) Matrine suppresses KRAS‐driven pancreatic cancer growth by inhibiting autophagy‐mediated energy metabolism. Mol Oncol 12, 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A (2005) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin‐proteasome system and onto human diseases and drug targeting. Cell Death Differ 12, 1178–1190. [DOI] [PubMed] [Google Scholar]
- Courivaud T, Ferrand N, Elkhattouti A, Kumar S, Levy L, Ferrigno O and Prunier C (2015) Functional characterization of a WWP1/Tiul1 tumor‐derived mutant reveals a paradigm of its constitutive activation in human cancer. J Biol Chem 290, 21007–21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui D, Xiong X and Zhao Y (2016) Cullin‐RING ligases in regulation of autophagy. Cell Div 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Xu T, Dayan S, Nicolson S and Kumar S (2019) Dpp regulates autophagy‐dependent midgut removal and signals to block ecdysone production. Cell Death Differ 26, 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo M, Antonioli M, Fusco C, Liu Y, Mari M, Orhon I, Refolo G, Germani F, Corazzari M, Romagnoli A et al (2019) Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63‐linked polyubiquitin chains. Sci Adv 5, eaau8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rita A, Peschiaroli A, D'Acunzo P, Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M, Melino G, Papaleo E et al (2018) HUWE1 E3 ligase promotes PINK1/PARKIN‐independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun 9, 3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Hachem N, Habel N, Naiken T, Bzioueche H, Cheli Y, Beranger GE, Jaune E, Rouaud F, Nottet N, Reinier F et al (2018) Uncovering and deciphering the pro‐invasive role of HACE1 in melanoma cells. Cell Death Differ 25, 2010–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Jia Y, Zhang Y, Ma F, Zhu Y, Hong X, Zhou Q, He R, Zhang H, Jin J et al (2019) Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 27, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU and Levine B (2017) The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti‐tuberculous host defense. Cell Host Microbe 21, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Wang J, Hu X, Zhou RR, Fu Y, Tang D, Kang R, Huang Y, Sun L, Li N et al (2018) Crosstalk between hepatitis B virus X and high‐mobility group box 1 facilitates autophagy in hepatocytes. Mol Oncol 2, 322–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco C, Mandriani B, Di Rienzo M, Micale L, Malerba N, Cocciadiferro D, Sjøttem E, Augello B, Squeo GM, Pellico MT et al (2018) TRIM50 regulates Beclin 1 proautophagic activity. Biochim Biophys Acta Mol Cell Res 1865, 908–919. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goiran T, Duplan E, Rouland L, El Manaa W, Lauritzen I, Dunys J, You H, Checler F and Alves da Costa C (2018) Nuclear p53‐mediated repression of autophagy involves PINK1 transcriptional down‐regulation. Cell Death Differ 25, 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC and Dikic I (2014) Autophagy in antimicrobial immunity. Mol Cell 54, 224–233. [DOI] [PubMed] [Google Scholar]
- Grumati P and Dikic I (2018) Ubiquitin signaling and autophagy. J Biol Chem 293, 5404–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Shen S, Song S, He S, Cui Y, Xing G, Wang J, Yin Y, Fan L, He F et al (2011) The E3 ligase Smurf1 regulates Wolfram syndrome protein stability at the endoplasmic reticulum. J Biol Chem 286, 18037–18047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta M, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐ mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J and Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyer‐Hansen M, Bastholm L, Mathiasen IS, Elling F and Jäättelä M (2005) Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1‐mediated autophagic cell death. Cell Death Differ 12, 1297–1309. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S and Howley PM (1995) A family of proteins structurally and functionally related to the E6‐AP ubiquitin‐protein ligase. Proc Natl Acad Sci USA 92, 2563–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SH, Wu H and Wu KJ (2018) Ubiquitination by HUWE1 in tumorigenesis and beyond. J Biomed Sci 25, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L and Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun 7, 12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH and Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- Kenific CM, Thorburn A and Debnath J (2010) Autophagy and metastasis: another double‐edged sword. Curr Op Cell Biol 22, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Choi TG, Park S, Yun HR, Nguyen NNY, Jo YH, Jang M, Kim J, Kim J, Kang I et al (2018) Mitochondrial ROS‐derived PTEN oxidation activates PI3K pathway for mTOR‐induced myogenic autophagy. Cell Death Differ 25, 1921–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS and Kim DH (2015) mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 57, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B and Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno O, Cuervo AM, Dednath J, Deretic V, Elazar Z, Eskelinen EL et al (2011) A comprehensive glossary of autophagy‐ related molecules and processes. Autophagy 7, 1273–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koganti P, Levy‐Cohen G and Blank M (2018) Smurfs in protein homeostasis, signaling, and cancer. Front Oncol 8, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, Niesman IR, Yang HJ, López-Otín C, Yang WY et al (2012) Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 8, e1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT and Ciechanover A (2017) The ubiquitin code in the ubiquitin‐proteasome system and autophagy. Trends Biochem Sci 42, 873–886. [DOI] [PubMed] [Google Scholar]
- Lee YR, Chen M, Lee JD, Zhang J, Lin SY, Fu TM, Chen H, Ishikawa T, Chiang SY, Katon J et al (2019) Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC‐WWP1 inhibitory pathway. Science 364, eaau0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B and Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang L, Zhou J, Luo S, Huang R, Zhao C and Diao A (2015) Nedd4 E3 ubiquitin ligase promotes cell proliferation and autophagy. Cell Prolif 48, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Dai Q, Meng H, Sun A, Wei J, Peng K, Childress C, Chen M, Shao G and Yang W (2017) The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J Cell Sci 130, 3839–3850. [DOI] [PubMed] [Google Scholar]
- Lindqvist LM, Frank D, McArthur K, Dite TA, Lazarou M, Oakhill JS, Kile BT and Vaux DL (2018) Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ 25, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF et al (2016) Cul3–KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell 61, 84–97. [DOI] [PubMed] [Google Scholar]
- Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB et al (2008) The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cells. J Clin Invest 118, 3917–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lu Y, Zhang S, Li Y, Huang J, Yin Z, Ren J, Huang K, Liu L, Yang K et al (2018) β‐Trcp ubiquitin ligase and RSK2 kinase‐mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ 25, 1473–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JH, Kim IJ, Wu D, Climent J, Kang HC and DelRosario R (2008) FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321, 1499–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina G, Zhang Y, Tang Y, Gottwein E, Vana ML, Bouamr F, Leis J and Carter CA (2005) The functionally exchangeable L domains in RSV and HIV‐1 Gag direct particle release through pathways linked by Tsg101. Traffic 6, 880–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melino G, Gallagher E, Aqeilan RI, Knight R, Peschiaroli A, Rossi M, Scialpi F, Malatesta M, Zocchi L, Browne G et al (2008) Itch: a HECT‐type E3 ligase regulating immunity, skin and cancer. Cell Death Differ 15, 1103–1112. [DOI] [PubMed] [Google Scholar]
- Montero J and Letai A (2018) Why do BCL‐2 inhibitors work and where should we use them in the clinic? Cell Death Differ 25, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrow L and Debnath J (2013) Autophagy as a stress‐response and quality‐control mechanism: implications for cell injury and human disease. Annu Rev Pathol 8, 105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM et al (2016) Fine‐tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol 215, 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM et al (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self‐association and function through AMBRA1 and TRAF6. Nat Cell Biol 15, 406–416. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Matsuda R, Takada N, Tomokiyo M, Yamamoto S, Shizukusihi S, Yamaji T, Yoshikawa Y, Yoshida M, Tanida I et al (2018) Molecular mechanisms of Streptococcus pneumoniae‐targeted autophagy via pneumolysin, Golgi‐resident Rab41, and Nedd4‐1‐mediated K63‐linked ubiquitination. Cell Microbiol 20, e12846. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M et al (2011) Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan E, Yu H and Deisenhofer J (2005) Mechanistic insight into the allosteric activation of a ubiquitin‐conjugating enzyme by RING‐type ubiquitin ligases. Proc Natl Acad Sci USA 102, 18890–18895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T (2007) P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- Pei G, Buijze H, Liu H, Moura‐Alves P, Goosmann C, Brinkmann V, Kawabe H, Dorhoi A and Kaufmann SHE (2017) The E3 ubiquitin ligase NEDD4 enhances killing of membrane‐perturbing intracellular bacteria by promoting autophagy. Autophagy 13, 2041–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Balatti V and Croce CM (2018) BCL2 and miR‐15/16: from gene discovery to treatment. Cell Death Differ 25, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentimalli F (2018) BCL2: a 30‐year tale of life, death and much more to come. Cell Death Differ 25, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart R, Rohner L, Wicki S, Fux M and Kaufmann T (2018) BH3 mimetics efficiently induce apoptosis in mouse basophils and mast cells. Cell Death Differ 25, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C and Martens S (2012) Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J 31, 4304–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra‐Martin MA, Bampton ET, Glynn P, Bonanno G, Knight RA et al (2009) Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci 122, 3330–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, Cavasotto CN, Knight RA, Ciechanover A et al (2014) High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis 5, e1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotin D and Kumar S (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 10, 398–409. [DOI] [PubMed] [Google Scholar]
- Sanarico AG, Ronchini C, Croce A, Memmi EM, Cammarata UA, De Antoni A, Lavorgna S, Divona M, Giacò L, Melloni GEM et al (2018) The E3 ubiquitin ligase WWP1 sustains the growth of acute myeloid leukaemia. Leukemia 32, 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane S, Hafner A, Srinivasan R, Masood D, Slunecka JL, Noldner CJ, Hanson AD, Kruisselbrink T, Wang X, Wang Y et al (2018) UBXN2A enhances CHIP‐mediated proteasomal degradation of oncoprotein mortalin‐2 in cancer cells. Mol Oncol 12, 1753–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS and Kehrl JH (2010) TRAF6 and A20 regulate lysine 63‐linked ubiquitination of Beclin‐1 to control TLR4‐induced autophagy. Sci Signal 3, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG and Macleod KF (2019) Autophagy, cancer stem cells and drug resistance. J Pathol 247, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L and Rotin D (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16, 6325–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M et al (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22, 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A and Vaux DL (2018) Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ 25, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A, Wei J, Childress C, Shaw JH 4th, Peng K, Shao G, Yang W and Lin Q (2017) The E3 ubiquitin ligase NEDD4 is an LC3‐interactive protein and regulates autophagy. Autophagy 13, 522–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S and Zhong Q (2011) The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem 286, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JW, Nagel J, Hoving S, Gerrits B, Bauer A, Thomas JR, Kirschner MW, Schirle M and Luchansky SJ (2014) Quantitative Lys‐ε‐Gly‐Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin‐mediated proteolysis of DNA damage‐inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. J Biol Chem 289, 28942–28955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W, You Z, Zhou L, Xu Y, Peng C, Zhou T, Yi C, Shi Y and Liu W (2018) mTORC1‐regulated and HUWE1‐mediated WIPI2 degradation controls autophagy flux. Mol Cell 72, 303–315. [DOI] [PubMed] [Google Scholar]
- Wan L, Zou W, Gao D, Inuzuka H, Fukushima H, Berg AH, Drapp R, Shalk S, Hu D, Lester C et al (2011) Cdh1 regulates osteoblast function through an APC/C‐independent modulation of Smurf1. Mol Cell 44, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun RQ, Camera D, Zeng XY, Jo E, Chan SM, Herbert TP, Molero JC and Ye JM (2016) Endoplasmic reticulum stress up‐regulates Nedd4‐2 to induce autophagy. FASEB J 30, 2549–2556. [DOI] [PubMed] [Google Scholar]
- Watt JE, Hughes GR, Walpole S, Monaco S, Stephenson GR, Bulman Page PC, Hemmings AM, Angulo J and Chantry A (2018) Discovery of Small Molecule WWP2 Ubiquitin Ligase Inhibitors. Chemistry 24, 17677–17680. [DOI] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D and Peng J (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Matsuda N and Tanaka K (2016) The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep 17, 300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z and Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T and Klionsky DJ (2005) Autophagy: molecular machinery for self‐eating. Cell Death Differ 2, 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Haraguchi S, Koda T, Hashimoto K and Nakagawara A (2011) Muscle atrophy and motor neuron degeneration in human NEDL1 transgenic mice. J Biomed Biotechnol 2011, 831092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Gao W, Du F and Wang X (2005) Mule/ARF‐BP1, a BH3‐only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl‐1 and regulates apoptosis. Cell 121, 1085–1095. [DOI] [PubMed] [Google Scholar]