

Abstract
The kainate receptors GluR6 and GluR7 differ considerably in their ion channel properties, despite sharing 86% amino acid sequence identity. When expressed in Xenopus oocytes GluR6 conducts large agonist-evoked currents, whereas GluR7 lacks measurable currents. In the present study, we localized the determinants that are responsible for the functional differences between GluR6 and GluR7 to the extracellular loop domain L3. In addition, we generated several GluR7 point mutants that are able to conduct currents that can be readily measured in Xenopus oocytes.
In GluR6, glutamate- and kainate-evoked maximal currents are of the same magnitude when desensitization is inhibited with the lectin concanavalin A. By contrast, all functional GluR7 mutants were found to have glutamate current amplitudes significantly larger than those evoked by kainate. We localized the domain that determines the relative agonist efficacies to the C-terminal half of the L3 domain of GluR7.
Our data show that EC50 values for glutamate (but not for kainate) in GluR7 mutants or chimeras tend to be increased in comparison to the EC50 values in GluR6. The high EC50 for wild-type GluR7 reported in the literature appears to be linked to the S1 portion of the agonist-binding domain.
Finally, we determined the C-terminal half of the L3 domain plus the far C-terminal domain of GluR7 to be responsible for the recently reported reduction of current amplitude seen when GluR7 is coexpressed with GluR6. We conclude that coexpression of GluR6 and GluR7 leads to nonstochastical assembly of heteromeric receptor complexes.
Keywords: GluR7, GluR6, glutamate receptors, kainate receptors, chimeras, point mutations, coexpression, ion channel function
Ionotropic glutamate receptors (GluRs) are the prevalent excitatory neurotransmitter receptors in the CNS of vertebrates. They can be pharmacologically classified in three distinct classes: AMPA receptors, kainate (KA) receptors, and NMDA receptors (Monaghan and Cotman, 1989; Hollmann, 1999). All ionotropic glutamate receptors have three transmembrane domains (TMD): A, B, and C, a pore-forming region (P) located between TMDs A and B, an extracellular N terminus (N), and an intracellular C terminus (C). Their ligand-binding sites are comprised of two interacting nonconsecutive domains: the S1 domain located upstream of TMD A and the S2 domain in the extracellular loop (L3) between TMDs B and C. Short stretches of amino acids connect the S1 domain to TMD A and the S2 domain to TMDs B and C; those sequences hereafter will be referred to as linker A, linker B, and linker C, respectively (Fig.1).
Fig. 1.

Schematic representation of the structure of a typical ionotropic glutamate receptor. The N terminus is extracellular, the C terminus intracellular, and there are three TMDs,A, B, and C. The pore-forming region (P) consists of a hairpin loop plus two small intracellular loops (L1 andL2). The hairpin loop inserts into the membrane from the inside and is thought to line the ion permeation pathway (the “pore”), whereas loops L1 and L2 connect the pore to TMDsA and B, respectively. A large, extracellular domain (L3) resides between TMDsB and C. S1 and S2 are two extracellular domains homologous to bacterial amino acid-binding proteins that are involved in the formation of the ligand-binding site. LinkersA (15 amino acids long in low-affinity kainate receptors), B (12 amino acids), and C (13 amino acids) are here defined as the connecting sequences between the S1 and S2 domains and TMDs A, B, andC, respectively (Fig. 4). FC comprises the C-terminal sequence downstream of TMD C. NruI,EcoRI, ClaI, and NaeI are the introduced or native restriction sites used for constructing chimeras between GluR6 and GluR7. The ClaI site splits the L3 domain into an N-terminal part (L3N) and a C-terminal part (L3C).
The kainate receptor family can be further divided into low-affinity (GluR5, GluR6, GluR7) and high-affinity (KA1, KA2) kainate receptor subunits (Hollmann and Heinemann, 1994). Although the kainate receptors GluR6 and GluR7 are 86% identical at the amino acid sequence level (Bettler et al., 1992) and belong to the same receptor subfamily, they have very different electrophysiological properties. Whereas GluR6 shows large, desensitizing currents in both transfected mammalian cells and Xenopus oocytes, GluR7 responds only to unphysiologically high concentrations of agonists (Schiffer et al., 1997), and responses are only detectable in mammalian cells, but not inXenopus oocytes (Dingledine et al., 1999; Hollmann, 1999). GluR7 and GluR6 have similar regional distributions in the brain, and recent coexpression experiments showed that in human embryonic kidney (HEK) 293 cells GluR7 can have a reducing effect on GluR6 currents through formation of heteromeric receptors (Cui and Mayer, 1999).
Our study was aimed at determining where in the GluR7 molecule the functional differences to GluR6 reside. We chose a domain transplantation and site-directed mutagenesis strategy to test candidate stretches of sequence. The experiments described in this study identified the L3 domain as the region responsible for the functional differences between GluR6 and GluR7. In addition, several point mutants of GluR7 and GluR6 provided insights into the pharmacological behavior of GluR7 as compared with GluR6. In particular, GluR7-based mutants and chimeras had higher glutamate-evoked than kainate-evoked currents, whereas for GluR6 both agonists were equally effective.
MATERIALS AND METHODS
Mutagenesis. Single nucleotide exchanges were introduced by PCR-mediated site-directed mutagenesis using mutagenic primers as described previously (Hollmann et al., 1994). For all constructs, we used the “Q” editing variants of GluR6 and GluR7, GluR6(Q) and GluR7(Q), respectively, which from here on will be referred to as GluR6 and GluR7.
All mutated clones were sequenced across the PCR-amplified regions. We used two introduced restriction sites at homologous positions in GluR6 and GluR7 to construct chimeras between these two receptors: NruI C-terminally of TMD A at amino acids 548–550 (FVI) in GluR6 and at amino acids 519–521 (FVI) in GluR7; EcoRI N-terminally of TMD B at amino acids 603–605 (RIV) in GluR6 and at amino acids 574–576 (RII) in GluR7. Numbering starts with the first codon of the mature protein. GluR6 with introduced NruI and EcoRI sites showed no functional differences compared with the wild-type receptor. Each chimera between GluR6 and GluR7 contains a chimeric ligand-binding site composed of either S1 from GluR6 and S2 from GluR7, or vice versa. The resulting clones were named GluR6N–GluR7PC, GluR6NP–GluR7C, GluR7N–GluR6PC, and GluR7NP-GluR6C (where N stands for N-terminal domain plus TMD A, P for pore-forming region, and C for the region from TMD B to the C-terminal end of the protein).
We constructed GluR6–GluR7FC, a GluR6 chimera containing the “far C-terminal” (FC) domain downstream of TMD C of GluR7 by using a restriction site (NaeI at amino acid 768) in TMD C that is present in both receptors. We also engineered the reverse construct, GluR7–GluR6FC. Additional chimeras contained the region between TMDs B and C (L3 domain) of GluR7 inserted into GluR6 (GluR6–GluR7L3), and, vice versa, the L3 domain of GluR6 was inserted into GluR7 (GluR7–GluR6L3). For construction of these chimeras, we used one introduced restriction site at homologous positions in GluR6 and GluR7 (EcoRI, see above) and the native NaeI site in TMD C.
To subdivide the L3 domain, a silent mutation was introduced in GluR6 to generate a ClaI site at amino acid 728, which is a native site in GluR7 at amino acid 698. By taking advantage of theClaI restriction site and the ClaI site in the vector pSGEM (Villmann et al., 1999), the C-terminal half of the L3 domain plus the far C-terminal domain of GluR6 could be transplanted into GluR7. The resulting clone was named GluR7–GluR6L3C/FC. To transplant the first half of the S2 domain plus the linker region connecting the S2 domain with TMD B, we used the introduced restriction site EcoRI (see above) and the ClaI site.
In addition to the chimeras, we generated the following reciprocal mutations, insertion mutants (ins), and deletions (▵): GluR7(K626E) and GluR6(E655K), GluR7(insR651a-K652R-P653Q) and GluR6(▵R681/R682K/Q683P), GluR7(I705V) and GluR6(V735I), GluR7(I732M) and GluR6(M762I), GluR7(N748S) and GluR6(S778N), GluR7(K758N) and GluR6(N788K). Also, the connecting region between the S2 domain and TMD C (Fig. 1, linker C) was completely exchanged by PCR. The resulting clones were named GluR6–GluR7(linkerC) and GluR7–GluR6(linkerC).
cRNA synthesis. cRNA synthesis was done as described earlier (Hollmann and Heinemann, 1994). Briefly, template DNA was linearized with a suitable restriction enzyme. cRNA was synthesized from 1 μg of linearized DNA using an in vitro transcription kit (Stratagene, La Jolla, CA) with a modified protocol that uses 800 μmm7GpppG (Pharmacia, Freiburg, Germany) for capping and an extended reaction time of 3 hr with T7 polymerase. Trace labeling was performed with [32P]UTP to allow calculation of yields and transcript quality check by agarose gel electrophoresis.
Electrophysiological measurements in Xenopusoocytes. Oocytes of stages V–VI were surgically removed from the ovaries of Xenopus laevis as described elsewhere (Everts et al., 1997). Oocytes were injected with 10 ng of cRNA for homomeric receptors and 5 ng of cRNA for each subunit of heteromeric receptors using a 10 μl Drummond (Broomall, PA) microdispenser. Two-electrode voltage-clamp recordings were performed 4–8 d after cRNA injection with a TurboTec 10CD amplifier (npi, Tamm, Germany) by superfusion of the oocyte with glutamatergic agonists (300 μm) prepared in normal frog Ringer's solution (in mm: NaCl, 115; CaCl2, 1,5; KCl, 2,5; and HEPES-NaOH, 10, pH 7.2). Current electrodes were filled with 3 m CsCl and had resistances of ≈0.5–1.5 MΩ. Voltage electrodes were filled with 3 m KCl and had resistances of ≈4 MΩ. Oocytes were held at −70 mV, and agonists (kainate and glutamate) were applied for 10 sec at a flow rate of 10–14 ml/min. To minimize receptor desensitization, bath pretreatment of oocytes with concanavalin A (ConA; 10 μm for 8 min) preceded agonist application (Everts et al., 1997). EC50 values were measured with 10 or 11 different agonist concentrations. Data from each oocyte were fitted separately with the SigmaPlot program (Jandel Scientific, San Rafael, CA) to the equation I = Imax/[1 + (EC50/A)nH], where Imax is the maximal current, EC50 is the agonist concentration giving half-maximal currents, and nH is the Hill coefficient. Each EC50 value obtained represents an average of three or four oocytes.
Labeling of cell surface protein using biotinylated ConA. To identify the fraction of receptor protein inserted in the plasma membrane, surface proteins were tagged with biotinylated ConA and isolated by streptavidin/Sepharose-mediated precipitation of the labeled protein. Briefly, intact oocytes were incubated in 10 μm biotinyl-ConA (Sigma, München, Germany) for 30 min at room temperature. After five 10 min washes in normal frog Ringer's solution, intact oocytes were homogenized with a Teflon pestle in H-buffer (20 μl/oocyte; 100 mmNaCl, 20 mm Tris-HCl, pH 7.4, 1% Triton X-100, and 1 mm phenylmethylsulphonyl fluoride) plus a mixture of proteinase inhibitors (Complete; Boehringer Mannheim, Mannheim, Germany) and were kept at 4°C for 1 hr on a rotating rod. After centrifugation for 60 sec at 16,000 × g, the supernatants were supplemented with 20 μl of washed streptavidin-Sepharose beads (Sigma) and incubated at 4°C for 3 hr on a rotating rod. The streptavidin-Sepharose beads were then pelleted by a 120 sec spin at 1600 × g and washed three times in H-buffer. The final pellets were boiled in 40 μl of SDS-PAGE loading buffer (0.8 m β-mercaptoethanol, 6% SDS, 20% glycerol, 25 mm Tris-HCl, pH 6.8, and 0.1% bromphenol blue).
Gel electrophoresis and Western blotting. Proteins were separated on 20 cm SDS-PAGE gels (Laemmli, 1970). Gels were blotted (Towbin et al., 1979) onto Hybond enhanced chemoluminescence (ECL) nitrocellulose membranes (Amersham, Braunschweig, Germany). For development of the blots, a previously described protocol was followed (Villmann et al., 1997). The antibody was kindly provided by Dr. Robert Wenthold (National Institutes of Health, Bethesda, MD) (polyclonal anti-GluR6/R7 antibody directed against the C terminus of GluR6).
[3H]kainate binding to HEK 293 cell membranes. To assay [3H]kainate binding to GluR6 and GluR6 mutants as well as GluR7 and GluR7 mutants, membranes of transfected HEK 293 cells were used. Adhesive HEK 293 cells (catalog #CRL 1573; American Type Culture Collection, Manassas, VA) transfected using the calcium phosphate precipitation technique were harvested in ice-cold 0.5 mm EDTA, 100 μm/ml 4–2-aminoethyl benzenesulfonyl fluoride (AEBSF), and PBS. After centrifugation at 4000 × g, the pelleted cells (from 10–30 plates; 10-cm-diameter) were homogenized with a Teflon-glass homogenizer in ice-cold 50 mm Tris-acetate buffer containing 10 mm EDTA, 100 μm/ml AEBSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 μg/ml pepstatin A and centrifuged at 8000 × g. The supernatant was collected and centrifuged at 600,000 × g. The latter step was repeated twice more in NaCl-free 50 mmTris-acetate buffer at pH 7.3. After suspension and homogenization of the pellets in NaCl-free 50 mm Tris-acetate buffer at pH 7.3, membranes were frozen and kept in liquid nitrogen until use for the [3H]kainate-binding assay. Displacement curves were constructed by incubating on ice membranes (50–150 μg of protein) with 80 nm[3H]kainate (58 Ci/mmol) in a total volume of 250 μl of 50 mm Tris-acetate buffer at pH 7.3 in the presence of increasing concentrations of unlabeled kainate (10 nm, 30 nm, 100 nm, 300 nm, 1 μm, 3 μm, and 10 μm). After 60 min, the membranes were centrifuged at 12,000 × g for 1 hr, and the pellet was resuspended in 100 μl of 0.5 m NaOH. After a 1 hr of incubation, 75 μl of 12% acetic acid were applied for neutralization. The samples were counted with scintillation fluid (Lumax-xylene). The specific binding of [3H]kainate was defined as the total binding minus the binding obtained in the presence of 1 mm kainate. All experiments were performed in triplicate. Binding data were analyzed using the Prism program (GraphPad, San Diego, CA).
Molecular modeling of GluR6 and GluR7. The three-dimensional model structures of the ligand-binding domains of GluR6 and GluR7 were constructed based on the experimental structure of the corresponding domain of GluR2 (Armstrong et al., 1998). The sequences of GluR6 and GluR7 are very similar to that of GluR2 showing 51.2 and 51% identity, respectively, for the 249 amino acids comprising the ligand-binding domains. There are three 1- and 2- residue deletions in the sequences of GluR6 and GluR7 compared with GluR2, all in loops. Two of these deletions occur in loops next to the cysteine residues, which were found to form a disulfide bridge in GluR2. The conformations of these loops were adjusted to preserve the S–S bond.
Initial models of GluR6 and GluR7 were built using the Homology module of MSI (San Diego, CA). These models were energy-minimized with the Discover module, using the CVFF force field parameters. The Cα atoms were constrained to their initial positions during minimization, which therefore served to relieve interatomic clashes where they occurred, but the overall folding of the proteins was not disrupted.
RESULTS
Chimeric GluR6–GluR7 constructs
To initially determine whether the functional differences between GluR6 and GluR7 are linked to the first or second half of the receptor protein, we constructed chimeras containing the N terminus plus the pore domain (NP) or just the N terminus (N) of GluR6, whereas the rest of the protein, the C-terminal part (C), or pore domain plus C-terminal part (PC), respectively, originated from GluR7 (Fig.2). We also engineered the respective reverse constructs. These four chimeras were named GluR6N–GluR7PC, GluR6NP–GluR7C, GluR7N–GluR6PC, and GluR7NP–GluR6C. Interestingly, all four chimeras gave functional ion channels that could be activated by kainate and glutamate (Fig. 2). Maximal current amplitudes of GluR6N–GluR7PC and GluR6NP–GluR7C were rather small compared with wild-type GluR6 (∼0.1–5.5%). However, they could be reproducibly measured in every oocyte tested provided that current desensitization was minimized by ConA pretreatment of oocytes (see Materials and Methods). The chimeras GluR7NP–GluR6C and GluR7N–GluR6PC, on the other hand, showed maximal glutamate- and kainate-evoked current amplitudes comparable with or even larger than wild-type GluR6 (138–235%) (Table 1). Based on these data, the functional differences between GluR6 and GluR7 appear to be linked to the sequence C-terminal of transmembrane domain B. By contrast, the origin of the ion pore (GluR6 or GluR7) does not seem to effect maximal current amplitudes. These data show that the two half sites of the ligand-binding domains S1 and S2 can be exchanged between GluR6 and GluR7 without loss of function. Thus, the huge differences in maximal current amplitudes between ConA-treated GluR7 (0 nA) and GluR6 (∼15,000 nA) is unlikely to be caused by a disruption of the interaction of S1 and S2 in GluR7.
Fig. 2.

Schematic structures of the chimeras and point mutants between GluR6 and GluR7. Domains originating from GluR6 are shown blank, and those derived from GluR7 are black. Sample current traces (recorded with 300 μm glutamate) are shown below the corresponding graphic representations. To minimize desensitization, all receptor-expressing oocytes have been treated with 10 μm ConA before recording. Current traces shown were recorded in different experiments. Note the different scales of the current traces.
Table 1.
Relative current amplitudes and EC50 values of chimeras between GluR6(Q) and GluR7(Q)
| Clone | KA current [%] | Glu current [%] | n | Ratio Glu/KA | EC50 KA [μm] | EC50Glu [μm] | n |
|---|---|---|---|---|---|---|---|
| GluR6(Q) | 100.0 ± 17.9 | 100.0 ± 21.6 | 34 | 0.9 ± 0.1 | 1.7 ± 0.7 | 45.6 ± 15.2 | 3 |
| GluR7(Q) | 0.0 ± 0.0 | 0.0 ± 0.0 | 3 | 0.0 ± 0.0 | |||
| GluR6N–GluR7PC | 0.09 ± 0.04 | 0.6 ± 0.1 | 3 | 9.1 ± 2.8 | ctl | 20.0 ± 3.5 | 3 |
| GluR6NP–GluR7C | 0.4 ± 0.1 | 5.5 ± 1.6 | 8 | 13.1 ± 0.9 | 3.7 ± 1.0 | 47.2 ± 4.2 | 3 |
| GluR7N–GluR6PC | 155.0 ± 76.1 | 137.0 ± 64.4 | 7 | 1.0 ± 0.4 | 1.7 ± 0.2 | 223.0 ± 65.8 | 3 |
| GluR7NP–GluR6C | 235.0 ± 40.6 | 228.0 ± 38.2 | 4 | 0.8 ± 0.1 | 1.7 ± 0.3 | 103.0 ± 12.5 | 3 |
| GluR6–GluR7FC | 83.7 ± 5.1 | 68.6 ± 4.2 | 3 | 1.10 ± 0.01 | 1.5 ± 0.1 | 45.3 ± 9.0 | 4 |
| GluR7–GluR6FC | 0.02 ± 0.02 | 0.1 ± 0.1 | 4 | 5.5 ± 1.3 | ctl | ctl | |
| GluR6–GluR7L3 | 0.07 ± 0.01 | 0.003 ± 0.002 | 6 | 6.7 ± 0.4 | ctl | ctl | |
| GluR7–GluR6L3 | 207.0 ± 39.3 | 166.3 ± 34.8 | 4 | 0.79 ± 0.05 | 2.4 ± 0.6 | 314.0 ± 92.5 | 3 |
| GluR7–GluR6L3C/FC | 7.9 ± 0.5 | 7.0 ± 0.7 | 9 | 1.07 ± 0.06 | 2.5 ± 0.1 | 129.1 ± 5.0 | 3 |
| GluR7–GluR6L3N | 0.01 ± 0.01 | 0.10 ± 0.03 | 5 | 12.5 ± 0.6 | ctl | 307.3 ± 16.1 | 3 |
Receptors expressed in Xenopus oocytes were probed with 300 μm glutamate and 300 μm kainate as agonists. All currents were measured after 8 min of pretreatment of oocytes with ConA to minimize desensitization. Relative currents were calculated by taking GluR6 currents of the same batch as 100%. EC50 values were determined using 11 different concentrations (0.03, 0.1, 0.3, 1, 3, 10, 30, 100, 300, 1000, and 3000 μm) of either kainate or glutamate. Data are shown as means ± SEM. For an explanation of the constructs, see Figure 1and Materials and Methods. ctl, Currents too low for EC50values to be calculated. Absolute currents for wild-type GluR6 are 28615 ± 2078 nA (KA) and 27593 ± 2134 nA (Glu).
To test whether the entire C-terminal half of GluR6 is required for efficient ion channel function of chimeras between GluR6 and GluR7, we engineered a chimeric receptor containing the FC domain of GluR7 transplanted into GluR6 to produce a construct we named GluR6–GluR7FC. We also engineered the reverse construct GluR7–GluR6FC as a control. The maximal current amplitudes of GluR6–GluR7FC were comparable with its parent clone, wild-type GluR6, whereas GluR7–GluR6FC gave only minimal responses, ∼0.02–0.12% of GluR6 (Table 1). Thus, the far C-terminal domain is unlikely to be responsible for the large currents seen in the chimeras GluR7NP–GluR6C and GluR7N– GluR6PC.
To test this further, we engineered chimeras containing the GluR7- derived L3 domain inserted into GluR6 (GluR6–GluR7L3), as well as the reverse construct (GluR7–GluR6L3). GluR6–GluR7L3 showed very reduced currents compared with wild-type GluR6 (0.003–0.07%). By contrast, maximal amplitudes for GluR7–GluR6L3 were comparable with wild-type GluR6 (166–207%; Table 1). These data support the conclusion that the region between transmembrane domains B and C (L3 domain) is the critical determinant for efficient channel function of GluR6 and GluR7.
To analyze the L3 domain further, we introduced a restriction site (ClaI) in the middle of this domain in GluR7 by silent mutation. Such a ClaI site occurs naturally at the homologous position in GluR6, facilitating the independent exchange of the N-terminal or C-terminal half of the L3 domain. Exchange of the sequence downstream of the ClaI site results in a construct (GluR7–GluR6L3C/FC) that is composed of GluR7 up to approximately the middle of the L3 domain. The remainder of this protein is derived from GluR6. Maximal current amplitudes in this construct were reduced compared with wild-type GluR6 (7–8%) as well as compared with GluR7–GluR6L3, which carries the entire L3 domain of GluR6 (3.8–4.2%; Table 1). By using the same ClaI site and theEcoRI site in TMD B (see Materials and Methods), it was possible to exchange the N-terminal part of the L3 domain. The resulting chimera GluR7–GluR6L3N (L3N = N-terminal part of the L3 domain) showed very reduced maximal current amplitudes compared with wild-type GluR6 (0.01–0.1%) and GluR7–GluR6L3 (0.005–0.06%).
Western blots showed that all chimeras were expressed (Fig.3T, lanes 1–8,11, 15) and incorporated into the plasma membrane (Fig.3P, lanes 1–8, 11, 15). Expression levels of chimeras were generally comparable with wild-type receptor subunits (Fig. 3, lanes 10, 17). GluR7–GluR6L3 and GluR6N–GluR7PC appeared to be somewhat less efficiently expressed; however, differences in membrane-inserted protein levels were small compared with differences in ion channel function and thus were unlikely to distort the interpretation of the functional data.
Fig. 3.
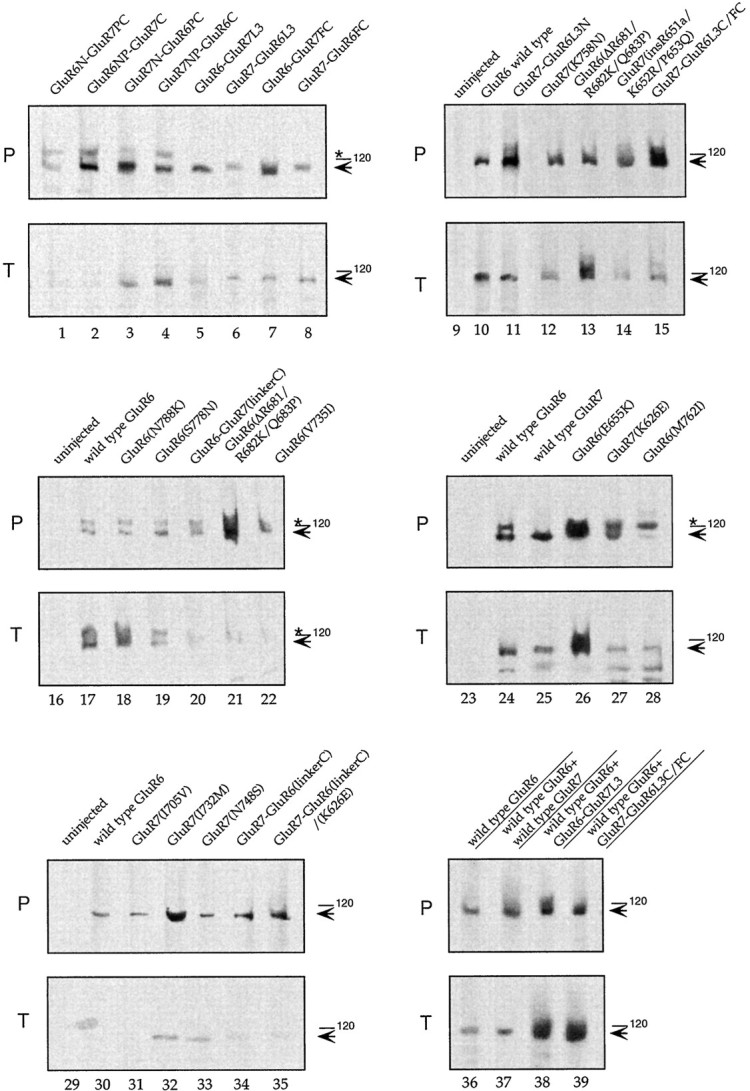
Western blots demonstrating protein expression of chimeric and mutated GluR6 and GluR7 receptors.P, Plasma membrane protein that was labeled with biotinyl-ConA and then streptavidin-precipitated (11 oocytes/lane).T, Total oocyte protein (1 oocyte/lane). Samples including controls from uninjected oocytes were separated on an SDS gel, Western-blotted, and probed with affinity-purified antibodies against C-terminal peptides of GluR6 that cross-react with GluR7 (Wenthold et al., 1994). Filled arrows point to the position of the ∼115 kDa band of wild-type GluR6, GluR6 mutants, and wild-type GluR7 and GluR7 mutants. The asterisk denotes an unidentified band cross-reacting with the GluR6/GluR7 antibody (Villmann et al., 1999).
We compared dose–response curves for kainate- and glutamate-evoked currents of wild-type GluR6(Q) and all chimeras. The chimeras containing at least the L3 region of GluR6 (GluR7NP–GluR6C, GluR7N–GluR6PC, and GluR7–GluR6L3) display a decrease in glutamate efficiency of twofold to sixfold compared with wild-type GluR6, whereas the efficiency for kainate is unchanged (Table 1). Similarly, increases in EC50 values for glutamate were noted for GluR7–GluR6L3C/FC and GluR7–GluR6L3N (2.8-fold and 6.7-fold, respectively), with no differences in kainate EC50 values. The increased EC50 values for glutamate of some chimeras compared with wild-type GluR6 (Table 1) appear to be linked to the N-terminal domain of GluR7, because none of the chimeras with GluR6-derived N-terminal domains have glutamate EC50 values different from wild-type GluR6. EC50 values for kainate were not significantly different among the chimeras.
Wild-type GluR6 and GluR7 have similarKD values for kainate binding (95 nm for GluR6, 77 nm for GluR7) (Bettler et al., 1992). However, it cannot be excluded offhand that chimeras between the two receptors might suffer from altered agonist-binding sites and thus have differentKD values. This in turn could affect maximal current amplitudes and render comparison of chimeras invalid. Therefore, GluR6, GluR7, and GluR7–GluR6L3, the GluR7-based chimera with the largest current amplitudes, were expressed in HEK 293 cells and assayed for [3H]kainate binding.KD values for kainate in our hands were 33 nm (wild-type GluR6), 47 nm (wild-type GluR7), and 61 nm (GluR7–GluR6L3). These results demonstrate that differences in agonist KD values are unlikely to account for the efficient ion channel function of chimeras such as GluR7–GluR6L3.
Mutations in GluR6 and GluR7
Our analysis of chimeras between GluR6 and GluR7 (see above) provided strong indications that the L3 domain is responsible for the functional differences between GluR6 and GluR7. We therefore focused on this domain that contains 20 amino acids that are different between GluR6 and GluR7 (Fig. 4). To gain further insight into the molecular basis of the functional differences between GluR6 and GluR7, we selected positions with the most extreme amino acid differences and tested reciprocal point mutations in GluR6 and GluR7. Some of the amino acids could be excluded from the analysis as another group working on a different project (Swanson et al., 1997) had already reported point mutations at positions E655, D672, R682, V685, S689, S701, and F705 in GluR6. Those mutants had been named R6(E686R), R6(D703E), R6(R713Q), R6(V716A), R6(S720N) R6(S731T), R6(F735L), respectively, using a different numbering scheme, and showed currents not significantly different from wild-type GluR6.
Fig. 4.
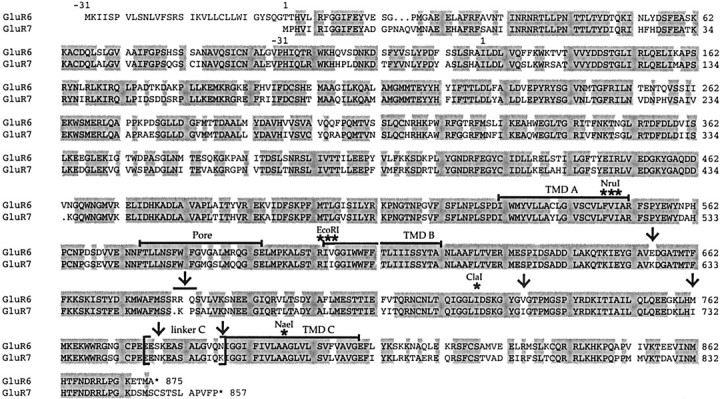
Amino acid sequence alignment of GluR6 and GluR7. Sequence identity between these receptors is 86%. The TMDsA, B, and C, and the pore domain are marked by black bars. Linker C, which is transplanted in the chimeras GluR6–GluR7(linkerC) and GluR7–GluR6-(linker C), is marked by brackets. Thearrows show the positions of the various reciprocal mutations GluR6(E655K) and GluR7 (K626E), GluR6(▵R681/ R682K/Q683P) and GluR7(insR651a/K652R/ P653Q), GluR6(V735I) and GluR7(I705V), GluR6(M762I) and GluR7(I732M), GluR6(S778N) and GluR7(N748S), and GluR6(N788K) and GluR7(K758N). Asterisks point to the native NaeI site in TMD C and to the positions where restriction sites were introduced either by silent mutation (ClaI) or by conservative amino acid exchange (NruI, EcoRI) for constructing chimeras between GluR6 and GluR7.
Here, the following reciprocal mutations were analyzed: GluR6(E655K) and GluR7(K626E), GluR6(▵R681/R682K/Q683P) and GluR7(insR651a-K652R-P653Q), GluR6(M760I) and GluR7(I731M), GluR6(V733I) and GluR7(I704V), GluR6(S778N) and GluR7(N748S), and GluR6(N788K) and GluR7(K758N). Among these mutants, all those based on GluR6 as well as the four GluR7 mutants GluR7(K626E), GluR7(insR651a-K652R-P653Q), GluR7(N748S), and GluR7(K758N) were capable of conducting currents in oocytes. However, maximal current amplitudes were quite different: for GluR6(E655K), GluR6(▵R681/R682K/Q683P), GluR6(V733I), and GluR6(S778N) current amplitudes were comparable with wild-type GluR6. By contrast, a reduction compared with wild-type GluR6 of ∼50% was observed for GluR6(N788K), and a decrease of ∼90% was observed for GluR6(M762I) (Table 2). Because GluR6(M762I) shows a reduction of incorporation into the plasma membrane (Fig.3P, lane 28), this might, at least partly, explain the enormous reduction of current. The four GluR7 mutants, GluR7(K626E), GluR7(N748S), GluR7(K758N), and GluR7(insR651a/K652R/P653Q), all gave glutamate- and/or kainate-evoked currents, with the largest, for GluR7(K758N), reaching ∼2% of wild-type GluR6 (Table 2).
Table 2.
Relative current amplitudes and EC50 values of several mutants of GluR6(Q) and GluR7(Q)
| Clone | KA current [%] | Glu current [%] | n | Ratio Glu/KA | EC50 KA [μm] | EC50Glu [μm] | n |
|---|---|---|---|---|---|---|---|
| GluR6(Q) | 100.0 ± 17.9 | 100.0 ± 21.6 | 34 | 0.91 ± 0.05 | 1.7 ± 0.7 | 45.6 ± 15.2 | 3 |
| GluR7(Q) | 0.0 ± 0.0 | 0.0 ± 0.0 | 3 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 3 |
| GluR6(E655K) | 101.5 ± 23.8 | 94.8 ± 22.2 | 3 | 0.91 ± 0.03 | 0.8 ± 0.1 | 23.4 ± 4.2 | 3 |
| GluR7(K626E) | 0.0 ± 0.0 | 0.02 ± 0.01 | 3 | 0.0 ± 0.0 | 0.0 ± 0.0 | ctl | |
| GluR6(N788K) | 41.6 ± 16.9 | 49.5 ± 21.6 | 14 | 1.13 ± 0.05 | 3.0 ± 0.3 | 71.6 ± 7.4 | 3 |
| GluR7(K758N) | 0.16 ± 0.06 | 1.8 ± 0.9 | 14 | 10.8 ± 1.3 | 1.2 ± 0.1 | 79.5 ± 2.1 | 3 |
| GluR6(S778N) | 77.8 ± 3.5 | 78.8 ± 1.9 | 3 | 0.95 ± 0.02 | 1.0 ± 0.2 | 31.0 ± 1.5 | 3 |
| GluR7(N748S) | 0.0 ± 0.0 | 0.03 ± 0.01 | 8 | 0.0 ± 0.0 | 0.0 ± 0.0 | 184.0 ± 12.8 | 3 |
| GluR6(V735I) | 151.0 ± 41.0 | 136.8 ± 38.4 | 3 | 0.93 ± 0.02 | 0.40 ± 0.06 | 13.9 ± 0.6 | 3 |
| GluR7(I705V) | 0.0 ± 0.0 | 0.0 ± 0.0 | 3 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | |
| GluR6(M762I) | 9.5 ± 2.7 | 7.3 ± 1.9 | 4 | 0.76 ± 0.02 | 1.60 ± 0.01 | 54.9 ± 2.5 | 3 |
| GluR7(I732M) | 0.0 ± 0.0 | 0.0 ± 0.0 | 4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | |
| GluR6(ΔR681/R682K/Q683P) | 112.6 ± 5.3 | 106.3 ± 9.3 | 3 | 0.94 ± 0.04 | 0.50 ± 0.07 | 77.8 ± 3.5 | 3 |
| GluR7(insR651a/K652R/P653Q) | 0.0 ± 0.0 | 0.02 ± 0.01 | 3 | 0.0 ± 0.0 | 0.0 ± 0.0 | ctl | 3 |
| GluR6–GluR7(linkerC) | 35.6 ± 6.1 | 30.2 ± 4.7 | 7 | 1.02 ± 0.02 | 1.9 ± 0.4 | 54.3 ± 4.9 | 3 |
| GluR7–GluR6(linkerC) | 0.03 ± 0.01 | 0.6 ± 0.1 | 8 | 12.4 ± 1.1 | ctl | 81.4 ± 4.2 | 3 |
| GluR7–GluR6(linkerC)/(K626E) | 0.82 ± 0.05 | 1.2 ± 0.4 | 4 | 8.2 ± 0.3 | 1.2 ± 0.2 | 61.3 ± 3.7 | 3 |
For legend, see Table 1.
The point mutations GluR7(N748S) and GluR7(K758N) both reside in L3 in the short linker sequence that connects the S2 domain to TMD C (linker C). This led us to test whether an exchange of the entire linker C region (which would include those two amino acid exchanges plus another one at position 756, I756V) could make GluR7 even more GluR6-like. The required construct was engineered by PCR and was named GluR7-GluR6(linkerC) (a GluR7 clone with linker region C derived from GluR6). Surprisingly, the simultaneous exchange of all three differential amino acids in this linker region did not lead to current amplitudes larger than the single amino acid mutant GluR7(K758N). Similarly, the reverse construct, GluR6–GluR7(linkerC) (a GluR6 clone with the linker region C derived from GluR7), was not different from the single amino acid mutant GluR6(N788K) (Table 2). Thus, effects of point mutations in linker C are not additive.
It seemed reasonable to combine GluR7–GluR6(linkerC) with the other functional GluR7 point mutation, GluR7(K626E). This construct was named GluR7–GluR6(linkerC)/(K626E). However, this combination mutant showed no differences in current amplitude compared with GluR7–GluR6(linkerC).
Western blot data prove that all mutants were expressed and incorporated into the plasma membrane (Fig. 3, lanes 12–14,18–22, 26–28, 31–35). EC50 values were either unchanged from wild type for both glutamate and kainate (GluR6(S778N), GluR6-GluR7(linkerC)), or slightly decreased for glutamate (GluR6(N788K)). For the GluR7 mutants, comparison to wild type is difficult because up to now it has not been possible to measure GluR7 in oocytes. However, an EC50 value of 6 mm (glutamate) has been reported for wild-type GluR7 expressed in HEK 293 cells (Swanson et al., 1997). Compared with this EC50 value, all current-conducting GluR7 mutants show considerably decreased EC50 values (1–5% of wild-type GluR7). However, compared with wild-type GluR6, all GluR7 mutants tested in our study displayed a tendency toward increased EC50 values for glutamate (twofold to fourfold). The EC50 values for kainate were not significantly different.
Comparison of mutant amino acid interactions in the three-dimensional model structures of the ligand-binding domains of GluR6 and GluR7
We have demonstrated that the L3 domain is responsible for the functional differences between GluR6 and GluR7. Division of this region such that the first half derives from GluR6 and the second from GluR7 resulted in a dramatic reduction of currents. Therefore, amino acids from both parts of the L3 domain presumably have a role in forming a highly functional kainate receptor channel.
The crystal structure of the ligand-binding domain of GluR2 bound to kainate has recently been solved (Armstrong et al., 1998). Therefore, it was possible to create models of the ligand-binding domains of GluR6 and GluR7 based on sequence alignments with GluR2. We used those models to look for amino acids in the two halves of the S2 domains (L3 domain minus linkers B and C) that might interact with each other and at the same time differ between GluR6 and GluR7. However, amino acids in the S2 domain that differ between GluR6 and GluR7 appear to be interacting only with amino acids that are identical in both receptors. Furthermore, most of those differing amino acids reside at the surface of the structure and are exposed to the solvent, and none of these amino acids are located near the ligand-binding site of kainate (Fig.5). Thus, interference with agonist binding by these amino acids is unlikely to explain the functional differences between GluR6 and GluR7.
Fig. 5.

Three-dimensional model structures of the ligand-binding domains of GluR6 (right) and GluR7 (left) based on the solved x-ray structure of the corresponding domains of GluR2. Marked amino acids in GluR7 were mutated to amino acids at homologous positions in GluR6 and vice versa. The bound agonist molecule kainate is shown as black lines at the center of the models. Note that mutated amino acids do not interact with the agonist-binding site or with each other.
Our data on mutations within linker C could not be evaluated in the context of the ligand-binding model as the linker region is not part of the S1–S2 crystal structure and therefore cannot be modeled.
One has to keep in mind that although the ligand-binding domains of GluR6 and GluR7 can be modeled, the actual structure might be different.
Coexpression of GluR7 mutants
In addition to modeling, we used a coexpression technique to look for interacting amino acids. This approach was based on the assumption that in a multi-subunit complex mutations in a single subunit might dominate the functional properties of the entire complex.
The cRNAs of constructs to be coexpressed were mixed in equal amounts before injection into oocytes, thereby creating receptor complexes with mixed mutated subunits. Expression of GluR7(insR651a/K652R/P653Q) with GluR7–GluR6L3C/FCgave 9.5% of wild-type GluR6 current for glutamate and 14.9% for KA. Similarly, expression of GluR7(K626E) with GluR7–GluR6L3C/FC showed 6.9% of wild-type GluR6 current for glutamate and 0.7% for KA. Thus, compared with GluR7–GluR6L3C/FC alone (7% of wild-type GluR6 current for glutamate, and 8% for KA) these combinations yielded no significant increase in relative current amplitudes (Table3). All other mutant subunit combinations tested (GluR7(insR651a/K652R/P653Q) + GluR7(K758N), GluR7(insR651a/K652R/P653Q) + GluR7(I731M), GluR7(K626E) + GluR7(I731M), and GluR7 (I731M) + GluR7(K758N)) expressed either no or only tiny currents. Thus, coexpression of mutant receptor subunits was not informative in identifying interacting amino acids involved. Interestingly, expression of GluR7–GluR6L3C/FC with GluR7–GluR6L3N also yielded no significant increase in current amplitude compared with homomeric currents of both mutants (Table 3). This was a somewhat surprising result because GluR7–GluR6L3, which comprises both these mutants combined in one subunit, showed currents like wild-type GluR6. Therefore, on coexpression of the two chimeras that separately contain the two halves of the L3 domain of GluR6 in GluR7, currents comparable with GluR7–GluR6L3 had been expected. However, this was not the case. Presumably, interacting determinants in the L3 domain involved in forming a highly functional ion channel have to reside on the same subunit rather than on different subunits.
Table 3.
Relative current amplitudes of coexpressed GluR7 mutants and chimeras between GluR6 and GluR7
| Clone | KA current [%] | Glu current [%] | n |
|---|---|---|---|
| GluR6(Q) + H2O | 100.0 ± 9.7 | 100.0 ± 8.6 | 7 |
| GluR7(insR651a/K652R/P653Q) + GluR7(K758N) | 0.2 ± 0.1 | 1.9 ± 1.0 | 5 |
| GluR7(insR651a/K652R/P653Q) + GluR7(I731M) | 0.8 ± 0.3 | 2.1 ± 0.7 | 5 |
| GluR7(K626E) + GluR7(I731M) | 0.0 ± 0.0 | 0.0 ± 0.0 | 3 |
| GluR7(I731M) + GluR7(K758N) | 0.09 ± 0.04 | 0.90 ± 0.04 | 6 |
| GluR7(insR651a/K652R/P653Q) + GluR7–GluR6L3C/FC | 14.9 ± 5.8 | 9.5 ± 3.6 | 4 |
| GluR7(K6262E) + GluR7-GluR6L3C/FC | 6.9 ± 4.0 | 6.7 ± 4.5 | 4 |
| GluR7–GluR6L3N + GluR7–GluR6L3C/FC | 0.8 ± 0.2 | 0.8 ± 0.2 | 3 |
Receptors expressed in Xenopus oocytes were probed with 300 μm glutamate and 300 μm kainate as agonists. All currents were measured after 8 min of pretreatment of oocytes with ConA to minimize desensitization. Relative currents were calculated by taking GluR6 currents of the same batch as 100%. Data are shown as means ± SEM.
The coexpression technique can therefore not be used to determine the interacting amino acids, but it gives us the important information that both determinants have to reside on the same subunit.
Coexpression of wild-type GluR6(Q) with GluR7 chimeras or mutants
Recently, it has been shown that coexpression of GluR7 with GluR6 dramatically reduces the amplitude of GluR6 responses to kainate (Cui and Mayer, 1999). Therefore, it was of interest to test whether any of the chimeras or mutants of GluR7 would show an altered inhibitory influence on wild-type GluR6. If the domain responsible for ion channel function is the same that is responsible for the inhibitory action of GluR7, the extent of the inhibitory action of GluR7 mutants is expected to be inversely related to their maximal current amplitudes: the more GluR6-like in function the GluR7 mutant is, the less inhibitory it should be after coexpression with GluR6.
Efficient incorporation into the oocyte plasma membrane of receptor subunits was verified by Western blot for wild-type GluR6, wild-type GluR6 plus wild-type GluR7, wild-type GluR6 plus GluR6–GluR7L3, and wild-type GluR6 plus GluR7–GluR6L3C/ FC (Fig. 3, lanes 36–39).
Wild-type GluR6 was mixed with H2O at a ratio of 1:1 before injection into oocytes. Therefore, wild-type GluR6 RNA concentration was 10 ng/oocyte, and the RNA concentration of GluR6 plus GluR7 mutant was 20 ng/oocyte. To verify that the maximal current amplitude depends on the injected RNA amount in a linear way, 10 and 20 ng of wild-type GluR6 were injected, and the maximal current amplitudes compared. This control demonstrated that indeed the maximal current amplitudes linearly depend on the injected RNA concentration.
All GluR7 mutants reduced wild-type GluR6 currents similar to wild-type GluR7 (to ∼0.4–12%). Interestingly, however, coexpression of the chimeric receptors GluR7–GluR6L3C/FC, GluR7N–GluR6PC, GluR7NP–GluR6C, or GluR6–GluR7FC with wild-type GluR6 gave no reduction in GluR6 current amplitudes, whereas the reverse chimeras (GluR6N–GluR7PC, GluR6NP–GluR7C, GluR7–GluR6FC, and GluR7–GluR6L3N) showed the same reduction as GluR7. Only a slight decrease in currents was seen for the coexpression of GluR7–GluR6L3 with wild-type GluR6 (to ∼60–80% of wild-type GluR6) and GluR6–GluR7L3 with wild-type GluR6 (to ∼40–50% of wild-type GluR6). The reduction of currents observed in some coexpression experiments is not caused by decreased expression of the receptor or decreased incorporation into the plasma membrane as shown by Western blot (see above).
This pattern suggests that the second half of the L3 domain as well as the intracellular C-terminal domain contribute to the GluR7-mediated current reduction at GluR6. Therefore, whereas the L3 domain alone is responsible for the functional differences between GluR6 and GluR7, the results of coexpression of GluR6 and GluR7 mutants indicate that the full reducing effect of GluR7 on GluR6 current amplitudes requires a combination of the L3 and FC domains.
GluR7 mutants show a significant increase in the ratio of glutamate- to kainate-evoked currents after treatment with concanavalin A
The ratio of glutamate- to kainate-evoked currents after treatment with concanavalin A was calculated for each mutant and chimeric receptor. This ratio is ∼1 for wild-type GluR6. All GluR6 mutants give similar values. The situation is different for the GluR7 mutants whose ratios are ∼10 (Table 2). For those chimeras that have at least the second half of the L3 domain of GluR7 (GluR6N–GluR7PC, GluR6NP–GluR7C, GluR7–GluR6FC,GluR6–GluR7L3, and GluR7–GluR6L3N), the ratio is similar to the ratio found for GluR7 mutants (5–10) (Tables 1, 2). The ratio for the reciprocal chimeras (GluR7N–GluR6PC, GluR7NP–GluR6C, GluR6–GluR7FC, and GluR7–GluR6L3) is ∼1, as for wild-type GluR6. The chimera GluR7–GluR6L3C/FC, where the first half of the L3 domain originates from GluR7 and the second half from GluR6, also showed a ratio of 1 (Table 1).
Interestingly, the ratio of glutamate- to kainate-evoked currents for wild-type GluR6 coexpressed with different chimeras or GluR7 mutants (see above) is always ∼1, suggesting a dominance of GluR6 over GluR7.
DISCUSSION
Chimeric GluR6–GluR7 constructs and GluR7 mutants
For a long time it was thought that the kainate receptor GluR7 is a nonfunctional glutamate receptor subunit, because no agonist-activated responses could be elicited in Xenopusoocytes or transfected cells (Bettler et al., 1992; Lomeli et al., 1992; Partin et al., 1993; Nutt et al., 1994). Agonist application in the millimolar concentration range to HEK 293 cells transfected with GluR7 then showed that GluR7 receptor subunits indeed can form functional receptors, albeit with an EC50 for glutamate of only ∼6 mm (Schiffer et al., 1997). Such unphysiologically high agonist concentrations cannot be used in the Xenopus oocyte expression system because they cause spurious responses seen also in noninjected control oocytes (Hollmann, 1999). This explains why GluR7 activation by high agonist concentrations went undetected in the early expression studies. An ion pore transplantation study later demonstrated that GluR7 indeed has a fully functional ion permeation pathway (Villmann et al., 1999). Taken together, the available data indicate that GluR7 is not a nonfunctional receptor but rather a receptor with a very low efficacy of pore opening. This is supported by our observation that several GluR7 mutants (GluR7(K626E), GluR7(insR651a-K652R-P653Q), GluR7(N748S),GluR7(K758N) as well as GluR7 chimeras with GluR6 form functional ion channels, which can easily be measured in oocytes because they are activated by agonists in the micromolar range (Tables1, 2).
Correlation of data with model of ligand-binding site
The recently published crystal structure of the soluble ligand-binding domain of the rat GluR2 “flop” isoform bound to kainate (Armstrong et al., 1998) can serve as a template to create models of homologous glutamate receptor ligand-binding domains based on amino acid sequence alignments. In the GluR2 S1–S2 crystal structure, kainate binds in a crevice that forms between the S1 and S2 domains. The crevice is comprised of four α helices, three of which contain a kainate-interacting residue (F, H, and I) whereas one helix (D) provides a connection between S1 and S2 (Armstrong et al., 1998).
Based on the crystal structure of the S2 domain of GluR2, the interactions between the homologous amino acids in the S2 domains of GluR6 and GluR7 can be compared (Fig. 5). It turned out that amino acids in the S2 domain that differ between GluR6 and GluR7 are only interacting with amino acids that are identical in both receptors. Thus, there is no evidence of compensatory changes in interacting amino acids for those amino acids differing between GluR6 and GluR7. In other words, amino acids in the S2 domain differing between GluR6 and GluR7 appear to be functionally independent of each other. However, because the GluR6 and GluR7 structures presented in Figure 5 are models based on the GluR2 S1–S2 crystal structure, it has to be kept in mind that the actual structure of the domains in their natural environment could be different. Nevertheless, the data on the chimeras GluR7–GluR6L3, GluR7–GluR6L3C/FC, and GluR7–GluR6L3N as well as the fact that four mutations at distinct sites within the L3 domain of GluR7 were each able to turn GluR7 into a functional receptor, suggest that a different folding of the L3 domain of GluR6 and GluR7 might be responsible for the differences in maximal current amplitudes. The interaction of at least two determinants, located in separate halves of the L3 domain, is different in GluR7 from that in GluR6 and results in loss of efficient opening of the pore.
As mentioned above, the EC50 for glutamate of wild-type GluR7 expressed in HEK 293 cells was reported to be 6 mm (Schiffer et al., 1997). Whereas our data show that EC50 values for glutamate in GluR7 mutants or chimeras tend to be increased in comparison to EC50 values in GluR6, those EC50 values do not reach the millimolar range, not even for GluR7 point mutants. The high EC50for glutamate of wild-type GluR7 appears to be linked to the S1 region of the ligand-binding domain as shown by the chimeras and GluR7 mutants (Table 1).
The L3 domain of GluR7 turned out to be responsible for the different ratios of glutamate- to kainate-elicited maximal currents (measured after ConA treatment) that were seen for some GluR7 mutants compared with GluR6. All mutants that contained at least the C-terminal half of the L3 domain of GluR7 showed increased glutamate to kainate current ratios.
Heteromeric subunit assembly
When two different subunits are coexpressed, homomeric receptors normally will be assembled according to the rules of binomial distribution. Thus, when the two subunits are present in equal amounts, there is a 6.25% probability for each homomeric receptor to be formed, assuming a tetrameric receptor stoichiometry. Surprisingly, some subunit combinations gave currents that were considerably smaller than the respective contributions of the two homomeric receptor populations present in those mixtures (Tables 1, 4). These unexpectedly low values were not caused by a reduction in protein expression. This conclusion is based on the assumption that the expression levels of subunits are comparable. We think, however, that this assumption is justified, because we could show that the expression levels of individually expressed subunits used in the coexpression studies show comparable protein expression levels (Fig. 3, lanes 5, 10, 15, 24, 25). Furthermore, coexpressed subunits produce similar levels of protein (Fig. 3, lanes 36–39) compared with individually expressed subunits. This observation suggests nonstochastical behavior of the receptor subunits during assembly. This means they are not assembling according to the rules of binomial distribution. For some subunit combinations, however, no significant differences between total current amplitudes (Table 4) were found compared with the estimated homomeric receptor current contributions calculated from measured homomeric receptor current amplitudes (Table 1). It is formally possible that, in coexpression, formation of a heteromeric complex was inhibited so that only homomeric receptor currents were seen. If, however, only homomeric receptors had formed, the currents would have been expected to be more than the 100% contributed by GluR6 alone (Table 4). Expected currents for GluR6 + GluR7N–GluR6PC in this case were ∼250%, for GluR6 + GluR7NP–GluR6C ∼300%, for GluR6 + GluR6–GluR7FC ∼200%, and for GluR6 + GluR7–GluR6L3 ∼300%. Because observed currents were ∼100%, we conclude that heteromeric receptors must have been formed in each of these cases. These data again suggest that coexpression of GluR6 and GluR7 may lead to nonstochastical assembly of subunits, probably favoring heteromeric receptors.
Table 4.
Relative current amplitudes of chimeras between GluR6 and GluR7, and GluR7 mutants coexpressed with wild-type GluR6
| Clone | Measured total KA current [%] | Measured total Glu current [%] | n | Ratio GLU/KA |
|---|---|---|---|---|
| GluR6(Q) + H2O | 100.0 ± 9.7 | 100.0 ± 8.6 | 7 | 1.6 ± 0.04 |
| GluR6(Q) + GluR7(Q) | 1.9 ± 0.9 | 4.0 ± 2.0 | 8 | 2.2 ± 0.09 |
| GluR6 + GluR6N–GluR7PC | 6.5 ± 1.0 | 11.9 ± 1.3 | 3 | 1.9 ± 0.09 |
| GluR6 + GluR6NP–GluR7C | 1.3 ± 0.4 | 3.8 ± 1.6 | 3 | 3.7 ± 0.3 |
| GluR6 + GluR7N–GluR6PC | 102.5 ± 7.6 | 88.8 ± 9.0 | 3 | 0.5 ± 0.02 |
| GluR6 + GluR7NP–GluR6C | 125.4 ± 34.4 | 85.6 ± 26.1 | 4 | 0.7 ± 0.06 |
| GluR6 + GluR6–GluR7FC | 104.4 ± 10.2 | 106.2 ± 10.2 | 4 | 1.00 ± 0.01 |
| GluR6 + GluR7–GluR6FC | 3.0 ± 0.9 | 2.7 ± 0.8 | 3 | 1.2 ± 0.02 |
| GluR6 + GluR6–GluR7L3 | 44.7 ± 5.4 | 49.8 ± 5.9 | 3 | 1.10 ± 0.01 |
| GluR6 + GluR7–GluR6L3 | 79.2 ± 8.9 | 62.9 ± 8.0 | 4 | 0.8 ± 0.03 |
| GluR6 + GluR7–GluR6L3C/FC | 108.0 ± 22.0 | 98.0 ± 21.8 | 6 | 0.8 ± 0.01 |
| GluR6 + GluR7–GluR6L3N | 0.4 ± 0.1 | 0.8 ± 0.2 | 4 | 2.1 ± 0.1 |
| GluR6 + GluR7(K626E) | 12.1 ± 3.4 | 12.1 ± 4.3 | 3 | 0.9 ± 0.1 |
| GluR6 + GluR7(K758N) | 7.3 ± 2.4 | 14.0 ± 5.4 | 8 | 2.0 ± 0.1 |
| GluR6 + GluR7(N748S) | 1.0 ± 0.4 | 1.5 ± 0.5 | 3 | 1.5 ± 0.1 |
| GluR6 + GluR7(I705V) | 2.5 ± 0.4 | 2.4 ± 0.4 | 3 | 1.0 ± 0.1 |
| GluR6 + GluR7(I732M) | 0.8 ± 0.2 | 1.3 ± 0.2 | 3 | 1.5 ± 0.1 |
| GluR6 + GluR7(insR651a/K652R/P653Q) | 0.4 ± 0.2 | 0.6 ± 0.3 | 4 | 1.7 ± 0.1 |
Receptors expressed in Xenopus oocytes were probed with 300 μm glutamate and 300 μm kainate as agonists. All currents were measured after 8 min of pretreatment of oocytes with ConA to minimize desensitization. Relative currents were calculated by taking GluR6 currents of the same batch as 100%. Data are shown as means ± SEM. Top part of table, Coexpression of GluR6 with chimeras; bottom part of table, coexpression of GluR6 with GluR7 point mutants. Absolute currents for wild-type GluR6 were 8915 ± 964 nA (KA) and 10009 ± 860 nA (Glu).
Recently, it was shown that the kainate receptor subunits GluR5, GluR6, and GluR7 exhibit promiscuous coassembly after coexpression in HEK 293 cells (Cui and Mayer, 1999). Similar to our findings in oocytes, coassembly of GluR7(R) with GluR6(Q) was reported to markedly decrease the amplitude of agonist responses. This was interpreted as indication that coassembly with GluR7(R) downregulates the high levels of functional expression typical for homomeric GluR6(Q). It was speculated that, although high concentrations of kainate are required to activate GluR7 channel gating (Schiffer et al., 1997), lower concentrations would produce desensitization (Cui and Mayer, 1999). In such a scenario, activation of GluR6 subunits, even after treatment with ConA, could be decreased by strong, lectin-insensitive desensitization mediated by GluR7 when combined with GluR6 (Cui and Mayer, 1999).
This latter interpretation, however, is in conflict with our finding that all GluR6–GluR7 chimeras (Table 1) could be potentiated by treatment with ConA, and the same was true for four GluR7 mutants (GluR7(K626E), GluR7(insR651a/K652R/P653Q), GluR7(N748S), and GluR7(K758N)) (Table 2). For GluR7(K758N), for example, the potentiation factor for glutamate (calculated as the current after ConA treatment/current before ConA treatment) is ∼1400. It appears highly unlikely that all of these mutants, altered at different sites in the receptor, would have their desensitization properties rendered lectin-insensitive. As we have previously shown for GluR6, lectin-mediated inhibition of desensitization is a rather unspecific effect that cannot be abolished by simple point mutations (Everts et al., 1997). Therefore, we conclude that when GluR6 is coexpressed with GluR7 in Xenopusoocytes, the reduction of current amplitude is not caused by a rapid GluR7-mediated desensitization of the receptor complex.
Taken together, our data from the coexpression studies of chimeras and wild-type GluR6 suggest that the C-terminal half of the L3 domain plus the FC domain (the C-terminal part of the receptor downstream of L3) cause the reduction of current amplitude when GluR7 is coexpressed with GluR6. The C-terminal half of the L3 domain is critical for the effect but is not the only factor. A GluR7 construct with the C-terminal half of the L3 domain plus the far C-terminal domain of GluR6 (GluR7–GluR6L3C/FC) when coexpressed with wild-type GluR6 does not show any reduction of currents. The same tendency is seen for other chimeras. Therefore, the second half of the L3 domain together with the FC domain of GluR7, which by itself does not seem to have much influence, is responsible for the observed current reduction when GluR6 is coexpressed with GluR7.
This raises the question whether there is any possible physiological relevance for GluR7 having a reducing effect on GluR6. GluR6 and GluR7 are codistributed in some but not all tissues (Hollmann and Heinemann, 1994; Bischoff et al., 1997) in vivo, which is compatible with the possibility that the physiological function of GluR7 indeed could be the regulation of GluR6 currents.
Footnotes
This work was supported by German–Israel Foundation Grant SFB 406 (M.H., V.I.T.), and by a grant of the Minerva Foundation (V.I.T.). We thank Dr. Robert Wenthold (National Institutes of Health, National Institute on Deafness and Other Communication Disorders, Bethesda, MD) for the generous donation of the affinity-purified anti-GluR6 antiserum used in this study.
Correspondence should be addressed to Prof. Dr. Michael Hollmann, Department of Biochemistry I, Receptor Biochemistry, Ruhr University Bochum, Level 6, Room 170, D-44780 Bochum, Germany. E-mail:michael.hollmann@ruhr-uni-bochum.de.
REFERENCES
- 1.Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 2.Bettler B, Egebjerg J, Sharma G, Pecht G, Hermans-Borgmeyer I, Moll C, Stevens CF, Heinemann S. Cloning of a putative glutamate receptor: a low affinity kainate binding subunit. Neuron. 1992;8:257–265. doi: 10.1016/0896-6273(92)90292-l. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff S, Barhanin J, Bettler B, Mulle C, Heinemann S. Spatial distribution of kainate receptor subunit messenger RNA in the mouse basal ganglia and ventral mesencephalon. J Comp Neurol. 1997;379:541–562. doi: 10.1002/(sici)1096-9861(19970324)379:4<541::aid-cne6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Cui C, Mayer ML. Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J Neurosci. 1999;19:8281–8291. doi: 10.1523/JNEUROSCI.19-19-08281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 6.Everts I, Villmann C, Hollmann M. N-glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol Pharmacol. 1997;52:861–873. doi: 10.1124/mol.52.5.861. [DOI] [PubMed] [Google Scholar]
- 7.Hollmann M. Structure of ionotropic glutamate receptors. In: Jonas P, Monyer H, editors. Ionotropic glutamate receptors in the CNS. Springer; Berlin: 1999. pp. 3–98. [Google Scholar]
- 8.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 9.Hollmann M, Maron C, Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 11.Lomeli H, Wisden W, Kšhler M, Keinänen K, Sommer B, Seeburg PH. High-affinity kainate and domoate receptors in rat brain. FEBS Lett. 1992;307:139–143. doi: 10.1016/0014-5793(92)80753-4. [DOI] [PubMed] [Google Scholar]
- 12.Monaghan DT, Cotman CW. The NMDA receptor. In: Watkins JC, Collingridge GL, editors. Regional variations in NMDA receptor properties. IRL; Oxford: 1989. pp. 53–64. [Google Scholar]
- 13.Nutt SL, Hoo KH, Rampersad V, Deverill RM, Elliott CE, Fletcher EJ, Adams SL, Korczak B, Foldes RL, Kamboj RK. Molecular characterization of the human EAA5 (GluR7) receptor: a high-affinity kainate receptor with novel potential RNA editing sites. Receptors Channels. 1994;2:315–326. [PubMed] [Google Scholar]
- 14.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 15.Schiffer HH, Swanson GT, Heinemann SF. Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron. 1997;19:1141–1146. doi: 10.1016/s0896-6273(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 16.Swanson GT, Gereau RW, Green T, Heinemann SF. Identification of amino acid residues that control functional behavior in GluR5 and GluR6 kainate receptors. Neuron. 1997;19:913–926. doi: 10.1016/s0896-6273(00)80972-1. [DOI] [PubMed] [Google Scholar]
- 17.Towbin H, Stähelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villmann C, Bull L, Hollmann M. Kainate binding proteins possess functional ion channel domains. J Neurosci. 1997;17:7634–7643. doi: 10.1523/JNEUROSCI.17-20-07634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villmann C, Strutz N, Morth T, Hollmann M. Investigation by ion channel domain transplantation of rat glutamate receptor subunits, orphan receptors and a putative NMDA receptor subunit. Eur J Neurosci. 1999;11:1765–1778. doi: 10.1046/j.1460-9568.1999.00594.x. [DOI] [PubMed] [Google Scholar]
- 20.Wenthold RJ, Trumpy VA, Zhu WS, Petralia RS. Biochemical and assembly properties of GluR6 and KA2, 2 members of the kainate receptor family, determined with subunit specific antibodies. J Biol Chem. 1994;269:1332–1339. [PubMed] [Google Scholar]