

Abstract
To date, none of the available antipsychotic drugs are curative, all have significant side-effect potential, and a receptor-binding profile predictive of superior therapeutic ability has not been determined. It has become increasingly clear that schizophrenia does not result from the dysfunction of a single neurotransmitter system, but rather from an imbalance between several interacting systems. Targeting neuropeptide neuromodulator systems that concertedly regulate all affected neurotransmitter systems could be a promising novel therapeutic approach for schizophrenia. A considerable database is concordant with the hypothesis that antipsychotic drugs act, at least in part, by increasing the synthesis and release of the neuropeptide neurotensin (NT). In this report, we demonstrate that NT neurotransmission is critically involved in the behavioral effects of antipsychotic drugs in two models of antipsychotic drug activity: disrupted prepulse inhibition of the acoustic startle response (PPI) and the latent inhibition (LI) paradigm. Blockade of NT neurotransmission using the NT receptor antagonist 2-[[5-(2,6-dimethoxyphenyl)-1-(4-(N-(3-dimethylaminopropyl)-N-methylcarbamoyl)-2-isopropylphenyl)-1H- pyrazole-3-carbonyl]-amino]-adamantane-2-carboxylic acid, hydrochloride (SR 142948A) prevented the normal acquisition of LI and haloperidol-induced enhancement of LI. In addition, SR 142948A blocked the PPI-restoring effects of haloperidol and the atypical antipsychotic drug quetiapine in isolation-reared animals deficient in PPI. We also provide evidence of deficient NT neurotransmission as well as a left-shifted antipsychotic drug dose–response curve in isolation-reared rats. These novel findings, together with previous observations, suggest that neurotensin receptor agonists may represent a novel class of antipsychotic drugs.
Keywords: prepulse inhibition, latent inhibition, haloperidol, quetiapine, SR 142948A, isolation rearing
Schizophrenia is a devastating psychiatric disease with a worldwide prevalence of ∼1%. Antipsychotic drugs have been shown repeatedly to be effective in reducing some of the cardinal symptoms of schizophrenia, and yet none of the available antipsychotic drugs are curative, all have significant side-effect potential, and the underlying neurochemical actions responsible for their clinical efficacy remain poorly understood. It has become increasingly clear, however, that schizophrenia does not result from the dysfunction of a single neurotransmitter system, but rather from an imbalance between several interacting systems (Weinberger and Lipska, 1995; Bachus and Kleinman, 1996). Targeting of neuropeptide neuromodulator systems capable of concomitantly regulating all affected transmitter systems may therefore be a promising approach for the development of increasingly effective and side-effect-free antipsychotic drugs.
Neurotensin (NT) is a neuropeptide modulator implicated in the pathophysiology of schizophrenia that specifically modulates neurotransmitter systems hypothesized to be dysregulated in this disorder (O'Connor et al., 1992; Tanganelli et al., 1994; Fuxe et al., 1995; Jolas and Aghajanian, 1997). The behavioral and biochemical effects of centrally administered NT remarkably resemble those of systemically administered antipsychotic drugs, and antipsychotic drugs increase NT neurotransmission (for review, see Kinkead et al., 1999), leading to the hypothesis that NT functions as an endogenous antipsychotic (Nemeroff, 1980).
Determination of whether increased NT neurotransmission mediates the clinically relevant behavioral effects of antipsychotic drugs requires the use of animal models relevant to the clinical efficacy of antipsychotic drugs. There is increasing evidence that deficits in sensorimotor gating are part of the diverse symptomatology of schizophrenia, and that these deficits are restored by effective antipsychotic drug treatment (McGhie and Chapman, 1961; Venables, 1966;Baruch et al., 1988; Freedman et al., 1991; Lubow and Gewirtz, 1995;Kumari et al., 1999). Sensorimotor gating can be assessed in humans and laboratory animals by measuring similar parameters: acquisition of latent inhibition (LI) and the prepulse inhibition of the acoustic startle reflex (PPI) (Geyer and Braff, 1987; Hemsley, 1993). Both have been shown to be disrupted in acutely ill schizophrenic patients and may be restored by clinically effective antipsychotic drug treatment (Lubow et al., 1987; Baruch et al., 1988; Grillon et al., 1990; Gray et al., 1995; Braff et al., 1999; Kumari et al., 1999; Perry et al., 1999).
Both antipsychotic drug-induced enhancement of LI and disrupted PPI have been shown to have predictive validity for antipsychotic drug activity (Weiner and Feldon, 1997; Swerdlow and Geyer, 1998). Using these two paradigms, we examined the effects of blocking NT neurotransmission using the selective NT receptor antagonist 2-[[5-(2,6-dimethoxyphenyl)-1-(4-(N-(3-dimethylaminopropyl)-N-methylcarbamoyl)-2-isopropylphenyl)-1H-pyrazole-3-carbonyl]-amino]-adamantane-2-carboxylic acid, hydrochloride (SR 142948A) (Gully et al., 1997) on the LI-enhancing effects of antipsychotic drugs and on the antipsychotic drug-induced restoration of isolation rearing-induced deficits in PPI. To determine whether disrupted PPI in isolation-reared rats and the restoration of PPI by antipsychotic drugs is associated with deficits in the NT system, we examined the NT system in these two rearing groups at baseline and in response to antipsychotic drug administration.
MATERIALS AND METHODS
Drugs. The NT receptor antagonist SR 142948A was a generous gift of Sanofi Recherche (Toulouse, France). SR 142948A was suspended in several drops of Tween 20 and brought to volume with 0.9% NaCl. Haloperidol (Sigma, St. Louis, MO) and quetiapine (Zeneca Pharmaceutical, Wilmington, DE) were dissolved in 0.3% tartaric acid (drug vehicle). All drugs were administered in a fixed volume of 1.0 ml/kg body weight.
Latent inhibition. NNIH (an outbred strain of animals developed at the National Institute of Health) animals were obtained from the local breeding facility and used between the ages of 60 and 80 d (150–180 gm). NNIH rats were used for this experiment because of the superior response of this strain to antipsychotic drug-induced enhancement of LI. Same-sex animals were housed three per cage on a reversed 12 hr light/dark cycle (lights on at 10:00 P.M. and off at 10:00 A.M.) in an environmentally controlled animal facility with access to food ad libitum. All components of the LI paradigm were performed in the dark phase between 12:00 P.M. and 5:00 P.M. The Emory Institutional Animal Care and Use Committee approved all animal protocols.
The LI procedure used house light as the pre-exposed and conditioned stimulus (CS), footshock as the unconditioned stimulus (UCS), water spout licking as the quantitated behavior, and conditioned response suppression as the behavioral baseline and index of LI (Fig.1). Animals were water-restricted during all stages of the experiment, receiving 30 min of access to waterad libitum per 24 hr. Before being subjected to the LI paradigm, animals commenced 4 consecutive days of daily training and acclimation to the lick task in a nonilluminated behavioral box (MED Associates, Georgia, VT) with a task criterion of 400 photobeam interruptions of the lick circuit. For stimulus pre-exposure, animals were placed in behavioral test boxes and received either 10 or 25 exposures to the light (pre-exposed group) or were placed in the unlit test cage for the same amount of time (non-pre-exposed group). Conditioning was initiated 5 min after the stimulus pre-exposure component. All animals were presented with the house light followed immediately by a 1 sec, 1.0 mA scrambled footshock. Three additional light-shock pairings occurred at 5 min intervals. LI testing was performed 24 hr later with animals returned to the unlit test cage with the water bottle present. (Stimulus pre-exposure and conditioning were performed in the absence of the water bottles.) After the 90th lick, the house light (CS) was presented and extinguished only after the completion of 10 more licks.
Fig. 1.
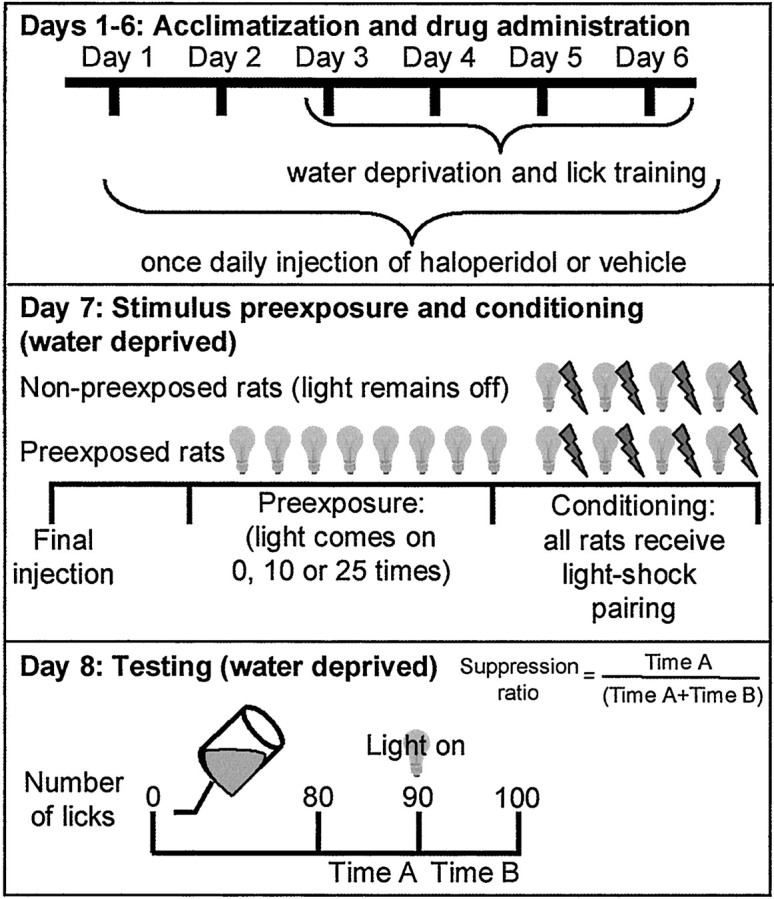
The latent inhibition paradigm.
The pre-CS interval from licks 80 to 90 (time A) and the post-CS interval between licks 91 and 100 (time B) were recorded to 0.01 sec. The LI effect, measured as the conditioned suppression of licking, is expressed as a suppression ratio [time A/(time A + time B)]. A suppression ratio approaching 0.0 is indicative of negligible LI, and a ratio of >0.5 is indicative of maximal LI.
Isolation rearing. Time-pregnant Long–Evans rats (Charles River Laboratories, Raleigh, NC) were housed individually on a reversed 12 hr light/dark cycle (lights on at 10:00 P.M. and off at 10:00 A.M.) in an environmentally controlled animal facility with access to food ad libitum. Three days after birth, male pups were removed, and the female pups were randomized to 10 per litter and allowed to mature with normal institutional care until weaning. At weaning (postnatal day 23), rats were randomly divided into two groups and housed either singly (isolation-reared animals) or in groups of three per cage (socially reared animals). To further enhance the difference in PPI between isolation-reared and socially reared animals, isolation-reared rats were allowed limited access to water (15 min twice each day) on days 2–4 after weaning, with water access returned on the afternoon of day 5. This stressor consistently enhances isolation rearing-induced PPI deficits in our laboratory (our unpublished observation). Animals weighed between 150 and 200 gm during PPI testing.
Prepulse inhibition of the acoustic startle reflex. PPI testing was completed in the dark phase between 11:00 A.M. and 5:00 P.M. PPI testing was performed in a San Diego Instruments (San Diego, CA) startle chamber. Startle amplitude was measured by converting the vibrations of a Plexiglas cylinder (resting on a platform) caused by whole-body response into analog signals using a piezoelectric unit attached to the platform. These signals were then digitized and stored in a personal computer. The testing session started with a 5 min acclimatization to the startle chamber in the presence of 65 dB background white noise. Testing consisted of nine 120 dB pulses alone and 18 pulses preceded (100 msec) by a prepulse of 4, 8, or 12 dB above background. Pulses were presented in a pseudorandom order with an average of 15 sec between pulses. Percent PPI for each rat at each prepulse intensity was calculated using the following formula: %PPI = 100 − (startle amplitude with prepulse × 100/startle amplitude with pulse alone).
NT radioimmunoassay. Rat brains were dissected based on the method of Glowinski and Iversen (1966). The brain regions dissected included the nucleus accumbens, the caudate–putamen, and the prefrontal cortex. NT tissue concentrations were measured as described previously (Bissette et al., 1984).
NT receptor autoradiography. Animals were deeply anesthetized with Euthanasia 5 solution (Schein, Port Washington, NY) and decapitated; brains were removed immediately, frozen on dry ice, stored at −70°C until they were sectioned in a cryostat at 20 μm thickness, mounted on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA), and again stored at −70°C. At the time of assay, slides were gradually brought to room temperature and desiccated. Sections were fixed for 2 min in 0.1% paraformaldehyde at 4°C and then preincubated for 15 min at room temperature in incubation buffer (50 mm Tris base, 10 mmMgCl2, 2 mm EGTA, 0.1% BSA, and 142 mg/l bacitracin, pH 7.4). Incubation with the radioligand (0.2 nm[125I]NT; DuPont NEN, Wilmington, DE) was performed in incubation buffer for 60 min at room temperature. Nonspecific binding was determined in the presence of 1.0 μm unlabeled NT (Bachem, Torrance, CA). The sections were washed at 4°C two times for 5 min in 50 mm Tris containing 0.1% BSA, rinsed twice in ice-cold H2O, and rapidly dried under a constant stream of cool air. Dried sections were then exposed to Kodak Biomax MR film (Eastman Kodak, Rochester, NY) for 3 d. Quantitative receptor autoradiography was performed by computerized densitometry (AIS Software; Imaging Research Inc., St. Catharines, Ontario, Canada). Optical density was calibrated with coexposed standards, revealing brain substance-like quench coefficients for iodinated isotopes (Amersham Pharmacia Biotech, Piscataway, NJ) and converted to nanocuries per milligram of protein.
NT mRNA in situ hybridization. Animals used forin situ hybridization analysis of NT mRNA expression were anesthetized using Euthanasia 5 solution (Schein) and transcardially perfused via the ascending aorta with ice-cold 0.9% saline (200 ml) followed by ice-cold 4% paraformaldehyde in 0.1m phosphate buffer (250 ml), pH 7.6. Brains were then removed and post-fixed in 4% paraformaldehyde for 24 hr at 4°C, followed by 20% sucrose for 48 hr at 4°C. After post-fixing, brains were rinsed in double-distilled H2O, dried, and stored at −70°C until use. Thirty-micrometer-thick sections were cut on a sliding microtome and collected in 24 well series. The level of the appearance of the anterior commisure was marked for future reference. Tissue sections were then stored in cryoprotectant solution (30% ethylene glycol and 20% glycerol in 25 mm phosphate buffer, pH 7.4) at −20°C. Before assaying, the tissue sections were rinsed in 50 mm phosphate buffer, pH 7.6, slide mounted, and stored at −20°C.
Template plasmid consisting of a 336 bpEcoRV-BglII fragment (nucleotides 626–961) of the rat NT/N gene in a BamHI-SmaI-digested pGEM4 (Promega, Madison, WI) was generously provided by P. Dobner (University of Massachusetts Medical Center, Worcester, MA).35S-labeled antisense riboprobes were generated using EcoRV-linearized plasmid, nucleotides,35S-uridine triphosphate, and T7 RNA polymerase (protocol adapted from the T7/T3 MAXIscript kit; Ambion, Austin, TX). Unincorporated nucleotides were removed from the reactions using Quick Spin Columns (Boehringer Mannheim, Indianapolis, IN). The35S-labeled riboprobes were then diluted to 1 × 105 cpm/μl in hybridization buffer (62.5% formamide, 12.5% dextran sulfate, 0.375m NaCl, 2.5% Denhardt's solution, 12.5 mm Tris, pH 8.0, and 1.25 mm EDTA, pH 8.0) and stored at −20°C until use. The protocol for in situ hybridization was adapted from the methods of Simmons et al. (1989). Briefly, the slide-mounted tissue underwent a proteinase K digestion followed by acetylation in acetic anhydride. The slides were then rinsed in 2× SSC buffer (NaCl–Na citrate) and quickly dehydrated in ascending concentrations of fresh ethanol. After drying at room temperature, 100 μl (1 × 106 cpm) of riboprobe mixture (riboprobe in hybridization solution with tRNA and DTT) was added to each slide. The slides were then covered with Parafilm and stored overnight at 60°C in covered trays. Tissues soaked with 50% formamide were placed in the bottom of the trays to prevent evaporation of the probe mixture. The next day, the slides were rinsed in 4× SSC before ribonuclease (RNase) digestion (1:500 dilution of 10.0 mg/ml RNase A) to remove nonspecifically bound riboprobe. The slides were then rinsed, gradually desalted, and incubated for 30 min at 60°C to decrease background signal. Slides were then quickly dehydrated in ethanol (containing salt and DTT) and exposed to Kodak Biomax MR film. Film autoradiographs were digitized and quantified using AIS Software (Imaging Research Inc.). A14C standard curve (Amersham Pharmacia Biotech) was included in each film cassette for use in standardizing quantification between films.
Statistical analyses. Data from the LI experiments were analyzed by two-way ANOVA (treatment × pre-exposure level) followed by a Student–Newman–Keuls multiple comparisons test. Data from verification of deficits in PPI in isolation-reared compared with socially reared animals were analyzed by two-way ANOVA (rearing × prepulse intensity) followed by Student–Newman–Keuls multiple comparisons test. Data from experiments examining the effects of antipsychotic drugs and the NT receptor antagonist SR 142948A on PPI in isolation-reared and socially reared rats were analyzed by three-way ANOVA (rearing × treatment × prepulse intensity). Because of the lack of interaction between treatment × prepulse intensity, rearing × prepulse intensity, or rearing × treatment × prepulse intensity, data were then collapsed across prepulse intensities and further analyzed by two-way ANOVA (rearing × treatment) followed by Student–Newman–Keuls multiple comparisons test. Baseline rearing differences in NT peptide concentrations, NT mRNA expression, and NT receptor binding were analyzed separately within each brain region by t test. The effects of quetiapine on NT mRNA expression were analyzed separately within each brain region by two-way ANOVA (rearing × treatment) followed by Student–Newman–Keuls multiple comparisons test.
RESULTS
Effects of the NT receptor antagonist on acquisition of latent inhibition
We first established a dose–response curve for the effects of the NT receptor antagonist SR 142948A on the acquisition of LI after a sufficient number of pre-exposures to the conditioned stimulus (25 pre-exposures). Pre-exposed and non-pre-exposed animals received 0, 0.1, 1.0, 10.0, or 100.0 μg/kg SR 142948A 1 hr before the pre-exposure and conditioning session. Two-way ANOVA (stimulus pre-exposure level × treatment) of suppression ratios revealed a significant pre-exposure effect (F(1,55) = 9; p < 0.005), treatment effect (F(3,55) = 3;p < 0.05), and no interaction between the two factors (F < 1). Post hoc analyses revealed that 25 pre-exposures to the conditioned stimulus induced LI in vehicle-treated animals (Fig. 2a). The LI effect of 25 pre-exposures was blocked by 0.1, 1.0, and 10.0 μg/kg SR 142948A but not by 100.0 μg/kg SR 142948A.
Fig. 2.

Effect of the NT receptor antagonist SR 142948A on the acquisition of LI. Data are expressed as suppression ratio (mean ± SEM). a, Dose–response curve for the effects SR 142948A on the acquisition of LI after 25 pre-exposures to the conditioned stimulus. *p < 0.05 compared with the 25 pre-exposure control group. +p < 0.05 compared with the 0 pre-exposure group at the same dose of NT receptor antagonist (n = 5 or 6). b, Effect of the NT receptor antagonist SR 142948A on haloperidol-induced enhancement of latent inhibition in pre-exposed (10 pre-exposures) rats. *p < 0.05 compared with all other treatment groups (n = 5 or 6 mixed male and female).
The effects of the NT receptor antagonist SR 142948A on the LI-enhancing effect of haloperidol were then examined. Adult rats were injected subchronically with haloperidol (0.3 mg/kg, i.p.) or vehicle for 7 consecutive days, beginning 6 d before stimulus pre-exposure–conditioning day. On stimulus pre-exposure–conditioning day, animals received a single injection of SR 142948A (30 μg/kg, i.p.) or vehicle 1 hr before the last haloperidol or vehicle injection. Haloperidol or vehicle was administered 45 min before stimulus pre-exposure. Two-way ANOVA (stimulus pre-exposure level × treatment) of suppression ratios revealed a significant pre-exposure effect (F(1,51) = 13;p < 0.001), treatment effect (F(3,51) = 4; p < 0.05), and interaction between the two factors (F(3,51) = 3; p < 0.05). Haloperidol significantly enhanced the subthreshold LI effect of 10 stimulus pre-exposures (Fig. 2b). Pretreatment with SR 142948A (30 μg/kg), which had no significant effect on LI by itself, significantly decreased the effect of haloperidol in this paradigm.
Effects of the NT receptor antagonist SR 142948A in the PPI paradigm
In each of the experiments using isolation rearing, the presence of PPI deficits in isolation-reared animals was confirmed 3 weeks after weaning. In all cases, there was a significant rearing effect (p < 0.001) and prepulse intensity effect (p < 0.001) but no interaction between the two factors. Figure 3 represents a typical set of data obtained from initial PPI testing.
Fig. 3.

Isolation rearing-induced disruption of PPI. Data are expressed as percent PPI (mean ± SEM) and calculated separately for each prepulse intensity (n = 40–43 females). Data shown are the verification of phenotype in animals subsequently used to determine the effects of haloperidol and the NT receptor antagonist on PPI; these effects are shown in Figure5a.
We first established a dose–response curve for the effects of quetiapine on PPI. One week after the establishment of the desired phenotype, isolation-reared and socially reared animals received either a single injection of quetiapine (0.5, 2.0, 4.0, or 5.0 mg/kg) or vehicle 30 min before PPI testing. There was a significant rearing effect (F(1,1131) = 10;p < 0.001), dose effect (F(4,1131) = 21; p < 0.001), and interaction between the two factors (F(4,1131) = 3; p < 0.05). Quetiapine (2.0, 4.0, and 5.0 mg/kg, but not 0.5 mg/kg) restored PPI deficits in isolation-reared animals compared with socially reared animals (Fig. 4). In addition, quetiapine (4.0 and 5.0 mg/kg) significantly enhanced PPI in socially reared animals.
Fig. 4.

Dose–response curve for the effect of quetiapine (0, 0.5, 2.0, 4.0, or 5.0 mg/kg) on PPI in both isolation-reared and socially reared animals (n = 6 or 7 females). *p < 0.05 compared with socially reared animals within a treatment group. +p < 0.05 compared with socially reared control animals.
Three separate but similar experiments were then conducted to investigate the effects of coadministration of the NT receptor antagonist (100.0 μg/kg) with haloperidol (0.1 mg/kg) or quetiapine (2.0 or 5.0 mg/kg) in this paradigm. One hour before PPI testing, animals received a single intraperitoneal injection of the NT receptor antagonist (100.0 μg/kg) or vehicle. Thirty minutes before PPI testing, animals received a single injection of the antipsychotic drug (haloperidol intraperitoneally or quetiapine subcutaneously) or vehicle. Raw startle amplitudes from all PPI studies are presented in Table 1.
Table 1.
Startle values for pulse alone and prepulse–pulse trials in isolation-reared and socially reared animals for PPI experiments shown in Figures 3-5
| Treatment | Socially reared animals | Isolation-reared animals | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pulse alone | 4 dB | 8 dB | 12 dB | Pulse alone | 4 dB | 8 dB | 12 dB | ||
| Experiment 1 | None | 50.7 (1.8) | 32.3 (1.5) | 27.5 (1.3) | 22.6 (1.1) | 41.5 (1.5) | 30.6 (1.1) | 27.7 (1.1) | 24.5 (1.0) |
| Experiment 2 | Control | 49.0 (4.0) | 27.1 (2.46) | 24.0 (2.1) | 23.9 (2.8) | 50.1 (3.8) | 35.2 (3.6) | 30.3 (2.5) | 26.2 (30) |
| Haloperidol (0.1 mg/kg) | 53.4 (3.3) | 31.9 (2.2) | 24.1 (1.5) | 21.1 (1.5) | 57.5 (6.6) | 28.1 (2.4) | 22.65 (2.5) | 18.1 (1.3) | |
| NTR antagonist (100 μg/kg) | 59.3 (4.0) | 29.8 (2.5) | 20.7 (1.4) | 26.5 (3.3) | 35.61-160 (2.2) | 21.2 (1.3) | 21.4 (1.4) | 18.8 (1.5) | |
| Haloperidol + NTR antagonist | 45.8 (3.5) | 25.4 (2.3) | 19.1 (1.4) | 16.43 (1.9) | 43.5 (3.7) | 27.2 (1.5) | 25.9 (2.4) | 23.8 (2.8) | |
| Experiment 3 | Quetiapine (0 mg/kg) | 38.7 (3.5) | 23.1 (1.7) | 24.8 (2.1) | 16.8 (1.2) | 35.2 (4.3) | 26.5 (2.2) | 21.9 (2.6) | 21.6 (2.5) |
| Quetiapine (0.5 mg/kg) | 36.0 (3.6) | 19.2 (2.5) | 13.3 (1.4) | 13.9 (1.4) | 41.5 (3.4) | 25.9 (2.9) | 23.6 (2.5) | 21.1 (2.1) | |
| Quetiapine (2.0 mg/kg) | 36.3 (3.5) | 18.5 (1.2) | 15.7 (1.5) | 13.9 (1.3) | 30.2 (3.6) | 19.8 (2.1) | 15.8 (1.7) | 14.7 (1.7) | |
| Quetiapine (4.0 mg/kg) | 32.4 (3.4) | 16.3 (2.1) | 15.7 (2.1) | 11.0 (1.5) | 41.8 (3.5) | 19.7 (1.7) | 19.2 (2.3) | 17.8 (1.7) | |
| Quetiapine (5.0 mg/kg) | 45.3 (4.1) | 20.1 (2.3) | 13.9 (1.3) | 14.7 (1.8) | 40.6 (4.4) | 19.2 (2.1) | 13.3 (1.3) | 13.9 (1.6) | |
| Experiment 4 | Control | 81.1 (13.3) | 39.5 (3.9) | 34.7 (2.4) | 29.4 (2.5) | 50.3 (3.8) | 43.7 (4.0) | 33.2 (2.9) | 33.5 (3.8) |
| Quetiapine (2.0 mg/kg) | 35.5* (3.8) | 21.7 (2.2) | 18.7 (2.0) | 18.2 (1.8) | 51.6 (4.6) | 28.1 (3.0) | 22.4 (2.0) | 23.0 (2.1) | |
| NTR antagonist (100 μg/kg) | 52.0* (4.5) | 35.6 (3.4) | 28.3 (2.0) | 22.5 (2.5) | 52.4 (3.6) | 41.0 (4.2) | 37.1 (4.2) | 28.7 (2.8) | |
| Quetiapine + NTR antagonist | 54.6* (5.9) | 30.0 (3.7) | 24.3 (2.9) | 20.8 (2.6) | 55.2 (12.1) | 39.5 (5.9) | 26.8 (3.8) | 27.0 (4.8) | |
| Experiment 5 | Control | 56.0 (4.5) | 32.9 (2.3) | 30.6 (2.5) | 25.4 (2.1) | 51.9 (4.5) | 33.6 (2.9) | 31.0 (2.9) | 26.2 (2.5) |
| Quetiapine (5.0 mg/kg) | 45.3 (6.2) | 20.1 (2.2) | 14.5 (1.6) | 13.2 (1.4) | 40.6 (4.1) | 19.2 (2.1) | 13.4 (1.4) | 13.9 (1.6) | |
| NTR antagonist (100 μg/kg) | 76.6 (14.4) | 38.3 (4.8) | 34.1 (4.3) | 26.5 (3.3) | 47.4 (4.3) | 31.3 (3.4) | 25.5 (2.7) | 23.9 (2.8) | |
| Quetiapine + NTR antagonist | 56.7 (7.6) | 26.5 (2.7) | 21.5 (1.8) | 17.3 (1.8) | 32.6 (3.9) | 20.6 (2.5) | 17.9 (2.1) | 13.9 (1.5) | |
Data are arbitrary units expressed as mean ± SEM. All animals in the PPI studies were female.
p < 0.05 compared with pulse alone amplitude in socially reared, vehicle-treated animals.
F1-160: p < 0.05 compared with pulse alone amplitude in isolation-reared, vehicle-treated animals. NTR, NT receptor.
The effects of the NT receptor antagonist (100.0 μg/kg) on haloperidol (0.1 mg/kg)-induced restoration of PPI in isolation-reared animals were first examined (Fig.5a). There was a significant rearing effect (F(1,1460) = 41;p < 0.001), treatment effect (F(3,1460) = 5; p < 0.001), and interaction between the two factors (F(3,1460) = 7; p < 0.001). Although none of the treatments had any effect on PPI in socially reared animals, haloperidol significantly enhanced PPI in isolation-reared animals, restoring it to levels equivalent to socially reared animals. The NT receptor antagonist alone had no effect on PPI but blocked haloperidol restoration of PPI in isolation-reared animals.
Fig. 5.

Effects of the NT receptor antagonist SR 142948A on restoration of PPI by haloperidol or quetiapine in isolation-reared animals. a–c, Data shown are percent PPI (data from three prepulse intensity levels combined) expressed as mean ± SEM. *p < 0.05 compared with socially reared animals within a treatment group. **p < 0.05 compared with control, NT receptor antagonist, and NT receptor antagonist plus haloperidol isolation-reared animals. +p < 0.05 compared with socially reared control animals. a, Effect of SR 142948A (0.1 mg/kg) on haloperidol (0.1 mg/kg)-induced restoration of PPI in isolation-reared rats (n = 10 or 11 females). b, Effect of SR 142948A on quetiapine (2.0 mg/kg)-induced restoration of PPI in isolation-reared animals (n = 8 or 9 females). c, Effect of SR 142948A on quetiapine (5.0 mg/kg)-induced restoration of PPI in isolation-reared animals (n = 7 or 8 females).
Similar to haloperidol, quetiapine (2.0 mg/kg) significantly enhanced PPI in isolation-reared animals, restoring it to levels equivalent to socially reared animals, although not effecting PPI in socially reared animals (Fig. 5b). The NT receptor antagonist (100.0 μg/kg) alone had no significant effect on PPI in isolation-reared or socially reared animals but blocked the quetiapine (2.0 mg/kg)-induced restoration of PPI in isolation-reared animals. Overall there was a significant rearing effect (F(1,1073)= 26; p < 0.001), treatment effect (F(3,1073) = 7; p < 0.001), and interaction between the two factors (F(3,1073) = 9; p < 0.001).
In contrast to 2.0 mg/kg quetiapine, 5.0 mg/kg quetiapine significantly enhanced PPI in socially reared animals, and the NT receptor antagonist did not block this effect (Fig. 5c). In isolation-reared animals, 5.0 mg/kg quetiapine enhanced PPI to levels significantly higher than PPI levels in socially reared control animals. The NT receptor antagonist partially blocked this effect. Two-way ANOVA revealed a significant rearing effect (F(1,1026) = 8; p < 0.005), treatment effect (F(3,1026) = 16; p < 0.001), and interaction between the two factors (F(3,1026) = 3;p < 0.05).
In contrast to the LI paradigm, blockade of NT neurotransmission after systemic administration of SR 142948A had no effect on the basal expression of PPI. (A total of 0.01–1000 μg/kg administered between 30 min and 12 hr before PPI testing had no effect; data not shown.)
Differences in the NT system in isolation-reared versus socially reared animals
To assess basal differences in the NT system, we quantified NT peptide tissue concentrations by radioimmunoassay, NT receptor binding by 125I-NT autoradiography, and NT mRNA expression by in situ hybridization. At baseline, NT peptide concentrations did not significantly differ between the rearing groups in any brain region examined (data not shown). NT mRNA expression and NT receptor binding were quantified in the nucleus accumbens (shell and core) and the caudate–putamen. NT receptor binding was significantly higher (21%) in the nucleus accumbens shell of isolation-reared animals compared with socially reared animals but did not differ in the core of the nucleus accumbens or the caudate–putamen (Fig. 6a; data for caudate–putamen not shown). In addition, NT mRNA expression in the shell of the nucleus accumbens was significantly lower (45%) in isolation-reared than in socially reared animals (Fig.6a,b). NT mRNA expression in the nucleus accumbens core was not significantly different between the rearing groups.
Fig. 6.
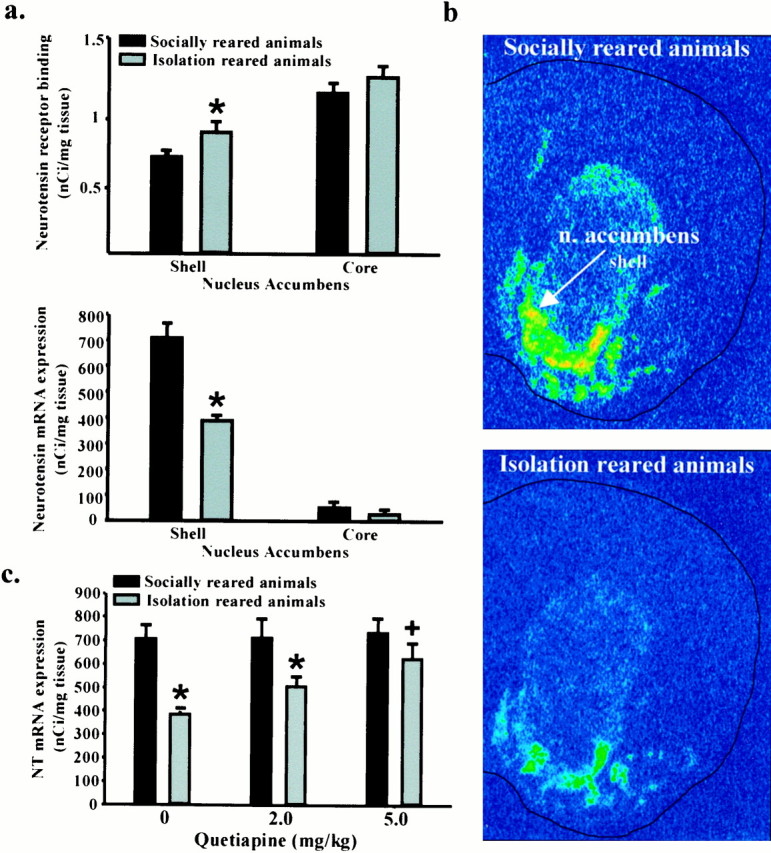
NT neurotransmission is altered in isolation-reared animals. a, NT receptor binding and NT mRNA expression in the shell of the nucleus accumbens in isolation-reared and socially reared animals. Deficits in PPI in isolation-reared animals were confirmed immediately before killing the animals. Data are expressed as mean ± SEM NT receptor binding (in nanocuries per milligram of tissue) and mean ± SEM NT mRNA expression (in nanocuries per milligram of tissue). *p < 0.05 compared with socially reared rats within a brain region (n = 5–7 females).b, NT mRNA in situ hybridization in the nucleus accumbens of socially reared and isolation-reared animals.c, Effect of quetiapine on NT mRNA expression in the nucleus accumbens shell. Data are expressed as mean ± SEM NT mRNA expression (in nanocuries per milligram of tissue). *p < 0.05 compared with socially reared animals within a treatment group. +p < 0.05 compared with vehicle treated isolation-reared animals (n = 5 or 6 females).
We subsequently examined NT mRNA expression in isolation-reared and socially reared animals 4 hr after a single injection of vehicle or quetiapine (2.0 or 5.0 mg/kg). Increasing doses of quetiapine significantly increased NT mRNA expression in the shell subdivision of the nucleus accumbens in isolation-reared animals but had no effect on NT mRNA expression in socially reared animals or in the nucleus accumbens core (Fig. 6c).
DISCUSSION
In this report, we demonstrate that disruption of NT neurotransmission blocks the behavioral effects of antipsychotic drugs in two distinct tests of sensorimotor gating, PPI and LI. LI is defined as a reduction in associative learning (i.e., the association of the pre-exposed conditioned stimulus with an UCS) because of repeated inconsequential exposure (pre-exposure) to a “to be conditioned stimulus” (Lubow and Moore, 1959). In laboratory animals, all classes of antipsychotic drugs, but not other clinically used psychoactive drugs, enhance the LI effect of a subthreshold number of pre-exposures (Christison et al., 1988; Dunn et al., 1993; Weiner et al., 1996). PPI is defined as a decrease in the startle reflex induced by a strong acoustic stimulus when preceded by a weak prepulse (Swerdlow et al., 1994). Isolation rearing is a nonpharmacological means of disrupting PPI (Geyer et al., 1993). In contrast, isolation rearing does not disrupt LI (Wilkinson et al., 1994). Typical and atypical antipsychotic drugs restore isolation rearing-induced deficits in PPI (Geyer et al., 1993; Varty and Higgins, 1995; Bakshi et al., 1998). Because it is independent of primary pharmacological manipulations, isolation rearing-induced disruption of PPI may therefore represent a superior animal model for investigating the neural circuits involved in antipsychotic activity. To date, all tested antipsychotic drugs have been shown to restore isolation rearing-induced disruptions in PPI (Swerdlow and Geyer, 1998). These results are the first demonstration that an NT receptor antagonist blocks potentially clinically relevant behavioral effects of antipsychotic drugs and strongly suggest that intact NT neurotransmission is necessary for the effects of antipsychotic drugs in both of these paradigms.
The NT receptor antagonist blocked the effects of haloperidol, a typical antipsychotic drug, and quetiapine, an atypical antipsychotic drug, suggesting that increased NT neurotransmission may be a common component involved in the behavioral effects of all clinically effective antipsychotic drugs. Haloperidol and quetiapine have very different receptor-binding profiles. Haloperidol has high affinity for the D2 dopamine (DA) receptor and the ς receptor and relatively lower affinity for serotonergic receptors. Quetiapine, however, has lower affinity for the DA D2 receptor than serotonergic or histaminergic receptors (Wirshing, 1998). A direct interference of SR 142948A with haloperidol or quetiapine at the receptor level can be ruled out, because SR 142948A does not bind to any receptor targeted by haloperidol or quetiapine (Gully et al., 1997).
Despite these pharmacological differences, both haloperidol and quetiapine specifically affect the NT system and restore isolation rearing-induced PPI deficits in an NT-dependent manner. SR 142948A therefore likely blocks the behavioral effects of antipsychotic drugs by blocking the neurobiological consequences of antipsychotic drug-stimulated NT release. In addition, the NT receptor antagonist alone did not affect PPI, in contrast to other PPI-disrupting compounds such as DA agonists and NMDA receptor antagonists, which may also interfere with the effects of antipsychotic drugs (Swerdlow and Geyer, 1998).
The U-shaped dose–response curve for SR 142948A in the LI paradigm is similar to the bimodal dose–response curves previously reported with SR 142948A and seems to characterize responses to NT receptor activation as well as antagonism (Gully et al., 1997). The bimodal effects of NT and SR 142948A may stem from the opposing effects of presynaptic versus postsynaptic NT receptors on DA neurotransmission. For example, at low doses, local in vivo application of NT decreases DA release and increases GABA release in a TTX-sensitive manner (O'Connor et al., 1992). Coadministration of the GABAA antagonist bicuculine prevented an NT-induced decrease in DA release, suggesting that this effect is mediated by NT receptors located on GABAergic neurons. At higher doses, intra-accumbens NT increases DA release in the nucleus accumbens (Chapman et al., 1992; Ferraro et al., 1997). There is some anatomical evidence for NT receptors located presynaptically on DA terminals, especially in the nucleus accumbens core (Dilts and Kalivas, 1989), indicating that the increases in DA release could be because of the effects of NT at a presynaptically located NT receptor with a lower affinity for NT than the postsynaptic receptor. There is also pharmacological evidence for distinct presynaptic versus postsynaptic NT receptor subtypes within the striatum with differing affinities for SR 142948A (for review, see Le et al., 1996; Rostène et al., 1997). In addition, SR 142948A has been shown to bind with equal affinity to both the NT1 and NT2 receptors. The LI-antagonizing effect of SR 142948A, however, is most likely mediated by its action at the high-affinity NT1 NT receptor. Similarly, studies with SR 48692, an NT receptor antagonist with relative selectivity for NT1 (Gully et al., 1993), showed a dose-dependent disruption of LI (Lambert et al., 1995), suggesting that NT2 activity is less crucial for LI expression. Effects on NT2 have to be considered, however, because SR 142948A has high affinity for this receptor and NT2 mRNA is present in non-DAergic cells in the midbrain as well as in accumbal neurons (Walker et al., 1998).
In humans, explicitly antipsychotic effects are only observed in schizophrenic patients but not in normal controls, suggesting that the neurochemical effects of antipsychotic drugs may be different in disrupted versus intact systems. Isolation-reared animals have been shown to have elevated basal DA levels, decreased serotonin metabolite 5-hydroxyindole-3-acetic acid, and increased release of DA in the nucleus accumbens in response to amphetamine administration compared with socially reared animals (Hall et al., 1998). The establishment of a dose–response curve for the effects of quetiapine on PPI in both rearing groups showed that, although lower doses (2.0 mg/kg) of quetiapine specifically affected isolation-reared animals, higher doses (4.0 and 5.0 mg/kg) had PPI-enhancing effects in both groups of animals. The NT receptor antagonist partially blocked the effects of quetiapine (5.0 mg/kg) in isolation but not in socially reared animals. NT neurotransmission appears, therefore, to be preferentially involved in antipsychotic drug-induced restoration of deficits in PPI but not in the enhancement of PPI above levels seen in socially reared control animals, and perhaps by extension, the therapeutically relevant effects of antipsychotic drugs in a disrupted system.
The hypothesized deficit in gating or internal screening of sensory input in schizophrenic patients is viewed as leading to an involuntary flooding of unfiltered sensory data, likely contributing to the cognitive fragmentation and thought disorder characteristic of this disease (McGhie and Chapman, 1961; Venables, 1966; Freedman et al., 1991). Similar to the absence of LI seen in schizophrenic patients, the NT receptor antagonist prevented the normal acquisition of LI, suggesting that intact NT neurotransmission may be necessary for some aspects of normal sensorimotor gating. This is also supported by our findings in isolation-reared animals. The environmentally induced deficit in sensorimotor gating in isolation-reared animals was paralleled by an apparent decrease in NT neurotransmission in the nucleus accumbens shell, a brain region thought to be critical for the antipsychotic effects of antipsychotic drugs. Conversely, antipsychotic drugs likely enhance sensorimotor gating by increasing NT neurotransmission. This is supported not only by the fact that antipsychotic drug-induced restoration of PPI and enhancement of LI are prevented by pretreatment with the NT receptor antagonist but also by the fact that the NT system of isolation-reared rats was found to be hyper-responsive to antipsychotic drug administration. It has been demonstrated previously that higher doses of antipsychotic drugs (both typical and atypical) increase NT neurotransmission in the nucleus accumbens shell of socially reared animals (Kinkead and Nemeroff, 1994;Huang and Hanson, 1997; Radke et al., 1998; Kinkead et al., 2000). It appears therefore, that the dose–response curve for the effects of antipsychotic drugs on NT mRNA expression in isolation-reared animals is shifted to the left.
These findings indicating an involvement of NT in sensorimotor gating and in the mechanism of action of antipsychotic drugs are supported by clinical studies. Subgroups of schizophrenic patients have repeatedly been shown to have subnormal NT CSF concentrations that normalize after effective antipsychotic drug treatment (Widerlöv et al., 1982;Lindström et al., 1988; Nemeroff et al., 1989; Garver et al., 1991; Breslin et al., 1994; Sharma et al., 1997). It is also evident that a subgroup of schizophrenic patients, not yet clearly defined, exhibit deficits in sensorimotor gating as assessed by LI and PPI, and that these deficits are restored by antipsychotic drug treatment (Baruch et al., 1988; Grillon et al., 1992; Lubow and Gewirtz, 1995;Braff et al., 1999; Kumari et al., 1999; Perry et al., 1999). Our data are consistent with the possibility that deficits in NT neurotransmission may be causally linked to deficits in sensorimotor gating and that normalization of NT neurotransmission after antipsychotic drug treatment may lead to restoration of these deficits. The fact that NT neurotransmission mediates the restoration or enhancement of sensory stimulus filtering by antipsychotic drugs supports the development of NT receptor agonists as a novel class of antipsychotic drugs specifically targeting these deficits.
Footnotes
This work was supported by National Institutes of Health Research Grant MH-39415. E.B.B. was supported by a stipend from the Austrian Academy of Sciences.
E.B.B. and B.K. contributed equally to this work.
Correspondence should be addressed to Charles B. Nemeroff, Laboratory of Neuropsychopharmacology, Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine, Suite 4000 WMRB, 1639 Pierce Drive, Atlanta, GA 30322. E-mail: cnemero@emory.edu.
REFERENCES
- 1.Bachus SE, Kleinman JE. The neuropathology of schizophrenia. J Clin Psychiatry. 1996;[Suppl 11] 57:72–83. [PubMed] [Google Scholar]
- 2.Bakshi VP, Swerdlow NR, Braff DL, Geyer MA. Reversal of isolation rearing-induced deficits in prepulse inhibition by Seroquel and olanzapine. Biol Psychiatry. 1998;43:436–445. doi: 10.1016/s0006-3223(97)00246-1. [DOI] [PubMed] [Google Scholar]
- 3.Baruch I, Hemsley DR, Gray JA. Differential performance of acute and chronic schizophrenics in a latent inhibition task. J Nerv Ment Dis. 1988;176:598–606. doi: 10.1097/00005053-198810000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Bissette G, Richardson C, Kizer JS, Nemeroff CB. Ontogeny of brain neurotensin in the rat: a radioimmunoassay study. J Neurochem. 1984;43:283–287. doi: 10.1111/j.1471-4159.1984.tb06711.x. [DOI] [PubMed] [Google Scholar]
- 5.Braff DL, Swerdlow NR, Geyer MA. Symptom correlates of prepulse inhibition deficits in male schizophrenic patients. Am J Psychiatry. 1999;156:596–602. doi: 10.1176/ajp.156.4.596. [DOI] [PubMed] [Google Scholar]
- 6.Breslin NA, Suddath RL, Bissette G, Nemeroff CB, Lowrimore P, Weinberger DR. CSF concentrations of neurotensin in schizophrenia: an investigation of clinical and biochemical correlates. Schizophr Res. 1994;12:35–41. doi: 10.1016/0920-9964(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 7.Chapman MA, See RE, Bissette G. Neurotensin increases extracellular striatal dopamine levels in vivo. Neuropeptides. 1992;22:175–183. doi: 10.1016/0143-4179(92)90160-x. [DOI] [PubMed] [Google Scholar]
- 8.Christison GW, Atwater GE, Dunn LA, Kilts CD. Haloperidol enhancement of latent inhibition: relation to therapeutic action? Biol Psychiatry. 1988;23:746–749. doi: 10.1016/0006-3223(88)90060-1. [DOI] [PubMed] [Google Scholar]
- 9.Dilts RP, Kalivas PW. Autoradiographic localization of mu-opioid and neurotensin receptors within the mesolimbic dopamine system. Brain Res. 1989;488:311–327. doi: 10.1016/0006-8993(89)90723-3. [DOI] [PubMed] [Google Scholar]
- 10.Dunn LA, Atwater GE, Kilts CD. Effects of antipsychotic drugs on latent inhibition: sensitivity and specificity of an animal behavioral model of clinical drug action. Psychopharmacology (Berl) 1993;112:315–323. doi: 10.1007/BF02244927. [DOI] [PubMed] [Google Scholar]
- 11.Ferraro L, O'Connor WT, Antonelli T, Fuxe K, Tanganelli S. Differential effects of intrastriatal neurotensin(1–13) and neurotensin(8–13) on striatal dopamine and pallidal GABA release: a dual-probe microdialysis study in the awake rat. Eur J Neurosci. 1997;9:1838–1846. doi: 10.1111/j.1460-9568.1997.tb00750.x. [DOI] [PubMed] [Google Scholar]
- 12.Freedman R, Waldo M, Bickford-Weimer P, Nagamoto H. Elementary neuronal dysfunctions in schizophrenia. Schizophr Res. 1991;4:233–243. doi: 10.1016/0920-9964(91)90035-p. [DOI] [PubMed] [Google Scholar]
- 13.Fuxe K, Li XM, Tanganelli S, Hedlund P, O'Connor WT, Ferraro L, Ungerstedt U, Agnati LF. Receptor-receptor interactions and their relevance for receptor diversity: focus on neuropeptide/dopamine interactions. Ann NY Acad Sci. 1995;757:365–376. doi: 10.1111/j.1749-6632.1995.tb17495.x. [DOI] [PubMed] [Google Scholar]
- 14.Garver DL, Bissette G, Yao JK, Nemeroff CB. Relation of CSF neurotensin concentrations to symptoms and drug response of psychotic patients. Am J Psychiatry. 1991;148:484–488. doi: 10.1176/ajp.148.4.484. [DOI] [PubMed] [Google Scholar]
- 15.Geyer MA, Braff DL. Startle habituation and sensorimotor gating in schizophrenia and related animal models. Schizophr Bull. 1987;13:643–668. doi: 10.1093/schbul/13.4.643. [DOI] [PubMed] [Google Scholar]
- 16.Geyer MA, Wilkinson LS, Humby T, Robbins TW. Isolation rearing of rats produces a deficit in prepulse inhibition of acoustic startle similar to that in schizophrenia. Biol Psychiatry. 1993;34:361–372. doi: 10.1016/0006-3223(93)90180-l. [DOI] [PubMed] [Google Scholar]
- 17.Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. J Neurochem. 1966;13:665–669. doi: 10.1111/j.1471-4159.1966.tb09874.x. [DOI] [PubMed] [Google Scholar]
- 18.Gray NS, Pilowsky LS, Gray JA, Kerwin RW. Latent inhibition in drug naive schizophrenics: relationship to duration of illness and dopamine D2 binding using SPET. Schizophr Res. 1995;17:95–107. doi: 10.1016/0920-9964(95)00034-j. [DOI] [PubMed] [Google Scholar]
- 19.Grillon C, Courchesne E, Ameli R, Geyer MA, Braff DL. Increased distractability in schizophrenic patients. Arch Gen Psychiatry. 1990;47:171–179. doi: 10.1001/archpsyc.1990.01810140071010. [DOI] [PubMed] [Google Scholar]
- 20.Grillon C, Ameli R, Charney DS, Krystal J, Braff D. Startle gating deficits occur across prepulse intensities in schizophrenic patients. Biol Psychiatry. 1992;32:939–943. doi: 10.1016/0006-3223(92)90183-z. [DOI] [PubMed] [Google Scholar]
- 21.Gully D, Canton M, Boigegrain R, Jeanjean F, Molimard JC, Poncelet M, Gueudet C, Heaulme M, Leyris R, Brouard A, Peleprat D, Labbe-Jullie C, Mazella J, Soubrie P, Maffrand J-P, Rostene W, Kitabgi P, Le Fur G. Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc Natl Acad Sci USA. 1993;90:65–69. doi: 10.1073/pnas.90.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gully D, Labeeuw B, Boigegrain R, Oury-Donat F, Bachy A, Poncelet M, Steinberg R, Suaud-Chagny MF, Santucci V, Vita N, Pecceu F, Labbé-Jullié C, Kitabgi P, Soubrié P, Le Fur G, Maffrand JP. Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J Pharmacol Exp Ther. 1997;280:802–812. [PubMed] [Google Scholar]
- 23.Hall FS, Wilkinson LS, Humby T, Inglis W, Kendall DA, Marsden CA, Robbins TW. Isolation rearing in rats: pre- and postsynaptic changes in striatal dopaminergic systems. Pharmacol Biochem Behav. 1998;59:859–872. doi: 10.1016/s0091-3057(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 24.Hemsley DR. A simple (or simplistic?) cognitive model for schizophrenia. Behav Res Ther. 1993;31:633–645. doi: 10.1016/0005-7967(93)90116-c. [DOI] [PubMed] [Google Scholar]
- 25.Huang W, Hanson GR. Differential effect of haloperidol on release of neurotensin in extrapyramidal and limbic systems. Eur J Pharmacol. 1997;332:15–21. doi: 10.1016/s0014-2999(97)01032-7. [DOI] [PubMed] [Google Scholar]
- 26.Jolas T, Aghajanian GK. Neurotensin and the serotonergic system. Prog Neurobiol. 1997;52:455–468. doi: 10.1016/s0301-0082(97)00025-7. [DOI] [PubMed] [Google Scholar]
- 27.Kinkead B, Nemeroff CB. The effects of typical and atypical antipsychotic drugs on neurotensin-containing neurons in the central nervous system. J Clin Psychiatry. 1994;55:30–32. [PubMed] [Google Scholar]
- 28.Kinkead B, Binder EB, Nemeroff CB. Does neurotensin mediate the effects of antipsychotic drugs? Biol Psychiatry. 1999;46:340–351. doi: 10.1016/s0006-3223(99)00070-0. [DOI] [PubMed] [Google Scholar]
- 29.Kinkead B, Shahid S, Owens MJ, Nemeroff CB. Effects of acute and subchronic administration of typical and atypical antipsychotic drugs on the neurotensin system of the rat brain. J Pharmacol Exp Ther. 2000;295:67–73. [PubMed] [Google Scholar]
- 30.Kumari V, Soni W, Sharma T. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999;156:1046–1051. doi: 10.1176/ajp.156.7.1046. [DOI] [PubMed] [Google Scholar]
- 31.Lambert PD, Gross R, Nemeroff CB, Kilts CD. Anatomy and mechanisms of neurotensin-dopamine interactions in the central nervous system. Ann NY Acad Sci. 1995;757:377–389. doi: 10.1111/j.1749-6632.1995.tb17496.x. [DOI] [PubMed] [Google Scholar]
- 32.Le F, Cusack B, Richelson E. The neurotensin receptor: is there more than one subtype? Trends Pharmacol Sci. 1996;17:1–3. doi: 10.1016/0165-6147(96)81561-6. [DOI] [PubMed] [Google Scholar]
- 33.Lindström LH, Widerlöv E, Bissette G, Nemeroff C. Reduced CSF neurotensin concentration in drug-free schizophrenic patients. Schizophr Res. 1988;1:55–59. doi: 10.1016/0920-9964(88)90040-0. [DOI] [PubMed] [Google Scholar]
- 34.Lubow RE, Gewirtz JC. Latent inhibition in humans: data, theory, and implications for schizophrenia. Psychol Bull. 1995;117:87–103. doi: 10.1037/0033-2909.117.1.87. [DOI] [PubMed] [Google Scholar]
- 35.Lubow RE, Moore AU. Latent inhibition: the effect of non reinforced pre-exposure to the conditioned stimulus. J Comp Physiol Psychol. 1959;52:415–419. doi: 10.1037/h0046700. [DOI] [PubMed] [Google Scholar]
- 36.Lubow RE, Weiner I, Schlossberg A, Baruch I. Latent inhibition and schizophrenia. Bull Psychon Soc. 1987;25:464–467. [Google Scholar]
- 37.McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 38.Nemeroff CB. Neurotensin: perchance an endogenous neuroleptic? Biol Psychiatry. 1980;15:283–302. [PubMed] [Google Scholar]
- 39.Nemeroff CB, Bissette G, Widerlöv E, Beckmann H, Gerner R, Manberg PJ, Lindström L, Prange AJ, Gattaz WF., Jr Neurotensin-like immunoreactivity in cerebrospinal fluid of patients with schizophrenia, depression, anorexia nervosa-bulimia, and premenstrual syndrome. J Neuropsychiatry Clin Neurosci. 1989;1:16–20. doi: 10.1176/jnp.1.1.16. [DOI] [PubMed] [Google Scholar]
- 40.O'Connor WT, Tanganelli S, Ungerstedt U, Fuxe K. The effects of neurotensin on GABA and acetylcholine release in the dorsal striatum of the rat: an in vivo microdialysis study. Brain Res. 1992;573:209–216. doi: 10.1016/0006-8993(92)90765-2. [DOI] [PubMed] [Google Scholar]
- 41.Perry W, Geyer MA, Braff DL. Sensorimotor gating and thought disturbance measured in close temporal proximity in schizophrenic patients. Arch Gen Psychiatry. 1999;56:277–281. doi: 10.1001/archpsyc.56.3.277. [DOI] [PubMed] [Google Scholar]
- 42.Radke JM, Owens MJ, Ritchie JC, Nemeroff CB. Atypical antipsychotic drugs selectively increase neurotensin efflux in dopamine terminal regions. Proc Natl Acad Sci USA. 1998;95:11462–11464. doi: 10.1073/pnas.95.19.11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rostène W, Azzi M, Boudin H, Lepee I, Souaze F, Mendez-Ubach M, Betancur C, Gully D. Use of nonpeptide antagonists to explore the physiological roles of neurotensin: focus on brain neurotensin/dopamine interactions. Ann NY Acad Sci. 1997;814:125–141. doi: 10.1111/j.1749-6632.1997.tb46151.x. [DOI] [PubMed] [Google Scholar]
- 44.Sharma RP, Janicak PG, Bissette G, Nemeroff CB. CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorders. Am J Psychiatry. 1997;154:1019–1021. doi: 10.1176/ajp.154.7.1019. [DOI] [PubMed] [Google Scholar]
- 45.Simmons D, Arriza J, Swanson L. A complete protocol for in situ hybridization of messenger RNAs in brain and other tissues with radiolabeled single-stranded RNA probes. J Histotechnol. 1989;12:169–181. [Google Scholar]
- 46.Swerdlow NR, Geyer MA. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr Bull. 1998;24:285–301. doi: 10.1093/oxfordjournals.schbul.a033326. [DOI] [PubMed] [Google Scholar]
- 47.Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–154. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- 48.Tanganelli S, O'Connor WT, Ferraro L, Bianchi C, Beani L, Ungerstedt U, Fuxe K. Facilitation of GABA release by neurotensin is associated with a reduction of dopamine release in rat nucleus accumbens. Neuroscience. 1994;60:649–657. doi: 10.1016/0306-4522(94)90493-6. [DOI] [PubMed] [Google Scholar]
- 49.Varty GB, Higgins GA. Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology (Berl) 1995;122:15–26. doi: 10.1007/BF02246437. [DOI] [PubMed] [Google Scholar]
- 50.Venables PH. Psychphysiological aspects of schizophrenia. Br J Med Psychol. 1966;39:289–297. [Google Scholar]
- 51.Walker N, Lépée-Lorgeoux I, Fournier J, Betancur C, Rostène W, Ferrara P, Caput D. Tissue distribution and cellular localization of the levocabastine-sensitive neurotensin receptor mRNA in adult rat brain. Mol Brain Res. 1998;57:193–200. doi: 10.1016/s0169-328x(98)00074-6. [DOI] [PubMed] [Google Scholar]
- 52.Weinberger DR, Lipska BK. Cortical maldevelopment, anti-psychotic drugs, and schizophrenia: a search for a common ground. Schizophr Res. 1995;16:87–110. doi: 10.1016/0920-9964(95)00013-c. [DOI] [PubMed] [Google Scholar]
- 53.Weiner I, Feldon J. The switching model of latent inhibition: an update of neural substrates. Behav Brain Res. 1997;88:11–25. doi: 10.1016/s0166-4328(97)02314-0. [DOI] [PubMed] [Google Scholar]
- 54.Weiner I, Shadach E, Tarrasch R, Kidron R, Feldon J. The latent inhibition model of schizophrenia: further validation using the atypical neuroleptic, clozapine. Biol Psychiatry. 1996;40:834–843. doi: 10.1016/0006-3223(95)00573-0. [DOI] [PubMed] [Google Scholar]
- 55.Widerlöv E, Lindström LH, Besev G, Manberg PJ, Nemeroff CB, Breese GR, Kizer JS, Prange AJ., Jr Subnormal CSF levels of neurotensin in a subgroup of schizophrenic patients: normalization after neuroleptic treatment. Am J Psychiatry. 1982;139:1122–1126. doi: 10.1176/ajp.139.9.1122. [DOI] [PubMed] [Google Scholar]
- 56.Wilkinson LS, Killcross SS, Humby T, Hall FS, Geyer MA, Robbins TW. Social isolation in the rat produces developmentally specific deficits in prepulse inhibition of the acoustic startle response without disrupting latent inhibition. Neuropsychopharmacology. 1994;10:61–72. doi: 10.1038/npp.1994.8. [DOI] [PubMed] [Google Scholar]
- 57.Wirshing WC. The new antipsychotic compounds: is a clinical choice algorithm possible? West J Med. 1998;169:43–44. [PMC free article] [PubMed] [Google Scholar]