

Abstract
To clarify the mechanisms of interaction between adenosineA1 receptor (A1-R) and adenosine A2 receptor (A2-R) on neurotransmitter release, this study determined the functional interactions among adenosine receptors (AD-Rs), voltage-sensitive Ca2+ channels (VSCCs), protein kinases (PKs), and synaptic proteins [N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptors] on hippocampal serotonin release usingin vivo microdialysis in freely moving rat. Basal serotonin release was regulated by two functional complexes: N-type VSCC (N-VSCC)/calcium-phospholipid-dependent protein kinase (PKC)/syntaxin (major pathway) and P-type VSCC (P-VSCC)/cyclic AMP-dependent protein kinase (PKA)/synaptobrevin (minor pathway). However, K+-evoked serotonin release was regulated by N-VSCC/PKC/syntaxin (minor pathway) and P-VSCC/PKA/synaptobrevin (major pathway). A1-R antagonists increased basal serotonin release, which was reduced by inhibitors of N-VSCC, PKC, and syntaxin predominantly and by inhibitors of PKA and synaptobrevin weakly, but was not affected by P-VSCC inhibitor. In the presence of A1-R antagonist, A2-R agonists increased basal serotonin release, which was inhibited by inhibitors of P-VSCC, PKA, and synaptobrevin predominantly and reduced by inhibitors of N-VSCC, PKC, and syntaxin weakly. Under the condition of activation of adenylate cyclase in the absence of A1-R antagonists, A2-R agonists increased basal serotonin release. A1-R antagonist and A2-R agonist enhanced K+-evoked serotonin release, which was inhibited by inhibitors of P-VSCC, PKA, and synaptobrevin predominantly. These results suggest that an activation of A1-R suppresses serotonin release via inhibition of both N-VSCC/PKC/syntaxin and P-VSCC/PKA/synaptobrevin pathways, and an activation of A2-R stimulates serotonin release via enhancement of the P-VSCC/PKA/synaptobrevin pathway. Therefore, PKA activity plays an important role in the interaction between A1-R and A2-R on hippocampal serotonin release.
Keywords: adenosine, serotonin, microdialysis, voltage-sensitive Ca2+ channel, protein kinase, SNARE
Adenosine is a potent modulator of synaptic transmission at various synapses (Linden, 1994). Recently, four major classes of adenosine receptor (AD-R) subtypes have been pharmacologically identified and cloned (Linden, 1994; Olah and Stiles, 1995). Many lines of evidence suggest that an activation of adenosine A1 receptor (A1-R) suppresses neurotransmitter release (Barraco and Stefano, 1990; Zetterstrom and Fillenz, 1990;Ambrosio et al., 1997; Okada et al., 1997, 1999a,b; Satoh et al., 1997;Ribeiro, 1999; Von Lubitz, 1999) primarily by presynaptic mechanisms, including the voltage-sensitive Ca2+channel (VSCC) and the K+ channel (Yawo and Chuhma, 1993; Ambrosio et al., 1997; Wu and Saggau, 1997; Wu et al., 1999). Activation of the adenosine A2receptor (A2-R) enhances the evoked release of various neurotransmitters (Popoli et al., 1995; Ambrosio et al., 1997; Satoh et al., 1997) that may involve P-type VSCC (P-VSCC) (Umemiya and Berger, 1994; Satoh et al., 1997). It has been recognized that the response to adenosine is a balance between A1-R and A2-R activations because monoamine release is reduced and enhanced by agonists of A1-R and A2-R, respectively. The stimulatory effects of A2-R can be masked by activation of A1-R (Correia-de-Sa et al., 1996; Okada et al., 1997,1999a,b). However, the mechanisms of this interaction between A1-R and A2-R have not been clarified yet.
A mechanistic model for neurotransmitter release, known as the soluble [N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor] (SNARE) hypothesis, proposes that vesicle membrane SNAREs bind to cognate proteins on the target membrane SNARE to form complexes that are recognized and dissociated by the general membrane-trafficking factors α-SNAP and NSF (Sollner et al., 1993;Sudhof, 1995). After arrival of an action potential at presynaptic terminals and the resulting entry of Ca2+via VSCC, the local level of Ca2+ rises abruptly to submillimolar levels (Sollner et al., 1993; Sudhof, 1995;Sheng et al., 1998). Biophysical and pharmacological analyses have led to the identification of N-type VSCC (N-VSCC) and P-VSCC as being involved in neurotransmitter release in the mammalian CNS (Dunlap et al., 1995; Bergquist et al., 1998; Okada et al., 1998a,b; Wu et al., 1998, 1999). These findings are consistent with observations that neurotransmitter release at many central synapses is blocked by N-VSCC or P-VSCC inhibitors (Takahashi and Momiyama, 1993; Wheeler et al., 1994; Bergquist et al., 1998; Okada et al., 1998a; Wu et al., 1998). Second-messenger-activated protein kinases (PKs), including calcium-phospholipid-dependent protein kinase (PKC) and cyclic AMP-dependent protein kinase (PKA), regulate neurotransmission via modulation of the interactions of SNARE apparatus (Sudhof, 1995;Zamponi et al., 1997; Kaneko et al., 1998; Hamid et al., 1999; Turner et al., 1999; Yoshihara et al., 1999). Moreover, our previous study (Okada et al., 1998a) demonstrated that the basal and K+-evoked striatal dopamine release was predominantly regulated by N-VSCC and P-VSCC, respectively. Together with previous evidence that clarifies the mechanisms of interaction between A1-R and A2-R on neurotransmitter release in spontaneous and depolarization states, this study determined the effects of AD-Rs, VSCCs, PKs, and SNARE complexes on hippocampal basal and K+-evoked serotonin releases usingin vivo microdialysis in freely moving rats.
MATERIALS AND METHODS
All experiments described in this report were performed in accordance with the specifications of the Animal Research Committee of Hirosaki University and met the Guideline Animal Experimentation of Hirosaki University. Male Wistar rats (Clea, Tokyo, Japan), weighing 250–300 gm, were housed under conditions of constant temperature (22 ± 2°C) with a 12 hr light/dark cycle.
Microdialysis system preparation. Each rat was placed in a stereotaxic frame and kept under halothane anesthesia (1.5% mixture of halothane and O2 with N2O). Before the microdialysis probe was inserted, each rat was pretreated with a microinfusion of 0.3 μl of modified Ringer's solution (MRS) with or without 0.03, 0.3, or 3 ng of botulinum toxins (BoNTs) (Capogna et al., 1997; Pierce and Kalivas, 1997) because the molecular weight of BoNTs (>100,000) is beyond the cutoff for diffusion through the dialysis membrane. A concentric I-type dialysis probe (0.22 mm diameter; 3 mm exposed membrane) (Eicom, Kyoto, Japan) was implanted in the hippocampus (anterior, −5.8 mm; lateral, 4.8 mm; ventral, −4.0 mm, relative to bregma), and the perfusion experiments were started 18 hr after the rats had recovered from anesthesia (Okada et al., 1998a). The perfusion rate was always 1 μl/min. The MRS contained (in mm): 145 Na+, 2.7 K+, 1.2 Ca2+, 1.0 Mg2+, and 154.4 Cl−; the pH was adjusted to 7.40 with 2 mm phosphate buffer and 1.1 mm Tris buffer (Okada et al., 1998b). To study the effects of an increase in the extracellular K+ level (K+-evoked stimulation) on the hippocampal extracellular serotonin level, MRS containing 50 mm K+ (HKMRS) was perfused for 20 min (Okada et al., 1998a). The ionic composition was modified, and isotonicity was maintained by an equimolar decrease of Na+ (Okada et al., 1998b). Each hippocampal dialysate was injected every 10 min into a high-performance liquid chromatography (HPLC).
HPLC system preparation. The HPLC system used for determination of the extracellular serotonin levels was equipped with an electrochemical detector (ECD-300; Eicom) with pump (EP-30; Eicom) and a graphite carbon electrode set at +450 mV (vs an Ag/AgCl reference electrode). The analytical column (100 × 1.5 mm, internal diameter) was packed with Mightysil RP-18 (particle size, 5 μm) (gift from Kanto Chemicals, Tokyo, Japan) by Masis Inc. (Hirosaki, Japan). The mobile phase was composed of 0.1 m phosphate buffer containing 20% (v/v) methanol, 900 mg/l octansulfonic sodium, and 50 mg/l EDTA-2Na; the final pH was 5.9, and the column temperature was maintained at 25°C with the flow rate set at 200 μl/min (Okada et al., 1998a).
Chemical agents. The summary of chemical agents used in this study is described in Table 1. The chemical agents were adenosine (Nacalai Tesque, Osaka, Japan); caffeine (Nacalai Tesque); the A1-R agonist, 2-chloro-N6-cyclopentyladenosine (CCPA; Research Biochemicals, Natick, MA); the A1-R antagonist, 8-cyclopentyl-1,3-dimethylxanthine (CPT; Research Biochemicals); the A2-R agonist, N6-[2-(3,5-dimethoxyphenyl)-2-(methyl-phenyl)-ethyl]adenosine (PD125944; Research Biochemicals); the A2-R antagonist, 3,7-dimethyl-1-propargylxanthine (DMPX; Research Biochemicals); the N-VSCC inhibitor, ω-conotoxin GVIA (GVIA; Peptide Institute, Osaka, Japan); the P-VSCC inhibitor, ω-agatoxin IVA (IVA; Peptide Institute); the PKA inhibitor, H-89 (Calbiochem, San Diego, CA); the PKC activator, phorbol 12-myristate 13-acetate (PMA; Nacalai Tesque); the PKC inhibitor, chelerythrine (CHR; Calbiochem); the adenylate cyclase activator, forskolin (Nacalai Tesque); the SNAP-25 inhibitor, BoNT type A (BoNT/A; Calbiochem); the synaptobrevin inhibitor, BoNT/B (Calbiochem); and the syntaxin inhibitor, BoNT/C (Calbiochem).
Table 1.
Summary of chemical agents and their action sites
| Chemical agents | Abbreviation | Main target site and action | Other target sites and action | Reference |
|---|---|---|---|---|
| Voltage-sensitive Ca2+channels | VSCCs | |||
| ω-Conotoxin GVIA | GVIA | N-type VSCC inhibition (IC50 = 100 nm) | (1) | |
| ω-Agatoxin IVA | IVA | P-type VSCC inhibition (IC50 = 10 nm) | N-type VSCC inhibition (IC50 > 500 nm) | (1) |
| Protein kinases | PKs | |||
| H-89 | PKA inhibition (Ki = 0.048 μm) | PKC inhibition (Ki = 31.7 μm) | (2) | |
| Chelerythrine | CHR | PKC inhibition (IC50 = 0.66 μm) | PKA inhibition (IC50 = 170 μm) | (3), (4) |
| Phorbol 12-myristate 13-acetate | PMA | PKC activation (nmorders) | Ca2+-ATPase activation | (5), (6) |
| Forskolin | Adenylate cyclase activation (EC50 = 4 μm) | (7) | ||
| SNAREs | ||||
| Botulinum toxin type A | BoNT/A | SNAP-25 inhibition | (8) | |
| Botulinum toxin type B | BoNT/B | Synaptobrevin inhibition | (8) | |
| Botulinum toxin type C | BoNT/C | Syntaxin inhibition | (8) | |
| Adenosine receptors | AD-Rs | |||
| 2-Chloro-N6-cyclopentyladenosine | CCPA | A1-R activation (Ki = 0.4 nm) | A2-R activation (Ki = 3.9 μm) | (9) |
| 8-Cyclopentyl-1,3-dimethylxanthine | CPT | A1-R inhibition (Ki = 11 nm) | A2-R inhibition (Ki = 1.4 μm) | (9) |
| N6-[2-(3,5-Dimethoxyphenyl)-2- (methylphenyl)ethyl]adenosine | PD125944 | A2-R activation (Ki = 4.4 nm) | A1-R activation (Ki = 142 nm) | (9) |
| 3,7-Dimethyl-1-propargylxanthine | DMPX | A2-R inhibition (Ki = 11 nm) | A1-R inhibition (Ki = 45 μm) | (9) |
(1) Takahashi and Momiyama, 1993; (2) Chijiwa et al., 1990; (3)Herbert et al., 1990; (4) Barg et al., 1992; (5) Oishi and Yamaguchi, 1994; (6) Liu and Heckman, 1998; (7) Seamon et al., 1981; (8) Niemann et al., 1995; (9) Williams, 1991.
Drug administration. All rats were pretreated with a microinfusion of 0.3 μl of MRS with or without 0.03, 0.3, or 3 ng of BoNTs before insertion of the dialysis probe. Perfusion was commenced with MRS. At least 6 hr after the perfusion started, the hippocampal extracellular serotonin level was measured. When the coefficients of variation of hippocampal extracellular serotonin level reached <5% over 60 min (stabilization) (Okada et al., 1998a), control data were obtained for an additional 60 min; then the perfusion medium (MRS) was switched to MRS containing the various agents (pretreatment period).
To study the effects of target agents on basal hippocampal serotonin release, after confirming that the hippocampal extracellular serotonin level had reached a plateau (stabilization), we switched the perfusion medium for pretreatment to the same perfusion medium added to the target agent for 120 min.
To study the effects of target agents on hippocampal K+-evoked serotonin release, after the confirming stabilization, we switched the pretreatment perfusion medium to HKMRS containing the same agents for 20 min (K+-evoked stimulation). After this K+-evoked stimulation, the perfusion medium was switched to the pretreatment perfusion medium for an additional 100 min.
RESULTS
The mean serotonin level in hippocampal perfusate during the spontaneous stage (basal serotonin level) was 3.6 ± 0.4 fmol/10 μl (10 min). In the pilot study, perfusion with MRS containing tetrodotoxin (TTX) and Ca2+-depletion MRS decreased the extracellular serotonin level to <0.7 fmol/10 μl (data not shown). Furthermore, an elevation of the K+ level in the perfusate from 2.7 to 50 mm increased the extracellular serotonin levels (K+-evoked release, 13.7 ± 1.6 fmol/10 μl) (see Fig. 3A). Therefore, these experiments demonstrate that under the microdialysis conditions that are presently used, the serotonin levels (basal serotonin release) in hippocampal perfusates were primarily of neuronal origin because the extracellular serotonin level was TTX-sensitive, Ca2+-dependent, and K+-sensitive (Westerink et al., 1989).
Fig. 3.

Effects of VSCC inhibitors (A), PK inhibitors (B), PK activators (C), and BoNTs (D) on hippocampal K+-evoked serotonin release. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl), and abscissas show the time in minutes. The open barsindicate perfusion with VSCC inhibitors, PK inhibitors, and PK activators, and striped bars indicate K+-evoked stimulation for 20 min.A–C, To study the effects of VSCC inhibitors, PK inhibitors, and PK activators on K+-evoked serotonin release, the perfusion medium was switched from MRS containing GVIA (1 μm), IVA (1 μm), H-89 (1 μm), CHR (1 μm), PMA (10 μm), or forskolin (10 μm) to HKMRS containing the same agents for 20 min. D, To compare the effects of BoNTs on K+-evoked serotonin release, the microinfusion of 0.3 ng of BoNT/A, BoNT/B, or BoNT/C is shown. The mean values obtained before and after K+-evoked stimulation were compared using repeated-measure one-way ANOVA and Dunnett's multiple comparison test.
Effects of VSCCs, PKs, and SNAREs on basal serotonin release
The perfusion with GVIA and IVA reduced the hippocampal basal serotonin release in a concentration-dependent manner (p < 0.01) (Figs.1A,2A), and the inhibitory effect of GVIA was more predominant than that of IVA (p < 0.01) (Fig. 2A). The perfusion with CHR and H-89 reduced the basal serotonin release in a concentration-dependent manner (p < 0.01) (Figs. 1B, 2B), and the inhibitory effect of CHR was more predominant than that of H-89 (p < 0.05) (Fig. 2B). The perfusion with PMA and forskolin increased the extracellular serotonin level in a concentration-dependent manner (p < 0.01) (Figs. 1C, 2C), and this stimulatory effect of PMA was more predominant than that of forskolin (p < 0.01) (Fig. 2C). The microinfusion of BoNT/B and BoNT/C decreased the basal serotonin release in a concentration-dependent manner (p< 0.01) (Figs. 1D, 2D). However, the microinfusion of BoNT/A decreased (p < 0.05) the basal serotonin release independent of concentration (data not shown). The inhibitory effect of BoNT/C was more predominant than that of BoNT/A and BoNT/B (p < 0.05) (Figs. 1D, 2D).
Fig. 1.
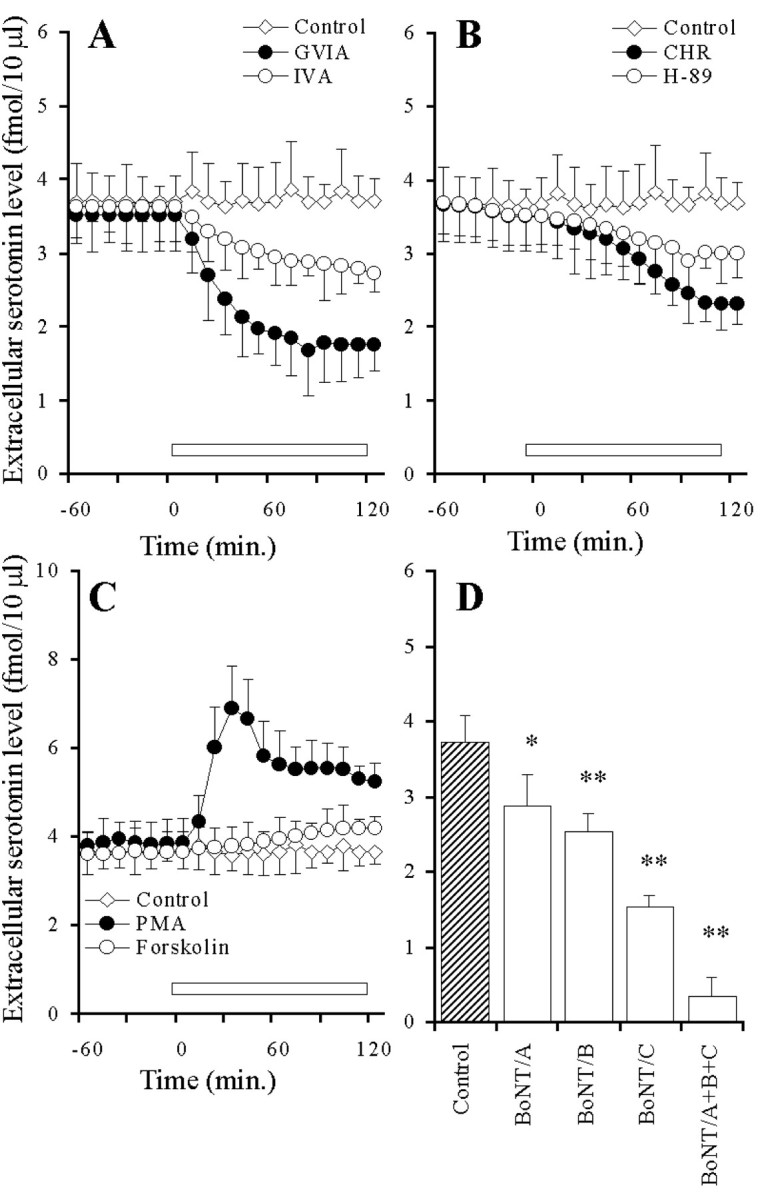
Effects of VSCC inhibitors (A), PK inhibitors (B), PK activators (C), and BoNTs (D) on hippocampal basal serotonin release.A–C, The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl), and abscissas show the time in minutes. The open barsindicate perfusion with VSCC inhibitors, PK inhibitors, or PK activators. To study the effects of VSCC inhibitors, PK inhibitors, and PK activators on basal serotonin release, the perfusion medium was switched from MRS to MRS containing GVIA (1 μm), IVA (1 μm), H-89 (1 μm), CHR (1 μm), PMA (10 μm), or forskolin (10 μm) for 120 min. The mean values obtained before and during perfusion with VSCC inhibitors, PK inhibitors, and PK activators were compared using repeated-measurements one-way ANOVA and Dunnett's multiple comparison test. D, The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). To compare the effects of BoNTs on basal serotonin release, the microinfusion of 0.3 ng of BoNT/A, BoNT/B, and BoNT/C is shown. The mean values obtained by microinfusion without (Control) and with BoNTs were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Fig. 2.

Concentration-dependent effects of VSCC inhibitors (A), PK inhibitors (B), PK activators (C), and BoNTs (D) on hippocampal basal serotonin release. The ordinates indicate the mean ± SD (n = 6) of area under the curve (AUC) values of extracellular serotonin level (fmol/10 μl), and abscissas show the concentration of VSCC inhibitors, PK inhibitors, and PK activators (log μm), or the dose of BoNTs (log ng). A–C, To study the effects of VSCC inhibitors, PK inhibitors, and PK activators on basal serotonin release, the perfusion medium was switched from MRS to MRS containing GVIA, IVA, H-89, CHR, PMA, or forskolin for 120 min. The mean values obtained by perfusion without (Control) and with each agent were compared by one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).D, To compare the effects of BoNTs on basal serotonin release, the microinfusion of BoNT/B and BoNT/C is shown. The mean values obtained by microinfusion without (Control) and with BoNTs were compared by one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effects of VSCCs, PKs, and SNAREs on hippocampal K+-evoked serotonin releases
Preperfusion with GVIA and IVA reduced the K+-evoked serotonin release in a concentration-dependent manner (p < 0.01) (Figs. 3A,4A), and the inhibitory effect of IVA was more predominant than that of GVIA (p < 0.01) (Fig. 4A). Preperfusion with CHR and H-89 reduced the K+-evoked serotonin release in a concentration-dependent manner (p < 0.01) (Figs. 3B, 4B), and the inhibitory effect of H-89 was more predominant than that of CHR (p< 0.01) (Fig. 4B). Preperfusion with PMA and forskolin increased the K+-evoked serotonin release in a concentration-dependent manner (p < 0.01) (Figs. 3C,4C), and this stimulatory effect of forskolin was more predominant than that of PMA (p < 0.01) (Fig.4C). The microinfusion of BoNT/B and BoNT/C reduced the K+-evoked serotonin release in a concentration-dependent manner (p < 0.01) (Figs. 3D, 4D). BoNT/A decreased the K+-evoked serotonin release weakly (p < 0.05), but this inhibition was not concentration-dependent (data not shown). The inhibitory effects of BoNT/B were more predominant than those of BoNT/A and BoNT/C (p < 0.05) (Fig. 4D).
Fig. 4.

Concentration-dependent effects of VSCC inhibitors (A), PK inhibitors (B), PK activators (C), and BoNTs (D) on hippocampal K+-evoked serotonin release. The ordinates indicate the mean ± SD (n = 6) of maximal change of extracellular serotonin level (fmol/10μl) induced by K+-evoked stimulation (for 20 min), and abscissas show the concentration of VSCC inhibitors, PK inhibitors, and PK activators (logμm) or the dose of BoNTs (log ng).A–C, To study the effects of VSCC inhibitors, PK inhibitors, and PK activators on K+-evoked serotonin release, the perfusion medium was switched from MRS containing GVIA, IVA, H-89, CHR, PMA, or forskolin to HKMRS containing the same agents for 20 min. The mean values obtained by perfusion with or without (Control) each agent were compared using two-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01). D, To compare the effects of BoNTs on K+-evoked serotonin release, the microinfusion of 0.3 ng of BoNT/B or BoNT/C is shown. The mean values obtained by microinfusion without (Control) and with BoNTs were compared by two-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effects of VSCC inhibitors and BoNTs on PK activator-induced serotonin releases
The 10 μm PMA-induced elevation of the extracellular serotonin level was decreased by perfusion with 1 μm GVIA (p < 0.01) but was not affected by perfusion with 1 μm IVA (Fig.5A), whereas the 10 μm forskolin-induced elevation of extracellular serotonin level was decreased by perfusion with 1 μm IVA (p < 0.01) but was not affected by perfusion with 1 μm GVIA (Fig. 5B). The 10 μm PMA-induced elevation of the extracellular serotonin level was decreased by microinfusion of 0.3 ng of BoNT/C (p < 0.01) but was not affected by that of BoNT/B (Fig. 5A), whereas the 10 μm forskolin-induced elevation of the extracellular serotonin level was decreased by 0.3 ng of BoNT/B (p < 0.01) but was not affected by BoNT/C (Fig.5B).
Fig. 5.

Effects of VSCC inhibitors and BoNTs on PK activator-induced elevation of extracellular serotonin level.A, B, The effects of VSCC inhibitors and BoNTs on the 10 μm PMA (A) or 10 μm forskolin (B) -induced elevation of extracellular serotonin level. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). To study the effects of VSCC inhibitors on PK activator-induced elevation of serotonin level, after the microinfusion of 0.3 μl of MRS, the perfusion medium was switched from MRS without (Control) or with 1 μm GVIA or 1 μm IVA (open columns) to the same MRS containing 10 μm PMA (A) or 10 μm forskolin (B) (closed columns) for 120 min. To study the effects of BoNTs on PK activator-induced elevation of serotonin level, after the microinfusion without or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS (open columns) to the same MRS containing 10 μm PMA (A) or 10 μm forskolin (B) (closed columns) for 120 min. C, D, The effects of VSCC inhibitors and BoNTs on the 10 μm PMA (C) or 10 μm forskolin (D) -induced elevation of K+-evoked serotonin release. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10μl) induced by K+-evoked stimulation for 20 min. To study the effect of interaction between VSCC inhibitors and PK activators on K+-evoked serotonin release, the perfusion medium was switched from MRS without (Control) or with 1 μm GVIA or 1 μm IVA added to 10 μm PMA (C) or forskolin (D) (open columns) to HKMRS containing the same agents (closed columns) for 20 min. To study the effect of interaction between BoNTs and PK activators on K+-evoked serotonin release, after the microinfusion without (Control) or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS containing 10 μm PMA (C) or forskolin (D) (open columns) to HKMRS containing the same agents (closed columns) for 20 min. The mean values obtained with the control (no treatment with VSCC inhibitors or BoNTs) and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
The 10 μm PMA-induced elevation of K+-evoked serotonin release was reduced by perfusion with 1 μm GVIA (p < 0.01) but was not affected by perfusion with 1 μm IVA (Fig. 5C), whereas the 10 μm forskolin-induced elevation of K+-evoked serotonin release was reduced by perfusion with 1 μm IVA (p < 0.01) but was not affected by perfusion with 1 μm GVIA (Fig. 5D). The 10 μm PMA-induced elevation of K+-evoked serotonin release was decreased by microinfusion of 0.3 ng of BoNT/C (p < 0.05) but was not affected by that of BoNT/B (Fig. 5C), whereas the 10 μm forskolin-induced elevation of K+-evoked serotonin release was decreased by microinfusion of 0.3 ng of BoNT/B (p < 0.01) but was not affected by microinfusion of 0.3 ng of BoNT/C (Fig.5D).
Effect of interaction between VSCCs and SNAREs on basal and K+-evoked serotonin releases
The microinfusion of 0.3 ng of BoNT/C and BoNT/B as well as perfusion with 1 μm GVIA and IVA reduced basal and K+-evoked serotonin releases (Fig.6A,B). However, under the condition of cleavage of syntaxin and synaptobrevin with pretreatment of 0.3 ng of BoNT/C and BoNT/B, 1 μm GVIA and IVA, respectively, could not affect basal serotonin release (Fig. 6A). Similar to basal release under the condition of cleavage of syntaxin and synaptobrevin with pretreatment of 0.3 ng of BoNT/C and BoNT/B, 1 μm GVIA and IVA, respectively, did not affect K+-evoked serotonin release (Fig.6B).
Fig. 6.
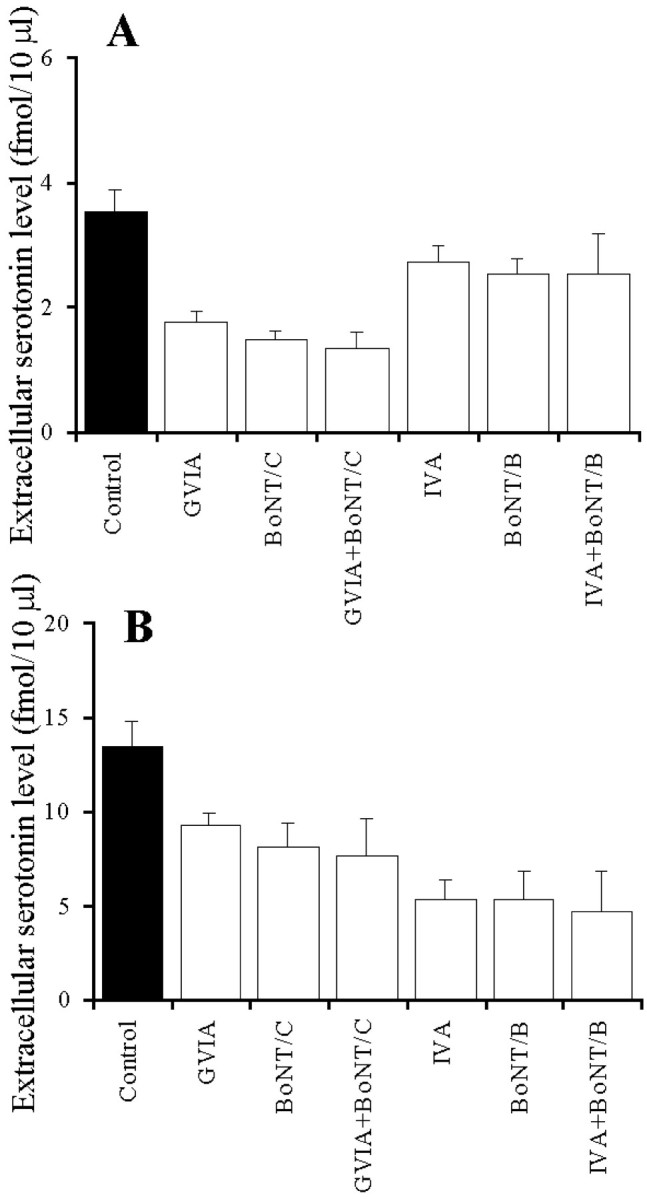
Effect of interaction between VSCC inhibitors and BoNTs on basal (A) and K+-evoked (B) serotonin release. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). After the microinfusion with or without 0.3 ng of BoNT/B and BoNT/C, the perfusion medium was switched from MRS to MRS without (Control) or with 1 μm GVIA or 1 μm IVA. After confirmation of the stabilization of basal release, the perfusion medium was switched from the same MRS to HKMRS containing the same agents for 20 min. The mean values obtained with the control (no treatment with VSCC inhibitor or BoNTs) and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test.
Effects of AD-R subtypes on basal serotonin release
Adenosine and CCPA decreased the basal serotonin release in a concentration-dependent manner (p < 0.01), whereas both caffeine and CPT increased it in a concentration-dependent manner (p < 0.01) (Fig.7A,B). Neither PD125944 nor DMPX affected the extracellular serotonin level (Fig. 7A,B). Under the condition of A1-R blockade by 10 μm CPT, adenosine and PD125944 increased the basal serotonin release in a concentration-dependent manner (p < 0.01), whereas caffeine and DMPX decreased it in a concentration-dependent manner (p < 0.01) (Fig.7C,D).
Fig. 7.

A, Effects of AD-R ligands on hippocampal basal serotonin release. The ordinate indicates the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl), and the abscissa shows the time in minutes. The open barindicates perfusion with 10 μm adenosine (AD), caffeine (CF),CPT, PD125944, DMPX, or 0.1 μmCCPA. The mean values obtained before and during perfusion with AD-R ligands were compared using repeated-measure one-way ANOVA and Dunnett's multiple comparison test.B, Concentration-dependent effects of AD-R ligands on the hippocampal basal serotonin release. The ordinate indicates the mean ± SD (n = 6) of percentage of control of the AUC value of extracellular serotonin level, and the abscissa shows the concentration of agents (log μm). The mean values obtained by control and during perfusion with AD-R ligands were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).C, Effects of AD-R ligands, under the condition of A1-R blockade by 10 μm CPT, on the hippocampal basal serotonin release. The ordinate indicates the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl), and the abscissa shows the time in minutes. The open barindicates perfusion with 10 μm CPT, and thestriped column indicates perfusion with 10 μmAD, CF,PD125944, or DMPX. The mean values obtained before and during perfusion with AD-R ligands were compared using repeated-measure one-way ANOVA and Dunnett's multiple comparison test. D, Concentration-dependent effects of AD-R ligands, under the condition of A1-R blockade by 10 μmCPT, on the hippocampal basal serotonin release. The ordinate indicates the mean ± SD (n = 6) of percentage of control of AUC of extracellular serotonin level, and the abscissa shows the concentration of agents (log μm). The mean values obtained by control and during perfusion with AD-R ligands were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effect of interaction between A1-R subtypes as well as VSCCs, PKs, and SNAREs on basal serotonin release
The elevation of basal serotonin release induced by both 10 μm PMA and forskolin was inhibited by perfusion with 0.1 μm CCPA (p < 0.01) (Fig.8A,B). The stimulatory effect of 10 μm CPT on the basal serotonin release was inhibited by perfusion with 1 μm GVIA (p < 0.01), 1 μm CHR (p < 0.01), and 1 μm H-89 (p < 0.05) but was not affected by perfusion with 1 μm IVA (Fig.9A,B). Microinfusion of 0.3 ng of both BoNT/B (p < 0.05) and BoNT/C (p < 0.01) decreased the elevation of basal serotonin release induced by perfusion with 10 μm CPT (Fig. 9C).
Fig. 8.

Effects of A1-R agonist on PK activator-induced elevation of extracellular serotonin level. A,B, The effects of 0.1 μm CCPA on the 10 μm PMA (A) or 10 μmforskolin (B) -induced elevation of extracellular serotonin level. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). To study the effects of A1-R agonist on PK activator-induced elevation of serotonin level, the perfusion medium was switched from MRS without (Control) or with 0.1 μm CCPA (open columns) to the same MRS containing 10 μm PMA (A) or 10 μm forskolin (B) (closed columns) for 120 min. The mean values obtained by control and treatment with CCPA were compared using Student's ttest (*p < 0.05, **p < 0.01).
Fig. 9.

Effects of VSCC inhibitors (A), PK inhibitors (B), and BoNTs (C) on A1-R antagonist-induced elevation of extracellular serotonin level. The ordinate indicates the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). A, B, The effects of VSCC inhibitors (A) and PK inhibitors (B) on the 10 μm CPT-induced elevation of extracellular serotonin level. The perfusion medium was switched from MRS without (Control) or with 1 μm GVIA, IVA, CHR, or H-89 (open columns) to the same MRS containing 10 μm CPT (closed columns) for 120 min. C, The effects of BoNTs on the 10 μm CPT-induced elevation of extracellular serotonin level. After the microinfusion without (Control) or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS (open columns) to MRS containing 10 μm CPT (closed columns) for 120 min. The mean values obtained by control and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effect of interaction between A2-R agonist as well as VSCCs, PKs, and SNAREs on basal serotonin release, under the condition of A1-R blockade
The stimulatory effect of perfusion with 10 μmPD125944, under the condition of A1-R blockade by perfusion with 10 μm CPT, on the basal serotonin release was inhibited by 1 μm GVIA (p < 0.05), 1 μm IVA (p < 0.01), 10 μm CHR (p < 0.05), and 10 μm H-89 (p < 0.01) (Fig.10A,B). The inhibitory effects of 1 μm IVA and 10 μm H-89 on the action of 10 μm PD125944 were more predominant than those of 1 μm GVIA and 10 μm CHR (p < 0.01), respectively (Fig.10A,B). The stimulatory effect of perfusion with 10 μm PD125944 on the basal serotonin release was decreased by microinfusion of 0.3 ng of BoNT/B (p < 0.01) and BoNT/C (p< 0.05) (Fig. 10C). The inhibitory effect of BoNT/B on the action of PD125944 was more predominant than that of BoNT/C (p < 0.01) (Fig. 10C).
Fig. 10.

Effects of VSCC inhibitors (A), PK inhibitors (B), and BoNTs (C) on A2-R agonist-induced elevation of extracellular serotonin level, under the condition of A1-R blockade. The ordinate indicates the mean ± SD (n = 6) of extracellular serotonin level (fmol/10μl). A, B, The effects of VSCC inhibitors (A) and PK inhibitors (B) on the 10 μm PD125944-induced elevation of extracellular serotonin level, under the condition of A1-R blockade by 10 μm CPT. The perfusion medium was switched from MRS containing 10 μm CPT without (Control) or with 1 μm GVIA, IVA, CHR, or H-89 (open columns) to the same MRS containing 10 μm PD125944 (closed columns) for 120 min. C, The effects of BoNTs on the 10 μmPD125944-induced elevation of extracellular serotonin level, under the condition of A1-R blockade by 10 μm CPT. After the microinfusion without (Control) or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS containing 10 μm CPT (open columns) to the same MRS containing 10 μm PD125944 (closed columns) for 120 min. The mean values obtained by control and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effect of interaction between AD-R subtypes as well as VSCCs, PKs, and SNAREs on K+-evoked serotonin release
Perfusion with both 10 μm CPT and PD125944 enhanced the K+-evoked serotonin release in a concentration-dependent manner (p < 0.01), whereas perfusion with both 0.1 μm CCPA and 10 μm DMPX reduced it in a concentration-dependent manner (p < 0.01) (Fig.11A,B). The stimulatory effect of perfusion with 10 μmCPT on the K+-evoked serotonin release was inhibited by perfusion with 1 μm IVA (p < 0.01), 10 μm CHR (p < 0.05), and 10 μmH-89 (p < 0.01) but was not affected by perfusion with 1 μm GVIA (Fig.12A). The stimulatory effect of perfusion with 10 μm PD125944 on the K+-evoked serotonin release was inhibited by perfusion with 1 μm IVA (p < 0.01) and 1 μmH-89 (p < 0.01), but was not affected by perfusion with 1 μm GVIA or 1 μm CHR (Fig. 12B). The stimulatory effect of perfusion with 10 μm CPT on the K+-evoked serotonin release was inhibited by microinfusion of 0.3 ng of BoNT/B (p < 0.01) and BoNT/C (p< 0.05) (Fig. 12A). The stimulatory effect of perfusion with 10 μm PD125944 on the K+-evoked serotonin release was inhibited by microinfusion of 0.3 ng of BoNT/B (p < 0.01) and BoNT/C (p < 0.05) (Fig.12B).
Fig. 11.

A, Effects of AD-R ligands on the hippocampal K+-evoked serotonin release. The ordinate indicates the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl), and the abscissa shows the time in minutes. The open bar indicates perfusion with 0.1 μm CCPA, 10 μm CPT, PD125944, or DMPX, and the striped bar indicates K+-evoked stimulation for 20 min. The mean values obtained before and after K+-evoked stimulation were compared using repeated-measure one-way ANOVA and Dunnett's multiple comparison test. B, Concentration-dependent effects of AD-R ligands on the hippocampal K+-evoked serotonin release. The ordinate indicates the mean ± SD (n = 6) of percentage of control (pre-evoked stimulation) of AUC of extracellular serotonin level induced by K+-evoked stimulation (for 20 min), and the abscissa shows the concentration of agents (logμm). The concentration-dependent effect of each AD-R ligand on K+-evoked serotonin release was analyzed using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Fig. 12.

Effect of interaction between AD-R ligands and inhibitors of VSCC, PK, and SNAREs on K+-evoked serotonin release. A, The effects of VSCC inhibitors, PK inhibitors, and BoNTs on the stimulatory effects of 10 μmCPT on K+-evoked serotonin release. After the microinfusion without (Control) or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS containing 10 μm CPT without (Control) or with 1 μm GVIA, IVA, CHR or H-89 to HKMRS containing the same agents for 20 min. B, The effects of VSCC inhibitors, PK inhibitors, and BoNTs on the stimulatory effects of 10 μmPD125944 on K+-evoked serotonin release. After the microinfusion without (Control) or with 0.3 ng of BoNT/B or BoNT/C, the perfusion medium was switched from MRS containing 10 μm PD125944 without (Control) or with 1 μm GVIA, IVA, CHR, or H-89 to HKMRS containing the same agents for 20 min. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl). The mean values obtained by control (nontreatment with inhibitors of VSCC, PK, or SNARE) and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
Effect of interaction between A2-R agents and forskolin on basal and K+-evoked serotonin release
Neither 10 μm PD125944 nor DMPX affected the basal serotonin release; however, preperfusion with 10 μmforskolin produced both the stimulatory effects of PD125944 (p < 0.01) and the inhibitory effects of DMPX (p < 0.01) on basal serotonin release in the absence of A1-R antagonist (Fig.13A). The stimulatory effects of perfusion with 10 μm forskolin on the K+-evoked serotonin release were reduced by perfusion with 0.1 μm CCPA (p < 0.05) and 10 μmDMPX (p < 0.05) (Fig. 13B).
Fig. 13.

Effect of interaction between AD-R ligands and forskolin on basal (A) and K+-evoked (B) serotonin release. The ordinates indicate the mean ± SD (n = 6) of extracellular serotonin level (fmol/10 μl).A, The effects of PD125944 and DMPX on basal serotonin release, under the condition of activation of adenylate cyclase by 10 μm forskolin. The perfusion medium was switched from MRS (Control) to MRS containing 10 μmforskolin without or with 10 μm PD125944 or DMPX for 120 min. The mean values obtained by forskolin and forskolin with PD125944 or DMPX were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01). B, The effect of interaction between forskolin and AD-R ligands on K+-evoked serotonin release. The perfusion medium was switched from MRS containing 10 μm forskolin without (Control) or with 0.1 μm CCPA or 10 μm DMPX to HKMRS containing the same agents for 20 min. The mean values obtained by control and treatment with each agent were compared using one-way ANOVA and Tukey's multiple comparison test (*p < 0.05, **p < 0.01).
DISCUSSION
Mechanisms of hippocampal serotonin release
This study demonstrated that hippocampal basal and K+-evoked serotonin releases require specific mechanisms for exocytosis. The basal serotonin release was regulated by N-VSCC, PKC, and syntaxin predominantly and by P-VSCC, PKA, and synaptobrevin weakly. In contrast to the basal release, K+-evoked serotonin release was regulated by P-VSCC, PKA, and synaptobrevin predominantly, but by N-VSCC, PKC, and syntaxin weakly. In addition, the stimulatory effect of the PKC activator on basal serotonin release was more strongly inhibited by GVIA and BoNT/C than by IVA and BoNT/B, respectively. The stimulatory effect of an adenylate cyclase activator (forskolin) on basal serotonin release was more strongly inhibited by IVA and BoNT/B than by GVIA and BoNT/C, respectively. In contrast, the stimulatory effect of PKC activator on K+-evoked serotonin release was more strongly inhibited by GVIA and BoNT/C than by IVA and BoNT/B, respectively. The stimulatory effect of adenylate cyclase activator on K+-evoked serotonin release was more strongly inhibited by IVA and BoNT/B than by GVIA and BoNT/C, respectively. Therefore, the present study proves the existence of two major functional complexes for hippocampal serotonin release; the first is the N-VSCC/PKC/syntaxin pathway, which is the major pathway for basal serotonin release, and the other is the P-VSCC/PKA/synaptobrevin pathway, which is the major pathway for K+-evoked serotonin release.
Mechanisms of N-type VSCC-related exocytosis
Several electrophysiological experiments have demonstrated that P-VSCC is present in high density at central synapses and transmitter release primarily requires P-VSCC, with N-VSCC playing a secondary role (Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994). However, this study demonstrated that the basal hippocampal serotonin release required the N-VSCC/PKC/syntaxin pathway predominantly.
Interaction of the synaptic protein interaction (synprint) site (Sheng et al., 1997) of N-VSCC with syntaxin and SNAP-25 has a biphasic Ca2+ dependence with maximal binding at a range of 10–30 μm Ca2+, which is near the threshold for neurotransmitter release (Sheng et al., 1996; Kim and Catterall, 1997). This interaction is regulated by PKC but not PKA (Yokoyama et al., 1997; Turner et al., 1999). The synprint site of N-VSCC binds to synaptotagmin in a Ca2+-independent manner (Sheng et al., 1996; Kim et al., 1997), but does not bind to synaptobrevin (Sheng et al., 1996). The synprint site of N-VSCC binds the dimetric complex of syntaxin/SNAP-25 and the trimetric complex syntaxin/SNAP-25/synaptobrevin in a Ca2+-dependent manner, with strongest binding in 4–18 μm Ca2+(Sheng et al., 1996). The synprint site of N-VSCC and synaptotagmin competitively interacts with syntaxin (Sheng et al., 1997). However, maximum binding of syntaxin to synaptotagmin requires a higher concentration of Ca2+ in the range of 100 μm[en]1 mm (Chapman et al., 1995; Li et al., 1995). When the Ca2+concentration increases beyond 30 μm, the interaction of syntaxin with the synprint site of N-VSCC is weakened, and interaction with synaptotagmin is strengthened. These previous observations can explain how the mechanisms of the basal hippocampal serotonin release are regulated by the N-VSCC/PKC/syntaxin pathway.
Mechanisms of P-type VSCC-related exocytosis
The α1A subunit of P-VSCC exists in two isoforms, designated rbA and BI when they were initially cloned (Mori et al., 1991; Starr et al., 1991). Binding of the synprint peptide from the rbA and BI isoforms of the P-VSCC shows different dependencies on Ca2+ concentration from the synprint site of N-VSCC (Kim and Catterall, 1997). The BI isoform binds to syntaxin, SNAP-25, and synaptotagmin Ca2+independently. By contrast, the rbA isoform binds to synaptotagmin Ca2+ dependently and to SNAP-25 Ca2+ independently, but it does not bind to syntaxin. The interaction between synaptobrevin, which does not bind to synaptotagmin (Schiavo et al., 1997), and P-VSCC has not been clarified; however, the cleavage of synaptobrevin decreased K+-evoked dopamine release but did not affect the dopamine release evoked by Ca2+ionophore ionomycin using rat brain synaptosomes (Fassio et al., 1999). In our previous study (Okada et al., 1998a), the basal striatal dopamine release was also regulated by N-VSCC predominantly; however, the K+-evoked striatal dopamine release was regulated by P-VSCC predominantly. This previous evidence, taken together with the present results, therefore suggests that the requirements for neurotransmitter release of the depolarization and the spontaneous stage mechanisms may be different. Furthermore, the depolarization-induced neurotransmitter release requires an unknown Ca2+-dependent process or some other influence such as synaptic membrane phosphorylation induced by depolarization (Sheng et al., 1998; Turner et al., 1999). Therefore, for an activation of the P-VSCC/PKA/synaptobrevin pathway, the synaptic membrane phosphorylation induced by depolarization or an unknown Ca2+-dependent process may be needed.
Effect of interaction between AD-R subtypes and synprint site on hippocampal basal serotonin release
This study demonstrated that the hippocampal basal serotonin release, which was N-VSCC/PKC/syntaxin high-sensitive and P-VSCC/PKA/synaptobrevin low-sensitive, was reduced by A1-R but was not affected by A2-R. The stimulatory effects of the PKC activator, PMA, and the adenylate cyclase activator, forskolin, on basal serotonin release were inhibited by the A1-R agonist, CCPA. The stimulatory effect of the A1-R antagonist, CPT, on basal serotonin release was inhibited by GVIA, CHR, and BoNT/C drastically, and H-89 and BoNT/B weakly, but was not affected by IVA. These results indicate that an activation of A1-R inhibits the N-VSCC/PKC/syntaxin pathway drastically, whereas A1-R inhibits actions of both PKA and synaptobrevin without affecting P-VSCC activity.
Under the condition of A1-R blockade, an activation of A2-R increased the basal serotonin release. The stimulatory effect of the A2-R agonist, PD125944, was inhibited by IVA, H-89, and BoNT/B drastically, and by GVIA, CHR, and BoNT/C weakly. Moreover, under the condition of A1-R being functional, activation of adenylate cyclase activity produced the inhibitory effect of the A2-R antagonist and the stimulatory effect of the A2-R agonist on the basal serotonin release. These results indicate that the PKA activity plays an important role in the mechanisms of A2-R synaptic modulation. Therefore, the inhibition of PKA activity induced by endogenous adenosine via A1-R may be the major mechanism for abolishment of the stimulatory effect of A2-R on basal serotonin release.
Effect of interaction between AD-R subtypes and synprint site on hippocampal K+-evoked serotonin release
The hippocampal K+-evoked serotonin release, which was N-VSCC/PKC/syntaxin low-sensitive and P-VSCC/PKA/synaptobrevin high-sensitive, was modulated by both A1-R and A2-R because the K+-evoked serotonin release was inhibited by CCPA and DMPX and enhanced by CPT and PD125944. Stimulatory effects of both CPT and PD125944 on K+-evoked serotonin release were inhibited by IVA, H-89, and BoNT/B predominantly, and by GVIA, CHR, and BoNT/C weakly. The stimulatory effect of forskolin on K+-evoked serotonin release was inhibited by both CCPA and DMPX. These results indicate that an activation of A1-R inhibits both the N-VSCC/PKC/syntaxin and P-VSCC/PKA/synaptobrevin pathways. A2-R affects the P-VSCC/PKA/synaptobrevin pathway more than the N-VSCC/PKC/syntaxin pathway.
Effect of interaction between A1-R and A2-R on hippocampal serotonin release
Colocalization and coexpression of mRNA encoding for A1-R and A2-R are found in hippocampus (Chuha et al., 1994). The present study suggests that there are multiple forms of functional interaction between these receptor subtypes so that the excitatory responses of A2-R on hippocampal basal and K+-evoked serotonin release were observed in the presence and absence, respectively, of A1-R antagonist conditions. This indicates that there is cross talk between A1-R and A2-R in the hippocampus and suggests that A2-R-mediated actions may be attenuated if these are concomitant activations of A1-R by endogenous adenosine via inhibition of PKA activity. The A2-R mediates attenuation of A1-R-mediated actions (Chuha et al., 1994), and A1-R-mediated inhibition of A2-R actions has been suggested (Abbracchio et al., 1992) on the grounds that A1-R desensitization is accompanied by a time-dependent amplification of A2-R-mediated stimulation of adenylate cyclase activity (Ribeiro, 1999). Furthermore, the excitatory response of A2-R is increased in the presence of A1-R antagonists (Correia-de-Sa and Ribeiro, 1994). However, these previous findings can explain the interaction between A1-R and A2-R on basal serotonin release partially but not fully because the interaction must be mediated by the activities of presynaptic N-VSCC/PKC/syntaxin and P-VSCC/PKA/synaptobrevin pathways.
Conclusion
In this study, we showed that hippocampal serotonin release is composed of at least two types of processes, N-VSCC/PKC/syntaxin high-sensitive and P-VSCC/PKA/synaptobrevin low-sensitive basal release (spontaneous release), as well as N-VSCC/PKC/syntaxin low-sensitive and P-VSCC/PKA/synaptobrevin high-sensitive K+-evoked release (depolarization-related release). An activation of A1-R inhibits both N-VSCC/PKC/syntaxin and P-VSCC/PKA/synaptobrevin activities, resulting in reduction of both basal and K+-evoked serotonin releases. In contrast, an activation of A2-R enhances P-VSCC/PKA/synaptobrevin, resulting in elevation of basal serotonin release, under the condition of preventing the inhibition of PKA activity by A1-R. An activation of A2-R produces the enhancement of K+-evoked serotonin release, under the condition of A1-R being functional, via an activation of PKA induced by K+-evoked stimulation. Thus, this study suggests that the interaction between A2-R-mediated potentiation of P-VSCC/PKA/synaptobrevin and A1-R-mediated attenuation of N-VSCC/PKC/syntaxin plays an important role in hippocampal serotonergic transmission, and the stimulatory effects of A2-R on basal serotonin release may be masked by the inhibition of PKA induced by activation of A1-R.
Footnotes
This study was supported by Grants-in-Aid for Scientific Research 05454309 and 11770532 from the Japanese Ministry of Education, Science and Culture, a grant from the Hirosaki Research Institute for Neurosciences, a grant from the Pharmacopsychiatry Research Foundation, and a grant from the Japan Epilepsy Research Foundation.
Correspondence should be addressed to Dr. Motohiro Okada, Department of Neuropsychiatry, School of Medicine, Hirosaki University, Hirosaki 036-8216, Japan. E-mail:okadamot@cc.hirosaki-u.ac.jp.
REFERENCES
- 1.Abbracchio MP, Fogliatto G, Paoletti AM, Rovati GE, Cattabeni F. Prolonged in vitro exposure of rat brain slices to adenosine analogues: selective desensitization of adenosine A1 but not A2 receptors. Eur J Pharmacol. 1992;227:317–324. doi: 10.1016/0922-4106(92)90010-s. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosio AF, Malva JO, Carvalho AP, Carvalho CM. Inhibition of N-, P/Q- and other types of Ca2+ channels in rat hippocampal nerve terminals by the adenosine A1 receptor. Eur J Pharmacol. 1997;340:301–310. doi: 10.1016/s0014-2999(97)01451-9. [DOI] [PubMed] [Google Scholar]
- 3.Barg J, Belcheva MM, Coscia CJ. Evidence for the implication of phosphoinositol signal transduction in μ-opioid inhibition of DNA synthesis. J Neurochem. 1992;59:1145–1152. doi: 10.1111/j.1471-4159.1992.tb08357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barraco RA, Stefano GB. Pharmacological evidence for the modulation of monoamine release by adenosine in the invertebrate nervous system. J Neurochem. 1990;54:2002–2006. doi: 10.1111/j.1471-4159.1990.tb04904.x. [DOI] [PubMed] [Google Scholar]
- 5.Bergquist F, Jonason J, Pileblad E, Nissbrandt H. Effects of local administration of L-, N-, and P/Q-type calcium channel blockers on spontaneous dopamine release in the striatum and the substantia nigra: a microdialysis study in rat. J Neurochem. 1998;70:1532–1540. doi: 10.1046/j.1471-4159.1998.70041532.x. [DOI] [PubMed] [Google Scholar]
- 6.Capogna M, McKinney RA, O'Connor V, Gahwiler BH, Thompson SM. Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ. The alpha-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron. 1995;14:591–597. doi: 10.1016/0896-6273(95)90315-1. [DOI] [PubMed] [Google Scholar]
- 8.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 9.Chuha RA, Johansson B, van der Ploeg I, Sebastiao AM, Ribeiro JA, Fredholm BB. Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res. 1994;649:208–216. doi: 10.1016/0006-8993(94)91066-9. [DOI] [PubMed] [Google Scholar]
- 10.Correia-de-Sa P, Ribeiro JA. Evidence that the presynaptic A2A-adenosine receptor of the rat motor nerve endings is positively coupled to adenylate cyclase. Naunyn Schmiedebergs Arch Pharmacol. 1994;350:514–522. doi: 10.1007/BF00173021. [DOI] [PubMed] [Google Scholar]
- 11.Correia-de-Sa P, Timoteo MA, Ribeiro JA. Presynaptic A1 inhibitory A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol. 1996;76:3910–3919. doi: 10.1152/jn.1996.76.6.3910. [DOI] [PubMed] [Google Scholar]
- 12.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 13.Fassio A, Sala R, Bonanno G, Marchi M, Raiteri M. Evidence for calcium-dependent vesicular transmitter release insensitive to tetanus toxin and botulinum toxin type F. Neuroscience. 1999;90:893–902. doi: 10.1016/s0306-4522(98)00475-8. [DOI] [PubMed] [Google Scholar]
- 14.Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- 15.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 16.Kaneko S, Akaike A, Satoh M. Differential regulation of N- and Q-type Ca2+ channels by cyclic nucleotides and G-proteins. Life Sci. 1998;62:1543–1547. doi: 10.1016/s0024-3205(98)00104-0. [DOI] [PubMed] [Google Scholar]
- 17.Kim DK, Catterall WA. Ca2+-dependent and -independent interactions of the isoforms of the α1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc Natl Acad Sci USA. 1997;94:14782–14786. doi: 10.1073/pnas.94.26.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Ullrich B, Zhang JZ, Anderson RGW, Brose N, Südhof TC. Ca2+-dependent and -independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 19.Linden J. Purinergic system. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic neurochemistry: molecular, cellular, and medical aspects. Raven; New York: 1994. pp. 401–416. [Google Scholar]
- 20.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 21.Luebke JI, Dunlap K, Turner TJ. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 1993;11:895–902. doi: 10.1016/0896-6273(93)90119-c. [DOI] [PubMed] [Google Scholar]
- 22.Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 23.Niemann H, Blasi J, Jahn R. Clostridial neurotoxins: new tools for dissecting exocytosis. Trends Cell Biol. 1995;4:179–185. doi: 10.1016/0962-8924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 24.Oishi K, Yamaguchi M. Effect of phorbol 12-myristate 13-acetate on Ca2+-ATPase activity in rat liver nuclei. J Cell Biochem. 1994;55:168–172. doi: 10.1002/jcb.240550203. [DOI] [PubMed] [Google Scholar]
- 25.Okada M, Kawata Y, Kiryu K, Mizuno K, Wada K, Tasaki H, Kaneko S. Effects of adenosine receptor subtypes on hippocampal extracellular serotonin level and serotonin reuptake activity. J Neurochem. 1997;69:2581–2588. doi: 10.1046/j.1471-4159.1997.69062581.x. [DOI] [PubMed] [Google Scholar]
- 26.Okada M, Wada K, Kiryu K, Kawata Y, Mizuno K, Tasaki H, Kaneko S. Effects of Ca2+ channel antagonists on striatal dopamine and DOPA release, studied by in vivo microdialysis. Br J Pharmacol. 1998a;123:805–814. doi: 10.1038/sj.bjp.0701675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada M, Kawata Y, Mizuno K, Wada K, Kondo T, Kaneko S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br J Pharmacol. 1998b;124:1277–1285. doi: 10.1038/sj.bjp.0701941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada M, Kawata Y, Murakami T, Wada K, Mizuno K, Kondo T, Kaneko S. Differential effects of adenosine receptor subtypes on release and reuptake of hippocampal serotonin. Eur J Neurosci. 1999a;11:1–9. doi: 10.1046/j.1460-9568.1999.00415.x. [DOI] [PubMed] [Google Scholar]
- 29.Okada M, Kawata Y, Murakami T, Wada K, Mizuno K, Kaneko S. Interaction between purinoceptor subtypes on hippocampal serotonergic transmission using in vivo microdialysis. Neuropharmacology. 1999b;38:707–715. doi: 10.1016/s0028-3908(98)00226-3. [DOI] [PubMed] [Google Scholar]
- 30.Olah ME, Stiles GL. Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- 31.Pierce RC, Kalivas PW. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. J Neurosci. 1997;17:3254–3261. doi: 10.1523/JNEUROSCI.17-09-03254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popoli P, Betto P, Reggio R, Ricciarello G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol. 1995;287:215–217. doi: 10.1016/0014-2999(95)00679-6. [DOI] [PubMed] [Google Scholar]
- 33.Ribeiro JA. Adenosine A2A receptor interactions with receptors for other neurotransmitters and neuromodulators. Eur J Pharmacol. 1999;375:101–113. doi: 10.1016/s0014-2999(99)00230-7. [DOI] [PubMed] [Google Scholar]
- 34.Satoh Y, Hirashima N, Tokumaru H, Kirino Y. Activation of adenosine A1 and A2 receptors differentially affects acetylcholine release from electric organ synaptosomes by modulating calcium channels. Neurosci Res. 1997;29:325–333. doi: 10.1016/s0168-0102(97)00104-1. [DOI] [PubMed] [Google Scholar]
- 35.Schiavo G, Stenbeck G, Rothman JE, Sollner TH. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc Natl Acad Sci USA. 1997;94:997–1001. doi: 10.1073/pnas.94.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- 38.Sheng ZH, Yokoyama CT, Catterall WA. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci USA. 1997;94:5405–5410. doi: 10.1073/pnas.94.10.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng ZH, Westenbroek RE, Catterall WA. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J Bioenerg Biomembr. 1998;30:335–345. doi: 10.1023/a:1021985521748. [DOI] [PubMed] [Google Scholar]
- 40.Sollner T, Whiteheart SW, Brunner M, Erdjument Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 41.Starr TVB, Prystay W, Snutch TP. Primary structure of a calcium channel that is highly expressed in the rat cerebellum. Proc Natl Acad Sci USA. 1991;88:5621–5625. doi: 10.1073/pnas.88.13.5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sudhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 44.Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- 45.Umemiya M, Berger AJ. Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in rat brainstem. Neuron. 1994;13:1439–1446. doi: 10.1016/0896-6273(94)90429-4. [DOI] [PubMed] [Google Scholar]
- 46.Von Lubitz DK. Adenosine and cerebral ischemia: therapeutic future or death of a brave concept? Eur J Pharmacol. 1999;365:9–25. doi: 10.1016/s0014-2999(98)00788-2. [DOI] [PubMed] [Google Scholar]
- 47.Westerink BH, Damsma G, de Vries JB. Effect of ouabain applied by intrastriatal microdialysis on the in vivo release of dopamine, acetylcholine, and amino acids in the brain of conscious rats. J Neurochem. 1989;52:705–712. doi: 10.1111/j.1471-4159.1989.tb02512.x. [DOI] [PubMed] [Google Scholar]
- 48.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 49.Williams M. Adenosine receptor agonists and antagonists. In: Stone T, editor. Adenosine in the nervous system. Academic; San Diego: 1991. pp. 137–171. [Google Scholar]
- 50.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 51.Wu LG, Borst JG, Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci USA. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yawo H, Chuhma N. Preferential inhibition of toxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y. Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J Neurosci. 1999;19:2432–2441. doi: 10.1523/JNEUROSCI.19-07-02432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha-1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 57.Zetterstrom T, Fillenz M. Adenosine agonists can both inhibit and enhance in vivo striatal dopamine release. Eur J Pharmacol. 1990;180:137–143. doi: 10.1016/0014-2999(90)90601-2. [DOI] [PubMed] [Google Scholar]