

Abstract
Modulation of the release probability of releasable vesicles in response to Ca2+ influx (ProbCa) is involved in mediating several forms of synaptic plasticity, including short-term depression, short-term augmentation, and potentiation induced by protein kinases. Given such an important role, however, the mechanism underlying modulation of the ProbCais unclear. We addressed this question by investigating how the activation of protein kinase C modulates the ProbCa at a calyx-type nerve terminal in rat brainstem. Various lengths of step depolarization were applied to the nerve terminal to evoke different amounts of Ca2+ currents and capacitance jumps, the latter of which reflect vesicle release. The relationship between the capacitance jump and the Ca2+ current integral was sigmoidal and was fit well with a Hill function. The sigmoidal relationship was shifted significantly to the left during the application of the PKC activator 12-myristate 13-acetate (PMA), suggesting that PMA increases the apparent affinity of the release machinery to Ca2+. This effect was blocked in large part by the application of the PKC inhibitor bisindolylmaleimide, suggesting that the effect is mediated mainly by the activation of PKC. We also found that PMA increased the rate of miniature EPSCs evoked by the application of hypertonic sucrose solution, which triggers release downstream of the Ca2+ influx. Taken together, our results suggest that PKC enhances the apparent affinity of the release machinery to Ca2+ by a mechanism downstream of the binding between Ca2+ and its sensor. These results have provided the first example of the mechanisms underlying modulation of the ProbCa.
Keywords: protein kinase C, calyx of Held, release probability, capacitance measurement, releasable pool, vesicle mobilization
Activation of protein kinase C (PKC) enhances synaptic transmission at a variety of synapses (Malenka et al., 1986; Shapira et al., 1987; Minota et al., 1991; Bachoo et al., 1992; Capogna et al., 1995; Redman et al., 1997; Stevens and Sullivan, 1998; Hori et al., 1999; Yawo, 1999; Honda et al., 2000; Oleskevich and Walmsley, 2000) and may underlie certain forms of synaptic plasticity (Byrne and Kandel, 1996; Son and Carpenter, 1996). Evidence suggests that the enhancement is mediated by an increase of transmitter release (Malenka et al., 1987; Shapira et al., 1987; Capogna et al., 1995;Gillis et al., 1996; Redman et al., 1997). Although the increased transmitter release might be caused in part by modulation of voltage-gated ion channels leading to an increase of presynaptic Ca2+ influx during action potentials (Byrne and Kandel, 1996; Majewski and Iannazzo, 1998; Honda et al., 2000), it is caused in large part by mechanisms downstream of the Ca2+ influx at many synapses (Stevens and Sullivan, 1998; Hori et al., 1999; Yawo, 1999; Honda et al., 2000). After the Ca2+ influx, transmitter release depends on two factors, the size of a pool of vesicles immediately available for release (called releasable pool or readily releasable pool) and the averaged release probability of vesicles in this pool in response to the Ca2+ influx (ProbCa). Which of these two factors is upregulated by PKC? By measurements of the membrane capacitance in chromaffin cells, it was found that PKC enhances vesicle exocytosis by increasing the releasable pool size (Gillis et al., 1996). A similar result was observed at cultured hippocampal synapses, in which the releasable pool size is defined as release evoked by 4–5 sec of hypertonic sucrose application (Stevens and Sullivan, 1998). In contrast, by an examination of EPSCs evoked by trains of electrical stimulation at calyx-type synapses, it was suggested that PKC may enhance transmitter release by increasing the ProbCa (Yawo, 1999; Oleskevich and Walmsley, 2000). In these studies, however, the conclusion in large part was based on the result that the EPSC reached a maximum at a high extracellular Ca2+ solution and that PKC did not increase the EPSC further in this condition. Under high release conditions a maximal EPSC may be reached because postsynaptic receptors are saturated and desensitized (Sun and Wu, 2001). Thus it remains inconclusive whether PKC enhances transmitter release by increasing the releasable pool size or the ProbCa in these studies (Yawo, 1999; Oleskevich and Walmsley, 2000).
In the present work we studied how PKC enhances synaptic transmission at a calyx-type synapse in the rat medial nucleus of the trapezoid body (MNTB). To study more directly how activation of PKC enhances transmitter release, we measured transmitter release by monitoring the change in the membrane capacitance at the nerve terminal (Sun and Wu, 2001), which is proportional to the surface area of the membrane and thus proportional to the number of vesicles released (Albillos et al., 1997; Von Gersdorff et al., 1998; Sun and Wu, 2001). This approach avoids the complication of saturation and desensitization of postsynaptic receptors. With this approach we found that activation of PKC enhanced release not by increasing the releasable pool size but by increasing the ProbCa.
Modulation of the ProbCa is often a pathway by which synaptic strength is regulated. For example, modulation of the ProbCa is involved in mediating short-term synaptic depression (Wu and Borst, 1999; Burrone and Lagnado, 2000), short-term synaptic augmentation (Stevens and Wesseling, 1999), and enhancement of transmitter release that is induced by the activation of protein kinase A (Trudeau et al., 1996). Although modulation of the ProbCa plays an important role in regulating synaptic strength, its underlying mechanisms remain unclear. The present work was aimed at investigating the mechanisms underlying modulation of the ProbCa. Three potential mechanisms may modulate the ProbCa: (1) modulation of the affinity of the Ca2+sensor to Ca2+, (2) modulation of the number of Ca2+ ions required to bind the Ca2+ sensor and trigger release, and (3) modulation of any step at the release machinery downstream of the Ca2+ sensor. By examining the relationship between the capacitance jump and the Ca2+influx, we found that PKC increased the apparent affinity of the release machinery to Ca2+. By applying hypertonic sucrose solution to trigger release independently of the binding between Ca2+ and its sensor (Rosenmund and Stevens, 1996), we found that PKC enhanced transmitter release by a mechanism downstream of the binding between Ca2+ and its sensor. These results suggest that enhancement of the release machinery downstream of the Ca2+ sensor is involved in increasing the apparent affinity of the release machinery to Ca2+ and thus the ProbCa.
MATERIALS AND METHODS
Recordings of presynaptic Ca2+currents and capacitance.Wistar rats (8–12 d old) were decapitated. Parasagittal slices 200 μm thick were cut from the auditory brainstem with a vibratome. Recordings were made at room temperature in a solution that pharmacologically isolated Ca2+ currents (Borst et al., 1995). This solution contained (in mm) 105 NaCl, 20 TEA-Cl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 25 dextrose, 0.4 ascorbic acid, 3 myo-inositol, 2 sodium pyruvate, 0.001 tetrodotoxin (TTX), and 0.1 3,4-diaminopyridine, pH 7.4, when bubbled with 95% O2/5% CO2. The presynaptic pipette (3.5–5 MΩ) solution contained (in mm) 125 Cs-gluconate, 20 CsCl, 4 Mg-ATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, and 0.05 BAPTA, pH-adjusted to 7.2 with CsOH. Presynaptic whole-cell recordings were made with an EPC-9 amplifier (HEKA Electronics, Lambrecht, Germany). The series resistance (<15 MΩ) was compensated by 60%. Holding potential was −80 mV, and the potential was corrected for a liquid junction potential of −11 mV between the extracellular and the pipette solutions (also applies to postsynaptic recordings). Currents were low-pass filtered at 5 kHz and digitized at 20 kHz (also applies to postsynaptic recordings) with a 16 bit analog-to-digital converter (Instrutech, Great Neck, NY).
The selection of calyces and capacitance recordings has been described in detail in our recent study (Sun and Wu, 2001). Briefly, calyces that had a basal capacitance <22 pF and showed approximately a single exponential decay in passive relaxation current, as judged by eye, were used. The capacitance was measured with the EPC-9 amplifier together with the software lock-in amplifier (PULSE, HEKA Electronics). A sinusoidal stimulus was applied in addition to the DC holding potential (−80 mV). The peak-to-peak voltage of the sine wave was <60 mV to avoid activation of the Ca2+ currents (Borst et al., 1995; Wu et al., 1998). The resulting current was processed via the Lindau–Neher technique (Lindau and Neher, 1988;Gillis, 1995) to give estimates of the membrane capacitance, membrane conductance, and the series conductance. The sine wave frequency was 1000 Hz. The reversal potential of the measured DC current was assumed to be 0 mV (Gillis, 1995). During step depolarization the capacitance was not measured. The capacitance jump was measured as the difference between the averaged capacitance value in 30–60 msec after stimulation and the baseline value. The capacitance jump returned to the baseline in a few seconds to tens of seconds (Sun and Wu, 2001). Therefore, measurements of capacitance jumps were not affected by the relatively slow endocytosis. The interval between two voltage commands was at least 30 sec to avoid short-term synaptic plasticity induced by the previous voltage command. All capacitance traces shown in the figures were taken from single recordings and were low-pass filtered at 100 Hz.
Recordings of EPSCs and mEPSCs. Wistar rats (8–12 d old) were decapitated. Transverse slices 200 μm thick were cut from the auditory brainstem with a vibratome. Because the axon that connects the calyx is approximately parallel to the surface of the transverse slice, there are calyces connected with a longer axon that can be stimulated electrically with a bipolar electrode positioned at the midline of the trapezoid body. Thus to induce an EPSC electrically (see Fig. 6), we applied a brief electrical stimulus (5–20 V, 0.1 msec) to induce an action potential in the axon and thus an EPSC at the postsynaptic neuron. [For the method of identifying such postsynaptic neurons, see Borst et al. (1995) and Wu et al. (1999).] To induce trains of mEPSCs with hypertonic solution, we positioned glass pipettes (1.5–2.5 MΩ) containing 2 m sucrose plus the bath solution described below close (<5 μm) to the surface of postsynaptic cells. The sucrose solution was pressure injected (0.2–10 sec, 5 psi) onto the surface of the synapse with a pneumatic picopump (PV830, World Precision Instruments, Sarasota, FL). The resulting mEPSCs were analyzed by a program (Jaejin Software, Leonia, NJ) with a threshold amplitude for detection set at 10 pA.
Fig. 6.

Release evoked by hypertonic sucrose solution and nerve stimulation share the same vesicle pool. A, A train of nerve stimulations (20 V, 0.1 msec at 100 Hz for 200 msec) was applied, followed at 300 msec after the train by a puff application of sucrose solution (2 m in the pipette) for 500 msec. The EPSC evoked by the train was truncated to see the mEPSCs clearly. Calibration also applies to B. B, The EPSC is evoked by a puff the same as in A but without a conditioning train of stimulation. C, Shown are the EPSCs evoked by two identical electrical trains (20 V, 0.1 msec at 100 Hz for 200 msec) applied to the nerve with an interval of 1 sec (left). The EPSCs evoked by the second train (right, top) were smaller than those evoked by the first train (right, bottom). The stimulation artifacts were blanked.
Whole-cell voltage-clamp recordings of EPSCs and mEPSCs at postsynaptic neurons were made at room temperature in a bath solution containing (in mm) 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 25 dextrose, 0.4 ascorbic acid, 3 myo-inositol, 2 sodium pyruvate, 0.05d-APV (NMDA receptor blocker), 0.01 bicuculline (GABAA receptor blocker), and 0.01 strychnine (glycine receptor blocker), pH 7.4, when bubbled with 95% O2/5% CO2. The postsynaptic pipette (2–3 MΩ) solution contained (in mm) 125 K-gluconate, 20 KCl, 4 Mg-ATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, and 0.5 EGTA, pH-adjusted to 7.2 with KOH. Whole-cell recordings were made with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). The holding potential was −80 mV. The series resistance (<12 MΩ) was compensated by 95% (lag, 10 μsec).
Reagents and statistical tests. Phorbol 12-myristate 13-acetate (PMA) was purchased from Sigma (St. Louis, MO) and bisindolylmaleimide (BIS) from Calbiochem (La Jolla, CA). PMA and BIS were dissolved first in DMSO and then diluted. The final concentration of DMSO did not exceed 0.1% and by itself had no effect on the capacitance jump and the EPSC. Data were expressed as means ± SE. The statistical test for calculation of p values was the Student's t test.
RESULTS
Measurement of the releasable pool size
We have found recently that a voltage step command of 10 msec from −80 to +10 mV evoked a maximal release the same as a 30 msec step to +10 mV in an extracellular solution containing 2 mmCa2+ [Sun and Wu (2001), their Fig. 4]. This result suggests that a pool of releasable vesicles can be depleted in 10 msec. To confirm this suggestion further, we determined whether release evoked by a 10 msec step to +10 mV was increased further when the extracellular Ca2+ concentration was increased. The calyx of Held was whole-cell voltage clamped in a bath solution that pharmacologically isolates Ca2+ currents. A step depolarization of 2 or 10 msec from the holding potential of −80 to +10 mV was applied alternately to the calyx to trigger vesicle release. Vesicle release was measured as the capacitance jump after the step depolarization. When the extracellular Ca2+ concentration was increased from 2 to 4 mm, the peak Ca2+ current at the end of a 2 or 10 msec step depolarization increased by 27 ± 4 and 33 ± 3% (n = 4) (Fig. 1), respectively. The capacitance jump evoked by the 2 msec step depolarization increased by 74 ± 4%, whereas the capacitance jump evoked by the 10 msec step depolarization was increased only by 8 ± 4% (n = 4; p = 0.06). This result further supports our hypothesis that a 10 msec step depolarization in normal (2 mm) extracellular Ca2+ is sufficient to deplete the releasable pool.
Fig. 1.

The capacitance jumps recorded in different extracellular Ca2+ concentrations. A, The Ca2+ current (top) and the capacitance jump (bottom) evoked by a 2 msec step from −80 to +10 mV in extracellular solutions containing 2 mm(solid trace) and 4 mm (dotted trace) Ca2+. Both the Ca2+ current and the capacitance jump were increased significantly in 4 mm Ca2+.B, The Ca2+ current (top) and the capacitance jump (bottom) evoked by a 10 msec step from −80 to +10 mV in extracellular solutions containing 2 mm (solid trace) and 4 mm (dotted trace) Ca2+. Only the Ca2+ current, but not the capacitance jump, was increased significantly in 4 mmCa2+. Data in A and Bwere obtained from the same calyx.
When the duration of the step depolarization was increased from 10 to 100–200 msec, the capacitance jump was increased by >50% (our unpublished results), suggesting that part of the releasable pool can be replenished rapidly in 100–200 msec (see also Fig. 4). This result argues against the possibility that the similar capacitance jumps observed during the 10–30 msec depolarization (Sun and Wu, 2001) or during a 10 msec depolarization in 2 and 4 mmextracellular Ca2+ (Fig. 1) are caused by adaptation of the Ca2+ sensor (Hsu et al., 1996) or by an unknown mechanism that is turned on to inhibit the release machinery during prolonged stimulation. We conclude that the releasable pool size can be estimated as the capacitance jump evoked by a 10 msec step depolarization.
Fig. 4.
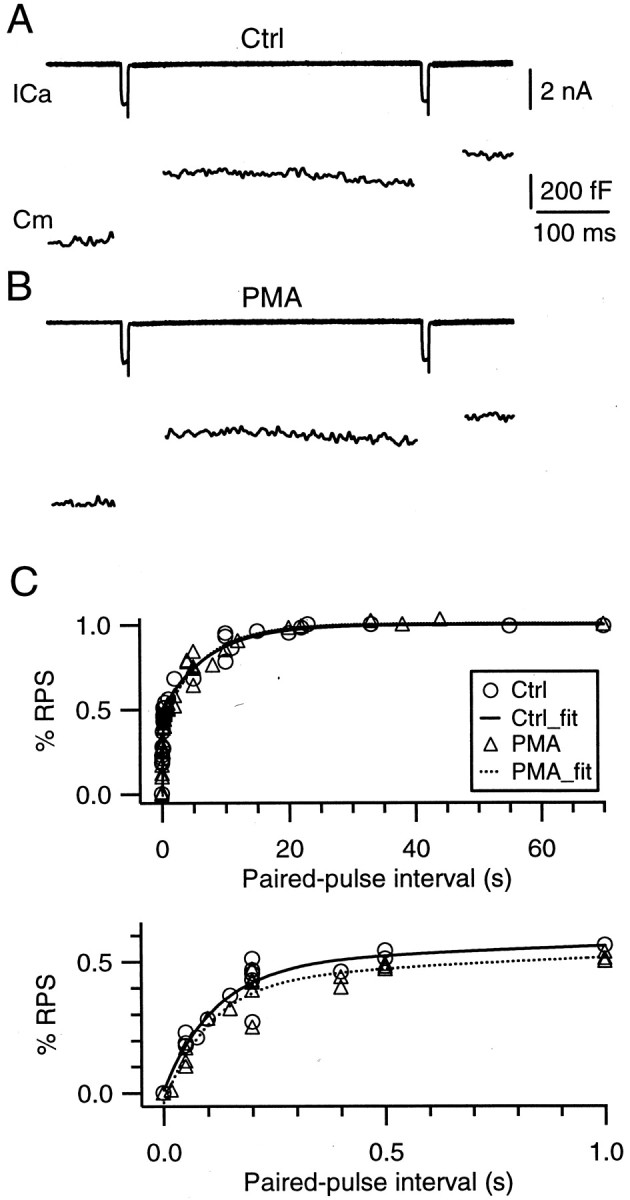
PMA does not affect the rate of vesicle mobilization. A, B, Sample recordings of Ca2+ currents (top) and capacitance jumps (bottom) induced by a pair of 10 msec step depolarizations (from −80 to +10 mV) with an interval of 400 msec before (A, Ctrl) and during (B) the application of 100 nm PMA. The labels and scales in A apply to B.C, The ratio between the second and the first ΔCm as a function of the paired pulse interval obtained in control and in the presence of PMA (100 nm). Data were obtained from experiments similar to those shown inA and B from 11 synapses. In control, the data were fit with a double-exponential function with time constants of 0.11 and 7.14 sec, respectively (solid curve). PMA does not affect these time constants and the relative contribution of these two components (dotted curve). The topand bottom panels show the same data in different scales.
A PKC activator PMA increases the apparent affinity of the release machinery to Ca2+
Bath application of the PKC activator PMA (100 nm) for 10 min increased the capacitance jump evoked by the 2 msec step depolarization by 82 ± 18% (n = 6) (Fig.2A). This effect reached the steady state in a <10 min application of the drug (data not shown) (but see Hori et al., 1999) and was not accompanied by any significant change in the amplitude or the integral of the Ca2+ current (p > 0.4, t test; n = 6) (see Figs.2A,B, 4A,B). In contrast, 100 nm PMA did not increase significantly the capacitance jump evoked by a 10 msec step depolarization (2 ± 2%; n = 6; p = 0.25, ttest) (Fig. 2B). Thus PMA enhanced the transmitter release evoked during a 2 msec step depolarization not by increasing the releasable pool size but by increasing the fraction of the releasable pool being released, i.e., the release probability of releasable vesicles. Because PMA did not affect the presynaptic Ca2+ current, the enhancement must occur downstream of the Ca2+ influx. In other words, PMA increased the ProbCa.
Fig. 2.
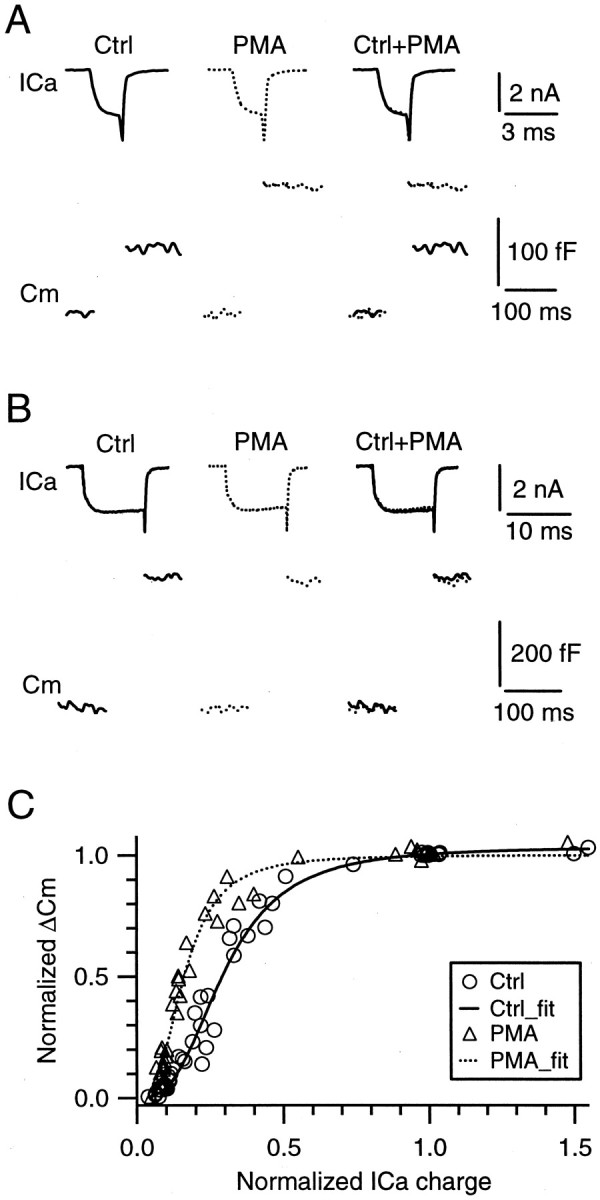
PMA increases the capacitance jump by increasing the apparent affinity of the release machinery to Ca2+, but not the releasable pool size.A, B, Sample traces of Ca2+ currents (top) and capacitance jumps (bottom) induced by a 2 msec (A) and a 10 msec (B) step depolarization from −80 to +10 mV before (left, solid trace) and during the application of 100 nm PMA for 10 min (middle, dotted trace). They are superimposed on the right for comparison. C, The relationship between the capacitance jump (ΔCm) and the charge of the Ca2+ current (ICa) obtained before (Ctrl, circles) and during the application of PMA (triangles) for at least 10 min (n = 12 synapses). Before the data were pooled from different synapses, the data were normalized to the value obtained during the 10 msec step depolarization to +10 mV in the control condition. Both the data in control and in the presence of PMA were fit with a Hill equation (see Eq. 1 in Results). The application of PMA did not change two parameters in the equation, theRPS and the n, but decreased the EC50 from 0.30 (solid curve,Ctrl_fit) to 0.16 (dotted curve,PMA_fit).
The increased ProbCa may be caused by either an increased apparent affinity of the release machinery to Ca2+ or a decrease in the number Ca2+ ions required to bind the Ca2+ sensor that triggers release. To identify which of these two mechanisms increases the ProbCa, we applied various lengths of step depolarizations to various voltages (0–20 mV) to the calyx to induce different amounts of Ca2+ influx and thus different amounts of vesicle release. The resulting capacitance jumps were plotted versus the Ca2+ current integral (integrated for 10 msec) in the control condition (Fig.2C, circles) and during the application of 100 nm PMA for at least 10 min (Fig. 2C,triangles). Before the data were pooled from different synapses into Figure 2C, the data were normalized to the capacitance jump and the Ca2+ current evoked by the 10 msec step depolarization in control at the same synapse. Both the data in control and in the presence of PMA were fit with the Hill function:
| Equation 1 |
where ΔCm is the normalized capacitance jump,RPS is the normalized releasable pool size or the maximal capacitance jump, ICaintegral is the integral of the Ca2+ current normalized to that evoked by a 10 msec step depolarization in control at the same synapse, EC50 is a value ofICaintegral at which ΔCm is 50% of RPS, and n is the Hill coefficient. In control theRPS, EC50, and n were 1.03, 0.30, and 2.4, respectively. In the presence of PMA these parameters were 1.02, 0.16, and 2.6, respectively. Evidently, PMA decreased the EC50 to approximately one-half of the control value without significantly affecting the Hill coefficient, the latter of which is interpreted widely as the number of Ca2+ ions required to bind the Ca2+ sensor to trigger release (Reid et al., 1998; Schneggenburger et al., 1999). Thus these results suggest that PMA enhanced transmitter release by increasing the apparent affinity of the release machinery to Ca2+, but not by modifying the number of Ca2+ions required to bind the Ca2+ sensor to trigger release.
A PKC inhibitor in large part blocks the effect of PMA on the capacitance jump
Phorbol esters may enhance synaptic transmission via two mechanisms, one by the activation of PKC and the other by the interaction with the synaptic protein Munc13-1 (Betz et al., 1998; Hori et al., 1999). Previous works at calyx-type synapses, including the MNTB synapse, indicate that phorbol esters (PMA and phorbol 1,2-dibutrate), but not their inactive forms (4α-PMA and 4α-phorbol 1,2-dibutrate), enhance the EPSC (Hori et al., 1999; Yawo, 1999;Oleskevich and Walmsley, 2000). The enhancement is blocked mainly or completely by PKC inhibitors, such as BIS, sphingosine, calphostin C, and a PKC inhibitor peptide (Hori et al., 1999; Yawo, 1999; Oleskevich and Walmsley, 2000). These results indicate that phorbol esters enhance synaptic transmission in large part by the activation of PKC at calyx-type synapses.
To confirm that PMA enhances the capacitance jump in large part by the activation of PKC, we applied a specific inhibitor of PKC, BIS (Toullec et al., 1991). The application of 1 μm BIS for 10 min slightly increased the ΔCm evoked by a 2 msec step (to +10 mV) by 10 ± 4% (n = 6; p = 0.04) (Fig. 3). The reason for the slight increase of the ΔCm is unclear.
Fig. 3.

A PKC antagonist BIS blocks in large part the effect of PMA on the capacitance jump. A, Sample recordings of presynaptic Ca2+ currents (ICa, top) and capacitance changes (Cm, bottom) evoked by a 2 msec presynaptic step from −80 to +10 mV in the control (left), in the presence of BIS (1 μm) for 10 min (middle left), and in the presence of BIS (1 μm) plus PMA (100 nm) for 10 min (middle right). These traces are superimposed on theright for comparison. B, Percentage of increase in the capacitance jump (ΔCm) evoked by a 2 msec step depolarization to +10 mV during the application of 1 μm BIS (n = 6), 1 μmBIS plus 100 nm PMA (n = 6), and 100 nm PMA (n = 6). The percentages refer to the changes in capacitance jumps during the application of the drug or drugs normalized to the value obtained before any drug application at the same synapse. Data are expressed as means ± SE.
In the presence of 1 μm BIS the application of PMA increased the ΔCm evoked by a 2 msec step to +10 mV by 27 ± 5% (n = 6; p = 0.02) (Fig.3) of the value obtained before the BIS application. This increase was significantly less than that (82 ± 18%; p = 0.02) (Fig. 3) measured in the absence of BIS, suggesting that PMA increases the capacitance jump in large part by the activation of PKC. This result is consistent with a recent study at MNTB synapses showing that phorbol esters enhance the EPSC mainly by the activation of PKC and partly by interaction with Munc13-1 (Hori et al., 1999).
PMA does not affect the rate of vesicle mobilization
Phorbol esters have been suggested to enhance the releasable pool size and the rate of vesicle mobilization from the reserve pool to the releasable pool at cultured hippocampal synapses and chromaffin cells (Stevens and Sullivan, 1998). Our results indicated that PMA did not increase the releasable pool size at the MNTB synapse (Fig. 2). In the following we examined whether PMA increases the rate of mobilization. A pair of 10 msec steps to +10 mV with intervals that varied from 0.05 to 70 sec were applied to the terminal, and the resulting capacitance jumps were recorded (Fig.4A). The first step depleted the releasable pool, and the second step depleted the vesicles that have been mobilized to the releasable pool during the interval between the two steps. Thus the ratio between the second and the first capacitance jump indicates the fraction of the releasable pool being replenished. Such ratios were plotted as a function of the interval and were fit well with the sum of two exponential functions. In the control the time constants were 0.11 and 7.14 sec, respectively, and the weights of the fast and the slow components were 48 and 52%, respectively (Fig. 4B). These results were consistent with the rate of mobilization estimated with measurements of the EPSC at the same synapse (Wu and Borst, 1999). After the application of 100 nm PMA for 10 min, the time constants were 0.10 and 6.67 sec, respectively, and the weights of the fast and the slow components were 46 and 54%, respectively (Fig. 4B). The results obtained in the presence and the absence of PMA were similar (Fig. 4), indicating that PMA did not affect the rate of vesicle mobilization.
PMA enhances transmitter release evoked by application of hypertonic sucrose solution
We have shown that PMA enhances transmitter release by increasing the apparent affinity of the release machinery to Ca2+ (Fig. 2). The increased apparent affinity may be caused by an increase in the affinity of the Ca2+ sensor to Ca2+ and/or by an enhancement of the release machinery at a step downstream of the Ca2+ sensor. To explore whether the latter mechanism is involved in mediating PMA-induced enhancement of the release, we applied hypertonic sucrose solution to trigger release. Release evoked by hypertonic sucrose solution is not affected when the voltage-dependent Ca2+ channels are blocked or when the Ca2+ buffer BAPTA is dialyzed to the nerve terminal to block effectively the action potential-evoked release (Rosenmund and Stevens, 1996). These results suggest that hypertonic sucrose solution triggers release independently of the binding between Ca2+ and its sensor (Rosenmund and Stevens, 1996). If PMA increases the release evoked by hypertonic solution, it suggests that PMA acts at a site downstream of the binding between Ca2+ and its sensor. Thus we determined whether PMA increases the release evoked by the application of hypertonic solution. For two reasons we monitored the release by measurements of the EPSC instead of the presynaptic capacitance jump. First, we applied the sucrose solution by puff application (see below), which generated an artifact in the capacitance recording that contaminated the capacitance signal (data not shown). Second, release evoked by puff application of hypertonic solution lasts for a few seconds (see below) during which significant endocytosis may occur (Stevens and Williams, 2000; Sun and Wu, 2001), which may result in a significant underestimate of exocytosis (Smith and Betz, 1996).
Postsynaptic MNTB cells on the surface (<10 μm from the top) of the slice were whole-cell voltage clamped at −80 mV. A glass pipette containing 2 m sucrose plus the bath solution was positioned close (<5 μm) to the cell. A pressure was applied for 0.2–10 sec to puff the sucrose solution (<1 μl) to the MNTB synapse. Such a puff induced a train of miniature EPSCs (mEPSCs) (Fig.5A). Consistent with the previous study (Rosenmund and Stevens, 1996), the frequency of mEPSCs was not affected by adding the nonspecific Ca2+ channel blocker CdCl2 (200 μm) into the bath solution (n = 6) (Fig. 5B). The application of the AMPA receptor blocker CNQX (10 μm) completely blocked the sucrose-induced mEPSCs (n = 4) (Fig. 5C).
Fig. 5.

Hypertonic sucrose application evokes mEPSCs.A, Shown are the mEPSCs induced by a 1 sec puff application of hypertonic sucrose solution (2 m sucrose plus the bath solution in the pipette). The puff application time is marked in C. B, The Ca2+ channel blocker CdCl2 (200 μm) did not block sucrose-induced mEPSCs.C, The non-NMDA glutamate receptor blocker CNQX (10 μm) blocked sucrose-induced mEPSCs. Calibration applies to all panels. Data in A–C were obtained from the same synapse.
The final concentration of sucrose at the synapse is unknown, because it depends on many factors such as the distance between the puff pipette tip and the synapse, the pipette tip diameter, the duration of application, and the geometry of the synapse. Thus the same concentration of sucrose in the pipette may induce quite different responses at different synapses. However, the results were repeatable at the same synapse (Fig. 5A,B). The mEPSCs continued to occur for ∼2–3 sec after puff application (Figs. 5A,B,6A,B), which may reflect the gradual decrease of sucrose concentration caused by diffusion of sucrose away from the synapse.
Hypertonic sucrose solution and nerve stimulation trigger release from the same releasable vesicle pool at cultured hippocampal synapses (Rosenmund and Stevens, 1996). To confirm this result at MNTB synapses, we positioned a bipolar electrode at the midline of the trapezoid body for stimulating the presynaptic axon. A train of nerve stimulations (20 V, 0.1 msec at 100 Hz for 200 msec) was applied to induce a train of EPSCs, followed at 300 msec after the end of the train by a puff application of hypertonic sucrose solution (2 m plus the bath solution) for 500 msec (Fig. 6A). The same puff also was applied without the conditioning train (Fig.6B). In control, the total number of mEPSCs in 3 sec during and after sucrose application was 206 ± 32 (n = 6). After the train of nerve stimulations the number was decreased to 129 ± 25 (or 63 ± 10%;p < 0.001; n = 6) (Fig.6A), and the mean amplitude did not change significantly (p = 0.47; n = 6). Similar results were observed in the presence of cyclothiazide that inhibits the desensitization of AMPA receptors (n = 2; data not shown). Consistently, when two identical electrical trains (20 V, 0.1 msec at 100 Hz for 200 msec) were applied to the nerve with an interval of 1 sec, the EPSCs evoked by the second train were depressed significantly (Fig. 5C). The sum of the amplitude of the EPSC evoked by each stimulus during the second train was 68 ± 9% (n = 4; p < 0.001) of that during the first train, similar to the decrease in the number of mEPSCs evoked by sucrose application. In addition, when the hypertonic sucrose solution (2 m in the pipette) was applied for 10 sec, the EPSC evoked by a single electrical stimulation or a train of stimulation (100 Hz for 200 msec) immediately after sucrose application was <25% of that evoked in control (n = 4; data not shown). Consistent with the previous study (Rosenmund and Stevens, 1996), these results suggest that vesicles released by hypertonic sucrose application and the electrical train share a common vesicle pool (Fig. 6) and that release evoked by hypertonic sucrose application is independent of the activation of voltage-gated Ca2+ channels (Fig. 5).
A 300 msec puff application of 2 m sucrose solution induced a train of ∼30–100 mEPSCs that occurred in ∼2–3 sec (Fig.7A). The application of 100 nm PMA for 10 min increased the number of mEPSCs (counted in 3 sec during and after puff application) by 154 ± 36% (p < 0.01; n = 5) (Fig.7A) without significantly affecting the amplitude of the mEPSC (p = 0.39; n = 5). This result suggests that PKC enhances release by a mechanism downstream of the binding between Ca2+ and its sensor.
Fig. 7.

PMA enhances the rate of mEPSCs evoked by hypertonic sucrose solution. A, The mEPSCs evoked by a puff application of hypertonic sucrose solution (2 m) for 300 msec in the control (top), in the presence of 100 nm PMA (middle), and in the presence of 10 μm CNQX (bottom). B, The mEPSC evoked by a puff application of hypertonic sucrose solution (2m) for 3 sec in the control (top), in the presence of 100 nm PMA (middle), and in the presence of 10 μm CNQX (bottom). The initial rise of the current is an artifact of the puff application because it was not blocked by CNQX (bottom trace). All data in this figure were obtained from the same synapse.
The percentage of increase in the number of mEPSCs decreased as the duration of the puff was prolonged. For example, a puff application of sucrose solution (2 m) for 3 sec induced a train of mEPSCs that persisted for ∼3 sec after the puff (Fig. 7B). The total number of mEPSCs that occurred during and 3 sec after puff application was increased by only 31 ± 9% (n = 5) (Fig. 7B), which is significantly lower than that (154 ± 36%; n = 5) obtained with a 300 msec puff application (p < 0.01). Because the frequency of mEPSCs was high (50–200 Hz) during long puff application, the number of mEPSCs may be underestimated because of simultaneous multiple quantal release. To avoid this potential problem, we compared the charge of the EPSC (integrated for 5–6 sec during and after sucrose application) before and after PMA application. A similar percentage of increase (36 ± 10%; n = 5) in the charge of the EPSC was observed, confirming that PKC-induced enhancement of release evoked by a long puff application is significantly smaller than that evoked by a brief puff application. These results are consistent with our finding that PMA enhanced release evoked by smaller stimulation to a larger extent than that evoked by larger stimulation, because PMA increased the apparent affinity of the release machinery to Ca2+, but not the releasable pool size (Fig. 2).
DISCUSSION
Modulation of the ProbCa versus modulation of the releasable pool size
By monitoring transmitter release with presynaptic membrane capacitance measurements at MNTB synapses, we provided direct evidence that PMA enhanced transmitter release at MNTB synapses. We found that PMA increased the ProbCa without affecting the releasable pool size (Fig. 2) or the rate of vesicle mobilization (Fig.4). This result is consistent with a previous study at the cholinergic synapse in the chick ciliary ganglion (Yawo, 1999), but it is in conflict with another study at hippocampal cultured synapses in which phorbol esters were suggested to increase the releasable pool size (Stevens and Sullivan, 1998). The reason for this apparent discrepancy is unclear. Because Hori et al. (1999) found that phorbol esters may enhance transmitter release by the activation of PKC and by interaction with another synaptic protein Munc13-1 at the MNTB synapse (Hori et al., 1999), differential distribution of PKC and Munc13-1 in different tissues has been postulated to account for the discrepancy (Hilfiker and Augustine, 1999). Combined with the result obtained by Hori et al. (1999), our result that PMA increased the ProbCabut not the releasable pool size at the same MNTB synapse argues against this hypothesis. This does not necessarily rule out the possibility that the discrepancy is attributable to a difference in synapses. However, as discussed below, the difference in the method used to define and estimate the releasable pool size might account for the discrepancy.
At cultured hippocampal synapses the releasable pool size is defined as the pool of vesicles released by a 4–5 sec application of hypertonic sucrose solution, subtracted by the number of vesicles that replenish the pool during sucrose application (Stevens and Sullivan, 1998). Because more than one-half of the releasable pool can be replenished in 4–5 sec (Dittman and Regehr, 1998; Stevens and Sullivan, 1998; Stevens and Wesseling, 1998; Wang and Kaczmarek, 1998; Wu and Borst, 1999), an accurate estimate of the releasable pool size relies on the estimate of replenishment. Vesicles that newly replenish the releasable pool may have a much lower release probability that takes >10 sec to recover (Wu and Borst, 1999; Burrone and Lagnado, 2000). In addition, in normal conditions the release probability of releasable vesicles is likely to be inhomogeneous with a fraction of vesicles at low release probability (Sakaba and Neher, 2001). It is possible that hypertonic sucrose application, which is far less efficient in triggering release than a direct depolarization at the nerve terminal, preferentially releases vesicles with a higher release probability. Activation of PKC may convert reluctant vesicles, such as vesicles with a low release probability in normal conditions and vesicles that just replenish the releasable pool during a 4–5 sec sucrose application, into vesicles ready for release by hypertonic solution, resulting in an apparent increase in the releasable pool size. For the same reason PKC may convert reluctant vesicles that have replenished the releasable pool during a pair of hypertonic shocks used to estimate the rate of mobilization, resulting in an apparent increase in the rate of mobilization. Our results suggest an approach to test whether PKC enhances the ProbCa at small synapses where the ProbCa is difficult to measure. This approach is to determine whether PKC increases release evoked by different intensities of hypertonic sucrose application to different degrees, as we show in Figure 7 for MNTB synapses.
Modulation of the ProbCa occurs at a step downstream of the binding between Ca2+ and its sensor
We found that PMA shifted the sigmoidal relationship between the capacitance jump and the Ca2+ current integral to the left (Fig. 2). When these sigmoidal curves were fit with the Hill function (see Eq. 1), the EC50 was reduced by approximately one-half by PMA (Fig. 2). These results suggest that PMA increases the ProbCa by increasing the apparent affinity of the release machinery to Ca2+. If we assume a linear relationship between the local Ca2+ concentration that triggers transmitter release and the Ca2+current integral, PMA would increase the affinity of the release machinery to Ca2+ to approximately two times the control value. However, whether this relationship is linear is currently unclear, making it difficult to estimate quantitatively the change in the apparent affinity of the release machinery to Ca2+. An alternative hypothesis that might account for the enhancement of the ProbCa is a rearrangement of Ca2+ channels such that a larger fraction of them is located physically closer to active zones where release occurs. This possibility is highly unlikely because PMA enhanced transmitter release that was induced by hypertonic solution (Fig. 7), which triggered release independently of activation of Ca2+ channels (Fig. 5) and the binding between Ca2+ and its sensor (Rosenmund and Stevens, 1996). In addition, release evoked by hypertonic sucrose solution shares a common pool of vesicles releasable by nerve stimulation or activation of Ca2+ channels (Fig. 6) (Rosenmund and Stevens, 1996). We conclude that PMA increased the apparent affinity of the release machinery to Ca2+ and thus the ProbCa at least partly by a mechanism downstream of the binding between Ca2+ and its sensor. Our results do not allow us to rule out completely the possibility that an increase in the affinity of the Ca2+ sensor also contributes to the increase in the ProbCa. To our knowledge this is the first example showing that modulation of the release machinery downstream of the Ca2+ sensor is involved in mediating modulation of the ProbCa and thus the synaptic strength. Such a mechanism may not be limited only to the PKC signaling pathway. Modulation of the ProbCamay be a common pathway by which synaptic strength is regulated during synaptic modulation by neurotransmitters and neuromodulators (Thompson et al., 1993; Wu and Saggau, 1997), during short-term synaptic depression (Wu and Borst, 1999; Burrone and Lagnado, 2000) and augmentation (Stevens and Wesseling, 1999), and during activation of second messenger pathways involving protein kinase A (Trudeau et al., 1996) and G-proteins (Blackmer et al., 2001). Modulation of the ProbCa in these conditions may be achieved by regulation of the release machinery downstream of the Ca2+ sensor.
Molecules that may be involved in modulation of the ProbCa
We found that the PKC inhibitor BIS inhibited in large part the PMA-induced enhancement of the capacitance jump (Fig. 3). This is consistent with previous works indicating that phorbol esters enhance synaptic transmission in large part by activation of PKC at calyx-type synapses (Hori et al., 1999; Yawo, 1999; Oleskevich and Walmsley, 2000). It would be of great interest to identify the substrate of PKC that is involved in modulation of the ProbCa. SNAP-25 and Munc18/nSec1 are two synaptic proteins phosphorylated by PKC (Fujita et al., 1996; Shimazaki et al., 1996). Phosphorylation of them reduces their interaction with syntaxin, which might enhance exocytosis by modifying dissociation or formation of SNARE complexes (Fujita et al., 1996; Shimazaki et al., 1996). Thus these two molecules are potential candidates that may mediate PKC-induced increase of the ProbCa, although it should be noted that there are many other transmitter release-related proteins that can be phosphorylated by PKC (Majewski and Iannazzo, 1998).
Our results show that BIS did not block PMA-induced enhancement of the capacitance jump completely (Fig. 3). This is consistent with a recent study at the same MNTB synapse showing that the enhancement of the EPSC by phorbol esters is blocked partly by high concentrations of PKC inhibitors, including BIS, calphostin C, and a PKC inhibitor peptide (Hori et al., 1999). In the same study (Hori et al., 1999) the enhancement also is blocked partly by presynaptic loading of a synthetic peptide with the sequence of the N-terminal domain of Doc2α interacting with Munc13-1. Thus, it is likely that the remaining effect of PMA on the capacitance jump in the presence of BIS (Fig. 3) involves interaction between Doc2α and Munc13-1. In other words, interaction between Doc2α and Munc13-1 may participate in regulation of the ProbCa.
Footnotes
This work was supported by the National Science Foundation (IBN-0076091) and by the Mcdonnell Center for Cellular and Molecular Neurobiology (Washington University, St. Louis, MO). We thank Drs. Gong Chen, Steve Mennerick, and Jian-yuan Sun for critical comments on this manuscript and Dr. Jian-yuan Sun for his help during the experiments.
Correspondence should be addressed to Ling-Gang Wu, Department of Anesthesiology, Campus Box 8054, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail:wul@morpheus.wustl.edu.
REFERENCES
- 1.Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M. The exocytotic event in chromaffin cells revealed by patch amperometry. Nature. 1997;389:509–512. doi: 10.1038/39081. [DOI] [PubMed] [Google Scholar]
- 2.Bachoo M, Heppner T, Fiekers J, Polosa C. A role for protein kinase C in long-term potentiation of nicotinic transmission in the superior cervical ganglion of the rat. Brain Res. 1992;585:299–302. doi: 10.1016/0006-8993(92)91223-2. [DOI] [PubMed] [Google Scholar]
- 3.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 4.Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G-protein βγ subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- 5.Borst JGG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol (Lond) 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrone J, Lagnado L. Synaptic depression and the kinetics of exocytosis in retinal bipolar cells. J Neurosci. 2000;20:568–578. doi: 10.1523/JNEUROSCI.20-02-00568.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capogna M, Gahwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Sudhof TC, Scheller RH, Takai Y. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C: its implication in regulating the interaction of Munc-18/n-Sec1/rbSec1 with syntaxin. J Biol Chem. 1996;271:7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- 11.Gillis KD. Techniques for membrane capacitance measurements. In: Sakmann B, Neher E, editors. Single-channel recording. Plenum; New York: 1995. pp. 155–198. [Google Scholar]
- 12.Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 13.Hilfiker S, Augustine GJ. Regulation of synaptic vesicle fusion by protein kinase C. J Physiol (Lond) 1999;515[Pt 1]:1. doi: 10.1111/j.1469-7793.1999.001ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda I, Kamiya H, Yawo H. Re-evaluation of phorbol ester-induced potentiation of transmitter release from mossy fibre terminals of the mouse hippocampus. J Physiol (Lond) 2000;529[Pt 3]:763–776. doi: 10.1111/j.1469-7793.2000.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hori T, Takai Y, Takahashi T. Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci. 1999;19:7262–7267. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu S-F, Augustine GJ, Jackson MB. Adaptation of Ca2+-triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- 17.Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflügers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- 18.Majewski H, Iannazzo L. Protein kinase C: a physiological mediator of enhanced transmitter output. Prog Neurobiol. 1998;55:463–475. doi: 10.1016/s0301-0082(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 19.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 20.Malenka RC, Ayoub GS, Nicoll RA. Phorbol esters enhance transmitter release in rat hippocampal slices. Brain Res. 1987;403:198–203. doi: 10.1016/0006-8993(87)90145-4. [DOI] [PubMed] [Google Scholar]
- 21.Minota S, Kumamoto E, Kitakoga O, Kuba K. Long-term potentiation induced by a sustained rise in the intra-terminal Ca2+ in bullfrog sympathetic ganglia. J Physiol (Lond) 1991;435:421–438. doi: 10.1113/jphysiol.1991.sp018517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oleskevich S, Walmsley B. Phosphorylation regulates spontaneous and evoked transmitter release at a giant terminal in the rat auditory brainstem. J Physiol (Lond) 2000;526:349–357. doi: 10.1111/j.1469-7793.2000.t01-1-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Redman RS, Searl TJ, Hirsh JK, Silinsky EM. Opposing effects of phorbol esters on transmitter release and calcium currents at frog motor nerve endings. J Physiol (Lond) 1997;501[Pt 1]:41–48. doi: 10.1111/j.1469-7793.1997.041bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid CA, Bekkers JM, Clements JD. N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J Neurosci. 1998;18:2849–2855. doi: 10.1523/JNEUROSCI.18-08-02849.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 26.Sakaba T, Neher E. Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci. 2001;21:462–476. doi: 10.1523/JNEUROSCI.21-02-00462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- 28.Shapira R, Silberberg SD, Ginsburg S, Rahamimoff R. Activation of protein kinase C augments evoked transmitter release. Nature. 1987;325:58–60. doi: 10.1038/325058a0. [DOI] [PubMed] [Google Scholar]
- 29.Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25 kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 30.Smith CB, Betz WJ. Simultaneous independent measurement of endocytosis and exocytosis. Nature. 1996;380:531–534. doi: 10.1038/380531a0. [DOI] [PubMed] [Google Scholar]
- 31.Son H, Carpenter DO. Protein kinase C activation is necessary but not sufficient for induction of long-term potentiation at the synapse of mossy fiber–CA3 in the rat hippocampus. Neuroscience. 1996;72:1–13. doi: 10.1016/0306-4522(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 32.Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- 33.Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 34.Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- 35.Stevens CF, Williams JH. “Kiss and run” exocytosis at hippocampal synapses. Proc Natl Acad Sci USA. 2000;97:12828–12833. doi: 10.1073/pnas.230438697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun JY, Wu LG. Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron. 2001;30:171–182. doi: 10.1016/s0896-6273(01)00271-9. [DOI] [PubMed] [Google Scholar]
- 37.Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- 38.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 39.Trudeau L-E, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 40.Von Gersdorff H, Sakaba T, Berglund K, Tachibana M. Submillisecond kinetics of glutamate release from a sensory synapse. Neuron. 1998;21:1177–1188. doi: 10.1016/s0896-6273(00)80634-0. [DOI] [PubMed] [Google Scholar]
- 41.Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 42.Wu LG, Borst JGG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 43.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 44.Wu LG, Borst JGG, Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci USA. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yawo H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]