

Abstract
Synapsins are major neuronal phosphoproteins involved in regulation of neurotransmitter release. Synapsins are well established targets for multiple protein kinases within the nerve terminal, yet little is known about dephosphorylation processes involved in regulation of synapsin function. Here, we observed a reciprocal relationship in the phosphorylation–dephosphorylation of the established phosphorylation sites on synapsin I. We demonstrate that, in vitro, phosphorylation sites 1, 2, and 3 of synapsin I (P-site 1 phosphorylated by cAMP-dependent protein kinase; P-sites 2 and 3 phosphorylated by Ca2+–calmodulin-dependent protein kinase II) were excellent substrates for protein phosphatase 2A, whereas P-sites 4, 5, and 6 (phosphorylated by mitogen-activated protein kinase) were efficiently dephosphorylated only by Ca2+–calmodulin-dependent protein phosphatase 2B–calcineurin. In isolated nerve terminals, rapid changes in synapsin I phosphorylation were observed after Ca2+entry, namely, a Ca2+-dependent phosphorylation of P-sites 1, 2, and 3 and a Ca2+-dependent dephosphorylation of P-sites 4, 5, and 6. Inhibition of calcineurin activity by cyclosporin A resulted in a complete block of Ca2+-dependent dephosphorylation of P-sites 4, 5, and 6 and correlated with a prominent increase in ionomycin-evoked glutamate release. These two opposing, rapid, Ca2+-dependent processes may play a crucial role in the modulation of synaptic vesicle trafficking within the presynaptic terminal.
Keywords: 4-aminopyridine, brain-derived neurotrophic factor (BDNF), Ca2+, calcineurin, cyclosporin A, glutamate, ionomycin, mitogen-activated protein (MAP) kinase, neurotrophins, okadaic acid, PD98059, phosphatases, synapsins, synaptosomes, neurotransmitter release
Neurotransmitter release from nerve terminals occurs by exocytosis of synaptic vesicles mediated by protein complexes ultimately regulated in a Ca2+-dependent manner. Synapsin I is a well characterized member of the synapsin family of neuron-specific proteins associated with the cytoplasmic surface of small synaptic vesicles (SSVs) (De Camilli et al., 1983a,b; Huttner et al., 1983;Greengard et al., 1993) and is one of the most prominent nerve terminal phosphoproteins regulated in response to changes in intraterminal Ca2+ concentrations. It was originally identified in the brain as a substrate for multiple protein kinases (Johnson et al., 1971; Krueger et al., 1977). Protein kinase A (PKA) and Ca2+–calmodulin-dependent protein kinase I (CaM kinase I) phosphorylate P-site-1 (Ser-9, numbering for rat synapsin I; Czernik et al., 1987) in response to activation of presynaptic second messenger cascades or Ca2+ influx, while CaM kinase II phosphorylates P-sites 2 and 3 (Ser-566 and Ser-603) in response to Ca2+ influx during nerve terminal activation (Huttner and Greengard, 1979; Sihra et al., 1989). P-sites 4 and 5 (Ser-62 and Ser-67) are phosphorylated by extracellular signal-regulated kinases (ERKs) 1 and 2, p44 and p42, of the mitogen-activated protein (MAP) kinase superfamily, whereas P-site 6 (Ser-549) is phosphorylated by MAP kinase, as well as by cyclin-dependent kinase (cdk) 1 and cdk 5, and, finally, P-site 7 (Ser-551) is phosphorylated only by cdk 5 (Jovanovic et al., 1996;Matsubara et al., 1996). MAP kinase-dependent phosphorylation of the synapsins has recently been characterized as a key step in the modulation of neurotransmitter release by brain-derived neurotrophic factor (BDNF) (Jovanovic et al., 2000). Although the role of synapsin I as a protein kinase substrate has been extensively characterized, little is known about the phosphatases involved in dephosphorylating synapsins.
In adult synapses, a variety of evidence suggests that synapsins tether a large proportion of synaptic vesicles to each other and to the actin-based cytoskeleton and thereby maintain a cluster of vesicles referred to as the “reserve pool” (Greengard et al., 1993; Hilfiker et al., 1999). A subset of vesicles, referred to as the “releasable pool”, is docked at the plasma membrane and is largely devoid of synapsin-like immunoreactivity (De Camilli et al., 1983a; Valtorta et al., 1988; Hirokawa et al., 1989; Torri-Tarelli et al., 1990, 1992;Pieribone et al., 1995). The site-specific phosphorylation of synapsins is associated with profound changes in affinity of synapsins for SSVs and for G- and F-actin (Benfenati et al., 1989). Thus, a proportion of synapsin I phosphorylated in response to Ca2+ dissociates from the vesicle membrane during sustained depolarization of synaptosomes (Sihra et al., 1989) or high-frequency stimulation in frog neuromuscular junction (Torri-Tarelli et al., 1992). Synapsin phosphorylation–dephosphorylation therefore likely represents a regulatory switch during SSV trafficking between these functionally distinct pools.
Here we demonstrate that Ca2+ influx after nerve terminal depolarization triggers a complex set of synapsin I phosphorylation–dephosphorylation reactions. Together with a rapid increase in a CaM kinase I and II-dependent phosphorylation of P-sites 1 and 2/3, a decrease in phosphorylation of P-sites 4, 5, and 6 was effected by the Ca2+–calmodulin-dependent phosphatase calcineurin. Moreover, the calcineurin inhibitor cyclosporin A (CsA) prevented the dephosphorylation of P-sites 4, 5, and 6 and facilitated ionomycin-triggered release of glutamate.
MATERIALS AND METHODS
Synaptosome preparation. Synaptosomes were prepared from the cerebral cortices of two-month old male Sprague Dawley rats as described previously (Sihra, 1996). Cerebral cortices were dissected and homogenized in 0.32 m sucrose at 4°C using a Potter–Elvehjem tissue grinder with a motor-driven pestle rotated at 900 rpm. The homogenate was centrifuged at 3000 × gfor 2 min at 4°C. The supernatants (S1) were centrifuged at 14,500 × g for 12 min at 4°C. The pellets (P2) were resuspended and loaded onto Percoll gradients consisting of three steps of (from bottom) 23, 10, and 3% Percoll in 0.32m sucrose additionally containing 1 μm EDTA and 250 μm DTT. Gradients were centrifuged at 32,500 × g for 6.5 min at 4°C. Synaptosomes were harvested from the interface between the 23 and 10% Percoll layers and washed in HEPES-buffered incubation medium (HBM) (in mm): NaCl, 140; KCl, 5; NaHCO3, 5; MgCl2 · 6H20, 1; Na2HPO4, 1.2; glucose, 10; and HEPES, 20, pH 7.4). Washed synaptosomes were sedimented at 27,000 × g for 10 min at 4°C. The protein concentration of the resuspended pellet was determined using the Bradford assay (Bio-Rad, Hercules, CA), with bovine serum albumin as standard. The resuspended synaptosomes were washed once again in HBM before final centrifugation at 3000 × gfor 10 min at 4°C. For synapsin I phosphorylation experiments, a pretreatment regimen schematically depicted below was followed (SchemeFS1), in which synaptosomal pellets were resuspended in HBM containing Ca2+, EGTA, Co2+–Cd2+, PD98059 (Parke-Davis, Ann Arbor, MI), cyclosporin A (Sigma, St. Louis, MO), or okadaic acid (Sigma), as noted in the legend to each figure, and each tube was placed at 37°C to start the reaction. 4-aminopyridine (4-AP) was added at a reaction time of 10 min, and subsequent incubations for various times proceeded as described in the individual figure legends.
Fig. FS1.

Immunoblot analysis. Synaptosomal samples were rapidly solubilized in 1–2% SDS (95°C), sonicated, and protein concentration was measured using BCA assay (Pierce, Rockford, IL), with bovine serum albumin as standard. Equal amounts of protein were subjected to SDS-PAGE and transferred onto nitrocellulose membranes. Immunoblots were done with 1:500 dilutions of the following phosphorylation state-specific antibodies: P-site 1 antibody (G-257), P-site 3 antibody (RU19), P-site 4/5 antibody (G-526), and P-site 6 antibody (G-555). The specificity of these antibodies for their respective sites has been characterized previously (Czernik et al., 1991; Jovanovic et al., 1996). Total synapsin I was detected by immunoblotting with synapsin I-specific antibody (G-486; 1:500 dilution). Primary incubations were followed by incubation with125I-labeled anti-rabbit IgG (1:500 dilution; Amersham Pharmacia Biotech, Little Chalfont, UK). Blots were exposed to a PhosphorImager screen, and quantification of immunoblots was accomplished using PhosphorImager scanning and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
MAP kinase assays. Synaptosomal MAP kinase activity was assayed either by using an in-gel kinase assay as described (Jovanovic et al., 1996) or by immunoblot analysis using dual-phosphorylation state-specific, anti-active p44 and p42 MAP kinase antibody (1:10,000 dilution; Promega, Southampton, UK), followed by incubation with125I-labeled anti-rabbit IgG and visualized by PhosphorImager scanning.
In vitro phosphorylation. Synapsin I was purified from bovine brain as described by Schiebler et al. (1986) and modified byBähler and Greengard (1987). MAP kinase, p44mpk (Sanghera et al., 1990), and the cyclin-dependent protein kinase (cdk1)–cyclin A complex (Labbe et al., 1989) were purified from sea star oocytes and assayed as described. The catalytic subunit of PKA was purified from bovine heart as described (Kaczmarek et al., 1980). CaM kinase II was purified from rat brain as described (McGuinness et al., 1985). Phosphorylation of synapsin I used the incubation conditions described for the catalytic subunit of PKA (Huttner et al., 1981), CaM kinase II (Kennedy et al., 1983; Bennett et al., 1983), MAP kinase, p44mpk, and cdk1-cyclin A (Jovanovic et al., 1996), in the presence of 150 μm ATP with trace amounts of [γ-32P]ATP, to yield a final stoichiometry of 0.7, 2.4, 1.3, and 0.8 molP/mol of synapsin I, respectively. Incorporation of 32P was measured using PhosphorImager scanning. The phosphorylated forms of synapsin I were repurified as described (Czernik et al., 1987). Dopamine- and cAMP-regulated phosphoprotein (Mr = 32,000) (DARPP-32) phosphorylated by PKA at Thr-34 to a stoichiometry of 0.5 molP/mol of protein (Girault et al., 1989) and phosphorylase a (Cohen et al., 1988a,b) were phosphorylated and repurified as described.
In vitro dephosphorylation. Catalytic subunits of PP1 (PP1c, Mr = 37,000) and PP2A (PP2Ac, Mr = 38,000) were purified from rabbit skeletal muscle (Cohen et al., 1988a,b) and calcineurin (Mr = 76,000) from rat brain (Nairn et al., 1995). Purified phosphatases were assayed in 50 mm Tris–HCl, pH 7.0, 15 mm2-mercaptoethanol, and 1 mg/ml BSA at 30°C, as described (Desdouits et al., 1998), in the presence of 0.3% Brij-35 and 0.3 mm EGTA in the case of PP1c and PP2Ac, or 100 μm CaCl2 and 1 μm calmodulin in the case of calcineurin. Reactions were started by the addition of substrate and terminated by the addition of 200 μl of 20% (w/v) trichloroacetic acid. After the further addition of 50 μl of 10 mg/ml bovine serum albumin, samples were centrifuged for 5 min at room temperature at 17,000 ×g, and the amount of 32P in the supernatant and the pellet was determined by measurement of Cerenkov radiation.
PP1c and PP2Ac activities were measured under initial rate conditions (the release of phosphate was linear with time and enzyme concentration and corresponded to <25% of the phosphate incorporated into the substrate), and used 1 μm[32P]phosphorylase a as substrate (Ingebritsen et al., 1983). For measurements of calcineurin activity, initial rate conditions were determined using 1 μm[32P]phospho-DARPP-32. Under the same conditions, PP1c-, PP2Ac- and calcineurin-catalyzed dephosphorylation of different [32P]-labeled phospho-forms of synapsin I (1 μm) was directly compared with dephosphorylation of the standard substrates (Table1). Data represent the means of two independent experiments, each done in duplicate.
Table 1.
Site-specific synapsin I dephosphorylation by purified protein phosphatases
| Dephosphorylation (% 32P removed) | |||
|---|---|---|---|
| PP1c | PP2Ac | Calcineurin | |
| P-Site 1 | 1.3 | 7.8 | 3.5 |
| P-Site 2,3 | 4.1 | 15.2 | 2.3 |
| P-Site 4,5,6 | 1.1 | 1.5 | 8.5 |
| P-Site 6 | 0.002 | 1.1 | 8.5 |
| Phosphorylase a | 11.2 | 10.2 | — |
| P-Thr34-DARPP32 | — | — | 25.5 |
The activities of purified PP1c, PP2Ac, and calcineurin were measured under initial rate conditions using 1 μm32P-labeled phospho-synapsin I, phospho-DARPP-32, and phosphorylase a as described (see Materials and Methods). Relative rates of dephosphorylation were expressed as a percentage of total32P released in the presence of a phosphatase in 2.5 min. Phospho-substrates incubated in the absence of phosphatases served as control. Data represent the mean of two independent experiments, done in duplicate.
For kinetic analysis, dephosphorylation assays were started by the addition of enzyme and performed with various concentrations of phospho-substrates; concentration of synapsin I phosphorylated at P-site 1 or at P-sites 4, 5, and 6, or at P-site 6 varied from 0.3–8 μm, concentration of synapsin I phosphorylated at P-sites 2 and 3 varied from 1–20 μm, and phospho-DARPP-32 concentration varied from 0.5–10 μm. The total amount of PP1c, PP2Ac, and calcineurin used per reaction was 100, 20, and 9 ng, respectively. The Km,kcat, andkcat/Kmvalues were calculated from linear regression analysis of Lineweaver–Burk transformations of data describing the initial rates of dephosphorylation as a function of substrate concentration and represent the means of two independent experiments, each done in duplicate.
Synaptosomal glutamate release assay. Glutamate release was assayed by on-line fluorimetry as described previously (Nicholls and Sihra, 1986). Pelleted synaptosomes were resuspended in HBM and incubated in a stirred and thermostated cuvette at 37°C in a Perkin-Elmer LS-3B spectrofluorimeter. NADP+ (1 mm), glutamate dehydrogenase (50 U/ml), CoCl2 (10 μm), and CdCl2 (10 μm), in the presence or absence of cyclosporin A (1 μm) were added at the start of incubation. CaCl2 (1 mm) was added 1 min after the start. The incubation was performed for 10 min when either ionomycin (5 μm) or KCl (30 mm) was added to trigger Ca2+-dependent glutamate release. Fluorescence was monitored at excitation and emission wavelengths of 340 and 460 nm, respectively, and data were accumulated at 2.2 sec intervals. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment, and the fluorescence response was monitored. The value of the fluorescence change produced by the standard addition was used to calculate the released glutamate as nanomoles of glutamate per milligram of synaptosomal protein. Unless otherwise indicated, release values stated in the text are levels attained at “steady-state” after 4 min of depolarization (nmol/mg protein/4 min). Cumulative data were analyzed using Lotus 1-2-3 software.
RESULTS
Synapsin I dephosphorylation by protein phosphatasesin vitro
The physiological implications of synapsin I phosphorylation at specific sites prompted us to identify the protein phosphatase or phosphatases responsible for dephosphorylation of these sites. In vitro analyses of the dephosphorylation of synapsin I by the purified catalytic subunits of PP1 (PP1c), PP2A (PP2Ac), and calcineurin were performed with four different phospho-forms of synapsin I: synapsin I phosphorylated at P-site 1 by PKA (P-site 1-phosphosynapsin I), synapsin I phosphorylated at P-sites 2 and 3 by CaM kinase II (P-site 2,3-phosphosynapsin I), synapsin I phosphorylated at P-sites 4, 5, and 6 by MAP kinase (P-site 4,5,6-phosphosynapsin I), and synapsin I phosphorylated at P-site 6 by cdk 1 (P-site 6-phosphosynapsin I). In these experiments the initial rates of dephosphorylation of various [32P]-phospho forms of synapsin I and [32P]-phospho-DARPP-32 or [32P]-phosphorylase a used as reference substrates, were assessed by measuring the release of phosphate (Table1). P-site 1-phosphosynapsin I was a good substrate for PP2Ac, yet a poor substrate for either PP1c or calcineurin. P-site 2,3-phosphosynapsin I was an excellent substrate for PP2Ac and a poorer substrate for either PP1c or calcineurin. P-site 4,5,6-phosphosynapsin I and P-site 6-phosphosynapsin I were most efficiently dephosphorylated by calcineurin. These initial results were extended by a more complete kinetic analyses of dephosphorylation of the four phospho-forms of synapsin I by PP1c, PP2Ac, and calcineurin (Table2). Three kinetic parameters,Km (the apparent affinity for substrate), kcat (the turnover number), andkcat/Km(the catalytic efficiency) were determined from linear regression analysis of Lineweaver–Burk transformations of data describing the initial rates of dephosphorylation as a function of substrate concentrations. Values obtained for phospho-DARPP-32 and phosphorylase a as standards were similar to those reported previously (King et al., 1984) (data not shown). P-site 1-phosphosynapsin I and P-site 2,3-phosphosynapsin I were high-affinity substrates for PP2Ac with very high turnover numbers (kcat) and catalytic efficiencies (kcat/Km). P-site 2,3-phosphosynapsin I was also a high-affinity substrate for PP1c, but with a significantly lowerkcat. In contrast, these phospho-forms of synapsin I were both poor substrates for calcineurin. P-site 4,5,6-phosphosynapsin I and P-site 6-phosphosynapsin I were efficiently dephosphorylated by calcineurin, withkcat values and catalytic efficiencies similar to those obtained for dephosphorylation of phospho-Thr34-DARPP-32. Previous studies of P-site 1-phosphosynapsin I and P-site 2,3-phosphosynapsin I dephosphorylation by purified protein phosphatases yielded qualitatively similar results; PP2Ac was the most effective phosphatase, calcineurin was active, although less effective at these sites, and neither phospho-form was dephosphorylated significantly by PP1c or PP2C (A. C. Nairn, H. C. Hemmings Jr, and P. Greengard, unpublished observations).
Table 2.
Kinetic analysis of site-specific synapsin I dephosphorylation by purified protein phosphatases
| PP1c | PP2Ac | Calcineurin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Km (μm) | kcat (sec−1) | kcat/Km (1/sec μm) | Km (μm) | kcat (sec−1) | kcat/Km (1/sec μm) | Km (μm) | kcat (sec−1) | kcat/Km (1/sec μm) | |
| P-Site 1 | * | * | * | 11.1 | 12000 | 1081 | 55 | 0.9 | 0.023 |
| P-Site 2,3 | 1.8 | 21 | 11.7 | 6.6 | 8780 | 1330 | 31 | 0.6 | 0.016 |
| P-Site 4,5,6 | * | * | * | * | * | * | 8.3 | 2.9 | 0.3 |
| P-Site 6 | * | * | * | * | * | * | 7.4 | 9.8 | 1.35 |
The activities of purified PP1c, PP2Ac, and calcineurin were measured under initial rate conditions for 5 min using various concentrations of 32P-labeled phospho-synapsin I as described (see Materials and Methods). Phospho-substrates incubated in the absence of phosphatases served as control. Kinetic parameters were calculated from linear regression of Lineweaver–Burk plots, each representing the mean of two independent experiments, each done in duplicate.
Indicates activities too low for accurate kinetic measurements.
The potential involvement of PP2C in dephosphorylation of various phosphorylated forms of endogenous synapsin I was investigated using synaptosomal lysates. Synaptosomes were incubated under standard conditions for 10 min and then depolarized by the addition of 1 mm 4-AP. Samples, taken before and 1 min after the addition of 4-AP, were lysed using hypotonic conditions in the presence of 0.5 μm okadaic acid and 2 mm EGTA for 10 min at 4°C to inhibit endogenous activities of PP1, PP2Ac, and calcineurin. Synaptosomal lysates were then incubated in the presence or absence of 10 mm MgCl2 for 10 min, under experimental conditions required for PP2C activity (Desdouits et al., 1998). Immunoblotting using phosphorylation state-specific antibodies showed no apparent change in the phosphorylation state of any of the P-sites in synapsin I in the presence of 10 mmMgCl2 (data not shown).
4-Aminopyridine-induced changes in synapsin I phosphorylation in intact synaptosomes
Site-specific changes in the phosphorylation state of synapsin I in an isolated nerve terminal preparation (synaptosomes) were monitored by immunoblot analysis using phosphorylation state-specific antibodies for P-sites 4/5 and 6 (Jovanovic et al., 1996) and P-sites 1 and 3 (Czernik et al., 1991). In these experiments synaptosomes were incubated in the presence of 1 mmCa2+ or 0.2 mm EGTA (in the absence of added Ca2+) for 10 min and then depolarized using 1 mm 4-AP for 1 min (Fig.1). In parallel incubations, PD98059 (50 μm), an inhibitor of MEK (MAP/ERK kinase) activation was used to inhibit the phosphorylation of synapsin I P-sites 4/5 and 6 by synaptosomal MAP kinases as a control. After depolarization by 4-AP and Ca2+ entry, phosphorylation of P-sites 4/5 and 6 in synapsin I was markedly decreased compared with that detected in the absence of extrasynaptosomal Ca2+(Fig. 1A,B). This was likely a result of a Ca2+-dependent dephosphorylation because the activities of both p44 and p42 MAP kinase isoforms, which were significantly inhibited by PD98059, remained unchanged in the presence or absence of Ca2+ or 4-AP (Fig.1C).
Fig. 1.
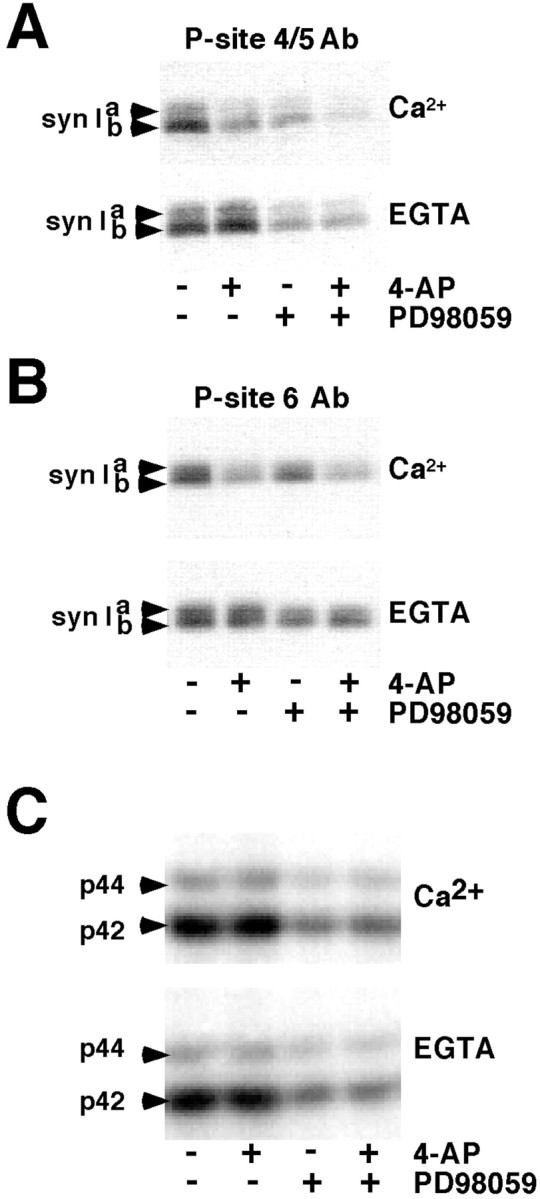
4-Aminopyridine-evoked depolarization and Ca2+ influx in synaptosomes result in a prominent dephosphorylation of MAP kinase-dependent P-sites 4/5 and 6 in synapsin I. Synaptosomes were incubated in the absence or presence of 50 μm PD98059 at 37°C, in HEPES-buffered medium containing either 1 mm CaCl2 (Ca2+) or 1 mm EGTA in the absence of added Ca2+(EGTA). After 10 min of incubation, 4-AP (1 mm) was added for an additional 1 min. Equal amounts of total protein (60 μg) were subjected to SDS-PAGE and immunoblot analysis using phosphorylation state-specific antibodies and 125I-labeled Protein A for detection. Results are representative of three independent experiments.A, Phosphorylation state of MAP kinase-specific P-sites 4/5 in synapsin I was analyzed using immunoblotting with P-site 4/5 antibody (G-526) (1:500 dilution). B, Phosphorylation state of MAP kinase/cdk5-dependent P-site 6 in synapsin I was analyzed using P-site 6 antibody (G-555) (1:500 dilution). C,Activities of MAP kinase isoforms ERK 1 and 2 were analyzed using immunoblotting with anti-active MAP kinase antibody (1:10,000 dilution; Promega).
Analysis of the time course of dephosphorylation after addition of 4-AP revealed that the decrease in phosphorylation of P-sites 4/5 (29.6 ± 2.4% of control; mean ± SEM; n = 4) (Fig.2A) and of P-site 6 (58.6 ± 2.9% of control; mean ± SEM; n = 4) (Fig. 2B) occurred rapidly, reaching a maximum within 1 min. In the presence of EGTA, this rapid dephosphorylation of P-sites 4/5 (Fig. 2A, EGTA) and of P-site 6 (Fig. 2B, EGTA) was inhibited.
Fig. 2.
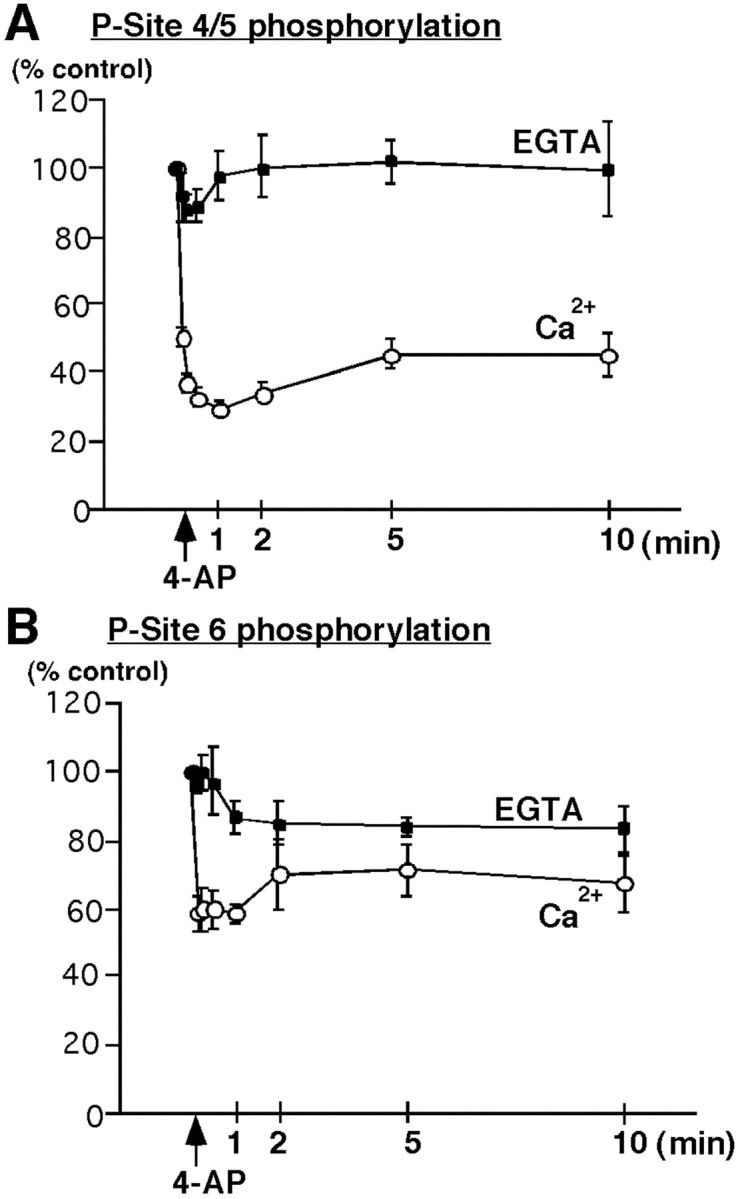
Time course of Ca2+-dependent dephosphorylation of synapsin I at MAP kinase-dependent P-sites 4/5 and 6 in synaptosomes. Synaptosomes were incubated for 10 min under standard conditions in the presence of 1 mmCa2+ (Ca2+) or 0.2 mm EGTA in the absence of added Ca2+ (EGTA). 4-AP (1 mm, arrow) was added, and samples were collected at various time points and lysed in 1% SDS. Equal amounts of total protein (60 μg) were analyzed using SDS-PAGE and immunoblotting with P-site 4/5 (G-526) or P-site 6 Ab (G-555) (1:500 dilution). Under Ca2+-free conditions no significant change in the level of phosphorylation of sites 4/5 or site 6 was observed.A, Time course of Ca2+-dependent dephosphorylation of synapsin I P-sites 4/5 occurs rapidly resulting in a decrease to 29.6 ± 2.4% (mean ± SEM;n = 4), 1 min after depolarization by 4-AP.B, Time course of Ca2+-dependent dephosphorylation of synapsin I P-site 6 results in a decrease to 58.6 ± 2.9% (mean ± SEM; n = 4), 1 min after depolarization by 4-AP.
The regulation of synapsin P-sites 4/5 and P-site 6 contrasted with that of synapsin I P-site 1 and 3. Phosphorylation of P-site 1, which was detected under basal conditions, showed a significant Ca2+-dependent increase in response to depolarization (Fig. 3A,P-site 1). Under basal conditions, P-site 3 was in a dephospho-state but underwent a dramatic increase in phosphorylation after nerve terminal depolarization (Fig. 3B, P-site 3). Maximal increases in the levels of phosphorylation of P-site 1 (Fig. 3A, Ca2+) and P-site 3 (Fig. 3B, Ca2+) were 2.1 ± 0.3-fold (mean ± SEM; n = 3) and 11.1 ± 2.4-fold (mean ± SEM; n = 3), respectively, and occurred with similar fast kinetics. In the presence of EGTA, the increase in phosphorylation of P-site 1 (Fig. 3A) and the rapid phosphorylation of P-site 3 (Fig. 3B) were completely inhibited. Thus, presynaptic Ca2+ entry has reciprocal effects on the phosphorylation state of specific P-sites of synapsin I, increasing phosphorylation at P-sites 1, 2, and 3, while decreasing phosphorylation on P-sites 4, 5, and 6.
Fig. 3.

Ca2+-dependent phosphorylation of synapsin I at CaM kinase-dependent P-sites 1 and 3 in synaptosomes. Synaptosomes were incubated for 10 min under standard conditions in the presence of 1 mm Ca2+(Ca2+) or 0.2 mm EGTA in the absence of added Ca2+ (EGTA). 4-AP (1 mm, arrow) was added, and samples were collected at various time points and lysed in 1% SDS. Equal amounts of total protein (60 μg) were analyzed using SDS-PAGE and immunoblotting with P-site 3 antibody (RU19) or P-site 1 antibody (G-257) (1:500 dilution). In Ca2+-free conditions no significant change in the level of phosphorylation of P-site 1 or P-site 3 was observed. A, Time course of Ca2+-dependent phosphorylation of synapsin I at P-site 1 results in an increase of 2.08 ± 0.25-fold (mean ± SEM; n = 3) in the level of phosphorylation of P-site 1, reaching a maximal increase 10 sec after depolarization by 4-AP. B, Time course of Ca2+-dependent phosphorylation of synapsin I at P-site 3 occurs rapidly resulting in an 11.1 ± 2.4-fold (mean ± SEM; n = 3) increase in the level of phosphorylation of P-site 3, reaching a maximal increase 1 min after depolarization by 4-AP.
Modulation of synapsin I phosphorylation by protein phosphatase inhibitors in intact synaptosomes
We used synaptosomes to investigate the effect of either okadaic acid-sensitive (inhibition of PP2A and PP1), or cyclosporin A-sensitive (inhibition of calcineurin) phosphatase activities on the phosphorylation of synapsin I at specific P-sites. Okadaic acid (0.5 μm) was added before the transition of synaptosomes to 37°C, and the incubation was performed for 10 min. Samples were collected before (control) and 1 min after the addition of 4-AP. A prominent increase in the state of phosphorylation of P-sites 1 and 3 was observed with okadaic acid treatment (Fig.4A, P-site 1and P-site 3) likely because of a specific inhibition of PP2A, given the concentration of okadaic acid used herein (Nishi et al., 1999). However, under the same conditions, an increase in phosphorylation state of P-sites 4/5 and 6 was also observed (Fig.4A, P-site 4/5 and P-site 6). The latter effect was likely attributable to an increase in MAP kinase activity itself, given that these sites were very poor substrates for PP1 and PP2A in vitro. To confirm this, we compared the activity of synaptosomal MAP kinases in the absence or presence of okadaic acid using an in-gel kinase assay with myelin basic protein as substrate (Jovanovic et al., 1996). A large increase in the activities of both p44 and p42 isoforms of MAP kinase was observed in the presence of okadaic acid as compared with controls (Fig. 4B). This effect likely reflected a direct inhibition of PP2A-dependent dephosphorylation/deactivation of MAP kinases (Alessi et al., 1995; Chajry et al., 1996).
Fig. 4.

Effect of okadaic acid on phosphorylation state of synapsin I at P-sites 1, 3, 4/5, and 6 in synaptosomes.A, Okadaic acid (OA; 0.5 μm) was added before the transition of synaptosomes to 37°C, and the incubation was performed under standard conditions (1 mm CaCl2) for 10 min followed by the addition of 1 mm 4-AP. Samples were collected before and 1 min after the addition of 4-AP and analyzed by SDS-PAGE and immunoblotting with phosphorylation state-specific antibodies followed by 125I-Protein A for detection. Results are representative of three independent experiments. Phosphorylation of synapsin I was analyzed using immunoblotting with P-site 1, 3, 4/5, and 6 phosphorylation state-specific antibodies (1:500 dilution). SDS-PAGE migration of the doublet of synapsin Ia and Ib (syn I) was analyzed using immunoblotting with synapsin I specific antibody (G-486) (1:500) (syn I). Hyperphosphorylation of synapsin I at all five P-sites in the presence of okadaic acid resulted in broad bands of higher apparent molecular mass. B, Synaptosomal samples were collected before or 10 min after the transition of synaptosomes to 37°C in the absence or presence of okadaic acid (0.5 μm). Activities of MAP kinase isoforms ERK 1 and 2 were analyzed by an in-gel kinase assay with myelin basic protein as a substrate incorporated within the gel and in the presence of 40 μm [γ -32P]ATP.
The ability of calcineurin to dephosphorylate various P-sites in synapsin I was tested using a specific inhibitor, cyclosporin A (1 μm). However, given previous reports of increased Ca2+ channel activity in the presence of calcineurin inhibitors (Sihra et al., 1995; Lukyanetz et al., 1998;Burley and Sihra, 2000), one potential complication in this experimental design is that an increase in CaM kinase-dependent phosphorylation of synapsin I may result from an increase in Ca2+ influx in the presence of cyclosporin A. To attempt to obviate this possibility, we incubated synaptosomes for 10 min in the presence of 0.2 mm EGTA in the absence or presence of cyclosporin A, and depolarized the synaptosomes with 4-AP before the addition of 1.2 mm CaCl2. Control samples were collected before the addition of 4-AP/Ca2+. The phosphorylation of synapsin I at specific sites was then determined 30 sec, 1 min, and 10 min after the addition of Ca2+ by immunoblotting with P-site-specific antibodies. Inhibition of calcineurin activity by cyclosporin A led to complete inhibition of Ca2+-dependent dephosphorylation of P-site 4/5 (Fig. 5A) and P-site 6 (Fig. 5B) with little or no effect on phosphorylation of P-site 1 (Fig. 5C) and P-site 3 (Fig. 5D). Cyclosporin A had no effect on basal levels of phosphorylation of synapsin I at specific sites before depolarization and Ca2+ entry.
Fig. 5.

Ca2+-dependent dephosphorylation of synapsin I at MAP kinase-dependent P-sites 4/5 and 6 in synaptosomes is completely blocked by the calcineurin inhibitor cyclosporin A (CsA). Synaptosomes were incubated for 10 min in the presence of 0.2 mm EGTA and in the absence or presence of cyclosporin A (1 μm) and then depolarized with 4-AP just before the addition of 1.2 mmCaCl2. Control samples were collected before the addition of 4-AP. The effect of Ca2+ entry was than followed at 30 sec, 1 min, and 10 min using SDS-PAGE and immunoblotting with P-site specific antibodies. A,Ca2+-dependent dephosphorylation of P-site 4/5 resulted in a decrease to 58.6 ± 4.8% of control (mean ± SEM; n = 4), 1 min after depolarization by 4-AP (control). This effect was completely inhibited in the presence of cyclosporin A resulting in a small increase to 119.4 ± 4.6% of control (mean ± SEM;n = 4), 1 min after depolarization by 4-AP (CsA). B,Ca2+-dependent dephosphorylation of P-site 6 resulted in a decrease to 77 ± 2.8% of control (mean ± SEM; n = 4), 1 min after depolarization by 4-AP (control). This effect was completely inhibited in the presence of cyclosporin A, resulting in a small increase to 108 ± 8.1% of control (mean ± SEM; n = 4), 1 min after depolarization by 4-AP (CsA). C, Ca2+-dependent phosphorylation of P-site 1 resulted in an increase to 155.5 ± 9.4% of control (mean ± SEM;n = 4), 1 min after depolarization by 4-AP (control). No significant effect (144 ± 9.6% of control; mean ± SEM; n = 4) was observed at 1 min in the presence of CsA. D,Ca2+-dependent phosphorylation of P-site 3 resulted in an increase to 1391 ± 305% of control (mean ± SEM;n = 4), 1 min after depolarization by 4-AP (control). No significant effect (1024 ± 165% of control; mean ± SEM; n = 4) was observed at 1 min in the presence of CsA.
Functional correlation of Ca2+-dependent dephosphorylation of synapsin I with the release of glutamate
To examine the possible functional significance of sustained high levels of phosphorylation of P-sites 4/5 and 6 in synapsin I produced by inhibition of calcineurin activity, we measured ionomycin-triggered glutamate release in the presence or absence of cyclosporin A using an on-line fluorometric assay (Nicholls and Sihra, 1986). Ionomycin causes a direct increase in intrasynaptosomal Ca2+ levels and triggers release of neurotransmitter without depolarization and Ca2+ channel activation (Sihra et al., 1992), therefore allowing us to detect only those modulatory influences directly affecting synaptic vesicle trafficking and exocytosis.
Incubations were performed at 37°C in the presence of CoCl2 (10 μm) and CdCl2 (10 μm) to completely block the activity of Ca2+ channels (Vickroy et al., 1992). The inhibition of Ca2+-channel activity by CoCl2 and CdCl2was complete and resulted in an abrogation of Ca2+-dependent glutamate release evoked by 30 mm KCl (Fig. 6,inset). Cyclosporin A (1 μm) was applied for 10 min, after which glutamate release was triggered by the addition of ionomycin in the presence of 1 mmCa2+. Ionomycin caused a Ca2+-dependent glutamate release of 11.5 ± 1.5 nmol/mg after 4 min (n = 3). In the presence of cyclosporin A, ionomycin-induced release of glutamate was potentiated by 35 ± 9% (n = 3;p < 0.05; Student's paired t test) to 15.2 ± 1.2 nmol/mg after 4 min (Fig. 6).
Fig. 6.

Inhibition of synaptosomal calcineurin activity by cyclosporin A correlates functionally with an increase in ionomycin-triggered glutamate release. Glutamate release was triggered from rat synaptosomes (0.3 mg/1.5 ml) incubated in the presence of CoCl2 (10 μm) and CdCl2 (10 μm) and in the absence or presence of cyclosporin A (1 μm) for 10 min and assayed by on-line fluorometry, as described in Materials and Methods. CaCl2 (1 mm) was added 3 min after the start of incubation. Inhibition of Ca2+ channel activity by the addition of Co2+/Cd2+ had no significant effect on ionomycin-triggered glutamate release (inset, KCl-evoked release). Ionomycin caused a Ca2+-dependent glutamate release (control) that was potentiated in the presence of cyclosporin A (+CsA). Data are means ± SEM values of three independent experiments, using synaptosomal preparations from three different animals. The SEM was computed for each time point (2.2 sec intervals), but error bars are shown every six time points for clarity. Inset, Traces showing on-line glutamate release stimulated by 30 mm KCl in the absence (control) or presence of 10 μmCo/Cd (+Co/Cd). Slow increase in fluorescence in the presence of Co/Cd reflected Ca2+-independent glutamate release.
DISCUSSION
We report here that nerve terminal stimulation and Ca2+ influx regulate the state of phosphorylation of the synaptic vesicle-associated protein synapsin I via two opposing mechanisms: (1) Ca2+-regulated kinase activities increase phosphorylation of synapsin I P-sites 1, 2, and 3, and (2) Ca2+-regulated phosphatase activity decreases phosphorylation of P-sites 4, 5, and 6 (SchemeFS2). These effects occurred with similarly fast kinetics and were consistent with the influx of Ca2+-activating CaM kinases I and II and calcineurin, respectively. We have also identified PP2A as the phosphatase that downregulates synapsin phosphorylation at P-sites 1, 2, and 3. In intact nerve terminals, calcineurin plays a role in regulating the phosphorylation state of synapsin I and concomitantly modulates glutamate release. Given the established importance of synapsin I-dependent regulation of synaptic vesicle trafficking from reserve pools of synaptic vesicles (Hilfiker et al., 1999), calcineurin appear to plays a key role in activity-dependent modulation of nerve terminal function.
Fig. FS2.

Synapsin I phosphorylation–dephosphorylation enzyme targets. A–F, synapsin I domains; Ia and Ib, synapsin I splice variants; PP2B, protein phosphatase 2B/calcineurin; PP2A,protein phosphatase 2A; CAMK I and CAMKII, Ca2+–calmodulin dependent protein kinases I and II; PKA, cAMP-dependent protein kinase; MAPK, mitogen-activated protein kinase; cdk1/5, cyclin-dependent protein kinase 1 and 5.
Nerve terminal phosphatase activities
Serine/threonine phosphatases have been extensively studied, but few studies have directly addressed their role in nerve terminal function (Sihra et al., 1992, 1995; Nichols et al., 1994; Steiner et al., 1996). Here we have characterized the role of the major serine–threonine phosphatases by examining their activity against a nerve terminal-specific substrate, synapsin I, both in vitro(using purified components) and in situ (within nerve terminals, using phosphorylation-state specific antibodies to various P-sites of synapsin I). Our results point to the presence and tonic activity of PP2A that limits the basal phosphorylation state of synapsin at P-sites 1, 2, and 3. After stimulation of nerve terminals, Ca2+ influx causes the rapid activation of calcineurin, to effect the dephosphorylation of synapsin at P-sites 4, 5, and 6. In contrast, none of the synapsin I P-sites appeared to be a good substrates for PP1, reflecting the primarily postsynaptic localization of this phosphatase and its established association with dendritic structures (Allen et al., 1997). Taken together, these data demonstrate that PP2Ac is the most likely phosphatase involved in the regulation of phosphorylation state of P-sites 1, 2, and 3 of synapsin I. However, calcineurin is the most likely phosphatase involved in regulation of the phosphorylation state of P-sites 4, 5, and 6, with a significantly lower activity for other synapsin I phospho-forms. Although both PP2A and calcineurin are ubiquitous enzymes, the latter is particularly enriched in the brain (Usuda et al., 1996) where it has been implicated in several forms of synaptic plasticity (Mansuy et al., 1998; Winder et al., 1998). Specific effects of calcineurin on presynaptic function may well be partly responsible for these effects on synaptic plasticity.
Phosphorylation–dephosphorylation of synapsin P-sites 1, 2, and 3
Synapsins interact dynamically with synaptic vesicles and actinin vitro and in situ. Phosphorylation of synapsin I at sites 2 and 3 by CaM kinase II results in a profound change in its conformation (Benfenati et al., 1990), decreases its affinity for synaptic vesicles (Schiebler et al., 1986), and almost completely inhibits its ability to interact with F- and G-actin (Bähler and Greengard, 1987; Petrucci and Morrow, 1987; Valtorta et al., 1992). Phosphorylation of site 3 is very low under basal conditions, probably, as shown here, because of a tonic activity of PP2A and also low basal activity of CaM kinase II, but increases rapidly during nerve terminal stimulation caused by activation of CaM kinase II by influx of Ca2+. Given that previous studies have demonstrated concomitant phosphorylation of P-sites 2 and 3 by CaM kinase II under all conditions examined (Czernik et al., 1987), it is likely that phosphorylation of P-site 2 was increased to a similar extent in parallel with P-site 3. Phosphorylation of synapsin P-site 1, 2, and 3 leads to dissociation of synapsin I from the vesicle membrane and the cytoskeletal matrix and its translocation into the cytosolic milieu of the synaptosomes (Sihra et al., 1989). The fraction of synapsin I that translocates to the cytosol consists of synapsin I stoichiometrically phosphorylated on P-sites 1, 2, and 3.
The fast rise in phosphorylation of P-sites 1, 2, and 3, as well as the relatively slow dephosphorylation of these sites during prolonged stimulation of nerve terminals (Fig. 3A,B) (5 and 10 min), are in agreement with our in vitro dephosphorylation data and point to regulation of these sites by a prolonged increase in CaM kinase activity concomitant with a tonic, Ca2+-independent activity of phosphatase PP2A. The latter activity is also consistent with the low basal levels of phosphorylation seen with these P-sites in resting terminals. The relatively high phosphorylation state of P-site 1 is of interest, and it likely results from a tonic PKA activity operating in nerve terminals (Chavez-Noriega and Stevens, 1994; Capogna et al., 1995; Trudeau et al., 1996; Chen and Regehr, 1997; Chavis et al., 1998;Hilfiker, 2001).
Phosphorylation–dephosphorylation of synapsin P-sites 4, 5, and 6
The basal activity of MAP kinases (Fig. 1C), together with their Ca2+-independent activation in unstimulated synaptosomes (J.N.J., T.S.S., A.J.C., and P.G., unpublished observations), resulted in relatively high levels of basal phosphorylation of synapsin I P-sites 4, 5, and 6. In vitro, phosphorylation of these sites caused a conformational change in synapsin I and a decrease in the ability of synapsin I to interact with actin without affecting the binding to synaptic vesicles (Jovanovic et al., 1996). Synapsin I phosphorylated at P-sites 4, 5, and 6 was in fact detected exclusively in synaptic vesicle-enriched synaptosomal fractions under all the conditions examined (our unpublished observations).
TrkB–MAP kinase-induced phosphorylation of P-sites 4, 5, and 6 has been implicated in the mechanism by which BDNF regulates neurotransmitter release (Jovanovic et al., 2000). This phosphorylation is likely to modulate the interaction between synapsin I-associated SSVs and the actin cytoskeleton (Jovanovic et al., 1996) and thereby alter a propensity of SSVs to undergo further steps leading to vesicle exocytosis. Overlying this regulation by synaptosomal MAP kinase, we demonstrate here that these sites are rapidly dephosphorylated by calcineurin after a rise in nerve terminal Ca2+ concentration, and this may function to acutely limit the in vivo activity of the MAP kinase cascade impinging on the synapsins. Thus, calcineurin-dependent dephosphorylation of P-sites 4, 5, and 6 of synapsin I may act as an inhibitory constraint on processes promoting neurotransmitter release. After inhibition of calcineurin activity therefore, facilitation of neurotransmitter release would occur by promoting the dissociation of phosphosynapsin from the actin-based cytoskeletal matrix and therefore enhancing the ability of vesicles to enter the releasable pool. Consistent with this hypothesis, we demonstrate here that inhibition of calcineurin by cyclosporin enhances the ionomycin-induced release of glutamate from synaptosomes. Although this effect could also be attributed to an inhibition of calcineurin-dependent dephosphorylation of substrates other than synapsin I, a recent study using synapsin I knock-out mice points to synapsin I being a major target for calcineurin in the regulation of neurotransmitter release (Zhang et al., 2000). Thus, in a mouse model for secondary hypertension in humans attributed to facilitated neurotransmitter release, inhibition of calcineurin by cyclosporin A precipitated the pathology in wild-type mice, but not in animals in which synapsin I had been ablated. This then is suggestive of a causal relationship between the phosphorylation state of the calcineurin-sensitive P-sites on synapsin I and its modulation of neurotransmitter release in response to nerve terminal Ca2+ entry.
Downstream of synapsin I-regulated recruitment of SSVs and the subsequent steps leading to exocytosis, a large number of recent studies have demonstrated that Ca2+-dependent activation of calcineurin is also required for clathrin-mediated synaptic vesicle recycling (Marks and McMahon, 1998; Lai et al., 1999). At least four proteins associated with the initiation of endocytosis are known substrates for calcineurin: amphiphysin I/II, dynamin, and synaptojanin. The demonstration that synapsin I is also a substrate for calcineurin, together with the fact that it has been shown to bind to some of these endocytic molecules in a phosphorylation-dependent manner (Onofri et al., 2000), suggests that dephosphorylation of synapsin at P-sites 4, 5, 6 may serve to create a pool of dephosphosynapsin that can regulate synaptic vesicle recycling back to the reserve pool of vesicles.
Footnotes
This work was supported by National Institutes of Health Grant MH-39327 (P.G.) and grants from the Wellcome Trust and Biotechnology and Biological Sciences Research Council (T.S.S). We thank Alexander Soukas for help with the protein phosphatase assays and Atsuko Horiuchi and Hsien-Bin Huang for providing purified protein phosphatases.
Correspondence should be addressed to Jasmina N. Jovanovic, Department of Pharmacology, University College, London, WC1E 6BT, UK. E-mail:j.jovanovic@ucl.ac.uk.
REFERENCES
- 1.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 2.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bähler M, Greengard P. Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature. 1987;326:704–707. doi: 10.1038/326704a0. [DOI] [PubMed] [Google Scholar]
- 4.Benfenati F, Valtorta F, Bahler M, Greengard P. Synapsin I, a neuron-specific phosphoprotein interacting with small synaptic vesicles and F-actin. Cell Biol Int Rep. 1989;13:1007–1021. doi: 10.1016/0309-1651(89)90016-7. [DOI] [PubMed] [Google Scholar]
- 5.Benfenati F, Neyroz P, Bahler M, Masotti L, Greengard P. Time-resolved fluorescence study of the neuron-specific phosphoprotein synapsin I. Evidence for phosphorylation-dependent conformational changes. J Biol Chem. 1990;265:12584–12595. [PubMed] [Google Scholar]
- 6.Bennett MK, Erondu NE, Kennedy MB. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J Biol Chem. 1983;258:12735–12744. [PubMed] [Google Scholar]
- 7.Burley JR, Sihra TS. A modulatory role for protein phosphatase 2B (calcineurin) in the regulation of Ca2+ entry. Eur J Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 8.Capogna M, Gahwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chajry N, Martin PM, Cochet C, Berthois Y. Regulation of p42 mitogen-activated-protein kinase activity by protein phosphatase 2A under conditions of growth inhibition by epidermal growth factor in A431 cells. Eur J Biochem. 1996;235:97–102. doi: 10.1111/j.1432-1033.1996.00097.x. [DOI] [PubMed] [Google Scholar]
- 10.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chavis P, Mollard P, Bockaert J, Manzoni O. Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron. 1998;20:773–781. doi: 10.1016/s0896-6273(00)81015-6. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen P, Alemany S, Hemmings BA, Resink TJ, Stralfors P, Tung HY. Protein phosphatase-1 and protein phosphatase-2A from rabbit skeletal muscle. Methods Enzymol. 1988a;159:390–408. doi: 10.1016/0076-6879(88)59039-0. [DOI] [PubMed] [Google Scholar]
- 14.Cohen P, Foulkes JG, Holmes CF, Nimmo GA, Tonks NK. Protein phosphatase inhibitor-1 and inhibitor-2 from rabbit skeletal muscle. Methods Enzymol. 1988b;159:427–437. doi: 10.1016/0076-6879(88)59042-0. [DOI] [PubMed] [Google Scholar]
- 15.Czernik AJ, Pang DT, Greengard P. Amino acid sequences surrounding the cAMP-dependent and calcium/calmodulin-dependent phosphorylation sites in rat and bovine synapsin I. Proc Natl Acad Sci USA. 1987;84:7518–7522. doi: 10.1073/pnas.84.21.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czernik AJ, Girault JA, Nairn AC, Chen J, Snyder G, Kebabian J, Greengard P. Production of phosphorylation state-specific antibodies. Methods Enzymol. 1991;201:264–283. doi: 10.1016/0076-6879(91)01025-w. [DOI] [PubMed] [Google Scholar]
- 17.De Camilli P, Cameron R, Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J Cell Biol. 1983a;96:1337–1354. doi: 10.1083/jcb.96.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Camilli P, Harris SM, Huttner WB, Jr, Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol. 1983b;96:1355–1373. doi: 10.1083/jcb.96.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desdouits F, Siciliano JC, Nairn AC, Greengard P, Girault JA. Dephosphorylation of Ser-137 in DARPP-32 by protein phosphatases 2A and 2C: different roles in vitro and in striatonigral neurons. Biochem J. 1998;330:211–216. doi: 10.1042/bj3300211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Girault JA, Hemmings HC, Williams KR, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32, a dopa. J Biol Chem. 1989;264:21748–21759. [PubMed] [Google Scholar]
- 21.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 22.Hilfiker S. Tonically active protein kinase A regulates neurotransmitter release at the squid giant synapse. J Physiol (Lond) 2001;531:141–146. doi: 10.1111/j.1469-7793.2001.0141j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huttner WB, Greengard P. Multiple phosphorylation sites in protein I and their differential regulation by cyclic AMP and calcium. Proc Natl Acad Sci USA. 1979;76:5402–5406. doi: 10.1073/pnas.76.10.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huttner WB, DeGennaro LJ, Greengard P. Differential phosphorylation of multiple sites in purified protein I by cyclic AMP-dependent and calcium-dependent protein kinases. J Biol Chem. 1981;256:1482–1488. [PubMed] [Google Scholar]
- 27.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingebritsen TS, Stewart AA, Cohen P. The protein phosphatases involved in cellular regulation. 6. Measurement of type-1 and type-2 protein phosphatases in extracts of mammalian tissues; an assessment of their physiological roles. Eur J Biochem. 1983;132:297–307. doi: 10.1111/j.1432-1033.1983.tb07362.x. [DOI] [PubMed] [Google Scholar]
- 29.Johnson EM, Maeno H, Greengard P. Phosphorylation of endogenous protein of rat brain by cyclic adenosine 3′,5′-monophosphate-dependent protein kinase. J Biol Chem. 1971;246:7731–7739. [PubMed] [Google Scholar]
- 30.Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- 32.Kaczmarek LK, Jennings KR, Strumwasser F, Nairn AC, Walter U, Wilson FD, Greengard P. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase enhances calcium action potentials of bag cell neurons in cell culture. Proc Natl Acad Sci USA. 1980;77:7487–7491. doi: 10.1073/pnas.77.12.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy MB, McGuinness T, Greengard P. A calcium/calmodulin-dependent protein kinase from mammalian brain that phosphorylates Synapsin I: partial purification and characterization. J Neurosci. 1983;3:818–831. doi: 10.1523/JNEUROSCI.03-04-00818.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King MM, Huang CY, Chock PB, Nairn AC, Hemmings HC, Chan KF, Jr, Greengard P. Mammalian brain phosphoproteins as substrates for calcineurin. J Biol Chem. 1984;259:8080–8083. [PubMed] [Google Scholar]
- 35.Krueger BK, Forn J, Greengard P. Depolarization-induced phosphorylation of specific proteins, mediated by calcium ion influx, in rat brain synaptosomes. J Biol Chem. 1977;252:2764–2773. [PubMed] [Google Scholar]
- 36.Labbe JC, Capony JP, Caput D, Cavadore JC, Derancourt J, Kaghad M, Lelias JM, Picard A, Doree M. MPF from starfish oocytes at first meiotic metaphase is a heterodimer containing one molecule of cdc2 and one molecule of cyclin B. EMBO J. 1989;8:3053–3058. doi: 10.1002/j.1460-2075.1989.tb08456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai MM, Hong JJ, Ruggiero AM, Burnett PE, Slepnev VI, De Camilli P, Snyder SH. The calcineurin-dynamin 1 complex as a calcium sensor for synaptic vesicle endocytosis. J Biol Chem. 1999;274:25963–25966. doi: 10.1074/jbc.274.37.25963. [DOI] [PubMed] [Google Scholar]
- 38.Lukyanetz EA, Piper TP, Sihra TS. Calcineurin involvement in the regulation of high-threshold Ca2+ channels in NG108–15 (rodent neuroblastoma x glioma hybrid) cells. J Physiol (Lond) 1998;510:371–385. doi: 10.1111/j.1469-7793.1998.371bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME. Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell. 1998;92:39–49. doi: 10.1016/s0092-8674(00)80897-1. [DOI] [PubMed] [Google Scholar]
- 40.Marks B, McMahon HT. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- 41.Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- 42.McGuinness TL, Lai Y, Greengard P. Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J Biol Chem. 1985;260:1696–1704. [PubMed] [Google Scholar]
- 43.Nairn AC, Sihra TS, Andjus P, Craig A-M, Miyawaki A, Kloppenburg P, Lin Z, Pouzat C. Rapid purification of protein phosphatase-2B (calcineurin) from rat forebrain. Neuroprotocols. 1995;6:105–107. [Google Scholar]
- 44.Nicholls DG, Sihra TS. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321:772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- 45.Nichols RA, Suplick GR, Brown JM. Calcineurin-mediated protein dephosphorylation in brain nerve terminals regulates the release of glutamate. J Biol Chem. 1994;269:23817–23823. [PubMed] [Google Scholar]
- 46.Nishi A, Snyder GL, Nairn AC, Greengard P. Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J Neurochem. 1999;72:2015–2021. doi: 10.1046/j.1471-4159.1999.0722015.x. [DOI] [PubMed] [Google Scholar]
- 47.Onofri F, Giovedi S, Kao HT, Valtorta F, Borbone LB, De Camilli P, Greengard P, Benfenati F. Specificity of the binding of synapsin I to Src homology 3 domains. J Biol Chem. 2000;275:29857–29867. doi: 10.1074/jbc.M006018200. [DOI] [PubMed] [Google Scholar]
- 48.Petrucci TC, Morrow JS. Synapsin I: an actin-bundling protein under phosphorylation control. J Cell Biol. 1987;105:1355–1363. doi: 10.1083/jcb.105.3.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- 50.Sanghera JS, Paddon HB, Bader SA, Pelech SL. Purification and characterization of a maturation-activated myelin basic protein kinase from sea star oocytes. J Biol Chem. 1990;265:52–57. [PubMed] [Google Scholar]
- 51. Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem 261 1986. 8383 8390 [Erratum (1986) 261:12428] [PubMed] [Google Scholar]
- 52.Sihra TS. Protein phosphorylation and dephosphorylation in isolated nerve terminals (synaptosomes). In: Hemmings HC, editor. Posttranslational modifications: techniques and protocols. Humana; Totowa: 1996. pp. 67–119. [Google Scholar]
- 53.Sihra TS, Wang JK, Gorelick FS, Greengard P. Translocation of synapsin I in response to depolarization of isolated nerve terminals. Proc Natl Acad Sci USA. 1989;86:8108–8112. doi: 10.1073/pnas.86.20.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sihra TS, Bogonez E, Nicholls DG. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J Biol Chem. 1992;267:1983–1989. [PubMed] [Google Scholar]
- 55.Sihra TS, Nairn AC, Kloppenburg P, Lin Z, Pouzat C. A role for calcineurin (protein phosphatase-2B) in the regulation of glutamate release. Biochem Biophys Res Commun. 1995;212:609–616. doi: 10.1006/bbrc.1995.2013. [DOI] [PubMed] [Google Scholar]
- 56.Steiner JP, Dawson TM, Fotuhi M, Snyder SH. Immunophilin regulation of neurotransmitter release. Mol Med. 1996;2:325–333. [PMC free article] [PubMed] [Google Scholar]
- 57.Torri-Tarelli F, Villa A, Valtorta F, De Camilli P, Greengard P, Ceccarelli B. Redistribution of synaptophysin and synapsin I during alpha-latrotoxin-induced release of neurotransmitter at the neuromuscular junction. J Cell Biol. 1990;110:449–459. doi: 10.1083/jcb.110.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torri-Tarelli F, Bossi M, Fesce R, Greengard P, Valtorta F. Synapsin I partially dissociates from synaptic vesicles during exocytosis induced by electrical stimulation. Neuron. 1992;9:1143–1153. doi: 10.1016/0896-6273(92)90072-l. [DOI] [PubMed] [Google Scholar]
- 59.Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 60.Usuda N, Arai H, Sasaki H, Hanai T, Nagata T, Muramatsu T, Kincaid RL, Higuchi S. Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons: immunohistochemistry on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J Histochem Cytochem. 1996;44:13–18. doi: 10.1177/44.1.8543776. [DOI] [PubMed] [Google Scholar]
- 61.Valtorta F, Greengard P, Fesce R, Chieregatti E, Benfenati F. Effects of the neuronal phosphoprotein synapsin I on actin polymerization. I. Evidence for a phosphorylation-dependent nucleating effect. J Biol Chem. 1992;267:11281–11288. [PubMed] [Google Scholar]
- 62.Valtorta F, Villa A, Jahn R, De Camilli P, Greengard P, Ceccarelli B. Localization of synapsin I at the frog neuromuscular junction. Neuroscience. 1988;24:593–603. doi: 10.1016/0306-4522(88)90353-3. [DOI] [PubMed] [Google Scholar]
- 63.Vickroy TW, Schneider CJ, Hildreth JM. Pharmacological heterogeneity among calcium channels that subserve acetylcholine release in vertebrate forebrain. Neuropharmacology. 1992;31:307–309. doi: 10.1016/0028-3908(92)90181-n. [DOI] [PubMed] [Google Scholar]
- 64.Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- 65.Zhang W, Li JL, Hosaka M, Janz R, Shelton JM, Albright GM, Richardson JA, Sudhof TC, Victor RG. Cyclosporine A-induced hypertension involves synapsin in renal sensory nerve endings. Proc Natl Acad Sci USA. 2000;97:9765–9770. doi: 10.1073/pnas.170160397. [DOI] [PMC free article] [PubMed] [Google Scholar]