

Abstract
The mammalian CNS contains an abundance of chelatable Zn2+ sequestered in the vesicles of glutamatergic terminals. These vesicles are particularly numerous in hippocampal mossy fiber synapses of the hilar and CA3 regions. Our recent observation of frequency-dependent Zn2+ release from mossy fiber synaptic terminals and subsequent entry into postsynaptic neurons has prompted us to investigate the role of synaptically released Zn2+ in the induction of long-term potentiation (LTP) in field CA3 of the hippocampus. The rapid removal of synaptically released Zn2+ with the membrane-impermeable Zn2+ chelator CaEDTA (10 mm) blocked induction of NMDA receptor-independent mossy fiber LTP by high-frequency electrical stimulation (HFS) in rat hippocampal slices. Mimicking Zn2+ release by bath application of Zn2+ (50–100 μm) without HFS induced a long-lasting potentiation of synaptic transmission that lasted more than 3 hr. Moreover, our experiments indicate the effects of Zn2+ were not attributable to its interaction with extracellular membrane proteins but required its entry into presynaptic or postsynaptic neurons. Co-released glutamate is also essential for induction of LTP under physiological conditions, in part because it allows Zn2+ entry into postsynaptic neurons. These results indicate that synaptically released Zn2+, acting as a second messenger, is necessary for the induction of LTP at mossy fiber→CA3 synapses of hippocampus.
Keywords: zinc, long-term potentiation, CA3, hippocampus, CaEDTA, mossy fiber, plasticity, Na-pyrithione, Newport Green, synaptic transmission
The mammalian CNS contains an abundance of chelatable Zn2+ sequestered in the vesicles of glutamatergic terminals. These vesicles are particularly numerous in hippocampal mossy fiber synapses of the hilar and CA3 regions (Haug, 1967; Perez-Clausell and Danscher, 1985;Frederickson, 1989; Frederickson et al., 2000; Li et al., 2001). A possible synaptic signaling role for Zn2+is suggested by its interactions with excitatory and inhibitory amino acid receptors such as NMDA, AMPA, and GABA receptors (Peters et al., 1987; Westbrook and Mayer, 1987). Zn2+accumulates in synaptic vesicles through a specific Zn2+ transporter, termed Zn transporter 3 (Palmiter et al., 1996). Vesicular Zn2+release can be elicited by electrical stimulation (Howell et al., 1984;Li et al., 2001) or membrane depolarization (by elevating extracellular K+ concentration; Assaf and Chung, 1984;Aniksztejn et al., 1987; Li et al., 2001). Characterization of this Zn2+ release has revealed that it is released in the same manner as neurotransmitters: the release is Ca2+-dependent and tetrodotoxin-sensitive (Li et al., 2001). Recently, it has been shown that extracellular Zn2+ enters neurons through glutamate receptors and voltage-dependent Ca2+channels (VDCCs; Weiss and Sensi, 2000). Despite a considerable amount of evidence suggesting that Zn2+ acts in concert with neurotransmitters in the CNS, a specific physiological role for synaptically released Zn2+ has yet to be identified.
In addition to having routes of entry into neurons that are activated during nerve transmission, Zn2+ is known to interact with the protein kinases and phosphatases of signal transduction pathways that affect changes in gene expression (Brewer et al., 1979; Hubbard et al., 1991; Weinberger and Rostas, 1991; Quest et al., 1992; Maret et al., 1999; Park and Koh, 1999; Lengyel et al., 2000). Our recent observation (Li et al., 2001) of frequency-dependent Zn2+ release from mossy fiber synaptic terminals and subsequent entry into postsynaptic neurons of the dentate gyrus has suggested to us that Zn2+ might play a role in the normal physiological function of these neurons. Detectable Zn2+ release varied over a range of frequencies (10–200 Hz), which included frequencies used to induce long-term potentiation (LTP). We hypothesized that translocation of Zn2+ across synapses might be an important physiological signal mediating some aspects of synaptic plasticity, such as LTP.
LTP is an important model for studying the cellular mechanisms of neuronal plasticity, learning, and memory. Zn2+-deficient rats and rhesus monkeys experience a learning and working memory deficit (Golub et al., 1995). Although the possibility that Zn2+released from the mossy fiber bouton might be involved in hippocampal LTP has been proposed by Weiss et al (1989) more than a decade ago, much is still unknown about the involvement of synaptically released Zn2+ in synaptic plasticity. Recently, two groups failed to alter the induction of mossy fiber LTP by removing synaptically released Zn2+ with the Zn2+ chelator CaEDTA (Lu et al., 2000;Vogt et al., 2000). Using fluorescence imaging, we show here that although a low concentration (1 mm) of CaEDTA was not sufficient to prevent synaptically released Zn2+ from reaching postsynaptic neurons after high-frequency stimulation (HFS), a higher concentration (10 mm) of Zn2+ chelator was. This treatment blocked the induction of LTP. Moreover, perfusion of slices with exogenous Zn2+ (50–100 μm) could also induce long-lasting potentiation of the EPSP in the absence of HFS. Finally, our experiments indicate that the effects of Zn2+ were not attributable to its interaction with extracellular membrane proteins but required its entry into presynaptic or postsynaptic neurons.
MATERIALS AND METHODS
Hippocampal slice preparation. Experiments were conducted according to the principles set forth in the Guide for Care and Use of Laboratory Animals (Institute of Animal Resources, National Research Council, National Institutes of Health publication 74-23). Male adult Sprague Dawley rats were anesthetized with ketamine hydrochloride and decapitated. The brain was quickly removed and immersed in ice-cold (1–4°C) artificial CSF (ACSF) with the composition of (in mm): NaCl, 124; KCl, 1.75; MgSO4, 1.3; CaCl2, 2.4; KH2PO4, 1.25; NaHCO3, 26; and dextrose, 10, continuously bubbled with 95% O2 and 5% CO2. Transverse hippocampal slices 400 μm in thickness were prepared using a McIlwain tissue chopper (Brinkmann Instruments, Westbury, NY) or Vibratome (Frederic Haer, Brunswick, ME). Slices were incubated in a 95% O2-5% CO2-saturated interface recording chamber for at least 1 hr before recording at 32°C.
Electrical stimulation and recordings. The mossy fiber→CA3 pyramidal neuron responses were induced by the stimulation of mossy fiber axons with a 100-μm-diameter monopolar Teflon-insulated stainless steel wire electrode, exposed only at the tip (Fig.1A). Extracellular recordings were obtained using glass micropipettes filled with 2 m NaCl, 2–6 MΩ resistance. The recording electrodes were placed at least 500 μm from the stimulating electrodes along the trajectory of the mossy fiber pathway. The recording electrodes were lowered to a distance of 80–100 μm beneath the slice surface. Paired pulse facilitation of the EPSP was conducted at 20 and 80 msec interpulse intervals. Slices were accepted for further study when the mossy fiber pathway showed facilitation at the 80 msec interval. Because of the complex circuitry of area CA3, the metabotropic Glu receptor (mGluR) II agonist 2-(2,3-dicarboxy-cyclopropyl)glycine (DCG-IV) was used at the end of the experiments to verify that the signal was generated by mossy fiber inputs (Kamiya et al., 1996). d-APV (50 μm) was added in ACSF to prevent contamination with the NMDA receptor-dependent pathway converging on CA3 neurons. For inducing mossy fiber→CA3 LTP, test stimuli were delivered to mossy fiber axons every 30 sec (0.03 Hz). The stimulus intensity was set to produce ∼30% of the maximum EPSP. HFS consisted of one train of 100 Hz lasting 2 sec at the intensity that induced the maximum EPSP. The maximal negative initial slope of the mossy fiber→CA3 EPSP was calculated and normalized to 30 min baseline value (defined as 100%).
Fig. 1.
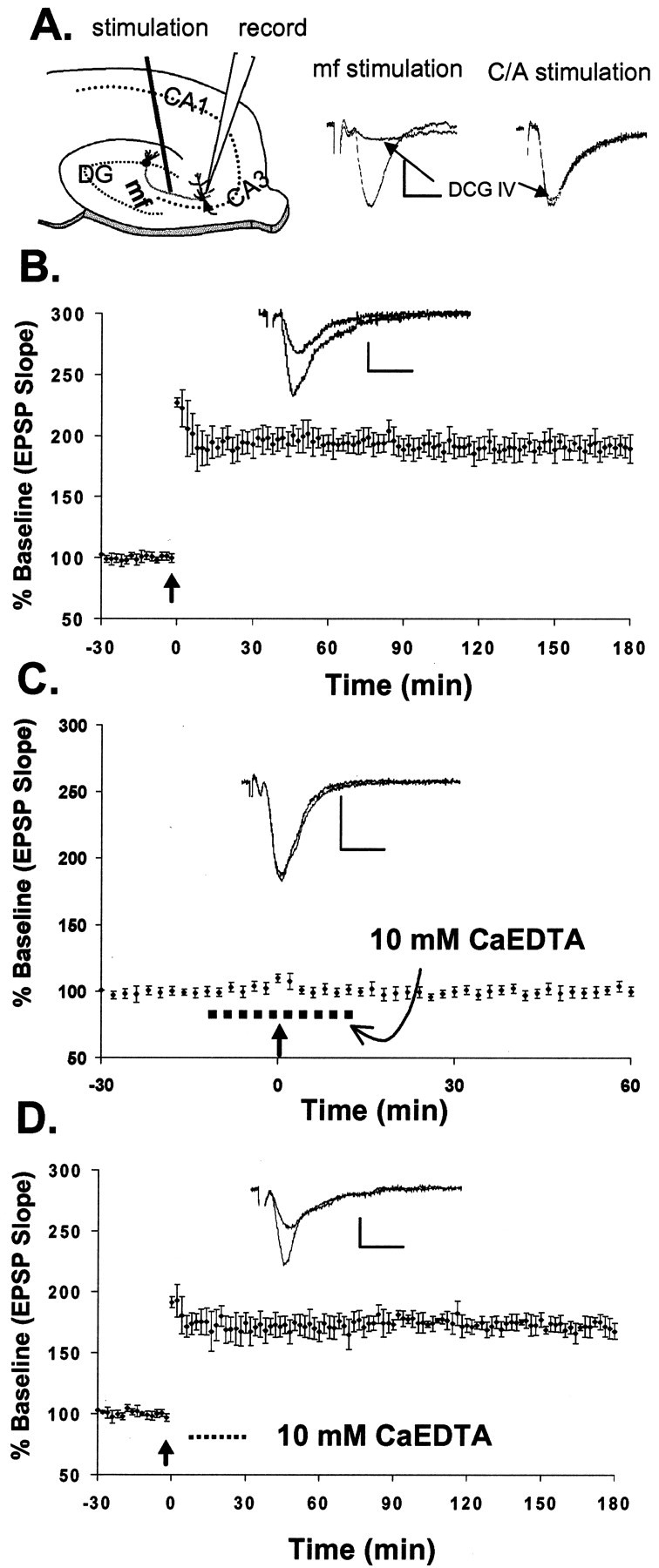
Rapid chelation of extracellular Zn2+ blocks induction of mossy fiber LTP.A, Schematic of a hippocampal slice showing stimulating and recording sites. The traces on theright show the field EPSPs evoked by stimulation of mossy fibers (mf) and the commissural–associational pathway. The group II mGluR agonist DCG-IV (5 μm) selectively blocked mossy fiber responses to 17.3 ± 2% (n = 6) of those before drug application but had little effect on the responses (to 97.7 ± 1.9%; n = 6) evoked by stimulating the commissural–associational fibers in the stratum radiatum (electrode not shown). B, Mossy fiber LTP (193 ± 10%; n = 9) induced by HFS after recording a 30 min baseline (see Materials and Methods).C, HFS failed to induce mossy fiber LTP (103 ± 4%; n = 11) in the presence of 10 mmCaEDTA (dashed line). D, Adding CaEDTA (10 mm;dashed line) 10 min after induction of LTP did not affect the late, or maintenance, phase of LTP (175 ± 8%; n = 3). Each point inB–D represents the averaged and normalized EPSP initial negative slope, and error bars indicate SEM. Arrowsindicate HFS (100 Hz, 2 sec). B–D, Insets, EPSP recorded during baseline and at the end of the recording period after HFS. Calibration: 1.0 mV, 5 msec.
Zn2+ imaging. Methods for imaging Zn2+ fluorescence have been published (Li et al., 2001). For extracellular Zn2+fluorescence imaging, hippocampal slices were preloaded with 20 μm Newport Green (Molecular Probes, Eugene, OR) dipotassium salt at room temperature in the dark for at least 30 min. For intracellular Zn2+ imaging, the slices were preloaded with 50 μm diacetate ester of Newport Green in 0.5% dimethylsulfoxide containing 0.1% pluronic acid for 1 hr and then washed with ACSF. The Zn2+-selective fluorescent dye Newport Green has a Kd of 1 μm for Zn2+. Newport Green fluorescence was minimally affected by the presence of Ca2+ and Mg2+at physiological concentrations (Li et al., 2001). Ca2+ or Mg2+(up to 10 mm), in the absence of Zn2+, had little effect on the dye fluorescence emission. All experiments were performed at 32°C under constant ACSF perfusion on the thermostatically heated stage of an inverted microscope (Axiovert 140; Zeiss, Oberkochen, Germany) coupled to a Delta Ram xenon light source (PTI, Manmouth Junction, NJ) and monochromator set to 506 nm. Emitted light images at 533 nm or greater were acquired at rates of 2–30 Hz through a 10× 0.1 numerical aperture objective with an intensified CCD camera (PTI IC-100) and digitized using ImageMaster software (PTI). Autofluorescence was below the detection limits of the camera, and photobleaching was negligible under these conditions; neither was subtracted from the data. Images in Figures 7 and 8 were captured by an Orca digital camera (Hamamatsu, Hamamatsu City, Japan) using Open Lab Software (Improvision). To induce the release of Zn2+ from mossy fiber terminals, bipolar electrodes 300–500 μm apart were used for electrical stimulation to excite mossy fiber axons. Trains of orthodromic stimuli (100 Hz, 200 μsec pulses at 500 μA unless otherwise noted) of various frequencies were delivered using an S44 stimulator and a PS1U6 photoelectric stimulus isolation unit (Grass Electronics, Quincy, MA).
Fig. 7.
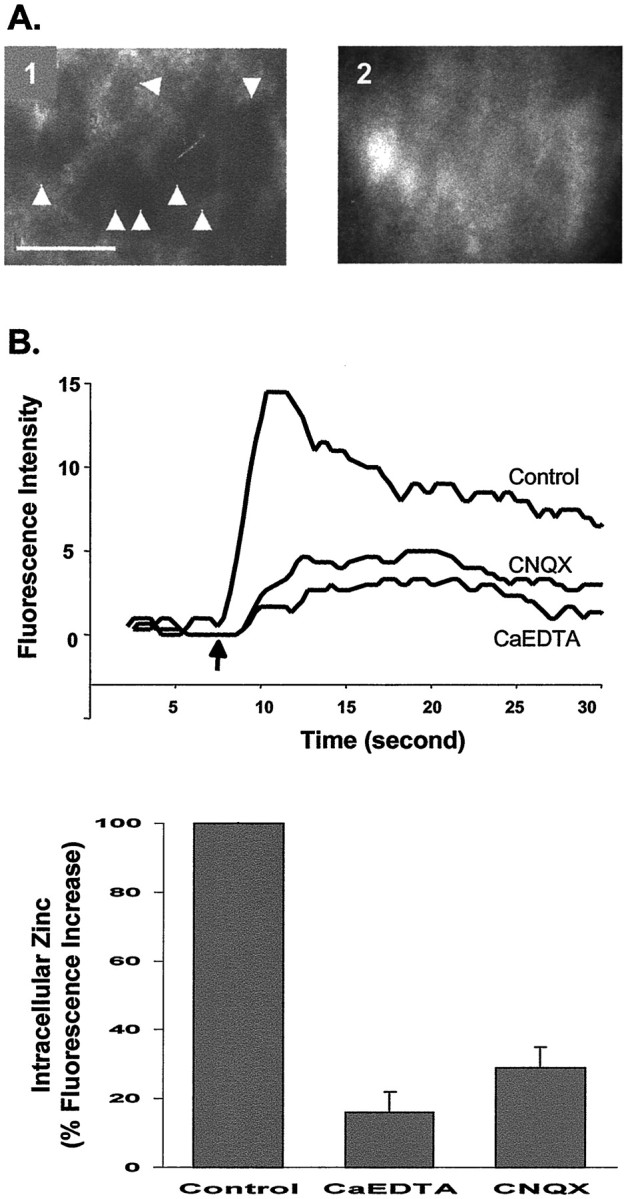
Increase of intracellular Zn2+after HFS. A, Paired images of the pyramidal layer of CA3 taken with a 63× water immersion objective lens. Electrical stimulation increased intracellular Zn2+ in the CA3 region of hippocampal slices loaded with Newport Green diacetate (cell-permeable). Before stimulation (1), no cell bodies were labeled; they became visible (2) after stimulation (100 Hz, 500 μA for 10 sec). Scale bar, 50 μm;arrowheads indicate pyramidal cell bodies.B, Intracellular Zn2+ was detected with the fluorescence indicator Newport Green diacetate in normal ACSF and ACSF containing CaEDTA (10 mm) or CNQX (10 μm). The results are plotted on the left. the arrow indicates the beginning of stimulation. Thebar graph on the right summarizes the effects of CaEDTA (n = 5) and CNQX (n = 3) on the increase in intracellular Zn2+ after electrical stimulation, expressed as percentage of control. Error bars indicate SEM.
Fig. 8.

Potentiation of the EPSP requires entry of Zn2+ into cells. A, Paired images of the pyramidal layer of CA3 (taken with a 63× objective lens) loaded with Newport Green diacetate before (1) and after (2) exposure to Zn2+ (100 μm), Na-pyrithione (50 μm), and CNQX (10 μm). Scale bar, 50 μm; arrowheadsindicate pyramidal cell bodies. B, Plot of the EPSP slope from a representative experiment to show that selectively increasing intracellular Zn2+ potentiates the EPSP (158 ± 12%; n = 4; filled diamonds). Zn2+ (100 μm) and Na-pyrithione (50 μm) were applied after the EPSP was blocked by CNQX (10 μm). In a similar experiment without Na-pyrithione, no EPSP potentiation was observed (93 ± 9%;n = 4; open squares).Lines indicate the duration of drug application.Inset, Summary of four experiments in which slices were treated with Na-pyrithione and Zn2+. Values of the EPSP amplitude were taken from baseline (1), in the presence of CNQX (2), and during EPSP potentiation (3). Numberscorrespond to those in B. *Significant difference (p < 0.05) between 1 and3.
RESULTS
Rapid chelation of synaptically released Zn2+blocks induction of mossy fiber LTP
To determine whether Zn2+ is involved in this specific form of synaptic plasticity, we examined the effects of applying CaEDTA, a cell-impermeable extracellular Zn2+ chelator (Kd = 10−16.4 M) that does not appreciably alter Ca2+ concentration (Fredens and Danscher, 1973; Dawson et al., 1986; Bers et al., 1994). Stimulation of the mossy fiber axons produced an extracellular field EPSP recorded in the dendritic region (stratum lucidum) of pyramidal neurons in field CA3 of hippocampal slices (Fig.1A). In control slices, brief HFS produced EPSP potentiation (mossy fiber LTP; Fig.1B). The averaged normalized EPSP slope 30 min after HFS was 193 ± 10% (mean ± SEM; n = 9) of baseline, and the potentiation was stable for >3 hr (maximum recording duration) after our standard recording procedure (Bramham and Sarvey, 1996). To chelate Zn2+ released by HFS and to prevent it from reaching postsynaptic neurons, we perfused slices with 10 mm CaEDTA for 10 min before and 10 min after HFS. This treatment blocked induction of LTP (103 ± 4%, mean ± SEM; n = 11; Fig. 1C). Although there was an initial small post-tetanic potentiation immediately after HFS, it decayed to baseline within 5–10 min. These results suggest that Zn2+ released during HFS plays an essential role in the induction of mossy fiber LTP. To determine whether chelation of Zn2+ could alter the maintenance of LTP, CaEDTA was applied 10 min after HFS. Figure1D shows that CaEDTA (10 mm) had no effect on established LTP, suggesting that the removal of Zn2+ after HFS did not influence the late, or maintenance, phase of LTP.
Kinetics of Zn2+ chelation by CaEDTA
To verify that Zn2+ released from mossy fiber terminals by HFS was adequately chelated by 10 mm CaEDTA, we loaded slices with the selective extracellular Zn2+ fluorescent indicator Newport Green dipotassium salt (cell-impermeable; Molecular Probes) and visualized Zn2+ release after stimulation in the presence and absence of CaEDTA. In a previous study (Li et al., 2001), we found that electrically stimulated Zn2+ release was frequency-dependent and could be detected with as little as 10 Hz stimulation at 500 μA. The degree of Zn2+ release also increased with increasing stimulus amplitudes ranging from 20 to -500 μA (100 Hz over 5 sec). Ten millimolar CaEDTA chelated 85% of the synaptically released Zn2+, as indicated by Newport Green fluorescence (Fig.2A). This result verifies that the effect of CaEDTA on LTP induction in mossy fiber→CA3 synapses was achieved by its selective chelation of the synaptically released Zn2+ from mossy fiber terminals. After bath perfusion of 10 mmCaEDTA for 10 min, followed by washout of the CaEDTA, we could still induce normal release of Zn2+ in normal ACSF (data not shown). On the other hand, a lower concentration of CaEDTA (1 mm) failed to reduce the extracellular Zn2+ after HFS, as evidenced by its inability to reduce Newport Green fluorescence (Fig.2A). Therefore, induction of LTP was essentially unaffected in 1 mm CaEDTA (Fig.2B).
Fig. 2.
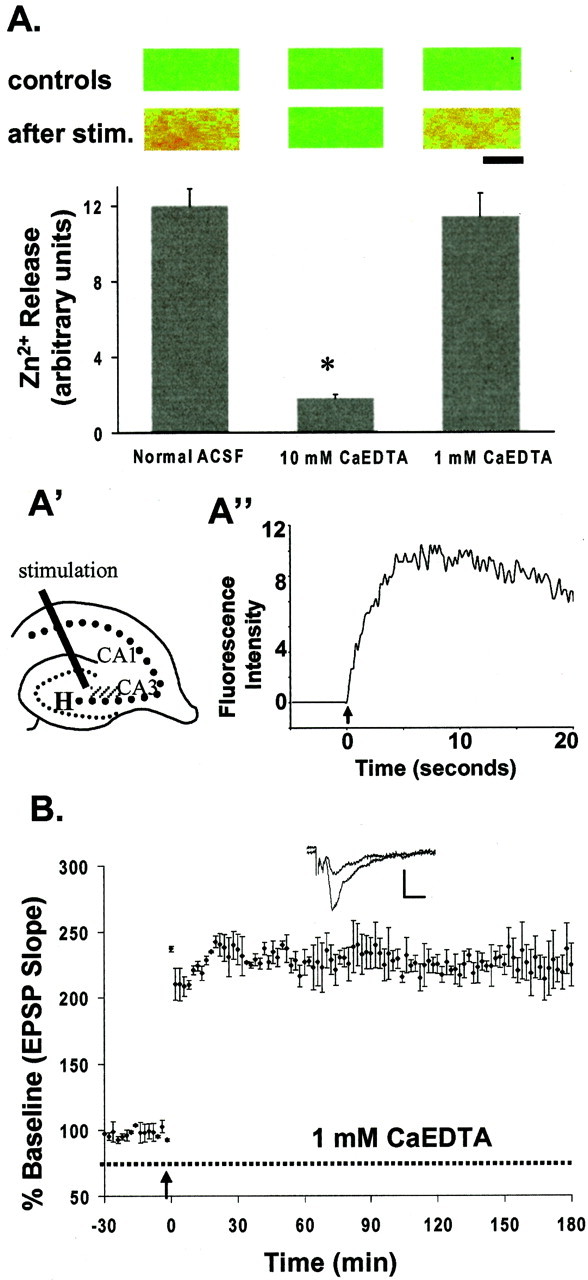
Zn2+ release with electrical stimulation and the effects of Zn2+ chelator.A, Extracellular Zn2+ released from terminals after electrical stimulation (stim.) was measured as the peak emission intensity of extracellular Newport Green fluorescence in the absence and presence of Zn2+chelator (3 determinations; error bars indicate SE). The paired images on top correspond with each condition of the bar graph. They were acquired before (controls in the first row) and after (second row) HFS (200 μsec, 0.5 mA pulses for 5 sec). In these false-color images, increasing intensity of Zn2+ fluorescence is represented by the spectrum ranging from blue (the lowest) to red(the highest). Scale bar, 100 μm. A', Thehatched area represents the region where images were acquired. H, Hilus. Values plotted are the mean ± SEM; n = 4; *p < 0.05.A“, Plot of electrical stimulation (100 Hz for 5 sec)-evoked rapid release of Zn2+ from neuronal terminals measured by changes in Newport Green fluo- rescence intensity (arbitrary units). Thearrow indicates the beginning of stimulation.B, A low concentration of CaEDTA (1 mm;dashed line) had little effect on mossy fiber LTP (227 ± 11%; n = 4) evoked by HFS. Eachpoint is the averaged and normalized EPSP initial negative slope, and error bars indicate SEM. Arrowsindicate the time giving HFS (100 Hz, 2 sec). Calibration: 1.0 mV, 5 msec.
These results are consistent with our calculations of the kinetics of Zn2+ chelation in ACSF containing CaEDTA. Our calculations predict that, in 10 mm CaEDTA, 100 μm extracellular Zn2+ would be reduced to 33 nm within 0.1 msec, whereas in 1 mm CaEDTA, this concentration of Zn2+ would only be reduced to 15 μm in 0.1 msec (Fig. 3; and). Thus, we suggest that 10 mm CaEDTA effectively removes Zn2+ released from nerve terminals by HFS before its physiological function can be performed (Basolo and Pearson, 1967; Davis et al., 1999). This explains why 1 mmCaEDTA failed to block induction of LTP in this study (Fig.2B) and in previous studies (Lu et al., 2000; Vogt et al., 2000).
Fig. 3.

Calculated kinetics of chelation of 100 μm Zn2+ in ACSF by 1 and 10 mm CaEDTA. The rate of chelation of 100 μmZn2+ by 1 and 10 mm CaEDTA was estimated using published and estimated forward and reverse rate constants as described in . Only the first 0.1 msec after the release of Zn2+ is given. Equilibrium conditions are assumed for 1 and 10 mm CaEDTA in ACSF in the absence of Zn2+ at time 0. Note that, under these conditions, equilibrium concentrations after introduction of 100 μmZn2+ are not achieved within this time frame.
Removal of synaptically released Zn2+ does not affect basal synaptic transmission
Disodium EDTA nominally saturated with equimolar Ca2+ (CaEDTA) has been used to add the chelator to physiological buffers such as ACSF without appreciably reducing the concentration of extracellular Ca2+, which is essential for normal synaptic transmission. To verify this, we measured the concentration of free Ca2+ in ACSF using a Ca2+ electrode. Addition of 1 mm CaEDTA did not alter the free Ca2+. Addition of 10 mm CaEDTA reduced the measured concentration of free Ca2+ from 2.25 ± 0.02 mm(mean ± SEM; n = 3) to 2.03 ± 0.03 mm (mean ± SEM;n = 3; Fig.4A). This was probably attributable to incomplete saturation of the EDTA with Ca2+ during its manufacture. When we added an extra 0.22 mm CaCl2 to ACSF containing 10 mm CaEDTA to compensate for the Ca2+ deficit, the concentration of free Ca2+ was 2.25 ± 0.01 mm (mean ± SEM; n = 3).
Fig. 4.
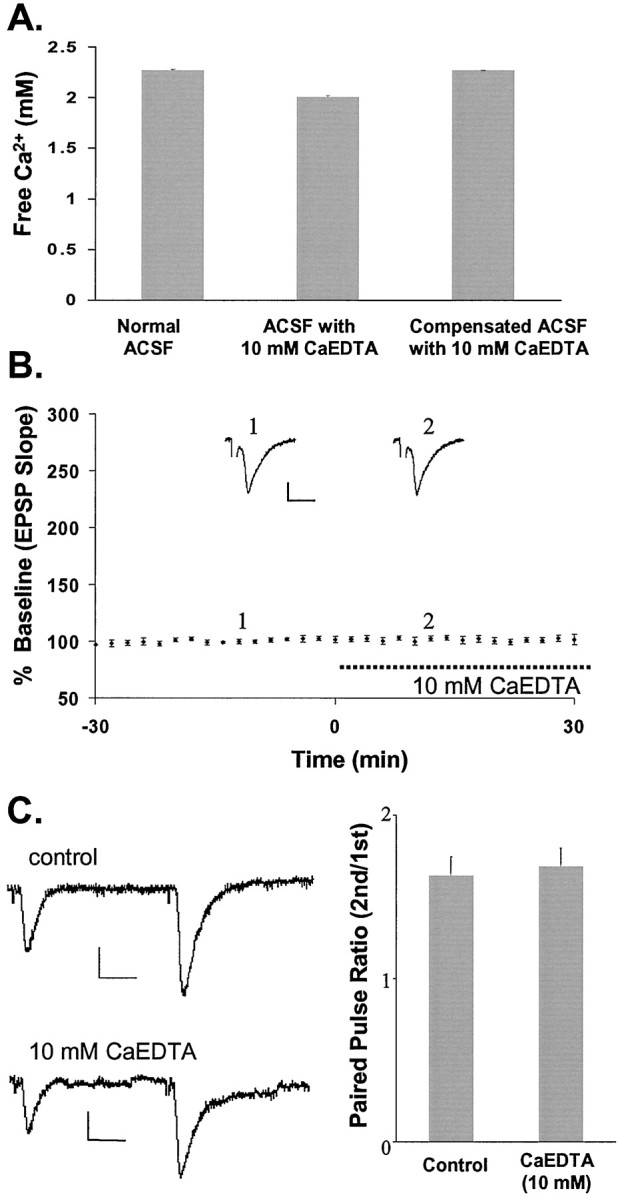
Exposure of slices to a high concentration of CaEDTA does not alter normal basal synaptic responses at mossy fiber→CA3 synapses with low-frequency stimulation. A, Free Ca2+ was measured in normal ACSF (left bar) and in ACSF containing 10 mm CaEDTA (middle bar). The right bar shows the concentration of Ca2+ measured in ACSF containing 10 mm CaEDTA with an additional 0.22 mmCaCl2 (2.4 mm CaCl2 in normal ACSF) added to restore free Ca2+ to the level in normal ACSF. B, Plot of the EPSP against time, with bath-applied CaEDTA (10 mm) indicated by a dashed line. Each point represents the averaged initial slope of evoked EPSP (n = 6). The averaged values were then normalized to the mean initial slope during 30 min baseline recording (percent ± SEM). The traces ontop represent recording before (1) and during (2) CaEDTA perfusion.C, The paired pulse ratio is unaffected by the addition of CaEDTA, as shown on the left. The bar graph demonstrates the average paired pulse ratios from six recordings in which two stimuli were given 40 msec apart in the presence and absence of CaEDTA. Calibration in B, C: 0.5 mV, 5 msec.
Ten millimolar CaEDTA had no effect on basal synaptic transmission elicited by low-frequency stimulation, as measured by the initial slope of the EPSP at mossy fiber→CA3 synapses (Fig. 4B). The chelator at this concentration also had no effect on paired pulse facilitation, a physiological property of presynaptic terminal function at mossy fiber→CA3 synapses (Fig. 4C). Our data demonstrate that basal synaptic function is not altered in the presence of 10 mm Zn2+chelator. Thus, the data suggest that the effects of CaEDTA we observed were attributable to changes in Zn2+concentration.
Mimicking Zn2+ release by adding exogenous Zn2+ induces a long-lasting potentiation of synaptic transmission
In the experiments described above, 10 mm CaEDTA apparently blocked the induction of LTP, because it chelated the Zn2+ released during HFS. We then hypothesized that the addition of exogenous Zn2+ to extracellular bathing solution in the absence of HFS would enhance the strength of synaptic transmission and would induce a long-lasting potentiation of the EPSP in field CA3. As shown in Figure 5A, the EPSP was gradually potentiated to 195 ± 17% of baseline (mean ± SEM; n = 8) during a 20 min exposure to 100 μm Zn2+ and remained potentiated for >3 hr. Zn2+ does not affect the afferent volley in our experimental conditions. Throughout this experiment, the only stimulation applied was low-frequency test stimuli. These data strongly suggest that Zn2+ is able to enhance synaptic strength at mossy fiber→CA3 synapses. Once long-lasting potentiation of the EPSP was established, washing away the exogenous Zn2+ with ACSF containing 10 mm CaEDTA (10 min) did not halt the potentiation (Fig. 5A). This result also indicates that the potentiating effect of Zn2+ was not attributable to its prolonged binding to plasma membrane components. Figure 5Bshows the frequency (given as a percentage) with which various concentrations of Zn2+ induced long-lasting potentiation. In these concentration–response studies, we could reliably induce long-lasting potentiation of the EPSP in concentrations of 50 μm (71%;n = 7) and 100 μm (100%;n = 9) Zn2+. Estimates of the concentration of Zn2+ released from the mossy fibers during HFS have ranged from 10 to 100 μm (Vogt et al., 2000; Li et al., 2001); Zn2+ concentration could reach as much as 300 μm under extreme conditions (Frederickson, 1989). In the presence of 300 μmZn2+, we observed long-lasting potentiation of the EPSP in less than half the slices tested (n = 7; Fig. 5B). This high failure rate might be caused by the neurotoxic effect of Zn2+ at this high concentration (Frederickson et al., 2000).
Fig. 5.
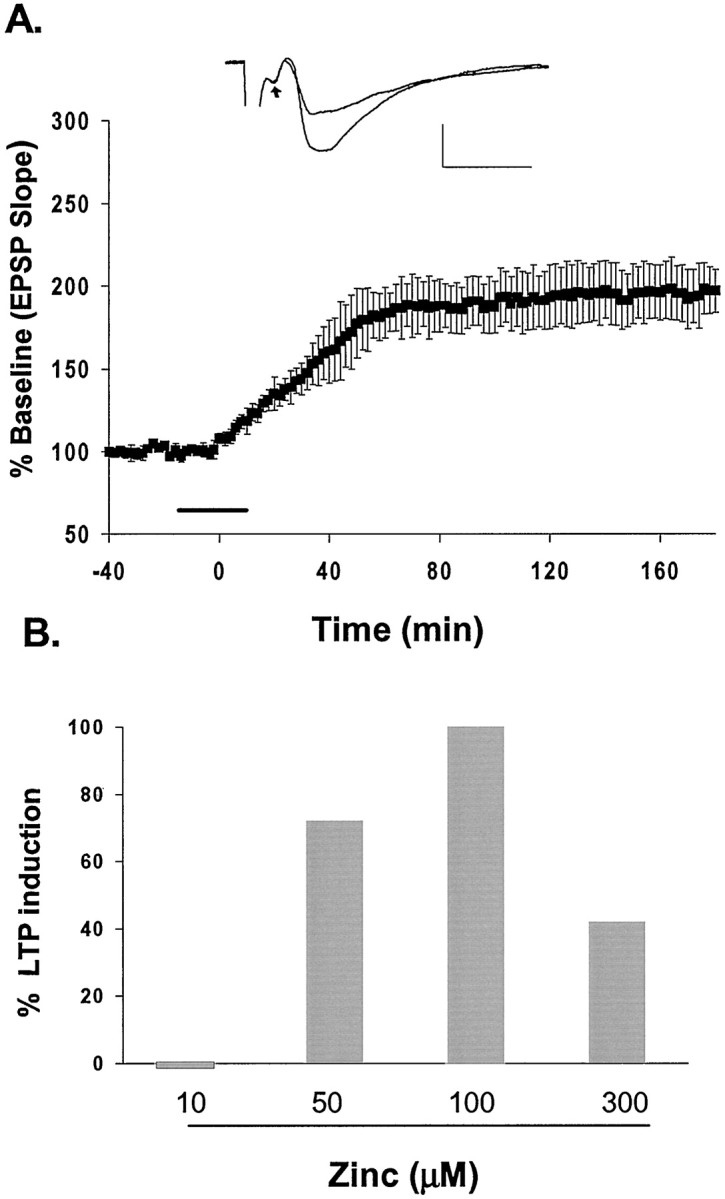
Exogenous Zn2+ induces long-lasting potentiation of the EPSP. A, Plot of exogenous Zn2+-induced long-lasting potentiation. Zn2+ (100 μm) was applied for 20 min (line) after recording a 30 min baseline. Application of Zn2+ was followed by wash with 10 mmCaEDTA. Inset, EPSP recorded before and after exposure to Zn2+. Note that the afferent volley (arrow) was not affected. B, Percentage of LTP induced by different concentrations of exogenous Zn2+. Calibration in A: 1.0 mV, 5 msec.
Glutamate facilitates the potentiating effect of Zn2+
Because glutamate is also released from mossy fiber terminals during electrical stimulation, it is reasonable to expect that, under physiological conditions, both glutamate and Zn2+ are required for LTP induction in these terminals. In our next set of experiments, we sought to verify that simultaneous application of exogenous glutamate and Zn2+ together is sufficient to induce long-lasting potentiation. In these experiments, co-perfusion of glutamate and Zn2+, to imitate the co-release of both from synaptic terminals, induced long-lasting potentiation (Fig. 6A) similar to that induced by Zn2+ alone (Fig. 5A). The mean potentiated EPSP slope was 241 ± 11% (mean ± SEM; n = 6) of baseline, and the potentiation was stable for >3 hr. Glutamate alone, however, in the presence of 10 mm CaEDTA induced only a transient EPSP potentiation, followed by an immediate return to baseline after glutamate washout (Fig. 6A). This result implies that glutamate itself could not induce a persistent EPSP potentiation. As summarized in Figure 6B, the effect of Zn2+ on induction of long-lasting potentiation was modulated by the level of glutamate in the bath. With the standard low-frequency (0.03 Hz) test stimulation, adding both glutamate and Zn2+ caused a more rapid EPSP potentiation than did Zn2+alone. These results indicate that glutamate enables the potentiating effect of Zn2+.
Fig. 6.
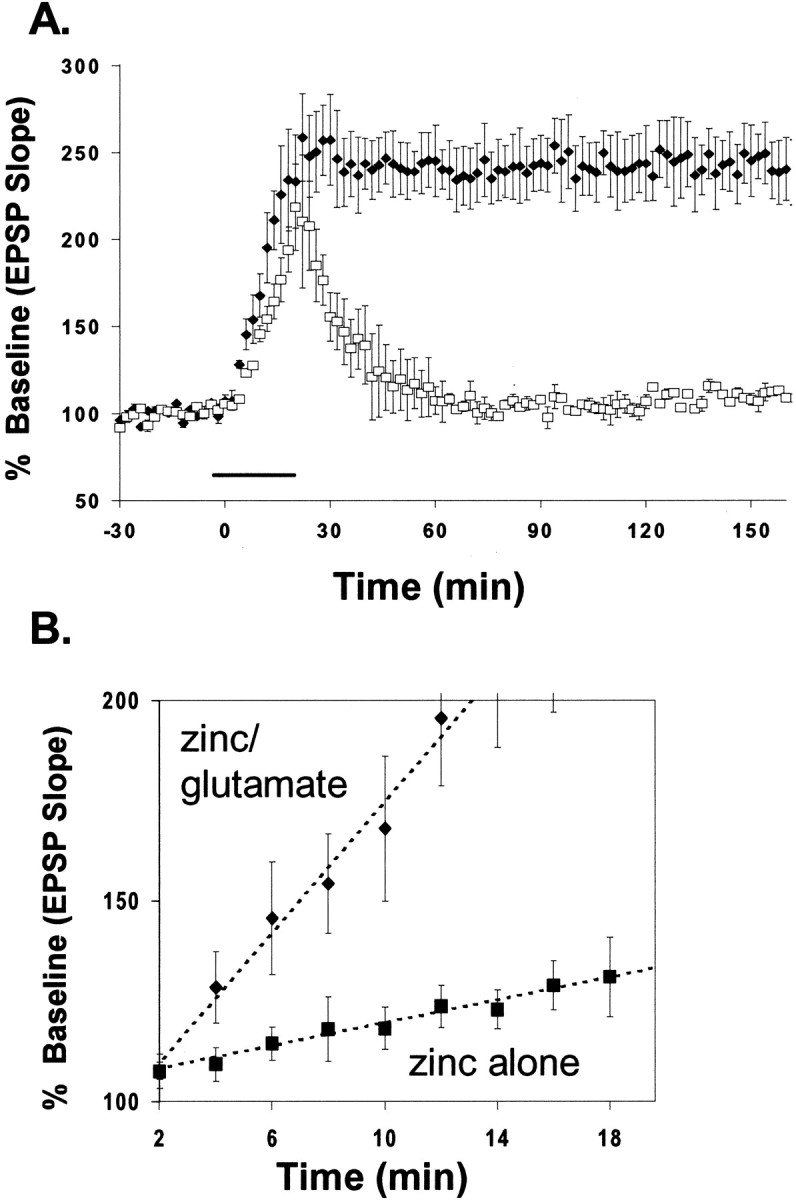
Glutamate facilitates the potentiating effect of Zn2+. A, Co-application of glutamate (100 μm) and Zn2+ (100 μm) expeditiously potentiated EPSPs; potentiation lasted several hours (filled diamonds). Glutamate alone (open squares; in the presence of CaEDTA) failed to induce long-lasting potentiation of the EPSP. Exogenous glutamate and Zn2+ were applied for 20 min (line) after recording a 30 min baseline. Application of glutamate or Zn2+ with glutamate was followed by wash with 10 mm CaEDTA. B, Comparison of the rate of onset of long-lasting potentiation induced by Zn2+alone and Zn2+ plus glutamate. Eachpoint represents the averaged, normalized EPSP initial slope ± SEM from data in Figure 5A andA, respectively.
Potentiation of the EPSP requires the entry of Zn2+ into neurons
One plausible explanation for this interaction is that glutamate is required to permit Zn2+ entry into pyramidal neurons. Glutamate allows Zn2+to enter both directly through Ca2+-permeable AMPA/kainate and NMDA receptors and indirectly, via its depolarizing effects, through VDCC (Choi and Koh, 1998; Weiss and Sensi, 2000). We directly tested whether glutamate mediates Zn2+ translocation across the postsynaptic membrane by the following experiments. Cells in the slice were loaded with Newport Green diacetate (the cell-permeable dye that, once inside the cell, is hydrolyzed by cytoplasmic esterases to become membrane-impermeable) and then thoroughly washed to eliminate unincorporated dye. Mossy fiber axons were then stimulated with HFS, and the entry of Zn2+ into cells in the CA3 pyramidal layer was imaged. In agreement with our previous findings, HFS caused an immediate influx of Zn2+ into cells (Fig.7A). The increase of intracellular Zn2+ fluorescence elicited by HFS was depressed by addition of either CaEDTA (10 mm) or CNQX (10 μm), an antagonist of AMPA/kainate receptors (Fig. 7B). The remaining fluorescence in the presence of 10 mmCaEDTA was likely caused by rapid entry of a small fraction of released Zn2+ into postsynaptic neurons or into presynaptic terminals. CNQX can block entry of Zn2+ not only through AMPA/kainate receptors but also through VDCC and NMDA receptors by preventing membrane depolarization by AMPA/kainate receptor channels. These results agree with our finding that CaEDTA blocks HFS-induced LTP by blocking Zn2+ entry into postsynaptic neurons.
Because CNQX blocked both Zn2+translocation and LTP induction, we tested whether the introduction of Zn2+ into neurons by a different route could restore LTP. In several experiments, Zn2+ entry via ionotropic glutamate receptors and VDCC was blocked by 10 μm CNQX and 50 μm AP-5 (the NMDA receptor antagonist AP-5 was present in the perfusate throughout these experiments) but allowed to enter via sodium pyrithione, a selective Zn2+ionophore (Zalewski et al., 1993), which directly increases intracellular Zn2+ (Fig.8A). After washout of CNQX, Zn2+, and Na-pyrithione, a long-lasting potentiation developed in the absence of HFS (158 ± 12%, mean ± SEM; n = 3). In contrast, without Na-pyrithione, addition of Zn2+ alone in the presence of CNQX failed to induce long-lasting potentiation of the EPSP (93 ± 9%, mean ± SEM; n = 4; Fig.8B). Likewise, pyrithione (50 μm) alone, in the absence of Zn2+, had no long-lasting effect on the EPSP (104 ± 5%, mean ± SEM; n = 5; data not shown). In slices loaded with intracellular Newport Green, which could also function as a selective intracellular Zn2+ chelator, the induction of LTP by HFS was blocked (110 ± 4.5%, mean ± SEM; Fig.9). These data indicate that the translocation of extracellular Zn2+ into postsynaptic neurons is critical for the induction of LTP in mossy fiber→CA3 synapses and suggest an intracellular site of action for Zn2+.
Fig. 9.

Intracellular Zn2+ chelator blocks the induction of LTP. HFS failed to induce mossy fiber LTP (110 ± 4.5%; n = 4) in slices loaded with intracellular Newport Green, which was used here as a selective intracellular Zn2+ chelator (dashed line). Each point represents the averaged and normalized EPSP initial negative slope, and error bars indicate SEM.Inset, EPSP recorded during baseline and at the end of recording after HFS. The arrow indicates HFS (100 Hz, 2 sec). Calibration: 1.0 mV, 5 msec.
DISCUSSION
Our results establish that synaptically released Zn2+ plays an essential role in the induction of LTP in mossy fiber→CA3 synapses. Effective Zn2+ chelation blocked the induction of LTP by HFS. Bath application of exogenous Zn2+ induced a long-lasting potentiation of the EPSP, apparently by acting at an intracellular site rather than at an extracellular site. Co-released glutamate is also essential for induction of LTP under physiological conditions, in part because it allows Zn2+ entry into postsynaptic neurons.
The mossy fiber→CA3 synapse contains high concentrations of Zn2+ in large synaptic boutons (Haug, 1967; Perez-Clausell and Danscher, 1985; Frederickson, 1989). This Zn2+ is released in the same manner as a neurotransmitter and is thought to be co-released with glutamate on stimulation of presynaptic terminals. We found that Zn2+ release is dependent on stimulation frequency (Li et al., 2001). Each mossy fiber bouton terminates on the proximal portion of apical CA3 dendrites with up to 35 release sites (Chicurel and Harris, 1992). Mossy fiber synapses show tremendous frequency facilitation that leads to plateau depolarizations during long stimulus trains (Vogt et al., 2000). Although we do not know the exact relationship between the prominent presence of Zn2+ and these unusual features, it indicates that these synapses are capable of rapidly releasing large amounts of Zn2+ during HFS. Therefore, it is not surprising that a high concentration of Zn2+ chelator was needed to buffer effectively the Zn2+ released during the HFS.
In this investigation of the possible role of Zn2+ in mossy fiber→CA3 synaptic plasticity, the removal of synaptically released Zn2+ with 10 mm CaEDTA blocked the induction of LTP. Direct fluorescence imaging showed that 10 mm CaEDTA chelated 85% of the synaptically released Zn2+. This result verifies that the effect of CaEDTA on LTP induction in mossy fiber→CA3 synapses was achieved by its selective chelation of the synaptically released Zn2+ from mossy fiber terminals. On the other hand, a lower concentration of CaEDTA (1 mm) failed to reduce the extracellular Zn2+ after HFS, as evidenced by its inability to reduce Newport Green fluorescence. We have calculated that, under equilibrium conditions, 1 and 10 mm CaEDTA should indeed reduce 10–100 μm Zn2+ to 0.04–4 nm. The presence of 2.4 mmCa2+ and 1.3 mmMg2+ in the medium and the slow dissociation rate of CaEDTA, however, limit the rate at which Zn2+ is chelated (see Materials and Methods and ). Kinetic calculations indicate that Zn2+ is chelated in a biphasic manner. The initial rate of chelation, limited by the initial concentration of free EDTA and rapidly dissociating MgEDTA, reaches completion in <0.1 msec. The slower phase, limited by the dissociation of CaEDTA, requires many seconds to reach completion at the equilibrium concentrations. Thus 1 mm CaEDTA was unable to prevent the micromolar accumulations of Zn2+ induced by HFS from mossy fiber terminals and entry into CA3 dendrites that we have observed to take place within milliseconds, because it provided a lower concentration of free EDTA and MgEDTA relative to the concentration of released Zn2+. Ten millimolar CaEDTA, on the other hand, provided enough free EDTA and MgEDTA to reduce the 10–100 μm Zn2+ released by the mossy fiber terminals to concentrations below the level readily detected by Newport Green fluorescence as well as those required for LTP.
In the present study, the addition of 50–100 μmexogenous Zn2+ to the solution bathing the slice was required to induce a reliable, long-lasting potentiation of the EPSP. These data suggest that Zn2+ is able to enhance synaptic strength at mossy fiber→CA3 synapses. Ten micromolar Zn2+ did not appreciably alter synaptic transmission. Hence, it can be assumed that under stimulation conditions necessary to induce LTP, concentrations of >10 μm Zn2+ are released in the synapse. In addition, this may explain why the removal of Zn2+ released into the synaptic cleft during basal conditions or low-frequency stimulation failed to alter synaptic transmission. However, Zn2+ is able to inhibit the NMDA receptor at concentrations as low as 50 nm (Chen et al., 1997) and may play an important role in shaping the NMDA receptor response at this synapse under normal physiological conditions. Our results confirm the previous observation that, although a deficiency of bouton Zn2+in rats resulted in the impairment of mossy fiber LTP, it does not appear to affect normal synaptic transmission (Lu et al., 2000). Thus, synaptically released Zn2+ appears to have little effect on basal synaptic function other than in modulating NMDA responses (Peters et al., 1987; Westbrook and Mayer, 1987; Chen et al., 1997) but is required for LTP induction by HFS in mossy fiber→CA3 synapses, which does not require NMDA receptor activation (Harris and Cotman, 1986). It is possible that Zn2+may act on GABAergic interneurons resulting in an indirect effect on the pyramidal cell. Zn2+ inhibits hippocampal postsynaptic GABA current with aKd of 11 μm(Mayer and Vyklicky, 1989). In our study, however, the removal of released Zn2+ or the addition of 10 μm exogenous Zn2+did not alter synaptic transmission in mossy fiber→CA3 synapses. Furthermore, Zn2+ did not affect the afferent volley in our experimental conditions. We have therefore concluded that the indirect effects of Zn2+ acting on interneurons did not inhibit LTP at this synapse.
Because glutamate is also released from mossy fiber terminals during electrical stimulation, it is reasonable to expect that, under physiological conditions, both glutamate and Zn2+ are required for LTP induction in these terminals. The results from the present study indicate that glutamate promotes Zn2+ entry into the neuron by opening Zn2+-permeable channels. It is known that glutamate allows Zn2+ to enter cultured neurons directly through Ca2+-permeable AMPA/kainate and NMDA receptors and indirectly, via its depolarizing effects, through VDCC (Choi and Koh, 1998; Weiss and Sensi, 2000). In the present study, the increase of intracellular Zn2+ elicited by HFS was depressed by addition of CNQX, which can block entry of Zn2+ not only through AMPA/kainate receptors but also through VDCC and NMDA receptors by preventing membrane depolarization through the activation of AMPA/kainate receptors. We can rule NMDA receptors out, because we included APV in the ACSF. Toth et al. (2000) have reported that Ca2+-permeable AMPA receptors are not expressed in CA3 principal neurons, raising doubt that these channels contribute significantly to Zn2+ entry in this region. Kainate receptors or VDCC remain as primary candidates. The possibility that these are the routes of Zn2+ entry in the proximal dendrites of CA3 pyramidal neurons in our experiments is also supported by the presence of a high density of VDCCs and putative Ca2+-permeable kainate receptors in mossy fiber→CA3 synapses (Westenbroek et al., 1990; Bortolotto et al., 1999; Sui and Ruan, 2000). Ca2+ can enter the CA3 pyramidal neurons through these same channels. An increase in intracellular Ca2+ in postsynaptic neurons during LTP induction has been established for all hippocampal synapses except the mossy fiber→CA3 synapse (Zalutsky and Nicoll, 1990). Whether Ca2+ is required in the induction of mossy fiber LTP is the subject of many debates. Other groups have provided evidence that there is an initial rise in postsynaptic intracellular Ca2+ during LTP induction at the mossy fiber synapse (Yeckel et al., 1999). In these reports, LTP was prevented by chelation of postsynaptic intracellular Ca2+. Interpretation of these results, however, is complicated by the fact that both the Ca2+ indicator and chelator used in those studies have higher affinities for Zn2+than for Ca2+. It is impossible to exclude in these studies the role of Zn2+ in LTP induction. A delineation of the separate roles of Zn2+ and Ca2+in LTP induction and the possible interactions between these two ions will require further investigation.
One of the hallmarks of the mossy fiber synapse is its apparent lack of NMDA receptor-dependent synaptic plasticity. Some reports have indicated that LTP at mossy fiber→CA3 synapses may be of presynaptic origin (Harris and Cotman, 1986; Nicoll and Malenka, 1999). However, in other studies, mossy fiber LTP has required both presynaptic and postsynaptic activation (Jaffe and Johnston, 1990). Many of these data could be explained by such factors as the type of LTP-inducing stimulus applied and the recording conditions, but the reasons for the conflicting results are still unclear. Although examining the presynaptic versus postsynaptic origin of LTP was not the goal of the present study, our results would support either a presynaptic or a postsynaptic origin of LTP. On release, Zn2+ could be taken up by selective high-affinity Zn2+ transporters in the mossy fiber terminals or could enter the postsynaptic neuron through glutamate and VDCC for induction of mossy fiber LTP. Our results cannot eliminate either mechanism for several reasons: First, CaEDTA prevented Zn2+ from entering both presynaptic terminals and postsynaptic neurons. Second, bath-applied Zn2+ could enter both mossy fiber terminals and CA3 neurons. Third, although CNQX blocked postsynaptic entry of Zn2+, there may still be a substantial amount of Zn2+ taken up by presynaptic terminals, as indicated by the difference in the intracellular Newport Green fluorescence obtained in the presence of CaEDTA compared with that obtained with CNQX blockade after HFS. These arguments do not negate our observation, however, that Zn2+ must enter cells to perform its role in LTP induction through interactions with kinases, phosphatases, and other intracellular signaling pathways.
Our results introduce the idea that Zn2+released from mossy fiber synapses acts as a presynaptically released second messenger or trans-synaptic factor. A presynaptically released factor that enhances synaptic strength could improve specificity and efficacy of synaptic transmission. In addition to its crucial role for gene expression and transcription, Zn2+has been shown to activate a number of protein kinases such as protein kinase C (Hubbard et al., 1991; Quest et al., 1992), Ca/calmodulin kinase II (Brewer et al., 1979; Weinberger and Rostas, 1991; Lengyel et al., 2000), and mitogen-activated protein kinase (Park and Koh, 1999), which are associated with establishing LTP (Feng, 1995; Soderling and Derkach, 2000; Sweatt, 2001). Mossy fiber boutons terminate on the proximal portion of apical dendrites of CA3 pyramidal neurons. This unusual structure may give Zn2+ direct access to modulate gene transcription. Additionally, nanomolar Zn2+ signals modulate protein-tyrosine phosphatases and, thus, the phosphorylation of myriad postsynaptic proteins (Maret et al., 1999). Therefore, the present study raises the intriguing possibility that entry of synaptically released Zn2+ modulates intracellular signaling pathways and gene transcription.
Calculations of Zn2+ chelation by CaEDTA in the presence of the ions in ACSF.
Our modeling of Zn2+ chelation by CaEDTA in the presence of the Ca2+ and Mg2+ in ACSF included both equilibrium and kinetics calculations. Although 1 mm CaEDTA may be adequate to remove released free Zn2+ by thermodynamic arguments, the kinetics of chelation may be too slow to achieve adequate removal within the time frame of a synaptic event. Equilibrium constants were obtained from Martell and Smith (1974) andBers et al. (1994). Kinetic constants were obtained from Davis et al. (1999) and Hering and Morel (1988) (Table1). In the absence of an experimentally derived on-rate for Zn2+ complexing with EDTA, we used the rate of diffusion (108m/sec) as the rate that limits Zn2+–EDTA complex formation. The off-rate constant was then calculated from theKd. The equations used for the equilibrium case are as follows:
Table 1.
Kinetic constants used in calculating the Zn2+concentration values in Figure 3
| Ion | kon(m/sec) | koff (/sec) |
|---|---|---|
| Mg2+ | 8.75 × 105 | 2.8 |
| Ca2+ | 2.20 × 107 | 0.7 |
| Zn2+ | 1 × 108 | 7 × 10−6 |
Let C, M, Z, and Erepresent the concentrations of the free ions of calcium, magnesium, zinc, and EDTA, respectively. CE, ME, andZE represent the concentrations of the EDTA complexes of Ca2+, Mg2+, and Zn2+, respectively.CT,MT,ZT, andET represent the total concentrations of Ca2+, Mg2+, Zn2+, and EDTA, respectively. KC,KM, andKZ represent the equilibrium dissociation constants for Ca2+, Mg2+, and Zn2+, respectively. Total concentration of any one metal ion is the sum of chelated and free ions. Thus:
| Equation 1 |
Total EDTA in solution is given by:
| Equation 2 |
Substituting the chelated forms for the expressions containing total concentrations in Equation 1 and rearranging, the total concentrations on the left:
| Equation 3 |
| Equation 4 |
Rearranging the definition of the dissociation constants and substituting Equation 1 for the chelated forms:
| Equation 5 |
we can derive three equations by substituting Equation 5 in Equation 4:
| Equation 6 |
| Equation 7 |
| Equation 8 |
Equations 6-8 are quadratic equations, each with three unknowns. They could be solved simultaneously, but we used an iterative calculation method using Microsoft Excel. Estimates were made of each ion concentration initially in the absence of Zn2+ and then including Zn2+, and a calculated value was determined using Equations 6-8, in order of increasing affinity for EDTA. With each calculation, the calculated value was substituted for the estimate made for that ion. A macro was created to do this iteratively until the difference between the calculated and estimated values for Mg2+ concentration was less than an arbitrary critical value (1 × 10−9). The results for Zn2+ concentration are shown in Table2. The equilibrium values for Ca2+, Mg2+, and Zn2+ agreed with those calculated by WEBMAXC version 2.10 (www.stanford.edu/∼cpatton/webmaxc2.htm) using the same equilibrium constants (Table 2). Our estimates of equilibrium concentrations assumed a temperature of 32°C, pH 7.40, and an ionic strength of 0.159 for ACSF.
Table 2.
Equilibrium constants and Zn2+ concentration in ACSF at two different concentrations of total Zn2+ and CaEDTA
| [Zn2+]T(μm) | [CaEDTA]T(mm) | [Zn2+] (m) | KZn2+(m) | KCa2+(m) | KMg2+ (m) |
|---|---|---|---|---|---|
| 100 | 10 | 3.68 × 10−11 | 4.16 × 10−14 | 3.01 × 10−8 | 1.20 × 10−6 |
| 1 | 3.55 × 10−9 | 4.00 × 10−14 | 2.9 × 10−8 | 1.1 × 10−6 | |
| 10 | 10 | 3.53 × 10−11 | 4.16 × 0−14 | 3.01 × 10−8 | 1.20 × 10−6 |
| 1 | 3.21 × 10−10 | 4.00 × 10−14 | 2.90 × 10−8 | 1.10 × 10−6 |
At 32°C, pH 7.4, ionic strength 0.159.
The kinetics of Zn2+ chelation were calculated as follows: Let koff andkon be the dissociation and association rate constants. Then:
| Equation 9 |
Substituting Equation 4 for E in Equation 9, the rate of zinc chelation can be calculated from the concentrations of the three metal ions and other constants:
| Equation 10 |
This equation is also quadratic. The equations for the other ions were derived in the same way and have the same form. Again, we used an Excel spreadsheet to calculate an approximation of the concentrations of each of the three metal ions over time from initial conditions using an appropriately small interval (1 × 10−7 sec). The initial conditions chosen were consistent with our experimental conditions. These take the concentrations of Ca2+ and Mg2+ in ACSF to be those at equilibrium with the given concentration of CaEDTA in the absence of Zn2+. The given concentration of Zn2+ was assumed to be released instantaneously into the medium at time 0. Figure 3 shows the time course of Zn2+ concentration change over the course of 0.1 msec, a time frame considered typical for neurotransmitter release.
Footnotes
This research was supported by grants from the Brain Injury Association and National Institutes of Health Grant NS23865 to J.M.S., generous support from Theodore and Vada Stanley to C.J.H., and in part by National Institutes of Health Grants NS40215 and NS38585 to C.J.F. We thank Richard Thompson for helpful discussions and Ajay Verma for assistance with measurement of free Ca2+.
The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as official or reflecting the views of the Uniformed Services University of the Health Sciences or the United States Department of Defense.
Correspondence should be addressed to John Sarvey, Department of Pharmacology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814. E-mail: jsarvey@usuhs.mil.
REFERENCES
- 1.Aniksztejn L, Charton G, Ben-Ari Y. Selective release of endogenous zinc from the hippocampal mossy fibers in situ. Brain Res. 1987;404:58–64. doi: 10.1016/0006-8993(87)91355-2. [DOI] [PubMed] [Google Scholar]
- 2.Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 3.Basolo F, Pearson RG. Mechanisms of inorganic reactions; a study of metal complexes in solution, Ed 2. Wiley; New York: 1967. [Google Scholar]
- 4.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- 5.Bortolotto ZA, Clarke VR, Delany CM, Parry MC, Smolders I, Vignes M, Ho KH, Miu P, Brinton BT, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402:297–301. doi: 10.1038/46290. [DOI] [PubMed] [Google Scholar]
- 6.Bramham CR, Sarvey JM. Endogenous activation of mu and delta-1 opioid receptors is required for long-term potentiation induction in the lateral perforant path: dependence on GABAergic inhibition. J Neurosci. 1996;16:8123–8131. doi: 10.1523/JNEUROSCI.16-24-08123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brewer GJ, Aster JC, Knutsen CA, Kruckeberg WC. Zinc inhibition of calmodulin: a proposed molecular mechanism of zinc action on cellular functions. Am J Hematol. 1979;7:53–60. doi: 10.1002/ajh.2830070107. [DOI] [PubMed] [Google Scholar]
- 8.Chen N, Moshaver A, Raymond LA. Differential sensitivity of recombinant N-methyl-d-aspartate receptor subtypes to zinc inhibition. Mol Pharmacol. 1997;51:1015–1023. doi: 10.1124/mol.51.6.1015. [DOI] [PubMed] [Google Scholar]
- 9.Chicurel ME, Harris KM. Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J Comp Neurol. 1992;325:169–182. doi: 10.1002/cne.903250204. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- 11.Davis JP, Tikunova SB, Walsh MP, Johnson JD. Characterizing the response of calcium signal transducers to generated calcium transients. Biochemistry. 1999;38:4235–4244. doi: 10.1021/bi982495z. [DOI] [PubMed] [Google Scholar]
- 12.Dawson RMC, Elliot DC, Elliot WH, Jones KM. Data for biochemical research, Ed 3. Oxford; New York: 1986. [Google Scholar]
- 13.Feng TP. The involvement of PKC and multifunctional CaM kinase II of the postsynaptic neuron in induction and maintenance of long-term potentiation. Prog Brain Res. 1995;105:55–63. doi: 10.1016/s0079-6123(08)63283-5. [DOI] [PubMed] [Google Scholar]
- 14.Fredens K, Danscher G. The effect of intravital chelation with dimercaprol, calcium disodium edetate, 1–10-phenantroline and 2,2′-dipyridyl on the sulfide silver stainability of the rat brain. Histochemie. 1973;37:321–331. doi: 10.1007/BF00274968. [DOI] [PubMed] [Google Scholar]
- 15.Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- 16.Frederickson CJ, Suh SW, Silva D, Thompson RB. Importance of zinc in the central nervous system: the zinc-containing neuron. J Nutr. 2000;130:1471.S–1483.S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 17.Golub MS, Keen CL, Gershwin ME, Hendrickx AG. Developmental zinc deficiency and behavior. J Nutr. 1995;125:2263.S–2271.S. doi: 10.1093/jn/125.suppl_8.2263S. [DOI] [PubMed] [Google Scholar]
- 18.Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl-d-aspartate antagonists. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- 19.Haug FM. Electron microscopical localization of the zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure. Histochemie. 1967;8:355–368. doi: 10.1007/BF00401978. [DOI] [PubMed] [Google Scholar]
- 20.Hering JG, Morel FMM. Kinetics of trace metal complexation: role of alkaline-earth metals. Environ Sci Technol. 1988;22:1469–1478. doi: 10.1021/es00177a014. [DOI] [PubMed] [Google Scholar]
- 21.Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 22.Hubbard SR, Bishop WR, Kirschmeier P, George SJ, Cramer SP, Hendrickson WA. Identification and characterization of zinc binding sites in protein kinase C. Science. 1991;254:1776–1779. doi: 10.1126/science.1763327. [DOI] [PubMed] [Google Scholar]
- 23.Jaffe D, Johnston D. Induction of long-term potentiation at hippocampal mossy-fiber synapses follows a Hebbian rule. J Neurophysiol. 1990;64:948–960. doi: 10.1152/jn.1990.64.3.948. [DOI] [PubMed] [Google Scholar]
- 24.Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol (Lond) 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lengyel I, Fieuw-Makaroff S, Hall AL, Sim AT, Rostas JA, Dunkley PR. Modulation of the phosphorylation and activity of calcium/calmodulin-dependent protein kinase II by zinc. J Neurochem. 2000;75:594–605. doi: 10.1046/j.1471-4159.2000.0750594.x. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Hough CJ, Suh SW, Sarvey JM, Frederickson CJ (2001) Rapid Translocation of Zn2+ from presynaptic terminals into postsynaptic hippocampal neurons following physiological stimulation. J Neurophysiol, in press. [DOI] [PubMed]
- 27.Lu Y, Taverna FA, Tu R, Ackerley CA, Wang Y-T, Roder J. Endogenous Zn2+ is required for the induction of long-term potentiation (LTP) at rat hippocampal mossy fiber-CA3 synapses. Synapse. 2000;38:187–197. doi: 10.1002/1098-2396(200011)38:2<187::AID-SYN10>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 28.Maret W, Jacob C, Vallee BL, Fischer EH. Inhibitory sites in enzymes: zinc removal and reactivation by thionein. Proc Natl Acad Sci USA. 1999;96:1936–1940. doi: 10.1073/pnas.96.5.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martell AE, Smith RM. Critical stability constants. Plenum; New York: 1974. [Google Scholar]
- 30.Mayer ML, Vyklicky L., Jr The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J Physiol (Lond) 1989;415:351–365. doi: 10.1113/jphysiol.1989.sp017725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann NY Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- 32.Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci USA. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park JA, Koh JY. Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- 34.Perez-Clausell J, Danscher G. Intravesicular localization of zinc in rat telencephalic boutons. A histochemical study. Brain Res. 1985;337:91–98. doi: 10.1016/0006-8993(85)91612-9. [DOI] [PubMed] [Google Scholar]
- 35.Peters S, Koh J, Choi DW. Zinc selectively blocks the action of N-methyl-d-aspartate on cortical neurons. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- 36.Quest AF, Bloomenthal J, Bardes ES, Bell RM. The regulatory domain of protein kinase C coordinates four atoms of zinc. J Biol Chem. 1992;267:10193–10197. [PubMed] [Google Scholar]
- 37.Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 38.Sui L, Ruan DY. Impairment of the Ca2+-permeable AMPA/kainate receptors by lead exposure in organotypic rat hippocampal slice cultures. Pharmacol Toxicol. 2000;87:204–210. doi: 10.1034/j.1600-0773.2000.d01-75.x. [DOI] [PubMed] [Google Scholar]
- 39.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 40.Toth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ. Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci. 2000;20:8279–8289. doi: 10.1523/JNEUROSCI.20-22-08279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 42.Weinberger RP, Rostas JA. Effect of zinc on calmodulin-stimulated protein kinase II and protein phosphorylation in rat cerebral cortex. J Neurochem. 1991;57:605–614. doi: 10.1111/j.1471-4159.1991.tb03791.x. [DOI] [PubMed] [Google Scholar]
- 43.Weiss JH, Sensi SL. Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci. 2000;23:365–371. doi: 10.1016/s0166-2236(00)01610-6. [DOI] [PubMed] [Google Scholar]
- 44.Weiss JH, Koh JY, Christine CW, Choi DW. Zinc and LTP [letter]. Nature. 1989;338:212. doi: 10.1038/338212b0. [DOI] [PubMed] [Google Scholar]
- 45.Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 46.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 47.Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zalewski PD, Forbes IJ, Betts WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II). Biochem J. 1993;296:403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]