

Abstract
The cyclooxygenases catalyze the rate-limiting step in the formation of prostaglandins from arachidonic acid and are the pharmacological targets of (NSAIDs). In brain, cyclooxygenase-2 (COX-2), the inducible isoform of cyclooxygenase, is selectively expressed in neurons of the cerebral cortex, hippocampus, and amygdala. As an immediate-early gene, COX-2 is dramatically and transiently induced in these neurons in response to NMDA receptor activation. In models of acute excitotoxic neuronal injury, elevated and sustained levels of COX-2 have been shown to promote neuronal apoptosis, indicating that upregulated COX-2 activity is injurious to neurons. COX-2 may also contribute to the development of Alzheimer's disease, for which early administration of NSAIDs is protective against development of the disease. To test the effect of constitutively elevated neuronal COX-2, transgenic mice were generated that overexpressed COX-2 in neurons and produced elevated levels of prostaglandins in brain. In cross-sectional behavioral studies, COX-2 transgenic mice developed an age-dependent deficit in spatial memory at 12 and 20 months but not at 7 months and a deficit in aversive behavior at 20 months of age. These behavioral changes were associated with a parallel age-dependent increase in neuronal apoptosis occurring at 14 and 22 months but not at 8 months of age and astrocytic activation at 24 months of age. These findings suggest that neuronal COX-2 may contribute to the pathophysiology of age-related diseases such as Alzheimer's disease by promoting memory dysfunction, neuronal apoptosis, and astrocytic activation in an age-dependent manner.
Keywords: transgenic mouse, COX-2, spatial memory, aversive behavior, TUNEL, GFAP
Cyclooxygenase-2 (COX-2), the inducible isoform of cyclooxygenase, is expressed at high basal levels selectively in pyramidal neurons of hippocampal and cortical circuits and in neurons of the amygdala (Yamagata et al., 1993; Breder et al., 1995). Within neurons, COX-2 immunoreactivity has been localized to dendritic spines (Kaufmann et al., 1996), the sites of NMDA receptor-mediated neurotransmission. COX-2 expression is rapidly and transiently upregulated in neurons in response to excitatory synaptic transmission mediated by NMDA receptor activation (Yamagata et al., 1993). COX-2 expression is also dramatically induced in paradigms of acute excitotoxic neuronal injury such as ischemia, and inhibition of COX-2 activity can prevent neuronal loss (Nogawa et al., 1997; Nakayama et al., 1998; Iadecola et al., 2001). Thus neuronal COX-2, like the NMDA receptor, is likely to function both physiologically in refining excitatory synaptic connections and pathologically in promoting neuronal injury, depending on the magnitude and regulation of COX-2 expression.
Recent studies have suggested that disruption of Ca2+ homeostasis resulting from increased activation of glutamate receptors may contribute to neuronal injury in degenerative diseases such as Alzheimer's disease (Greenamyre and Young, 1989; Khachaturian, 1989; Mattson, 1994). Activation of the NMDA receptor by glutamate induces the expression of COX-2, and increases in Ca2+ can activate phospholipase A2, which liberates arachidonic acid, the substrate for production of prostaglandins by COX-2. Prostaglandins in turn have been shown to stimulate astrocytic glutamate release into the synaptic cleft (Bezzi et al., 1998; Sanzgiri et al., 1999), and this release has the potential of leading to additional increases in COX-2 expression and prostaglandin production at dendritic spines (Drachman and Rothstein, 2000), resulting in an uncontrolled cycle of disrupted Ca2+ homeostasis at the spine. Thus COX-2, by virtue of its regulation by glutamate, may compromise the viability of synapses and neurons both in age-associated neurodegenerative diseases such as nonfamilial Alzheimer's disease (AD) and in acute excitotoxic insults such as ischemia, in which glutamate toxicity is thought to play a critical role.
Alzheimer's disease is a neurodegenerative disorder of unknown etiology that accounts for two-thirds of all dementia in the elderly. Although the inciting event responsible for initiation of the disease remains unclear, pathological studies of early to late stage AD demonstrate prominent neuronal and synaptic loss in concert with amyloid deposition and astrocytic and microglial activation. In AD, neuronal COX-2 levels have been found to be either elevated (Pasinetti and Aisen, 1998; Hoozemans et al., 2001) or unchanged (Yermakova et al., 2000). Recent studies suggest that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with a significantly lower risk of developing AD (McGeer et al., 1996; Stewart et al., 1997), raising the possibility that in normal aging populations, inhibition of cyclooxygenase activity may be protective against the development of AD.
In this study, we tested the possibility that constitutively increased COX-2 expression in neurons would lead to behavioral and pathological changes relevant to those observed in age-dependent neurodegenerative diseases such as AD. We have developed a transgenic mouse model in which the human isoform of COX-2 (hCOX-2) is overexpressed constitutively in neurons. We provide evidence that transgenic hCOX-2 mice develop age-related cognitive deficits that are associated with a parallel age-dependent increase in neuronal apoptosis and astrocytic activation.
MATERIALS AND METHODS
Generation of hCOX-2 mice
The human COX-2 open reading frame (ORF) was subcloned into the second exon of the Thy-1 promoter (Aigner et al., 1995). Both strands of the entire 1.8 kb human COX-2 ORF were sequenced, and the correct orientation of the clone within the Thy-1 promoter was assessed by restriction digestion and sequencing of the 5′ and 3′ junctional ends. Plasmid was purified by double banding on CsCl-ethidium bromide gradients. The 8.5 kb transgene–Thy-1 promoter fragment was purified away by restriction digest and gel electrophoresis followed by electroelution, phenol extraction, and passage through an ion exchange column (Elutip columns; Schleicher & Shuell, Keene, NH). Concentration of the transgene was adjusted to 500–1000 molecules/pl of embryo-tested 10 mm Tris and 0.01 mm EDTA. Pronuclear injections into C57B6/J male pronuclei were administered, and genomic DNA was isolated from tail segments of 4-week-old pups. Tail segments of potential founders were incubated in 1 ml each of 50 mm Tris, pH 8.0, 100 mm EDTA, 4 mmNaCl, 0.5% SDS, and 0.5 mg/ml proteinase K for 12–16 hr at 55°C. Protein was removed by adding 0.3 ml of a supersaturated NaCl solution followed by vigorous vortexing, incubation on ice, and centrifugation to remove the protein pellet. The supernatant was removed, and genomic DNA was recovered by adding 1 ml of 100% ethanol to each tube and inverting. DNA was washed twice with 70% ethanol and resuspended in TE, pH 8.0. Southern blot analysis was performed to identify founder mice carrying the transgene. Twenty micrograms of each DNA sample were digested with XhoI to liberate the 1.8 kb transgene, fractionated by gel electrophoresis, and transferred to nitrocellulose membranes. Transferred DNA was hybridized to the ORF of [32P]dCTP-labeled human COX-2 DNA probes, washed, and exposed to x-ray film. Subsequent genotyping of heterozygous progeny resulting from expansion of colonies was performed using PCR analysis of tail genomic DNA.
Western blot analysis
Brain regions were harvested and homogenized in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS, and protease inhibitors, fractionated by SDS-PAGE, and electrophoretically transferred to nitrocellulose membranes. Blots were probed with a polyclonal or monoclonal antibody that recognized both mouse and human COX-2 C-terminal peptide sequences (Cayman Chemicals Ann Arbor, MI), human full-length COX-2 protein (12A2; Pharmacia Corp., Peapack, NJ), actin (Sigma, St. Louis, MO), and GFAP (Dako, Carpinteria, CA). Immunoreactivity was detected using either sheep anti-rabbit or anti-mouse HRP-conjugated secondary antibody (Amersham Pharmacia Biotech) followed by enhanced chemoluminescence (Bio-Rad, Hercules, CA). Autoradiographic signals were analyzed using NIH Image.
Nissl staining and immunocytochemistry
Mice were perfused with 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4. Brains were removed, post-fixed for 4–16 hr, and either cryoprotected in 30% sucrose for free-floating sections and Nissl staining or processed for paraffin embedding for immunocytochemistry for COX-2 and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). Coronal free-floating sections 50 μm in thickness were collected at 500 μm intervals from the frontal to occipital cerebral cortical poles. Nissl surveys of neocortex, hippocampus, amygdala, limbic cortex, thalamus, and striatum were performed at 500 μm intervals. Paraffin sections (10 μm thick) were incubated in 0.3% Triton X-100 and 3% H2O2, washed, and microwaved in Tris-buffered saline, pH 9.0, to unmask the COX-2 antigen, blocked overnight at 4°C, and incubated with anti-human COX-2 antibody to visualize transgenic and not endogenous COX-2 protein. Sections were subsequently incubated with biotinylated horse anti-mouse secondary antibody, washed, incubated with avidin–biotin complex (Vector Laboratories, Burlingame, CA), and developed with nickel-cobalt diaminobenzidine (Pierce, Rockord, IL).
TUNEL
In situ end labeling of DNA fragments was performed on 10 μm paraffin sections through parietal cortex to identify cells dying by apoptosis or necrosis. TUNEL-positive cells were counted in a blinded manner in neocortical layers I–VI, excluding the periventricular zone, at intervals of 60–80 μm. DNA strand breaks were detected by enzymatically labeling the free 3′-OH ends of dying cells with the ApopTag peroxidase kit (Intergen, Purchase, NY). Sections were deparaffinized and pretreated with proteinase K (20 μg/ml), and endogenous peroxidase activity was quenched with a 5 min incubation in 3% H2O2. Sections were then incubated with terminal deoxynucleotidyl transferase activity and digoxigenin-labeled dUTP and incubated with anti-digoxigenin peroxidase conjugate, and the reaction product was visualized with DAB (Pierce). After TUNEL labeling, sections were stained with Hoechst (bis-benzamide, Sigma) for 10 min in PBS to label nuclei. TUNEL staining was visualized under light microscopy, and nuclear localization of the TUNEL label was confirmed by examining Hoechst staining under fluorescence. TUNEL-staining cells were identified by the characteristic condensation of nuclei. Nuclear morphology was further studied at high magnification (400–1000×) to observe condensed nuclei, chromatin margination, or apoptotic bodies. Only TUNEL- and Hoechst-positive cells that revealed these morphological signs of apoptosis were counted as TUNEL-positive apoptotic cells. TUNEL data were analyzed using two-way ANOVA (age × genotype) with the square root transformation of the number of apoptotic cells in cerebral cortex per section, with post hoc tests applied for the effect of genotype within each age group. For fluorescent double labeling of TUNEL-stained sections (n = 8 hCOX-2 andn = 8 nontransgenic sections for total of 23 Hoechst-positive TUNEL cells), sections were rehydrated, blocked with 10% donkey serum in PBS, and incubated with monoclonal anti-Neu N antibody (Chemicon, Temecula, CA) and polyclonal anti-GFAP antibody (Dako) overnight at 4°C. Sections were washed and incubated with donkey anti-rabbit FITC and donkey anti-mouse tetramethyl rhodamine isothiocyanate (Jackson ImmunoResearch, West Grove, PA) secondary antibodies, washed and mounted with Permafluor with 40 mg/ml 1,4-diazabicyclooctane to preserve fluorescence, and visualized with fluorescence microscopy.
Measurement of prostaglandin E2
Brain tissue was rapidly harvested and frozen in liquid N2, and the tissue was weighed and homogenized in 70% methanol and centrifuged at 12,000 rpm for 20 min. Supernatants were removed and evaporated under N2 gas and resuspended and assayed by ELISA for production of prostaglandin E2 (PGE2) as described previously (Smith et al., 1998).
Behavioral testing
To determine whether COX-2 overexpression would lead to specific deficits in cognition, three groups of hCOX-2 and nontransgenic control littermates were tested in a battery of behavioral tests at 7 months [n = 11 transgenic (Tg); and n = 9 non-Tg], 12 months (n = 10 Tg; and n = 9 non-Tg) and 20 months (n = 5 Tg; andn = 9 non-Tg) of age. All mice were female C57B6/J F1 or F2 generations and were naïve to the testing protocols. A wide range of behaviors were tested, including cognitive abilities (Morris water maze, active and passive avoidance, and spontaneous alternation), sensorimotor skills (inclined screen, wire suspension, bridge walking), and emotional and exploratory responses (plus maze and open field). The cross-sectional design of behavioral testing in this study ensured no loss of task sensitivity because of the retest effect. All behavioral testing was conducted in a blind coded manner.
Water maze apparatus
For place and visual discrimination tasks, a plastic ring 100 cm in diameter was placed in a large metal pool (180 cm in diameter) and filled with opaque water at 24 ± 2°C with a depth of 35 cm. A smaller ring (55 cm in diameter) or a straight plastic alley (10 × 70 cm) was placed in the large pool for swim pretraining or straight swim testing, respectively. An escape platform with its top made of Lucite (10 × 10 cm) was invisible to the mouse in its elevated position (0.5 cm below the water surface). In its lowered position (19 cm beneath the surface), the platform was unavailable for the mouse as an escape platform. During straight swim and visual discrimination tests, the tank was surrounded by a black curtain to prevent the use of spatial cues. During the place discrimination task, a set of distal visual cues was used on the walls around the water maze, and proximal cues were placed around the ring of the 180 cm tank. Performance in the water maze was recorded through a computer-based video tracking system (HVS Image Analysis VP-200; HVS Image, Hampton, England).
Straight swim test
On the first day, each mouse was pretrained to swim in the smaller ring of the water tank and climb onto and remain on the platform (Markowska et al., 1998). On the second day, mice were trained to swim the length of the narrow alley. This test assessed the swimming ability of the mice and was performed over two sessions consisting of five trials each.
Place discrimination task
This task was used to assess spatial memory, which is strongly dependent on an intact hippocampal system (Olton and Papas, 1979; Silva et al., 1998) as well as prefrontal cortex (Kolb et al., 1982; Compton et al., 1997) and is particularly sensitive to aging (Barnes, 1988;Markowska et al., 1989). Each daily session consisted of 12 trials with a random order of start positions, as described previously (Markowska et al., 1998). To prevent fatigue, the mice were run one after another in batches of four or five animals. Thus, after each trial the mouse was removed from the pool, dried with a towel to prevent alterations in thermoregulation, and placed in a waiting cage for ∼5–7 min (referred to later as “short delay”). During the platform trials, the platform was submerged but accessible to the mouse. During the probe trials conducted with either a short (5–7 min) or long (24 hr) delay after the platform trials (see Fig. 2A), the platform was collapsed for variable intervals (10–40 sec) to test the subject's preference of location. The collapsed platform was then returned to its raised position at the end of the probe trial to maintain the same response–reinforcement contingency as in the platform trials. If the mouse failed to locate the platform in 60 sec, the experimenter directed the mouse to the platform with his hand, and the mouse remained on the platform for 10 sec. Latency (the time in seconds to reach the platform from the start location), distance (path in centimeters from the start location to the platform), heading angle (angle between the actual and ideal directions of swimming to the platform, deg), and swim speed (average speed during a trial in centimeters per second) were measured during the platform trials. A lower score in platform measures (except for swim speed) indicated a better performance. In the probe trials, the measures recorded were annulus 20 time (percentage of time spent in an area 20 cm in diameter around the location of the platform), annulus 40 time (percentage of time spent in an area 40 cm in diameter around the location of the platform), and platform crossings (the number of swims over the platform location). Higher scores in the indexes of probe trials reflected a better performance. The order of platform and probe trials for 7-month-old mice was as for day 1 (see Fig. 2A). On the day after the 4 d of training, all mice were given a single probe trial with a 24 hr delay to assess the strength of memory traces.
Fig. 2.
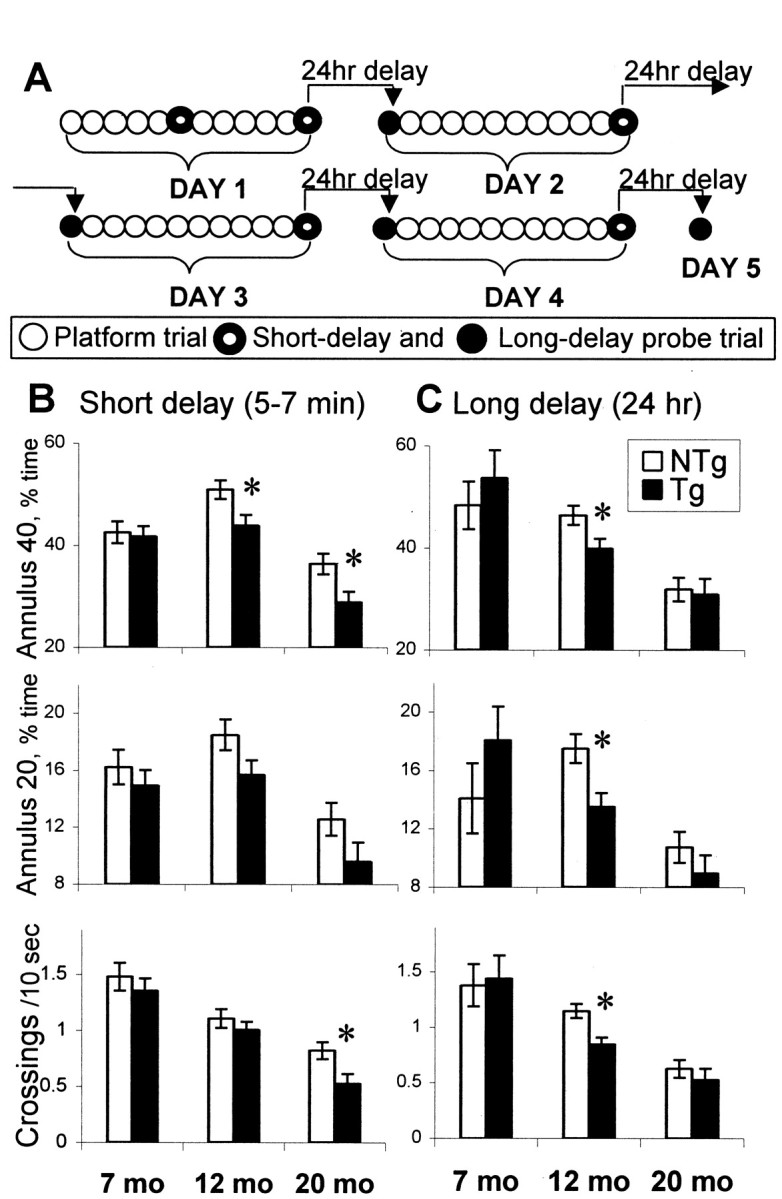
Performance of hCOX-2 and nontransgenic control littermates during probe trials in the Morris water maze.A, Order of platform and probe trials during training in the place discrimination task. Daily sessions consisted of 10 platform (open circles) and 2 probe trials. During the first day, the training started with five platform trials followed by a probe trial with short delay (5–7 min, ringed circle). The short-delay probe trial that was conducted at the end of the day's sessions was used as an index of memory for the platform location with a minimum retention interval. After the first day of training, probe trials with 24 hr of delay (filled circles) were included to assess the strength of long-term memory of the platform location and were performed on days 2–5. B, C, Preference for the platform location during short-delay (B) and long-delay (C) probe trials. Open bars, Nontransgenic (NTg) mice; filled bars, hCOX-2 transgenic (Tg) mice. At the age of 7 months, mice did not differ in any of the measures in both types of probe trials. At the age of 12 months, Tg mice performed similarly to NTg mice in an easy task, the short-delay probe, except for Annulus 40, and were significantly impaired in all measures in the more challenging task, the 24-hr-delay probe trial. Introduction of the 24 hr delay led to a more pronounced impairment of spatial memory in middle-aged Tg mice that was expressed both as a significant difference from NTg littermates of the same age as well as in a significant aging effect compared with younger Tg mice. At 20 months, the performance deficit of Tg mice had already occurred in short-delay probe trials. However, both Tg and NTg 20-month-old mice were significantly impaired in the more difficult 24-hr-delay probe trials. *Significant effect of genotype (simple main effect, p < 0.05). Data are mean ± SEM.
Visual discrimination task
On the day after place discrimination testing, mice were trained in a visual discrimination task in which the platform was made visible by the attachment of a high-contrast extension (0.5 cm above the water surface). The training consisted of two sessions, six trials each, with the visible platform located in different quadrants of the pool and at different distances from the wall. The start position was changed randomly for each trial. The measures of performance were the same as for the platform trials in the place discrimination task.
Spontaneous alternation task
This task assesses recent memory related to the optimal foraging strategies in the wild (Dember and Fowler, 1958) and depends on the integrity of prefrontal and hippocampal systems (Divac et al., 1975). Each arm of the Y maze was 22 × 7 cm. The mouse was placed in one arm and allowed to move freely through the maze for a 5 min test session. The sequence of arm entries was recorded. An alternation was defined as the number of triads containing entries into all three arms divided by the maximum possible alternations (Stone et al., 1992). For this and all subsequent tasks, the maze was cleaned with a diluted alcohol solution and dried with a paper towel.
T-maze foot shock active avoidance
Performance of this task depends on the integrity of the hippocampus (Flood et al., 1989, 1990). It also requires an association between the conditioned stimulus and a foot shock that depends on intact amygdala function (Davis, 1992; LeDoux, 1993). The T maze consists of a start box (15 × 20 cm) that is separated from two goal arms (15 × 40 cm) by a guillotine door. A scrambled foot shock (0.3 mA; Coulbourn Instrument) was delivered to the grid floor. The goal box opposite to that selected by the mouse on the initial trial was selected as the correct goal box for that mouse. At the start of the trial, the mouse was placed in the start box, and after 5 sec the guillotine door was opened, and a buzzer sounded simultaneously (75 dB white noise). A mouse succeeded in avoiding foot shock in the start box by entering the correct goal box within 5 sec after initiation of the buzzer. If the mouse entered the incorrect goal box, a foot shock was applied until it entered the correct goal box. If the mouse did not leave the start box within 5 sec after the initiation of the buzzer, a foot shock in the start box and incorrect goal box was applied until the mouse escaped the shock and entered the correct goal box. The maximal duration of shock was 30 sec. After a variable intertrial interval in the goal box (15 sec on average), the mouse was placed in the start box for the next trial. The pretraining session continued until the mouse made its first correct avoidance response. The following day, the mice were trained until they made five correct avoidance responses in six consecutive training trials (acquisition session). Immediately after, a reversal session was conducted with the correct goal box opposite to the one used during acquisition. During the reversal session, five correct avoidance responses in six consecutive training trials were required. The measures included the number of trials to the first correct avoidance, the number of trials to five correct avoidances in six consecutive trials, and the number of errors (entrances to the incorrect arm − during the pretraining session). To control for the animal's sensitivity to foot shock, the latency to escape from the stem of the T maze during the first pretraining trial was recorded. In addition, the thresholds for eliciting a burst activity to foot shock (Fanselow, 1982) were assessed in a separate box with a grid floor and a range of shock of 0.01–0.20 mA.
Inhibitory avoidance
The retention of inhibitory avoidance is dependent on an intact amygdala (Liang et al., 1982; Liang et al., 1994) as well as hippocampus and entorhinal cortex (Cammarota et al., 1995; Roozendaal et al., 1999; Walz et al., 2000). A rectangular box (29 × 10 × 50 cm) consisting of a start chamber (illuminated by a table light) and a dark testing chamber separated by a guillotine door was used for testing the step-through inhibitory avoidance. The scrambled foot shock (0.3 mA; Coulbourn Instrument) was delivered for 1 sec to the grid floor with a 1 cm gap in the middle. Testing was conducted with the lights off, and mice were acclimated to the dark for 30 min. Each mouse was then placed into the start chamber facing away from the guillotine door. After 15 sec the door to the dark area of the box was opened. When the mouse entered the dark testing chamber, the door was closed, and a foot shock was delivered. The mouse was then returned to its cage. The retention session was given 24 hr later in an identical manner to the acquisition session, except that no shock was delivered after the mouse entered the dark chamber.
Plus maze
This test has been pharmacologically, physiologically, and behaviorally validated as a measure of anxiety (Lister, 1987; Rodgers and Cole, 1993). The elevated plus maze consists of four arms extended from a central platform (10 × 10 cm), angled at 90° to each other, yielding a plus shape. Two opposing arms of the maze are left open (50 × 10 cm) without sidewalls, and the remaining two opposing arms are left closed (40-cm-high side and end walls). The maze was elevated on a tripod 70 cm above the floor. Each mouse was placed in the neutral area in the center of the maze, and the following measures were recorded during a single 5 min trial: (1) number of visits into the open and closed arms, (2) time spent in open and closed arms, (3) number of episodes of grooming, and (4) number of episodes of defecation.
Open field
Locomotor activity of the mice was tested using the open field paradigm. A square open field (Lucite, 60 × 60 cm) surrounded by walls (30 cm high) was divided into 36 squares. A central light source (25 W) on the ceiling gave invariant illumination in an otherwise dark room. One 5 min trial was conducted for each animal. The mouse was placed in the center of the open field and its performance in six measures was assessed: (1) crossings of outer squares near the walls, (2) crossings of inner squares, (3) number of rearings, (4) number of grooming episodes, (5) number of episodes of urination, and (6) number of episodes of defecation.
Sensorimotor tests
Sensorimotor testing (Markowska et al., 1998) assessed muscle strength, postural adjustments, and body coordination. The maximum latency for all tasks was 2 min, except for the fall from an inclined screen, which lasted 30 min. One trial for all tasks was performed for each mouse with an intertrial interval of ∼2 hr.
Turning in an alley. The mouse was positioned facing the back wall of an alley. The dependent measure was the amount of time (seconds) that elapsed before the mouse turned and faced the open end of the alley. In all the following tasks, the apparatus was raised 120 cm above a foam cushion.
Bridges (rectangular 2 cm wide and round 2 cm in diameter).At first, the mouse was placed on the escape platform of the bridge apparatus for 10 sec and then placed in the middle of the bridge. The latency to reach the platform or the latency to fall was recorded for each of the bridges.
Falling from a wire rod. The mouse was placed hanging by its forepaws from a wire, and the time to fall from the wire was recorded.
Falling from an inclined screen. Each mouse was placed facing upward in the center of a wire mesh screen, oriented at a 90° angle. The time to fall from the screen was recorded.
Data analyses
The data were analyzed using repeated measures ANOVA and nonparametric tests with the statistical package Statistica 8.0. The independent variables were genotype, a comparison between hCOX-2 and nontransgenic mice; and age, a comparison between 7, 12, and 20 months of age; and repeated measures were session, a comparison between means from trials during sessions; trial, a comparison between trials; and delay, a comparison between short-delay (5 min) and long-delay (24 hr) probe trials. Omnibus ANOVA (age × genotype) was used to assess the differences between transgenic and nontransgenic mice across different ages. Simple main effect ANOVAs were conducted to assess differences between hCOX-2 and nontransgenic mice at each age or an aging effect for each genotype separately. A Newman–Keuls post hoc test was applied to significant main effects and interactions to estimate differences between particular sets of means. For analysis of the sensorimotor performance, scores from the tests were standardized (Z scores) and ANOVA was applied to the averageZ score. For measures of the inhibitory avoidance task and the sensitivity to shock (the threshold of a burst reaction), the nonparametric tests were used, because the data distributions were not normal.
For estimation of genotype differences of GFAP levels, quantitative Western blot analysis was performed using two repetitions of the procedure in two batches of mice. For each genotype, four pairs (hCOX-2 and nontransgenic) of mice were used in the first batch, and three pairs of mice were used in the second batch. To avoid potential differences in intensities of signal between different runs of a procedure, the data were standardized (Z scores) within each run, and ANOVA was applied to Z scores of signal intensity.
RESULTS
Generation and characterization of transgenic mice that overexpress COX-2
To achieve elevated expression of COX-2 selectively in neurons, we generated transgenic C57BL6/J mice carrying the human isoform of COX-2 under the control of the mouse Thy-1 promoter (Aigner et al., 1995). The use of the inbred C57BL6/J strain of mice avoided the potential for artifactual results related to hybrid genetic backgrounds that could confound the cognitive performance of the mice (Gerlai, 1996). The choice of the strain C57BL6/J to create the transgenic lines was based on well studied behavioral traits of this strain in a wide range of cognitive tasks (Crawley et al., 1997; Owen et al., 1997; Lipp and Wolfer, 1998). The Thy-1 promoter was chosen for its high level of neuron-specific expression that begins in the second postnatal week (Vidal et al., 1990), a pattern that parallels the temporal and anatomical pattern of expression of endogenous COX-2. The human isoform of COX-2 can be detected with a species-specific antibody (anti-human COX-2 12A2 antibody), and its pattern of distribution and cellular localization can be distinguished from those of endogenous murine COX-2.
Western blot analysis demonstrated the presence of a ∼70 kDa band representing the transgenic hCOX-2 protein. The transgenic hCOX-2 protein was consistently overexpressed throughout the life span of the animals, indicating that the Thy-1 promoter remained active as the animals aged (Fig.1A,B). Immunocytochemistry demonstrated expression of hCOX-2 in hippocampal pyramidal neurons and cortical neurons in layers II/III and V and relatively lower expression in amygdala. At higher magnification, the hCOX-2 protein was appropriately localized to the perinuclear region and dendritic processes (Fig. 1C–E). We found that the temporal and anatomic expression of hCOX-2 protein driven by the Thy-1 promoter recapitulated the pattern of expression of endogenous mouse COX-2 in neurons, albeit at constitutively elevated levels.
Fig. 1.

Analysis of hCOX-2 expression in brains of transgenic and nontransgenic C57B6/J mice. A, Western blot analysis of murine COX-2 (mCOX-2) using species-specific monoclonal antibody 12A2, which detects human COX-2 and not murine COX-2. Human recombinant COX-2 is detected but not mouse COX-2 or mouse COX-1 (left panel). In 3-month-old hCOX-2 brains, a 70 kDa band (arrow) representing transgenic hCOX-2 is detected that is not present in nontransgenic brain. B, Western blot analysis of 20- to 24-month-old hCOX-2 mice using polyclonal anti-COX-2 antibody detects both human and murine COX-2 expression and demonstrates continued high expression of transgenic hCOX-2 in aged animals. Note the faint 70 kDa band consisting of endogenous murine COX-2 in nontransgenic (NTg) 20- to 24-month-old animals. C–E, Immunocytochemistry of hCOX-2 protein on paraffin sections of brain from nontransgenic and hCOX-2 mice stained with 12A2. C, Low-power (50×) magnification of hippocampus of a 14 month hCOX-2 brain (top panel) and a nontransgenic age-matched control (bottom panel) demonstrating the presence of transgenic hCOX-2 protein in pyramidal neurons of the hippocampus in transgenic hCOX-2 but not nontransgenic brain. Note the presence of hCOX-2 protein in the CA1 region (arrows) and CA3 region (asterisks) and the absence of hCOX-2 protein in dentate gyrus (d). D, Higher magnification (100×) of frontal cortex demonstrating hCOX protein in layers II/III and V. The hCOX-2 protein is present in the same subcellular patterns and anatomic patterns of expression as endogenous murine COX-2, which is present in a somatodendritic distribution in neurons and hippocampus and layers V and II/III of cortex. E, Higher magnification (400×) of pyramidal neurons of CA1 (top panel) and CA3 (bottom panel).Arrows point to hCOX-2 protein in apical dendritic processes in CA1 and CA3 pyramidal neurons. F, G, Determination of brain PGE2 levels in transgenic and nontransgenic mice. Data are mean ± SEM. F, PGE2 levels were significantly increased in transgenic (hCOX-2) versus nontransgenic (NTg) mice at 4, 6, and 9 months of age and were elevated on average ∼10- to 12-fold over endogenous PGE2 levels. G, PGE2 levels in hCOX-2 and nontransgenic mice can be rapidly reduced with administration of the COX-2 inhibitor celecoxib.
Because COX-2 is an enzyme with a measurable product, we were able to confirm its functional activity by measuring its downstream metabolite, PGE2 (Fig. 1F). PGE2 levels were increased on average ∼10- to 12-fold over endogenous levels in hCOX-2 mice. PGE2 levels remained elevated at different ages tested, including 4, 7, and 9 months of age in hCOX-2 mice compared with nontransgenic control littermates, indicating that hCOX-2 protein is enzymatically active. The increase in PGE2 levels from hCOX-2 enzymatic activity could be reduced almost completely by administration of the COX-2 inhibitor celecoxib (Fig. 1G; celecoxib, 30 mg/kg by gavage 4 hr before killing).
Morris water maze
Place discrimination task
In the present study, the new water maze protocol included regular platform trials, where the platform was submerged but accessible to the mouse, as well as two types of probe trials, which were designed to assess the strength of memory for the platform location after short (5–7 min) and long (24 hr) delays (Fig.2A).
In the water maze platform trials, hCOX-2 mice did not locate the submerged platform as quickly as nontransgenic age-matched littermates at 12 and 20 months of age; however, they showed no difference at 7 months of age (Fig. 3A). Omnibus ANOVA for latency (age × genotype × session) revealed significant effects of genotype (F(1,46) = 4.56; p < 0.05), age (F(2,46) = 60.09;p < 0.0001), session (F(3,138) = 67.18; p< 0.0001), and age × genotype interaction (F(2,46) = 3.77, p < 0.05), indicating that at 12 and 20 months of age, hCOX-2 mice showed significantly longer latency compared with nontransgenic mice (post hoc test, p < 0.05). A comparison of performance between the ages of 7, 12, and 20 months revealed that hCOX-2 mice showed a significant increase in time required to reach the hidden platform at the age of 12 months with additional deterioration at 20 months (simple main effect,F(2,23) = 50.01; p < 0.0001; post hoc test, p < 0.05), whereas nontransgenic mice performed similarly at 7 and 12 months of age and required a longer time to reach the platform only at 20 months of age (F(2,23) = 20.88; p < 0.0001; post hoc test, p < 0.001). Additional platform trial measures such as swim distance and heading angle also demonstrated similarly impaired performance in 12- and 20-month-old hCOX-2 mice compared with age-matched littermate controls (effect of age, F(1,46) = 9.15:32.62; 0.0001< p < 0.001; effect of age × genotype interaction, F(2,46) = 3.16:5.62; 0.05 < p < 0.01). Significantly, swim speed during the platform trials did not differ between genotypes (Fig.3B) but showed an age-related decrease (omnibus ANOVA,F(2,46) = 56.87; p < 0.0001) at ages of 12 and 20 months (post hoc test,p < 0.005). Considering the equal swim speed in both groups of mice, the observed increases in latency to find the platform in aging hCOX-2 mice in comparison with nontransgenic littermates could be interpreted as resulting from cognitive impairments rather than noncognitive effects.
Fig. 3.

Average latency (A) and swim speed (B) to reach the hidden platform during platform trials in the place discrimination task. Open bars, Nontransgenic (NTg) mice; filled bars, hCOX-2 transgenic (Tg) mice.A, Young Tg mice (7 months) did not differ from their age-matched littermates, whereas middle aged Tg mice (12 months) and old Tg mice (20 months) needed significantly more time to find the platform than their NTg counterparts. The latency increased with age in both groups of mice. However, the onset of this increase occurred earlier in Tg mice (at the age of 12 months) compared with NTg mice, in which it occurred later at age 20 months. B, Tg and NTg mice did not differ in swim speed at any age, suggesting that the longer latencies to the platform in Tg mice were not attributable to poor swimming abilities but rather to compromised cognition. *Significant effect of genotype (simple main effect,p < 0.05); #significant decline with age (post hoc test, p < 0.5). Data are mean ± SEM.
Figure 2, B and C, illustrates the performance of mice in two types of probe trials: short-delay probe trials conducted ∼5–7 min after the platform trials and the more challenging long-delay (24 hr) probe trials. Omnibus ANOVAs (age × genotype × type of probe) revealed significant effects of genotype (F(1,28) = 4.86/7.81; 0.05 < p < 0.01), age (F(1,28) = 30.80/55.35;p < 0.0001), and type of probe (F(1,28) = 5.79; p < 0.05 for annulus 40). No between-genotype differences were found in young mice (7 months, simple main effect, p > 0.3). At 12 months, the performance deficit of hCOX-2 mice compared with nontransgenic age-matched littermates was observed in only a single measure of short-delay probe trials (Fig. 2B,Annulus 40, simple main effect,F(1,17) = 8.75; p < 0.01) and in all measures of the more challenging 24 hr delay probe trials (Fig. 2C; F(1,17) = 4.70/8.57; 0.05 < p < 0.01). These data suggest that the introduction of longer delays and consequently greater demands on memory result in greater apparent differences between nontransgenic and hCOX-2 mice at 12 months. At 20 months of age, a significant impairment of performance in hCOX-2 mice also occurred in the majority of measures of short-delay probe trials compared with 20-month-old nontransgenic mice (Fig. 2B;F(1,11) = 5.20/5.60; p< 0.05). In the more difficult 24 hr delay probe trials (Fig.2C), both hCOX-2 and nontransgenic 20-month-old groups were impaired in all measures compared with their younger counterparts (post hoc tests applied to the significant effect of age, p < 0.05). These results indicate that at an advanced age, both groups showed impaired performance in the more challenging 24 hr probe trials; however, COX-2 mice were also impaired in the easy short-delay probe trials.
Straight swim test
The ability to swim as tested in the straight alley did not differ between hCOX-2 and nontransgenic littermates in all three age groups (latency to the platform, omnibus ANOVA, p > 0.7) (Fig. 4A). In the course of pretraining, the latency to reach the platform decreased in both hCOX-2 and nontransgenic mice (effect of session,F(1,46) = 24.60; p < 0.0001) reflecting an improvement in swimming. The significant effect of age (F(2,46) = 35.83;p < 0.0001) reflected an increase in time required to reach the platform at 12 months and further at 20 months of age (post hoc test, p < 0.001). No age × genotype interaction was observed (p> 0.5), indicating that an age-related decline in ability to swim occurred to the same extent in both hCOX-2 mice and nontransgenic littermates.
Fig. 4.
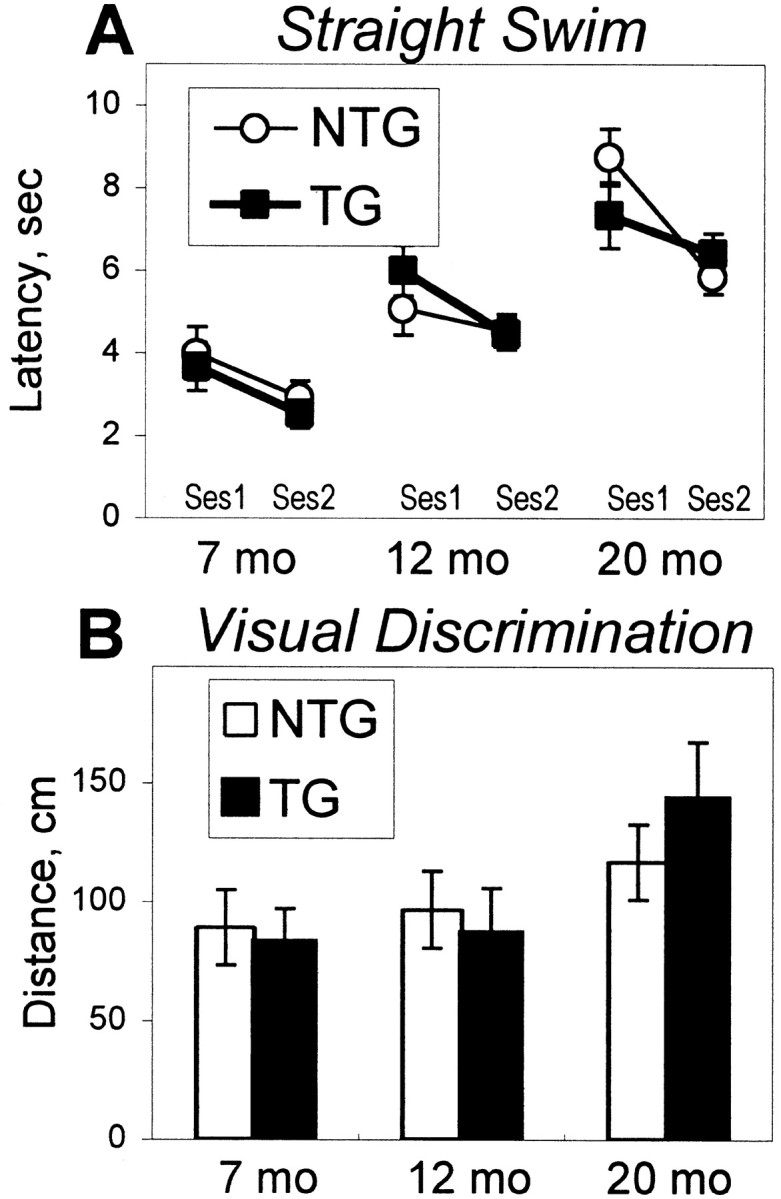
Performance of transgenic hCOX-2 (TG, open bars) and nontransgenic (NTG, filled bars) mice in straight alley (A) and visual discrimination (B) tasks. A, Average latency to reach the platform across sessions in the straight alley test. TG and NTG groups of mice did not differ at any of the ages tested. An age-related decline in the ability to swim was observed in both groups of mice at 12 months of age, with additional deterioration at 20 months of age. B, Average distance to reach a visible platform across all visual discrimination trials. Both groups of mice showed similar performance in all ages tested. Data are mean ± SEM.
Visual discrimination task
The time and distance to reach a visible platform did not show significant between-genotype differences (omnibus ANOVAs,p > 0.2), suggesting an equal visual ability in mice of both genotypes (Fig. 4B). Both hCOX2 and nontransgenic groups of mice improved their performance during training (effect of session, F(1,46) = 94.86; p < 0.0001). The significant age effect (F(2,46) = 12.09; p < 0.001) observed with strong age × session interaction (F(2,46) = 6.73; p < 0.005) demonstrated that age-related differences were apparent only in the first session (post hoc, p < 0.001) and were not reliable at the end of the training (post hoc for the second session, p> 0.1).
Spontaneous alternation task
hCOX-2 mice did not differ from nontransgenic littermates in alternation performance at any age tested (age × genotype ANOVA,p > 0.5). A significant age effect (F(2,46) = 16.69; p < 0.0001) was found in total arm visits, indicating a significant decrease of activity at the age of 12 months (post hoc test, p < 0.0001) with no additional decline at the age of 20 months. These data suggested that hCOX-2 mice did not differ from control counterparts in a task with relatively low demands on spatial memory.
Active and inhibitory avoidance
The results of T maze foot shock active avoidance and inhibitory avoidance tasks are summarized in Figure5. In the active avoidance task, aging hCOX-2 mice performed more poorly than younger hCOX-2 mice (Fig.5A). Two-way ANOVA (age × genotype) revealed a significant effect of age for all variables (F(2,46) = 6.20/15.89; 0.005 <p < 0.0001) consistent with a significant deterioration of performance in 20-month-old mice (post hoc test, p < 0.01). Simple main effect ANOVA demonstrated that the age-related decline in performance was significant only in hCOX-2 mice (F(2,23) = 5.93/8.13; 0.01 <p < 0.001) but not in nontransgenic control littermates.
Fig. 5.

Performance of transgenic hCOX-2 (Tg, shaded bars) and nontransgenic (NTg, open bars) mice in aversively motivated tasks. A, The learning in the T maze active avoidance task is represented as the number of trials to the first avoidance during the acquisition session. Higher scores indicate a poorer acquisition of avoidance reaction. Age-related alterations occurred only in 20-month-old Tg mice but not in 20-month-old NTg mice compared with their younger counterparts. *Bracketsindicate a significant effect of age in Tg mice (p < 0.01) as a result of simple main effect ANOVA. Data are mean ± SEM. B, Inhibitory avoidance task. Inhibitory avoidance reaction was measured by the latency to enter the previously shocked compartment. In this case, the higher latency reflects a better retention after a 24 hr delay. The age-related impairment occurred only in Tg mice, which showed a decreased latency at age of 20 months. This effect was not present in NTg mice. *Brackets indicate a significant effect of age in Tg mice (p < 0.01) as a result of χ2 tests. The latency for every group is presented as median ± interquartile range.
Aging hCOX-2 mice also demonstrated deficits in inhibitory avoidance compared with younger hCOX-2 mice (Fig. 5B). In both nontransgenic and hCOX-2 mice, the application of a single foot shock in the dark compartment of the inhibitory avoidance apparatus led to a significant increase in the latency to enter this compartment after a 24 hr delay (χ2 test, p< 0.005). Although there were no significant between-genotype differences at any age, the comparison of latency to enter the dark compartment between 7-, 12-, and 20-month-old mice revealed a significant effect of age in retention in hCOX-2 mice (Kruskal–Wallis ANOVA, H(2,24) = 10.41;p < 0.01). Transgenic mice showed a significant decline at the age of 20 months (χ2test, p < 0.01), whereas no age-related differences were revealed in nontransgenic mice. These data, together with those from the active avoidance task, support the observation that with advanced age, hCOX-2 mice demonstrated a significant deterioration in performance of aversively motivated tasks compared with their younger counterparts.
Experiments to control for sensitivity to shock between genotypes included estimation of the latency of the first escape reaction in the active avoidance procedure (Fig.6A) and estimation of the threshold for eliciting a burst activity to foot shock in a separate box. The sensitivity to shock did not differ between hCOX-2 and nontransgenic mice at any age (age × genotype ANOVA for latency of the first escape reaction, p > 0.1; Kruskal–Wallis ANOVAs for threshold of a burst reaction,p > 0.3). A significant effect of age was revealed for the threshold of a burst reaction consisting of a decrease of sensitivity to shock at age of 20 months and was reliable in transgenic mice (H(1,15) = 9.94;p < 0.005) as well as in nontransgenic mice (H(1,19) = 14.64; p < 0.005). This indicated that a between-genotype difference in age-related decline in active and inhibitory avoidance performance was not caused by a difference in sensitivity to shock.
Fig. 6.
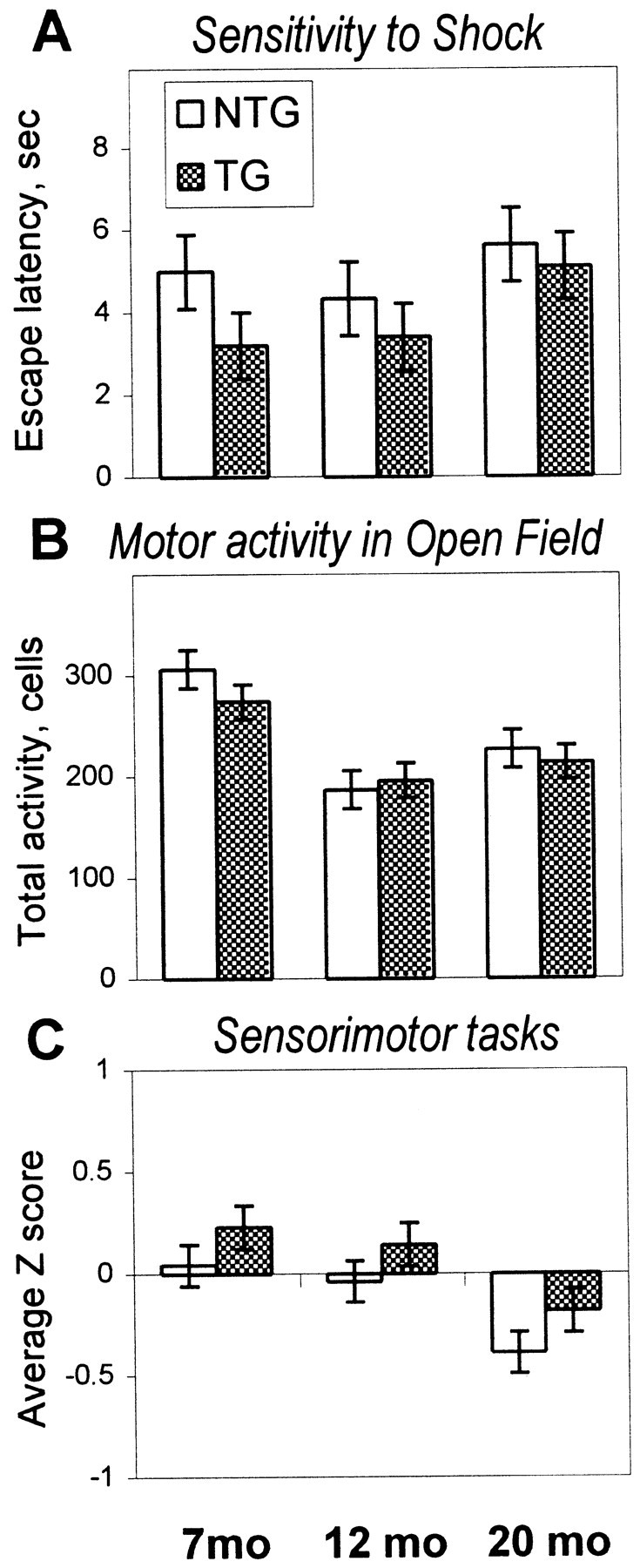
Performance of transgenic hCOX-2 (TG, shaded bars) and nontransgenic (NTG, open bars) mice in control tasks.A, Sensitivity to shock assessed as latency of the first escape reaction during the T maze active avoidance training did not differ between TG and NTG mice at any of the ages tested. No significant age-related changes were revealed for this index of sensitivity to shock (see Results for the results of burst activity measures). B, Total motor activity is expressed as the number of cells in the open field crossed in 5 min of testing. Mice of both genotypes did not differ for all ages tested. An age-related decline in exploratory activity was observed to the same extent in both TG and NTG mice at the age of 12 months. C, Performance in the sensorimotor tasks is shown as the average Zscore from a set of different tasks (turning in an alley, traversing bridges, wire hanging, and falling from a wire screen). There were no significant differences in sensorimotor skills between TG and NTG mice. A significant effect of age was observed in both groups of mice and consisted of a decline in performance at 20 months of age.
Locomotor activity, anxiety, and sensorimotor skills
Analysis of total motor activity in open field testing demonstrated the onset of an age-related decline in motor activity at 12 months of age in both genotypes of mice (effect of age,F(2,46) = 15.58; p < 0.0001), whereas no differences were identified between hCOX-2 and nontransgenic groups at any age (Fig. 6B). The proportion of activity in inner cells of the open field did not change with age (p > 0.1) and did not differ between genotypes (p > 0.3).
Analyses of behavior in the plus maze revealed a significant age-related decrease in the percentage of time spent in open arms (F(2,46) = 9.62; p < 0.001) but no between-genotype differences (p > 0.5) or age × genotype interaction (p > 0.3), indicating that the age-induced increase in the level of anxiety was observed to the same extent in hCOX-2 and nontransgenic mice.
Sensorimotor performance expressed as an average Zscore from a battery of sensorimotor tests (turning in an alley, traversing bridges, wire hanging, and falling from a wire screen) revealed a significant effect of age (F(2,46) = 3.26; p < 0.05) consisting of a deterioration in performance at the age of 20 months (post hoc test, p < 0.05). However, no significant effect of genotype (p > 0.3) or age × genotype interaction (p > 0.9) was observed, indicating that the magnitude of age-related alteration in sensorimotor performance was similar in both genotypes (Fig. 6C).
Thus, the behavioral tests applied to young, middle-aged, and old hCOX-2 and nontransgenic mice demonstrated an aging-related deterioration of performance in hCOX-2 mice with an initial onset of cognitive decline at the age of 12 months in spatial memory tasks. In addition to these marked deficits in spatial memory, aging hCOX-2 mice developed a less pronounced but significant deterioration in performance of nonspatial memory tasks at 20 months of age. In pilot studies using a second line of hCOX-2 mice, in which transgenic COX-2 is expressed primarily in hippocampus and yields PGE2 levels that are half as elevated as in the present line, we also find a similar but smaller impairment in performance in older hCOX-2 mice in spatial and nonspatial memory tasks.
GFAP quantitation
Prostaglandins have been shown to exert a number of effects on astrocytes in vitro, including promoting the proliferation of astrocytes (Lee et al., 1999; Sanzgiri et al., 1999), and upregulating GFAP synthesis (Morrison et al., 1985). Activated astrocytes in turn may exert injurious effects on neighboring neurons by elaboration of cytokines that are toxic to neurons. To determine whether there was evidence of astrocytic activation, proliferation, or both, we measured differences in astrocytic GFAP levels in aging hCOX-2 and nontransgenic mice using quantitative Western blot analysis (Fig.7A,B). Two-way ANOVA (genotype × procedure repetition) revealed a significant effect of genotype (F(1,12) = 5.82;p < 0.05) with a significantly increased GFAP level in aged hCOX-2 mice compared with nontransgenic age-matched control mice at 20–24 months. This increase appeared to be age-dependent and was not seen at younger ages, indicating an interaction between cumulative COX-2 expression and astrocytic activation.
Fig. 7.

Histopathological analysis of hCOX-2 and nontransgenic age-matched littermates. A, Levels of GFAP were compared with an assay for astrocytic activation, proliferation, or both in aged hCOX-2 mice. Quantitative Western blot analysis (50 μg of protein/lane) of GFAP and actin was performed at 20–24 months (n = 7 for each COX-2 and nontransgenic group). A representative set of four pairs of COX-2 and nontransgenic (NTg) controls demonstrates increases in GFAP in aged hCOX-2 brain compared with nontransgenic controls in three of four pairs, with the fourth pair displaying an equal amount of GFAP staining. B, Standardized GFAP of 20- to 24-month-old hCOX-2 and nontransgenic mice demonstrates a significant increase in GFAP in hCOX-2 mice compared with nontransgenic mice at 20–24 months of age. *Significant between-genotype difference as a result of ANOVA. Data are mean ± SEM. C, TUNEL analysis of 8-, 14-, and 22-month-old hCOX-2 and age-matched control littermates. Two-way (age × genotype) ANOVA with the square root transformation of the number of apoptotic cells in cerebral cortex per section showed significance for both effects (age,F(2,335) = 16.34; p< 0.0001; genotype, F(1,335) = 5.87;p < 0.02). Both groups of mice demonstrated a higher number of apoptotic cells at 14 and 22 months. However, the age-associated increase in apoptosis was more pronounced in hCOX-2 mice compared with nontransgenic littermates. #,##Significant age-related increase in the number of stained cells as a result ofpost hoc tests; #p < 0.01; ##p < 0.0001. *p < 0.01 for post hoc tests applied to the effect of genotype within each age group. Data are mean ± SEM. D–F, Determination of cellular phenotype of TUNEL-positive cells using double immunofluorescent staining. After TUNEL labeling, selected sections were doubly stained with Neu N monoclonal and anti-GFAP polyclonal antibodies to identify astrocytes and neurons, respectively. Apoptotic cells uniformly stained for Neu N and did not stain for GFAP, indicating that the TUNEL-positive cells are neurons. D, Light micrograph 630× magnification of a TUNEL-positive cell; note nuclear condensation and margination. E, Immunofluorescent staining of the same region with anti-Neu N antibody demonstrating staining of the nuclear compartment of a TUNEL-positive cell (vertical arrow); horizontal arrows in all panels demonstrate nonapoptotic neurons also stained with Neu N.F, Immunofluroscent staining with anti-GFAP antibody demonstrating the absence of GFAP staining of the TUNEL-positive cell; note GFAP-stained astrocyte (asterisks in all three panels).
TUNEL
Gross microscopic examination of Nissl-stained sections from young, middle aged, and old mice did not demonstrate overt changes in cellular density between genotypes in hippocampus and cortex. However, previous studies have implicated neuronally produced COX-2 in death of neurons in vivo and in vitro (Nogawa et al., 1997; Nakayama et al., 1998; Kelley et al., 1999; Hewett et al., 2000). The behavioral deficits observed in hCOX-2 mice occurred in an age-dependent manner beginning at 12 months of age. To identify whether elevated levels of hCOX-2 in neurons could result in neuronal injury that was similarly age-dependent, brains from selected groups of behaviorally tested hCOX-2 and nontransgenic control mice were harvested and processed for TUNEL immunocytochemistry (Fig.7C,D). TUNEL staining (ApopTag; Intergen) was performed at 8, 14, and 22 months of age in hCOX-2 and nontransgenic littermates that had been behaviorally tested to determine whether there was an increase in neuronal apoptosis in aging hCOX-2 mice compared with nontransgenic age-matched control littermates. Both hCOX-2 and nontransgenic littermate control brains for each age group were processed simultaneously under identical conditions to control for changes such as cell shrinkage that might occur during the process of paraffin embedding and immunostaining. Sections were stained with Hoechst to identify nuclei, and only nuclear profiles that were both Hoechst- and TUNEL-positive were scored. Because the appearance of TUNEL-positive cells is a relatively infrequent event, attributable in part to rapid clearance of these cells from the brain, the cerebral cortex was sampled because it contains more neuronal and nonneuronal cells than other structures, and the probability of identifying TUNEL-positive cells is increased. TUNEL-positive cells were counted only in layers I–VI of the cerebral cortex, and the periventricular zone was not included in the sample area surveyed. Apoptotic cells were counted by the examiner, who was blinded to the genotype of the sections. Because end-labeling methods can detect necrotic as opposed to apoptotic cells at a rate of up to 10%, TUNEL-positive cells were confirmed as apoptotic cells by examining nuclear morphology for nuclear condensation at high magnification (400–630×).
We found a statistically significant difference in the number of TUNEL-positive cells in aging hCOX-2 mice compared with age-matched nontransgenic mice. Although at 8 months of age there was no difference between hCOX-2 and nontransgenic littermates, at 14 months there was a significant increase in the number of TUNEL-staining cells, and this increase was more robust in hCOX-2 than in nontransgenic mice (Fig.7C). With advancing age, the difference in number of TUNEL-staining cells in neocortex became highly significant at 22 months of age between hCOX-2 and nontransgenic mice. A less pronounced age-associated increase in apoptotic cells was also found in nontransgenic control mice, a finding that may be of relevance to previous studies, which demonstrated neocortical atrophy in normal aging mice (Shimada et al., 1994; Zoli et al., 1999). In the present study, a small sampling bias arising from a potential difference in cell size between genotypes cannot be excluded. Although is it possible that a small bias could occur, we would conclude that this error will be small given the statistically significant differences seen in aging hCOX-2 and nontransgenic mice and the lack of difference in young hCOX-2 and nontransgenic mice.
To identify the cellular phenotype of the TUNEL-positive cells, double immunofluorescence was performed on selected TUNEL-labeled sections using Neu N (Chemicon) and anti-GFAP (Dako) antibodies to distinguish between neurons and astrocytes, respectively. Hoechst-positive TUNEL nuclei uniformly stained for Neu N and not anti-GFAP in both hCOX-2 and nontransgenic cerebral cortex, supporting the conclusion that these apoptotic cells are neurons (Fig. 7D–F). Taken together, these findings suggest that increased levels of COX-2 potentiate apoptotic injury in neurons in an age-dependent manner beginning at 14 months of age.
DISCUSSION
In this study, we demonstrate that transgenic mice overexpressing functional COX-2 in neurons of hippocampus, cortex, and amygdala develop cognitive deficits in an age-dependent manner. These behavioral deficits are associated with a parallel age-dependent increase in neuronal apoptosis and GFAP. The lack of behavioral differences between hCOX-2 and nontransgenic mice in a battery of control tasks ensures that the cognitive impairment observed in hCOX-2 mice was not caused by noncognitive factors, such as altered motor ability or sensory perception. Expression of transgenic COX-2 by the Thy-1 promoter begins postnatally; therefore, potential artifacts from abnormal fetal development are avoided. Importantly, the anatomic distribution of hCOX-2 protein parallels to a large extent that of endogenous murine COX-2 and colocalizes with structures that are involved in learning and memory (Morris et al., 1982; Kolb et al., 1983; DiMattia and Kesner, 1988; Davis, 1992; LeDoux, 1993; Moser et al., 1993).
We have developed a novel probe trial protocol in which the retention of spatial memory and the task demand can be parametrically manipulated: an easy task with a short delay after the last platform trial (5–7 min) with a low demand on memory and a more difficult task with a long delay (24 hr) and a high demand on memory. Young (7-month-old) transgenic animals demonstrate intact learning ability and memory retention, suggesting that upregulated COX-2 expression beginning postnatally does not affect the normal development of cognitive function. The onset of cognitive deficits appears in middle-aged (12-month-old) hCOX-2 mice in spatial memory tested in the Morris water maze. At this age, hCOX-2 mice were impaired compared with nontransgenic mice only in the more difficult task, the long-delay probe trials, but they were not different from nontransgenic control mice in the easy short-delay probe trials in most measures. At old age (20 months), both hCOX-2 and nontransgenic mice were severely impaired in the long-delay probe trials, performing equally poorly and close to the floor effect. However, at this advanced age, there was a dissociation in the performance of the easy short-delay probe trials between hCOX-2 and nontransgenic animals, with COX-2 mice performing significantly more poorly than controls in a majority of measures. The use of two distinct types of probe trials of varying levels of difficulty made possible the identification of a progressive age-associated memory decline. This decline began in middle age and was characterized initially by a deficit in long-delay probe trials but subsequently progressed at advanced ages to involve performance in short-delay probe trials.
The impairment in performance of aversively motivated tasks, which requires normal functioning of the amygdala, developed similarly with advancing age, albeit over a longer period. Although the group differences did not reach a level of significance in the active and inhibitory avoidance tasks, there was a significantly greater decline in performance with aging in hCOX-2 mice compared with nontransgenic littermates, suggesting a greater vulnerability to age-related cognitive impairment from hCOX-2 overexpression in amygdala. Considering that hCOX-2 transgenic protein was expressed in amygdala at a lower level than in cortex and hippocampus, it was not surprising that the deficit in the avoidance tasks became apparent only at a very advanced age. Notably, in the inhibitory avoidance task, old hCOX-2 mice demonstrated a significant deterioration in memory retention 24 hr after training. This is consistent with the impairment seen in long-delay probe trials in middle-aged hCOX-2 mice in the water maze. Therefore, hCOX-2 mice demonstrated deficits in long-term memory retention that were detected initially at middle age in the Morris water maze and later at advanced ages in the inhibitory avoidance task.
When considering the differences that aging hCOX-2 mice demonstrate in short- and long-term retention, it is interesting to note classic studies that have demonstrated a requirement for regulated protein synthesis in the establishment of long-term memory (Montarolo et al., 1986; Bailey et al., 1992; Frey et al., 1993). These studies have identified a requirement for a special group of proteins encoded by the class of “immediate-early genes” (IEGs, of which COX-2 is a member) that are rapidly and transiently synthesized in response to the conditioning stimulus. IEGs encode transcription factors and signaling molecules that can regulate successive cascades of gene transcription (Lanahan and Worley, 1998). In disrupting the normal regulation of COX-2 expression by constitutively expressing hCOX-2, we would anticipate a disruption in the regulatory effects of COX-2 metabolites on downstream transcription events (Lerea et al., 1997) important in the establishment of long-term memory. Significantly, no impairment in short- or long-term retention was evident in young mice, suggesting an interaction between the downstream effects of COX-2 metabolites and the aging process.
In parallel with the onset of cognitive impairment, we found an age-dependent increase in TUNEL-positive neurons beginning in middle age in both nontransgenic and hCOX-2 mice; however this increase was more robust in hCOX-2 mice. In counting only Hoechst- and TUNEL-positive cells with condensed nuclei, we tried to ensure that this increase was attributable to apoptotic rather than necrotic mechanisms. Although a sampling bias arising from differences in average cell size between genotypes cannot be excluded, we would conclude that on the basis of the large effect seen in aged hCOX-2 mice, this error should be small. It is also important to note that TUNEL staining alone is not a complete measure of cell death but reflects a single time point in a chronological process that leads to cell death. The complementary method of stereological analysis, currently beyond the scope of the present study, would be needed to document the longitudinal aspect of degeneration in this model. Although the TUNEL counts presented here are estimates of cell profiles per unit area rather than absolute cell number, they do provide insight into the mechanism of cell death in hCOX-2 transgenic mice, which we conclude here is apoptotic.
A precedent exists for neuronal COX-2-mediated apoptotic neuronal death in acute injury models such as ischemia and in vitro models of glutamate neurotoxicity. The identity of the COX-2 metabolite(s) that mediates excitotoxic neuronal damage remains undetermined and could include specific prostanoid metabolites or reactive oxygen species produced in the metabolism of arachidonic acid (Kukreja et al., 1986; Hanna et al., 1997). Neuronal injury in these paradigms follows severe disruptions in postsynaptic Ca2+homeostasis at dendritic spines, where ionotropic glutamate receptors are concentrated. It is hypothesized that just as in acute excitoxic events such as ischemia, Ca2+ homeostasis may also be perturbed by aberrant glutamatergic tone over long periods in aging and age-associated neurodegenerative diseases in populations of neurons that express high levels of glutamate ionotropic receptors (Mattson and Duan, 1999). It is interesting to note that COX-2 is normally expressed at robust basal levels selectively in pyramidal neurons of hippocampus and cerebral cortex and in neurons of the amygdala, populations of neurons that express high levels of ionotropic receptors and are coincidentally most vulnerable in AD.
Although COX-2 is induced in astrocytes and microglia in the inflammatory response to amyloid deposition, neuronal COX-2 may be playing an important pathological role early on, as suggested by the protective effect of NSAIDs in normal aging populations. By virtue of its subcellular localization at dendritic spines and its regulation by NMDA receptor activation, COX-2 activity may initiate a neurotoxic process at the dendritic spine, which could over time extend to the cell body, resulting in delayed neuronal loss. COX-2 levels have been shown to increase with aging in a number of tissues, including macrophages (Hayek et al., 1997), kidney (Kim et al., 2000), and neurons in AD (Oka and Takashima, 1997; Pasinetti and Aisen, 1998;Hoozemans et al., 2001), presumably because of increased oxidative stress and activation of nuclear factor-κB, an important regulatory element in COX-2 gene transcription (Lukiw and Bazan, 1998). Thus, in age-associated diseases such as AD, the cumulative effects of chronically upregulated COX-2 could lead to neuronal injury. Loss of neurons and synapses is a critical feature of AD and one that has been inconsistently observed in transgenic murine models of familial AD, in which abundant amyloid deposition and reactive inflammation have been demonstrated (Price and Sisodia, 1998; Holcomb et al., 1999; Moechars et al., 1999).
In summary, we have identified a cognitive deficit in spatial and nonspatial forms of memory that appears in middle-aged hCOX-2 mice beginning at 12 months. This memory deficit is characterized by an initial impairment in memory retention at 24 hr, with subsequent impairment of short-term memory retention at advanced ages. In parallel, hCOX-2 mice demonstrate an increase in apoptotic neurons beginning at middle age and astrocytic activation at old age. These behavioral and histological changes were not observed in younger hCOX-2 mice, suggesting an interaction between the aging process and the downstream effects of COX-2 metabolites. From the results of the current study, we propose an additional function for COX-2 in the pathogenesis of AD, distinct from its role in mediating the inflammatory response to amyloid deposition, in which upregulated neuronal COX-2 expression over time promotes neuronal injury and dysfunction. The hCOX-2 mice described in this study may provide a novel strategy in which to further explore the pathophysiology of AD.
Footnotes
This work was supported by National Institutes of Health Grant AG15799 (K.I.A.), Amersham Pharmacia Biotech (K.I.A. and A.L.M.), and Alzheimer's Association Grant IIRG-99-1608 (W.E.K.). We thank C. Thielemier, Y. Aguirre, J. L. Morton, K. W. Phelan, and G. D. Waters for technical assistance and P. Mouton and S. Dore for comments.
Correspondence should be addressed to Katrin I. Andreasson, Departments of Neurology and Neuroscience, Johns Hopkins School of Medicine, 600 North Wolfe Street, Meyer 5-119B, Baltimore, MD 21205. E-mail:kandreas@jhmi.edu.
REFERENCES
- 1.Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CH, Montarolo PG, Chen M, Kandel ER, Schacher S. Inhibitors of protein and RNA synthesis block the structural changes that accompany long-term facilitation in Aplysia. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- 3.Barnes CA. Aging and the physiology of spatial memory. Neurobiol Aging. 1988;9:563–568. doi: 10.1016/s0197-4580(88)80114-3. [DOI] [PubMed] [Google Scholar]
- 4.Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 5.Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995;355:296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cammarota M, Izquierdo I, Wolfman C, Levi de Stein M, Bernabeu R, Jerusalinsky D, Medina JH. Inhibitory avoidance training induces rapid and selective changes in 3[H]AMPA receptor binding in the rat hippocampal formation. Neurobiol Learn Mem. 1995;64:257–264. doi: 10.1006/nlme.1995.0008. [DOI] [PubMed] [Google Scholar]
- 7.Compton DM, Griffith HR, McDaniel WF, Foster RA, Davis BK. The flexible use of multiple cue relationships in spatial navigation: a comparison of water maze performance following hippocampal, medial septal, prefrontal cortex, or posterior parietal cortex lesions. Neurobiol Learn Mem. 1997;68:117–132. doi: 10.1006/nlme.1997.3793. [DOI] [PubMed] [Google Scholar]
- 8.Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 9.Davis M. The role of the amygdala in fear and anxiety. Annual Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 10.Dember WN, Fowler H. Spontaneous alternation behavior. Psychol Bull. 1958;55:412–427. doi: 10.1037/h0045446. [DOI] [PubMed] [Google Scholar]
- 11.DiMattia BD, Kesner RP. Spatial cognitive maps: differential role of parietal cortex and hippocampal formation. Behav Neurosci. 1988;102:471–480. doi: 10.1037//0735-7044.102.4.471. [DOI] [PubMed] [Google Scholar]
- 12.Divac I, Wikmark RGE, Gade A. Spontaneous alternation in rats with lesions in the frontal lobe: an extension of the frontal lobe syndrome. Physiol Psychol. 1975;3:39–42. [Google Scholar]
- 13.Drachman DB, Rothstein JD. Inhibition of cyclooxygenase-2 protects motor neurons in an organotypic model of amyotrophic lateral sclerosis. Ann Neurol. 2000;48:792–795. [PubMed] [Google Scholar]
- 14.Fanselow MS. The post-shock activity burst. Anim Learn Behav. 1982;10:448–454. [Google Scholar]
- 15.Flood JF, Baker ML, Hernandez EN, Morley JE. Modulation of memory processing by neuropeptide Y varies with brain injection site. Brain Res. 1989;503:73–82. doi: 10.1016/0006-8993(89)91706-x. [DOI] [PubMed] [Google Scholar]
- 16.Flood JF, Baker ML, Hernandez EN, Morley JE. Modulation of memory retention by neuropeptide K. Brain Res. 1990;520:284–290. doi: 10.1016/0006-8993(90)91717-u. [DOI] [PubMed] [Google Scholar]
- 17.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 18.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 19.Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer's disease. Neurobiol Aging. 1989;10:593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- 20.Hanna N, Peri K, Abran D, Hardy P, Doke A, Lachapelle P, Roy M-S, Orquin J, Varma D, Chemtob S. Light induces peroxidation in retina by activating prostaglandin G/H synthase. Free Radic Biol Med. 1997;23:885–897. doi: 10.1016/s0891-5849(97)00083-x. [DOI] [PubMed] [Google Scholar]
- 21.Hayek MG, Mura C, Beharka AA, Han SN, Paulson KE, Hwang D, Meydani SN. Enhanced expression of inducible cyclooxygenase with age in murine macrophages. J Immunol. 1997;159:2445–2451. [PubMed] [Google Scholar]
- 22.Hewett SJ, Uliasz T, Vidwans A, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-d-aspartate mediated neuronal cell death in primary cortical cultures. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- 23.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 24.Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol (Berl) 2001;101:2–8. doi: 10.1007/s004010000251. [DOI] [PubMed] [Google Scholar]
- 25.Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methyl-d-aspartate-mediated neurotoxicity in cyclooxygenase-2 deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. Cox-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci USA. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelley KA, Ho L, Winger D, Friere-Moar J, Borelli C, Aisen P, Pasinetti GM. Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol. 1999;155:995–1004. doi: 10.1016/S0002-9440(10)65199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khachaturian ZS. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging. 1989;1:17–34. doi: 10.1007/BF03323872. [DOI] [PubMed] [Google Scholar]
- 29.Kim HJ, Kim KW, Yu BP, Chung HY. The effect of age on cyclooxygenase-2 gene expression: NF-kappaB activation and IkappaBalpha degradation. Free Radic Biol Med. 2000;28:683–692. doi: 10.1016/s0891-5849(99)00274-9. [DOI] [PubMed] [Google Scholar]
- 30.Kolb B, Pittman K, Sutherland RJ, Whishaw IQ. Dissociation of the contributions of the prefrontal cortex and dorsomedial thalamic nucleus to spatially guided behavior in the rat. Behav Brain Res. 1982;6:365–378. doi: 10.1016/0166-4328(82)90018-3. [DOI] [PubMed] [Google Scholar]
- 31.Kolb B, Sutherland RJ, Whishaw IQ. A comparison of the contributions of the frontal and parietal association cortex to spatial localization in rats. Behav Neurosci. 1983;97:13–27. doi: 10.1037//0735-7044.97.1.13. [DOI] [PubMed] [Google Scholar]
- 32.Kukreja R, Kontos H, Hess H, Ellis E. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res. 1986;59:612–619. doi: 10.1161/01.res.59.6.612. [DOI] [PubMed] [Google Scholar]
- 33.Lanahan A, Worley P. Immediate-early genes and synaptic function. Neurobiol Learn Mem. 1998;70:37–43. doi: 10.1006/nlme.1998.3836. [DOI] [PubMed] [Google Scholar]
- 34.LeDoux JE. Emotional memory: in search of systems and synapses. Ann NY Acad Sci. 1993;702:149–157. doi: 10.1111/j.1749-6632.1993.tb17246.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee RK, Knapp S, Wurtmann RJ. Prostaglandin E2 stimulates amyloid precursor protein gene expression: inhibition by immunosuppressants. J Neurosci. 1999;19:940–947. doi: 10.1523/JNEUROSCI.19-03-00940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lerea LS, Carlson NG, Simonato M, Morrow J, Roberts JL, McNamara JO. Prostaglandin F 2alpha is required for NMDA receptor-mediated induction of c-fos mRNA in dentate gyrus neurons. J Neurosci. 1997;17:117–124. doi: 10.1523/JNEUROSCI.17-01-00117.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang KC, McGaugh JL, Martinez JL, Jensen RA, Vasquez BJ, Messing RB. Post-training amygdaloid lesions impair retention of an inhibitory avoidance response. Behav Brain Res. 1982;4:237–249. doi: 10.1016/0166-4328(82)90002-x. [DOI] [PubMed] [Google Scholar]
- 38.Liang KC, Hon W, Davis M. Pre- and posttraining infusion of N-methyl-d-aspartate receptor antagonists into the amygdala impair memory in an inhibitory avoidance task. Behav Neurosci. 1994;108:241–253. doi: 10.1037//0735-7044.108.2.241. [DOI] [PubMed] [Google Scholar]
- 39.Lipp HP, Wolfer DP. Genetically modified mice and cognition. Curr Opin Neurobiol. 1998;8:272–280. doi: 10.1016/s0959-4388(98)80151-7. [DOI] [PubMed] [Google Scholar]
- 40.Lister RG. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology. 1987;92:180–185. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- 41.Lukiw WJ, Bazan NG. Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer's disease superior temporal lobe neocortex. J Neurosci Res. 1998;53:583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Markowska AL, Stone WS, Ingram DK, Reynolds J, Gold PE, Conti LH, Pontecorvo MJ, Wenk GL, Olton DS. Individual differences in aging: behavioral and neurobiological correlates. Neurobiol Aging. 1989;10:31–43. doi: 10.1016/s0197-4580(89)80008-9. [DOI] [PubMed] [Google Scholar]
- 43.Markowska AL, Spangler EL, Ingram DK. Behavioral assessment of the senescence-accelerated mouse (SAM P8 and R1). Physiol Behav. 1998;64:15–26. doi: 10.1016/s0031-9384(98)00011-0. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP. Calcium and neuronal injury in Alzheimer's disease. Ann NY Acad Sci. 1994;747:50–76. [PubMed] [Google Scholar]
- 45.Mattson MP, Duan W. “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- 46.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 47.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 48.Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- 49.Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 50.Morrison RS, De Vellis J, Lee YL, Bradshaw RA, Eng LF. Hormones and growth factors induce the synthesis of glial fibrillary acidic protein in rat brain astrocytes. J Neurosci Res. 1985;14:167–176. doi: 10.1002/jnr.490140202. [DOI] [PubMed] [Google Scholar]
- 51.Moser E, Moser MB, Andersen P. Spatial learning impairment parallels the magnitude of dorsal hippocampal lesions but is hardly present following ventral lesions. J Neurosci. 1993;13:3916–3925. doi: 10.1523/JNEUROSCI.13-09-03916.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose M, Stetler RA, Isakson P, Chen J, Graham SH. Cyclooxygenase-2 inhibitions prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci USA. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nogawa S, Zhang F, Ross E, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oka A, Takashima S. Induction of cyclo-oxygenase 2 in brains of patients with Down's syndrome and dementia of Alzheimer type: specific localization in affected neurones and axons. NeuroReport. 1997;8:1161–1164. doi: 10.1097/00001756-199703240-00020. [DOI] [PubMed] [Google Scholar]
- 55.Olton DS, Papas BC. Spatial memory and hippocampal function. Neuropsychologia. 1979;17:669–682. doi: 10.1016/0028-3932(79)90042-3. [DOI] [PubMed] [Google Scholar]
- 56.Owen EH, Logue SF, Rasmussen DL, Wehner JM. Assessment of learning by the Morris water task and fear conditioning in inbred mouse strains and F1 hybrids: implications of genetic background for single gene mutations and quantitative trait loci analyses. Neuroscience. 1997;80:1087–1099. doi: 10.1016/s0306-4522(97)00165-6. [DOI] [PubMed] [Google Scholar]
- 57.Pasinetti GM, Aisen PS. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience. 1998;87:319–324. doi: 10.1016/s0306-4522(98)00218-8. [DOI] [PubMed] [Google Scholar]
- 58.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 59.Rodgers RJ, Cole JC. Influence of social isolation, gender, strain, and prior novelty on plus-maze behaviour in mice. Physiol Behav. 1993;54:729–736. doi: 10.1016/0031-9384(93)90084-s. [DOI] [PubMed] [Google Scholar]
- 60.Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci USA. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanzgiri R, Araque A, Haydon P. Prostaglandin E2 stimulates glutamate receptor dependent astrocyte neuromodulation in cultured hippocampal cells. J Neurobiol. 1999;41:221–229. [PubMed] [Google Scholar]
- 62.Shimada A, Hosokawa M, Ohta A, Akiguchi I, Takeda T. Localization of atrophy-prone areas in the aging mouse brain: comparison between the brain atrophy model SAM-P/10 and the normal control SAM-R/1. Neuroscience. 1994;59:859–869. doi: 10.1016/0306-4522(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 63.Silva AJ, Giese KP, Fedorov NB, Frankland PW, Kogan JH. Molecular, cellular and neuroanatomical substrates of place learning. Neurobiol Learn Mem. 1998;70:44–61. doi: 10.1006/nlme.1998.3837. [DOI] [PubMed] [Google Scholar]
- 64.Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 66.Stone WS, Rudd RJ, Gold PE. Glucose attenuation of scopolamine- and age-induced deficits in spontaneous alternation behavior and regional brain [3H]2-deoxyglucose uptake in mice. Psychobiology. 1992;20:270–279. [Google Scholar]
- 67.Vidal M, Morris R, Grosveld F, Spanopoulou E. Tissue-specific control elements of the Thy-1 gene. EMBO J. 1990;9:833–840. doi: 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walz R, Roesler R, Quevedo J, Sant'Anna MK, Madruga M, Rodrigues C, Gottfried C, Medina JH, Izquierdo I. Time-dependent impairment of inhibitory avoidance retention in rats by posttraining infusion of a mitogen-activated protein kinase kinase inhibitor into cortical and limbic structures. Neurobiol Learn Mem. 2000;73:11–20. doi: 10.1006/nlme.1999.3913. [DOI] [PubMed] [Google Scholar]
- 69.Yamagata K, Andreasson K, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 70.Yermakova AV, Chan DB, O'Banion MK. Changes in neuronal and glial expression of cyclooxygenase-2 in human Alzheimer's disease. Soc Neurosci Abstr. 2000;26:576.17. [Google Scholar]
- 71.Zoli A, Picciotto MR, Ferrari R, Cocchi D, Changeux J-P. Increased neurodegeneration during ageing in mice lacking high affinity nicotine receptors. EMBO J. 1999;18:1235–1244. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]