

Abstract
Antagonists at substance P receptors of the neurokinin 1 (NK1) type have been shown to represent a novel class of antidepressant drugs, with comparable clinical efficacy to the selective serotonin (5-HT) reuptake inhibitors (SSRIs). Because 5-HT1Areceptors may be critically involved in the mechanisms of action of SSRIs, we examined whether these receptors could also be affected in a model of whole-life blockade of NK1 receptors, i.e. knock-out mice lacking the latter receptors (NK1−/−). 5-HT1A receptor labeling by the selective antagonist radioligand [3H]N-[2-[4-(2-methoxyphenyl)1-piperazinyl]-ethyl]-N-(2-pyridinyl)-cyclohexanecarboxamide (WAY 100635) and 5-HT1A-dependent [35S]GTP-γ-S binding at the level of the dorsal raphe nucleus (DRN) in brain sections, as well as the concentration of 5-HT1A mRNA in the anterior raphe area were significantly reduced (−19 to −46%) in NK1−/− compared with NK1+/+ mice. Furthermore, a ∼10-fold decrease in the potency of the 5-HT1A receptor agonist ipsapirone to inhibit the discharge of serotoninergic neurons in the dorsal raphe nucleus within brainstem slices, and reduced hypothermic response to 8-OH-DPAT, were noted in NK1−/− versus NK1+/+ mice. On the other hand, cortical 5-HT overflow caused by systemic injection of the SSRI paroxetine was four- to sixfold higher in freely moving NK1−/− mutants than in wild-type NK1+/+ mice. Accordingly, the constitutive lack of NK1 receptors appears to be associated with a downregulation/functional desensitization of 5-HT1A autoreceptors resembling that induced by chronic treatment with SSRI antidepressants. Double immunocytochemical labeling experiments suggest that such a heteroregulation of 5-HT1A autoreceptors in NK1−/− mutants does not reflect the existence of direct NK1–5-HT1A receptor interactions in normal mice.
Keywords: 5-HT1A receptors, desensitization, dorsal raphe, NK1 receptors, electrophysiology, in vivomicrodialysis
The link between mood disorders and alterations in central serotoninergic neurotransmission has been the subject of numerous studies (Delgado et al., 1990; Maes and Meltzer, 1995). In particular, investigations in depressed patients revealed abnormalities in serotonin [5-hydroxytryptamine (5-HT)] metabolism in the CNS (Lloyd et al., 1974; Asberg et al., 1976). Furthermore, most antidepressant drugs, and especially the selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (Fuller et al., 1975) and paroxetine (Dechant and Clissold, 1991) are believed to exert their therapeutic effects through a facilitation of central serotoninergic neurotransmission (Hensler et al., 1991; Bel and Artigas, 1993; Jolas et al., 1994). Extensive neurobiological investigations have shown that prolonged blockade of the 5-HT transporter (5-HTT) by SSRIs induces a functional desensitization of somatodendritic 5-HT1A autoreceptors in the dorsal raphe nucleus (DRN) (Blier and De Montigny, 1983; Jolas et al., 1994; Le Poul et al., 1995). This adaptive change, which directly contributes to enhanced 5-HT neurotransmission, is currently thought to play a key role in the therapeutic efficacy of SSRIs (Blier and De Montigny, 1983; Artigas et al., 1996).
Although SSRIs are clinically effective, their clinical utility is limited by drug-induced adverse effects, and they do not alleviate depression in ∼30% of patients. Moreover, there is a delay of several weeks to achieve clinical benefit, and hence there remains a pressing need to develop novel antidepressant drugs.
Recently, therapeutic efficacy of the neurokinin-1 (NK1) substance P receptor antagonist MK-869 has been demonstrated in depressed patients (Kramer et al., 1998). In addition, behavioral studies suggested that NK1 receptor antagonists are as effective as currently used antidepressants to suppress psychological stress responses in guinea pigs and mice (Kramer et al., 1998; Rupniak et al., 2000) and restore responsiveness to rewarding stimuli in rats subjected to chronic mild stress (Papp et al., 2000).
Because functional interactions between substance P-containing neurons and 5-HT systems have been clearly demonstrated in brain (Pradhan et al., 1981; Walker et al., 1991; Shirayama et al., 1996), a key question to be addressed is whether the antidepressant action of NK1 receptor antagonists involves an alteration in central 5-HT neurotransmission. In this respect, 5-HT1A autoreceptors in the DRN are a key target to examine because of their role in both the homeostasis of central 5-HT systems (Blier and De Montigny, 1983) and the mechanisms of action of antidepressants, especially SSRIs.
Rather than investigating the fate of DRN 5-HT1Aautoreceptors after chronic blockade of NK1 receptors by an antagonist, we used the recently generated NK1 receptor knock-out mice (De Felipe et al., 1998), which can be considered as a model of whole-life treatment with such a drug. Autoradiographic studies with specific radioligands, quantitative determination of 5-HT1A receptor mRNA, recording of the electrophysiological responses of DRN serotoninergic neurons to 5-HT1A autoreceptor ligands, in vivomicrodialysis studies, assessment of 5-HT1Aagonist-evoked hypothermia, and immunocytochemical investigations were performed to thoroughly assess the functional properties of 5-HT1A receptors in NK1−/− mutants compared with wild-type mice.
MATERIALS AND METHODS
Animals. Wild-type and mutant mice used in the present study were the product of mating between heterozygous NK1+/− couples raised on 129/Sv × C57BL/6 genetic background (De Felipe et al., 1998). Genotyping was performed using Southern blot analysis as described (De Felipe et al., 1998). Males and females were separated at weaning (at 3 weeks after birth) and grouped at six per cage for their maintenance under standard conditions (12 hr light/dark cycle; 22 ± 1°C ambient temperature; 60% relative humidity; food and water ad libitum).
Procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national and international laws and policies [Council directive 87-848, October 19, 1987, Ministère de l'Agriculture et de la Forêt, Service Vétérinaire de la Santé et de la Protection Animale, permission no. 005037 to A.M.G., 75-116 to M.H., and 006269 to L.L.; and the UK Animals (Scientific Procedures) Act, 1986, and its associated guidelines].
Autoradiographic labeling experiments. Male mice (10–12 weeks old, 25–30 gm body weight) were decapitated, and their brains were rapidly removed, frozen in isopentane chilled at −30°C with dry ice, and stored at −80°C. Coronal sections (20 μm thick) were cut at −20°C, thaw mounted onto gelatin-coated slides, and then stored at −80°C until use.
Autoradiographic labeling of 5-HT1Areceptors by [3H]N-[2-[4-(2-methoxyphenyl)1-piperazinyl]-ethyl]-N-(2-pyridinyl)-cyclohexanecarboxamide.Autoradiographic labeling by the 5-HT1A antagonist radioligand [3H]N-[2-[4-(2-methoxyphenyl)1-piperazinyl]-ethyl]-N-(2-pyridinyl)-cyclohexanecarboxamide (WAY 100635) was performed according to slight modifications of the protocol previously described by Gozlan et al. (1995). Briefly, slides with brain sections were first brought to room temperature for 15 min and then preincubated for 15 min in 100 mm Tris-HCl, pH 7.4, at 25°C. Incubation proceeded for 1 hr at 25°C in the same but fresh buffer supplemented with 1 nm [3H]WAY 100635 (81 Ci/mmol). Nonspecific binding was estimated from adjacent sections incubated in the same medium supplemented with 10 μm 5-HT. Sections were then washed three times for 5 min each in Tris-HCl buffer at 4°C and briefly immersed in ice-cold distilled water. The slides were dried in a stream of cold air and apposed to a 3H-Fuji Imaging plate BAS-TR2040 (Fujifilm). After a 2 week exposure, the imaging plate was scanned using a phosphoimager FLA2000 (Fuji). The scanned image was transferred into a computerized imaged software (Aïda 2.1) and optical density was measured and converted to fmol [3H]WAY 100635 specifically bound per mg tissue according to a 3H-standard scale (Amersham Pharmacia Biotech, Buckinghamshire, UK).
Quantitative autoradiography of 5-HT1Areceptor-mediated [35S]GTP-γ-S binding. The protocol for autoradiographic measurement of 5-HT1A receptor-stimulated [35S]GTP-γ-S binding was adapted fromFabre et al. (2000). Briefly, brain sections were preincubated at room temperature for an initial 15 min period in 50 mmHEPES, pH 7.5, supplemented with 100 mm NaCl, 3 mm MgCl2, 0.2 mm EGTA, and 2 mmdithiothreitol, and then for another 15 min in the same buffer with 2 mm GDP and 10 μm8-cyclopentyl-1,3-dipropylxanthine (DPCPX; an A1adenosine receptor antagonist) to decrease background labeling (Fabre et al., 2000). Thereafter, sections were incubated for 1 hr at 30°C in the same buffer with 0.05 nm[35S]GTP-γ-S (1000 Ci/mmol) in either the absence (basal conditions) or presence (stimulated conditions) of 10 μm 5-carboxamido-tryptamine (5-CT). Nonspecific binding was determined in the presence of 10 μm WAY 100635 to block 5-HT1A receptors (Fletcher et al., 1996). The incubation was stopped by two 2 min washes in ice-cold 50 mm HEPES, pH 7.5, and a brief immersion in ice-cold distilled water. Sections were dried and exposed to β-max film (Amersham Pharmacia Biotech). Optical density was measured on autoradiographic films, using a computerized image system (Biocom, Les Ulis, France). 5-CT-stimulated [35S]GTP-γ-S binding is expressed as percentage over the baseline ([(stimulated-basal)/basal] × 100) ± SEM).
Quantitative determination of 5-HT1Areceptor mRNA. The method used to measure mRNAs was based on a competitive RT-PCR technique (Siebert and Larrick, 1992) in which mRNAs of analyzed gene are reverse-transcribed and amplified in the presence of a homologous deleted internal standard mRNA.
Quantitative determination of 5-HT1A receptor mRNA in the anterior raphe area was performed as described by Le Poul et al. (2000) using a RT-PCR Access System Kit (Promega, Madison, WI). Reverse transcription (45 min at 48°C) proceeded with 0.5 μg of total tissue RNA in the presence of standard deleted RNA at increasing dilutions (10−6 to 3 × 10−8). The sequences of the upstream and downstream oligonucleotide primers were 5′-CTCTACGGGCGCATCTTCAGA-3′ (nucleotides 762–782) and 5′-CCCAGAGTCTTCACCGTCTTC-3′ (nucleotides 1165–1145) (Albert et al., 1990). PCR amplification was performed with 1–2 U of Tfl DNA polymerase, 1 mm MgSO4, and 1 pg/μl of each primer for 30 cycles (1 min at 95°C, 1 min at 58°C, and 1 min at 72°C). After electrophoretic separation in 2% agarose gel stained with 4% ethidium bromide, both standard and tissue RT-PCR products were quantified with a gel analyzer software (NIH 1.6).
Imunohistochemistry. Male mice were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and perfused transcardially with ice-cold 4% paraformaldehyde in 0.1m sodium phosphate buffer (PB), pH 7.4. Brains were immediately removed and post-fixed overnight at 4°C, cryoprotected with 30% sucrose in PB containing 0.9% NaCl (PBS), and cut coronally at 30 μm on a sliding microtome. Serial transverse sections from the DRN were collected in PBS. The anteroposterior landmarks for the DRN were established between the disappearance of oculomotor nuclei (rostral DRN) and the presence of the dorsal tegmental nucleus of Gudden (caudal DRN) as described by Descarries et al. (1982). These landmarks corresponded approximately to plates 65 and 71 of the atlas of Franklin and Paxinos (1997).
For single immunoreactivity, free-floating sections were washed in PBS supplemented with 0.2% Triton X-100, then incubated successively in 3% hydrogen peroxide for 10 min and 10% normal goat serum for 60 min. Incubation with the primary antibodies, monoclonal mouse antiserum against serotonin (1:200) or polyclonal rabbit antiserum against NK1 receptor (1:10,000), was performed overnight at room temperature with 5% normal goat serum. Sections were then incubated in biotinylated goat anti-rabbit IgG (Vector, Burlingame, CA), diluted 1:500, for 2 hr, and in avidin–biotin peroxidase complex (Vector) for 1 hr, and finally treated with 0.001% H2O2in PB with 0.08% nickel ammonium sulfate and 0.02% 3,3′-diaminobenzidine (DAB; Sigma, St. Louis, MO) used here as a chromogen.
Dual antigen immunoreactivity was performed following two different standard techniques. For the first one, sections were stained for serotonin immunoreactivity as indicated above, yielding a dark purplish color. After several washes in PB, the same sections were processed again with the NK1 receptor antiserum using only DAB to yield a brown color (Moratalla et al., 1996). An immunofluorescence protocol was used for the second method. Briefly, sections were incubated simultaneously with the two different primary antibodies for 48 hr. Secondary fluorophore-labeled antibodies (Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 594 goat anti-rabbit IgG; Molecular Probes, Eugene, OR) were then used to visualize primary antibodies bound to sections. Sections were finally mounted on gelatin-coated slides, air-dried, and coverslipped with 50% glycerol in PBS containing 2% 1,4-diazabicyclo (2.2.2) octane, to be analyzed by confocal microscopy.
Electrophysiological experiments. Both male and female mice (10 weeks old, ∼25 gm body weight) were killed by decapitation, and their brains were rapidly removed and immersed in an ice-cold artificial CSF (aCSF) of the following composition (in mm): NaCl 126, KCl 3.5, NaH2PO4 1.2, MgCl2 1.3, CaCl2 2.0, NaHCO3 25, d-glucose 11, pH 7.3, continuously gassed with carbogen (95% O2/5% CO2).
A block of tissue containing the DRN was cut into sections (400 μm thick) in the same ice-cold aCSF using a vibratome. Brainstem slices were immediately incubated in oxygenated aCSF for ∼1 hr at room temperature. A single slice was then placed on a nylon mesh, completely submerged in the recording chamber, and continuously superfused with oxygenated aCSF (34°C) at a constant flow rate of 2–3 ml/min (Haj-Dahmane et al., 1991).
Extracellular recordings of the firing of DRN serotoninergic neurons were made using glass microelectrodes filled with 2 m NaCl (12–15 MΩ). Cells were identified as 5-HT neurons according to previously described criteria (Haj-Dahmane et al., 1991). Firing was evoked in the otherwise silent neurons by adding the α1-adrenoceptor agonist phenylephrine (3 μm) into the superfusing aCSF (VanderMaelen and Aghajanian, 1983).
Electrical signals were fed into a high-input impedance amplifier (VF 180, BioLogic, Claix, France), an oscilloscope, and an electronic rate meter triggered by individual action potentials, connected to an A/D converter and a personal computer (Haj-Dahmane et al., 1991). With use of dedicated software, the integrated firing rate was recorded, computed, and displayed on a chart recorder as consecutive 10 sec samples.
Baseline activity was recorded for at least 10 min before the infusion of increasing concentrations of the 5-HT1Areceptor agonist, ipsapirone (Hamon, 1997), into the chamber via a three-way tap system. Because complete exchange of fluids occurred within 2 min after the arrival of a new solution into the chamber, the duration of each application of ipsapirone was 3 min. The effects of ipsapirone were evaluated by comparing the mean discharge frequency during the 2 min before its application with that recorded at the peak action of the drug, i.e., 2–3 min after its removal from the perfusing aCSF. When ipsapirone was applied in the presence of the 5-HT1A receptor antagonist, WAY 100635, the effect of the agonist was compared with the baseline firing rate and with the discharge frequency recorded during superfusion with the antagonist alone. Data are expressed as percentage of the baseline firing rate ± SEM. Nonlinear regression fitting was performed using Prism 2.0 (Graph Pad) software for the calculation of EC50 values of ipsapirone.
Intracortical in vivo microdialysis in freely moving mice. Concentric dialysis probes were made of cuprophan fibers and constructed as described previously (Malagié et al., 1996). All probes (×0.30 mm outer diameter) presented an active length of 1.6 mm within the frontal cortex. Male mice (10–12 weeks old, 25–30 gm body weight) were anesthetized with chloral hydrate (400 mg/kg, i.p.) and implanted with the microdialysis probe into the right frontal cortex according to the mouse brain atlas of Franklin and Paxinos (1997) (coordinates from bregma: anterior = +2.0, lateral = +1.2, ventral = −1.6). The next day, after a ∼20 hr recovery from the surgery, the probe was perfused continuously with a microdialysis medium (composition in mm: NaCl 147, KCl 3.5, CaCl2 1.0, MgCl2 1.2, NaH2PO4 1.0, NaHCO3 25.0, pH 7.4) at a flow rate of 1.3 μl/min, using a CMA/100 pump (Carnegie Medicine, Stockholm, Sweden). Dialysate samples were collected every 15 min in small Eppendorf tubes for the measurement of their 5-HT contents using HPLC (XL-ODS column; 4.6 × 7.0 mm, particle size 3 μm; Beckman) coupled to amperometric detection (1049A; Hewlett-Packard, Les Ulis, France) as described previously (Malagié et al., 1996). Usually four fractions were collected to determine basal values (means ± SEM) before systemic administration of the drugs. The limit of sensitivity for 5-HT was ∼0.5 fmol per sample (signal-to-noise ratio = 2).
Paroxetine hydrochloride (1 mg/kg) was dissolved in 0.9% NaCl and administered intraperitoneally in a volume of 10 ml/kg. For interaction studies, WAY 100635 (0.5 mg/kg) was dissolved in 0.9% NaCl and administered subcutaneously 15 min before paroxetine. Control animals received two consecutive injections of 0.9% NaCl (10 ml/kg, by the same route) 15 min apart. Responses to drug administration were determined over a 180 min period. At the end of the experiments, placement of microdialysis probes was verified histologically.
8-OH-DPAT-induced hypothermia. Baseline body temperature was measured using a thermister probe inserted 2 cm into the rectum of 10- to 12-week-old male mice. Mice then received a subcutaneous injection of the 5-HT1A receptor agonist 8-OH-DPAT (0.25 or 0.5 mg/kg) or vehicle (0.9% NaCl), and body temperature was measured at 5 min intervals for up to 60 min. Data are presented as the difference in body temperature from baseline.
Statistical analyses. All data are given as means ± SEM. Extracellular recordings were analyzed by one-way ANOVA, and in case of significance (p < 0.05), the F test for significant treatment effects was followed by the unpaired two-tailed Student's t test to compare the experimental groups with their control.
5-HT1A receptor-mediated [35S]GTP-γ-S binding, [3H]WAY 10635 autoradiographic labeling, and 5-HT1A mRNA levels were analyzed by unpaired two-tailed Student's t test.
Microdialysis data were standardized by transforming dialysate 5-HT concentrations into percentages of baseline values based on averages of the first four fractions uncorrected for in vitro probe recovery. To compare [5-HT]ext with the respective basal value in each group of treated animals, statistical analysis was performed using a one-way ANOVA for repeated measures on time, followed by Fisher Protected Least Significance Differencepost hoc test. Furthermore, on the basis of percentage data relative to basal values, net changes in dialysate 5-HT levels were determined by calculating area under the curve (AUC; mean ± SEM) values for the amount of 5-HT outflow during the 0–180 min period after treatment. Statistical analyses were performed using the computer software StatView 4.02 (Abacus Concepts, Berkeley, CA). A two-way ANOVA on AUC values was performed with the drug treatment (NaCl/NaCl, NaCl/paroxetine 1 mg/kg, i.p., and WAY 100635 0.5 mg/kg, s.c./paroxetine 1 mg/kg, i.p.) and the mice genotype (wild-type or knock-out) as main factors. Then, statistical comparisons of the AUC values for each strain were performed by applying a one-way ANOVA with the treatment as main factor. Decreases in body temperature caused by 8-OH-DPAT or saline in wild-type mice and NK1−/− mutants were statistically analyzed using a two-way ANOVA followed by Student's Newman–Keuls multiple t test where appropriate. To determine whether the hypothermic response to 8-OH-DPAT was different in wild-type and NK1−/− mice, values obtained in both groups for each treatment were compared at the time of peak effect (30 min after treatment) using a two-way ANOVA followed by Student's Newman–Keuls multiple t test. In all cases, the significance level was set at p < 0.05.
Chemicals. [35S]GTP-γ-S and [3H]WAY 100635 were purchased from Amersham Pharmacia Biotech. Monoclonal mouse antibody against serotonin and DAB were from Sigma, goat anti-rabbit IgG were from Vector (Biovalley, Conches, France), Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 594 goat anti-rabbit IgG were from Molecular Probes, and NK1 receptor polyclonal antibodies were from Chemicon (Temecula, CA). Other compounds were WAY 100635 (Wyeth-Ayerst, Princeton, NJ), GDP dilithium salt (Boehringer Mannheim, Meylan, France), DPCPX, and 5-CT (Research Biochemicals International, Natick, MA), ipsapirone (Troponwerke Bayer, Cologne, Germany), and paroxetine hydrochloride (SmithKline Beecham, Harlow, UK).
RESULTS
5-HT1A receptor labeling in NK1−/− mutants and wild-type mice
Comparison between the two genotypes showed that the specific labeling of 5-HT1A receptors by [3H]WAY 100365 (Fig.1) was significantly less (−19%;p < 0.05) in the DRN of mutant mice (NK1−/−: 91.9 ± 4.1 fmol/mg tissue, mean ± SEM, n = 5; wild-type controls: 113.5 ± 7.4 fmol/mg tissue, mean ± SEM, n = 4). By contrast, in the hippocampus, 5-HT1A receptor labeling by [3H]WAY 100365 was not significantly different between the two groups (NK1−/−: 168.1 ± 4.4 fmol/mg tissue, mean ± SEM, n = 5; wild-type controls: 177.8 ± 8.7 fmol/mg tissue, mean ± SEM, n = 4) (Fig. 1).
Fig. 1.

Representative autoradiographic labeling of 5-HT1A receptors by [3H] WAY 100635 in brain sections from NK1−/− mutants compared with NK1+/+ wild-type mice. Coronal sections (20 μm) at the level of the DRN and the hippocampus were labeled with 1 nm [3H]WAY 100635. Similar autoradiograms were obtained from three to five mice per group. Cx, Cerebral cortex; DRN, dorsal raphe nucleus; Ent. Cx, entorhinal cortex;Hip, hippocampus; MRN, median raphe nucleus; Sup. Coll., superior colliculi.
5-HT1A receptor mRNA quantification in the anterior raphe area of NK1−/− mutants and wild-type mice
Quantitative determinations by competitive RT-PCR showed that 5-HT1A mRNA levels in the anterior raphe area were significantly less (−46%; p < 0.05) in NK1−/− mutant mice (1.10 ± 0.26 amol specific mRNA/μg of total RNA, mean ± SEM, n = 4) than in wild-type NK1+/+ mice (2.05 ± 0.37 amol specific mRNA/μg of total RNA, mean ± SEM, n = 4).
5-HT1A receptor-stimulated [35S]GTP-γ-S binding in NK1−/− mutants and wild-type mice
Optical density measurement of [35S]GTP-γ-S autoradiographic labeling was made within the DRN under three experimental conditions, nonspecific, basal, and 5-CT-evoked, and the percentage of 5-HT1A receptor-mediated stimulation was determined as described in Materials and Methods. In both groups of mice, 10 μm 5-CT induced an increase in [35S]GTP-γ-S labeling, which could be prevented by the selective 5-HT1A receptor antagonist WAY 100635 (10 μm) (Table1, Fig.2).
Table 1.
5-HT1A receptor-mediated increase in [35S]GTP-γ-S binding to the dorsal raphe nucleus and the hippocampus of NK1−/− mutants compared with NK1+/+ wild-type mice
| [35S]GTP-γ-S binding (%) | ||
|---|---|---|
| DRN | Hippocampus | |
| NK1+/+ | 100 ± 9 | 100 ± 10 |
| NK1−/− | 59 ± 3* (−41%) | 89 ± 19ns (−11%) |
Results, expressed as percentage of NK1+/+ values, are the means ± SEM of independent determinations in six to seven mice per group. 5-HT1A receptor-mediated increase in [35S]GTP-γ-S binding attributable to 5-CT (10 μm) reached +131% over baseline in the DRN and +201% in the hippocampus of brain sections from NK1+/+ mice. ns, Nonsignificant;
p < 0.05 as compared with NK1+/+ mice (unpaired Student's t test).
Fig. 2.

Representative autoradiograms of 5-HT1A receptor-mediated increase in [35S]GTP-γ-S binding to brain sections from NK1−/− mutants compared with NK1+/+ wild-type mice. Coronal sections (20 μm) were labeled by [35S]GTP-γ-S (50 pm) without (BASAL) or with 10 μm 5-CT at the level of the DRN and the hippocampus. Nonspecific (NS) labeling was obtained from adjacent sections exposed to 10 μm 5-CT plus 10 μmWAY 100635 (5-HT1A receptor antagonist). Similar autoradiograms were obtained from six mice per group.Cx, Cerebral cortex; DRN, dorsal raphe nucleus; Ent. Cx, entorhinal cortex; Hip, hippocampus; MRN, median raphe nucleus; Sup. Coll., superior colliculi.
Comparison between the two genotypes showed that the 5-CT-induced increase in [35S]GTP-γ-S binding within the DRN was significantly lower (−41%; p < 0.05) in NK1−/− mutants (+78 ± 4% over baseline; mean ± SEM; n = 6) than in wild-type mice (+131 ± 12% over baseline; mean ± SEM; n = 6) (Table 1, Fig.2). By contrast, in the hippocampus, 5-CT-stimulated [35S]GTP-γ-S binding was not significantly different in NK1−/− mutants (+201 ± 19% over baseline; mean ± SEM; n = 7) and paired wild-type mice (+178 ± 38% over baseline; mean ± SEM;n = 6).
Respective distribution of NK1 receptors and serotonin-containing neurons in the dorsal raphe nucleus of NK1+/+ wild-type mice
Sections taken throughout the extent of the DRN were doubly stained using either successively or simultaneously two different antibodies, one against serotonin and the other against the rat NK1 receptor. Both methods yielded similar results. As expected from the labeling of serotoninergic neurons (Descarries et al., 1982), serotonin-like immunoreactivity was found mainly in cell bodies and primary dendrites (Fig. 3A); however, more distal portions of the dendrites were sometimes labeled. By contrast, the immunoreactivity observed in the DRN with the NK1 receptor antibody was mainly confined to the neuropil with only a few labeled cell bodies (Fig. 3A′). NK1 receptor-expressing neurons were smaller than those stained with the serotonin antibody and not easily identified because of the strong label in the dendrites and neuropil. NK1 receptor-positive dendrites and neuropil were found throughout the DRN, but were especially abundant in its dorsomedial part, beneath the aqueduct of Sylvius. Double-stained sections revealed that 5-HT and NK1 receptor do not colocalize in DRN cell bodies and dendrites (Fig. 3B,C). In fact, these two antigens were found in cell types having a different morphology. Interestingly, NK1-positive neuropil was surrounding serotonin-labeled neurons and very often was basketing serotoninergic cells (Fig. 3C). As expected, no positive immunolabeling by NK1 receptor antibodies was observed in the DRN of NK1−/− mutants (data not shown).
Fig. 3.
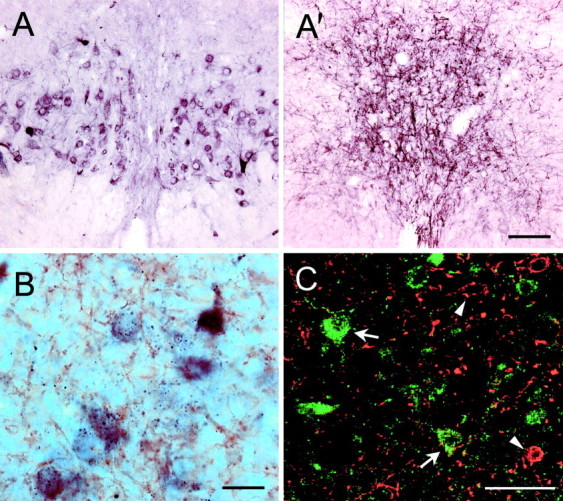
Immunohistochemical labeling of serotonin-positive neurons and NK1 receptors in the dorsal raphe nucleus.A, A′, Bright-field photomicrographs of close serial transverse sections through the DRN illustrating the distribution of serotoninergic neurons in A and NK1 receptors in A′. Scale bar (shown in A′ for A and A′), 100 μm. Bshows dual antigen immunocytochemical labeling (DAB technique), with serotonin-positive elements in purple and NK1 receptor-like immunoreactivity in brown. Serotonin antibody labels cell bodies and primary dendrites, whereas NK1 receptor antibody mainly labels dendrites and neuropil. Most serotonin neurons are surrounded by NK1-positive dendrites and neuropil. Scale bar, 20 μm. C shows a confocal photomicrograph of dual antigen immunocytochemistry developed with fluorescence secondary antibodies. Sections were stained for serotonin in green (examples at arrows) and for NK1 receptors in red(examples at arrowheads). Serotonin and NK1 receptors do not colocalize in the same neurons, but NK1-positive neuropil inred is intermingled with serotonin-positive cell bodies and primary dendrites. Scale bar, 50 μm.
Electrophysiology
Electrophysiological recordings under the various pharmacological conditions tested did not reveal any significant differences between males and females of both genotypes. Accordingly, both males and females were used indifferently in the experiments reported herein.
Basal firing rate
DRN serotoninergic neurons recorded in brainstem slices from wild-type NK1+/+ mice displayed the characteristic slow (1.93 ± 0.24 spikes/sec; mean ± SEM; n = 10) and regular pattern (Fig. 4) of discharge described previously for mice (Lanfumey et al., 1999) and rats (VanderMaelen and Aghajanian, 1983; Haj-Dahmane et al., 1991). No differences in both the frequency (1.95 ± 0.23 spikes/sec; mean ± SEM;n = 13) and pattern of discharge (Fig. 4) of DRN serotoninergic neurons were observed in NK1−/− mutants compared with paired wild-type mice.
Fig. 4.

Effect of ipsapirone on the firing of DRN 5-HT neurons in NK1−/− mutants compared with NK1+/+ wild-type mice.A, Ipsapirone-induced inhibition and prevention by WAY 100635. Integrated firing rate histograms (in spikes per 10 sec) show the effect of increasing concentrations of the 5-HT1Areceptor agonist ipsapirone on the electrical activity of a DRN 5-HT neuron in a NK1−/− mutant (right) compared with a paired NK1+/+ control (left). In both strains, ipsapirone-induced inhibition was markedly reduced by WAY 100635 (1 nm), a selective 5-HT1A receptor antagonist (Fletcher et al., 1996). B, Concentration–response curves of ipsapirone-induced inhibition of the firing of DRN 5-HT neurons in brainstem slices from NK1−/− compared with NK1+/+ mice. Ipsapirone-induced inhibition is expressed as percentage of the baseline firing rate. Each point is the mean ± SEM of data obtained from 10–13 individual cells. The dotted linesillustrate the EC50 values of ipsapirone (abscissa) in NK1−/− mutants and NK1+/+ wild-type mice. **p < 0.01, ***p < 0.001 as compared with the respective inhibition in wild-type mice.
Ipsapirone-induced inhibition of firing
Addition of the 5-HT1A receptor agonist ipsapirone (30–1000 nm) into the aCSF superfusing brainstem slices caused a concentration-dependent inhibition of the firing of DRN serotoninergic neurons, in both NK1+/+ and NK1−/− mice (Fig. 4A,B). However, this effect appeared significantly less pronounced in the NK1−/− mutants. Indeed, the concentration–response curve of ipsapirone in NK1−/− mutants was significantly shifted to the right as compared with that for wild-type mice (p < 0.01). Thus, the EC50 value of ipsapirone was ∼10 times higher in NK1−/− mutants (EC 50= 435.6 ± 38.4 nm; mean ± SEM; n = 13) than in wild-type mice (EC 50= 44.9 ± 1.9 nm; mean ± SEM; n = 10) (Fig. 4B). Complete inhibition of DRN 5-HT neuron firing required only 100 nm ipsapirone in NK1+/+ mice but up to 1 μm of the drug in NK1−/− mutants (Fig. 4A).
As expected from the action of ipsapirone through 5-HT1A autoreceptors, the inhibitory effect of the agonist was prevented by the selective 5-HT1Areceptor antagonist WAY 100635 (1 nm) in both NK1+/+ and NK1−/− mice (Fig. 4A).
Effects of paroxetine with or without WAY 100635 on 5-HT outflow in the frontal cortex of NK1−/− mutants and wild-type mice
Basal extracellular levels of 5-HT ([5-HT]ext) in the frontal cortex of NK1−/−mutants (2.20 ± 0.20 fmol/20 μl; mean ± SEM;n = 20) did not significantly differ from those in wild-type mice (2.34 ± 0.25 fmol/20 μl; mean ± SEM;n = 17).
Acute treatment with paroxetine (1 mg/kg, i.p.) increased [5-HT]ext in the frontal cortex in both wild-type (p < 0.01) and NK1−/− (p < 0.001) mice compared with the corresponding saline-treated control groups (Fig.5A,B). However, the maximal increase caused by this SSRI was more than twice as high in NK1−/− mutants (+474% over baseline) as in wild-type mice (+205% over baseline) (p < 0.001) (Fig.5B,D). A two-way ANOVA (genotype × treatment) on AUC values for 5-HT outflow in the frontal cortex revealed significant genotype factor (F(1,31) = 12.7; p < 0.01), significant treatment factor (F(2,31) = 34.43; p < 0.001), and significant interaction (F(2,31) = 3.75; p < 0.05).
Fig. 5.

Effects of paroxetine with or without the selective 5-HT1A receptor antagonist WAY 100635 on 5-HT outflow in the frontal cortex of NK1−/− mutants compared with NK1+/+ wild-type mice. Data are the means ± SEM of extracellular 5-HT levels expressed as percentages of baseline in NK1+/+ wild-type (open symbols) and NK1−/− knock-out (closed symbols) mice after exposure to saline (0.9% NaCl) (A), paroxetine (Prx) (B), and WAY 100635 given 15 min before paroxetine (C). The first arrowrepresents injection of saline (A, B) or WAY 100635 (0.5 mg/kg, s.c.) (C), and thesecond arrow represents injection of saline (A) or paroxetine (1 mg/kg, i.p.) (B, C). Basal [5-HT]extvalues (in femtomoles per 20 μl) were 2.20 ± 0.20 and 2.34 ± 0.25 in the frontal cortex of NK1+/+ and NK1−/− mice, respectively. D, AUC (mean ± SEM) values calculated for the amount of 5-HT collected during the 0–180 min period after treatment are expressed as percentages of basal values. Each value is the mean ± SEM of at least 12 independent determinations. **p < 0.01 and ***p < 0.001 compared with the corresponding control group (treated with NaCl/NaCl);†††p < 0.001 relative to wild-type;##p < 0.01 compared with wild-type treated with NaCl/paroxetine.
We also investigated the effects of previous administration (15 min before paroxetine) of the selective 5-HT1Areceptor antagonist, WAY 100635 (0.5 mg/kg, s.c.), on the cortical 5-HT overflow induced by paroxetine (1 mg/kg, i.p.) in wild-type and NK1−/− mice (Fig. 5C,D). AUC values for the amount of 5-HT collected in the frontal cortex of wild-type mice after administration of WAY 100635 and paroxetine were significantly higher than those found after paroxetine alone (p < 0.01), but similar to those found in NK1−/− mutants treated either with paroxetine alone or with WAY 100635 and paroxetine (Fig.5D). WAY 100635 alone did not alter [5-HT]ext in the frontal cortex of the two strains (data not shown).
8-OH-DPAT-induced hypothermia
In wild-type mice, the 5-HT1A receptor agonist 8-OH-DPAT (0.25 and 0.5 mg/kg, s.c.) caused a marked decrease in body temperature across the 60 min observation period, which peaked at 30 min with a maximal decrease of −2.57 ± 0.28°C after treatment with the 0.5 mg/kg dose (Fig.6). In NK1−/− mice, 8-OH-DPAT also induced hypothermia; however, the effect was approximately only half of that seen in wild-type mice, with a maximal decrease in body temperature of −1.61 ± 0.17°C (p < 0.05 compared with NK1+/+ mice) at the 0.5 mg/kg dose (Fig. 6).
Fig. 6.
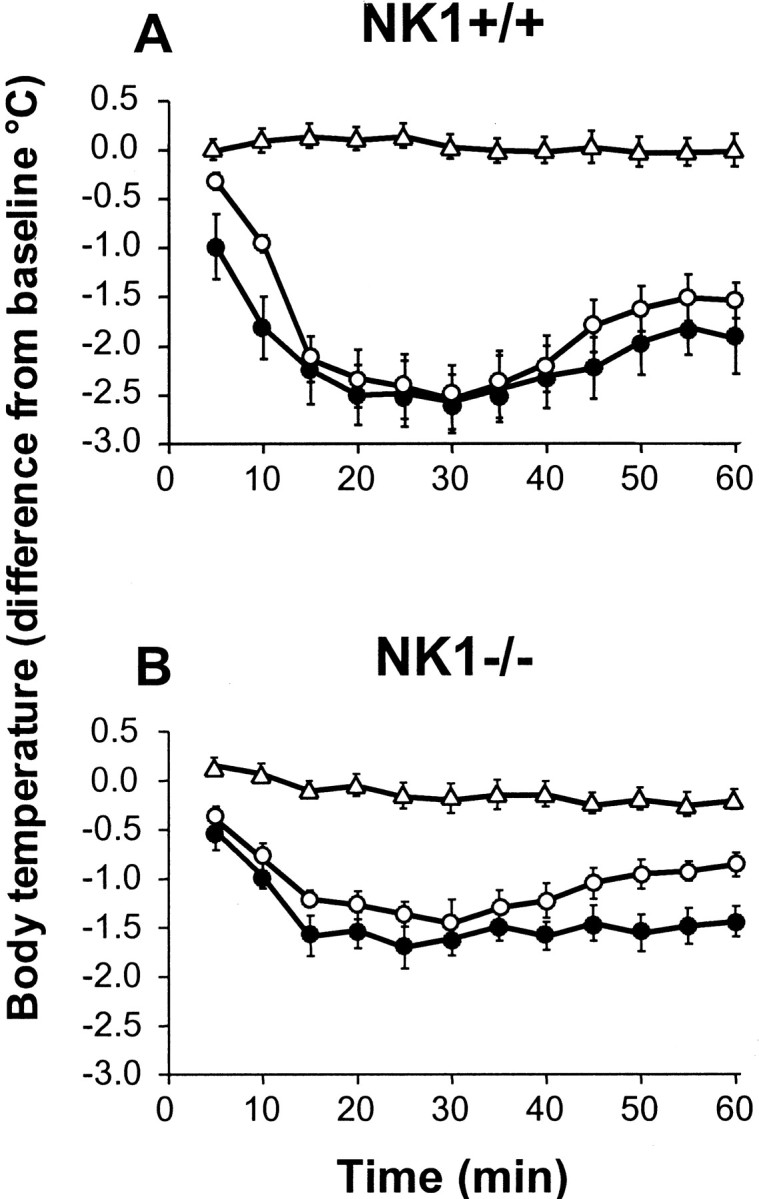
Time course of 8-OH-DPAT-induced hypothermia in NK1−/− mutants compared with NK1+/+ wild-type mice. Wild-type (A) and knock-out (B) mice received a subcutaneous injection of vehicle (▵, 10 ml of 0.9% NaCl/kg) or 8-OH-DPAT (○, 0.25 mg/kg; ●, 0.5 mg/kg), and body temperature was measured at 5 min intervals for up to 60 min. Change (in °C) in body temperature relative to baseline values (immediately before treatment, 0 on abscissa) is the mean ± SEM of determinations in six mice in each group. At both doses tested, body temperature was significantly (p < 0.05; Student's Newman–Keuls multiple t test) less in 8-OH-DPAT-treated than in vehicle-treated mice for the 10–60 min interval after injection in NK1+/+ as well as NK1−/− mice.
DISCUSSION
The present study aimed to assess the functional status of 5-HT1A autoreceptors within the DRN of NK1−/− mice compared with wild-type mice. Investigations using various complementary approaches showed that 5-HT1Aautoreceptor-mediated responses were downregulated in mutant mice devoid of NK1 receptors.
Within the anterior raphe area, a decrease in both the specific labeling by the 5-HT1A receptor antagonist radioligand [3H]WAY 100635 and the levels of 5-HT1A receptor mRNA was observed in NK1−/− versus NK1+/+ mice, suggesting a downregulation of 5-HT1A autoreceptors in mice lacking NK1 receptors. Because the DRN contains a large reserve of 5-HT1A autoreceptors (Meller et al., 1990), such a downregulation might not necessarily cause alterations in responses to 5-HT1A autoreceptor stimulation. Accordingly, we directly addressed the question of the functional status of 5-HT1A autoreceptors in NK1−/− compared with NK1+/+ mice using several complementary approaches.
The first series of experiments consisted of measuring the specific binding of [35S]GTP-γ-S to brain sections to assess the coupling of 5-HT1Areceptors to G-proteins (Fabre et al., 2000). Indeed, the 5-HT1A-mediated increase in [35S]GTP-γ-S binding by 5-CT was markedly less in the DRN of NK1−/− mutants compared with NK1+/+ mice. Similarly, in a model of whole-life treatment with SSRI antidepressants, the 5-HTT−/− knock-out mouse, which does not express the transporter responsible for 5-HT reuptake, DRN 5-HT1A autoreceptors were found to be downregulated and less efficiently coupled to G-proteins than in the paired wild-type mouse (Fabre et al., 2000).
Electrophysiological and in vivo microdialysis approaches were used in a second series of investigations. Consistent with findings reported previously in normal mice (Lanfumey et al., 1999), bath application of a 5-HT1A receptor agonist such as ipsapirone induced a concentration-dependent inhibition of DRN 5-HT neuron firing in brainstem slices from both NK1−/− and NK1+/+ mice. However, the potency of ipsapirone to inhibit this discharge was markedly reduced in NK1 −/− mice, being ∼10-fold lower in mutants than in wild-type animals. This difference suggests the occurrence of a functional desensitization of 5-HT1Aautoreceptors within the DRN as a consequence of the absence of NK1 receptors. Such a desensitization would be expected to alter 5-HT system homeostasis because 5-HT1A autoreceptors play key roles in the control of both synthesis and release of 5-HT in brain (Hamon, 1997). To assess directly how such changes in DRN 5-HT1A autoreceptors could affect the latter process, intracortical microdialysis was performed in awake freely moving mice of both genotypes. Our data show that the increase in cortical 5-HT outflow after acute blockade of 5-HT reuptake by paroxetine was four- to sixfold higher in NK1−/− mutants compared with NK1+/+ mice. Moreover, blockade of 5-HT1Aautoreceptors by the selective antagonist, WAY 100635, potentiated the effect of paroxetine treatment on extracellular 5-HT levels in wild-type mice but not in NK1−/− mutants. These data confirmed that in normal animals the stimulation of somatodendritic 5-HT1A autoreceptors by endogenous 5-HT limits the effects of the SSRI on extracellular 5-HT levels in projection areas (Malagié et al., 1996). By contrast, in NK1−/− mutants, the blockade of 5-HT1A autoreceptors by WAY 100635 did not affect [5-HT]ext levels, very probably because these receptors were markedly desensitized. In line with this conclusion, another response mediated by activation of 5-HT1A autoreceptors in mice, 8-OH-DPAT-induced hypothermia (Bill et al., 1991), was also significantly attenuated in NK1−/− versus wild-type mice. Altogether these data provided clear-cut evidence that the lack of NK1 receptors is associated with the desensitization/downregulation of 5-HT1Aautoreceptors, thereby producing marked changes in central 5-HT neurotransmission. Very recently, a similar conclusion was reported for NK1−/− mice generated on another genetic background (pure 129/SvEv) (Santarelli et al., 2001), indicating that 5-HT1Aautoreceptor desensitization/downregulation actually results from NK1 gene knock-out in these mutants.
The modifications in 5-HT1A autoreceptors observed in NK1−/− mice were very similar to those reported in two other experimental models. Thus, chronic impairment of 5-HT reuptake by long-term SSRI treatment also induces a marked functional desensitization of DRN 5-HT1A autoreceptors (Blier and De Montigny, 1983; Jolas et al., 1994; Le Poul et al., 1995). Although no downregulation of DRN 5-HT1Areceptor binding sites was observed in this model (Le Poul et al., 2000), both electrophysiological (Blier and De Montigny, 1983; Chaput et al., 1986; Le Poul et al., 1995) and microdialysis studies (Bel and Artigas, 1993; Gardier et al., 1996) demonstrated that chronic SSRI treatment leads to clear-cut hypofunctioning of 5-HT1A autoreceptors that is responsible for increased 5-HT availability at the postsynaptic targets (Bel and Artigas, 1993). Similarly, 5-HTT−/− knock-out mice (Bengel et al., 1998), which can be considered as a model of long-term (for the whole-life) 5-HT reuptake inactivation, have been reported to present identical 5-HT1A autoreceptor adaptation. In addition to a functional desensitization (Fabre et al., 2000; Mannoury la Cour et al., 2001), a downregulation of 5-HT1Aautoreceptors was also observed in the DRN of 5-HTT−/− knock-out mice (Fabre et al., 2000), like that found here in NK1−/− mice. Furthermore, after chronic SSRI antidepressant treatment in normal rats (Le Poul et al., 2000) as well as in 5-HTT−/− mice (Fabre et al., 2000; Mannoury la Cour et al., 2001), postsynaptic 5-HT1A receptors were shown to behave differently from 5-HT1A autoreceptors because no desensitization/downregulation was observed in the hippocampus, where 5-HT1A receptors are located postsynaptically (Hamon, 1997). Similarly, in the present study, measurement of [3H]WAY 100635 and 5-CT-evoked [35S]GTP-γ-S-specific binding demonstrated that hippocampal 5-HT1A receptors are functionally unaltered in NK1−/− mutants compared with paired wild-type mice. Such similarities in the respective fate of 5-HT1A autoreceptors and postsynaptic receptors suggest further that the neurobiological changes in NK1−/− mutants closely resemble those induced by chronic blockade of 5-HT reuptake by SSRI antidepressants.
Several hypotheses can be considered regarding the mechanisms by which the disruption of the NK1 receptor gene induces marked modifications in 5-HT homeostasis. Interactions between 5-HT1A and NK1 receptors might occur on the basis of their respective anatomical localizations. NK1 receptors are expressed in limbic areas, including amygdala, septum, hippocampus, and hypothalamus (McLean et al., 1991), which are all innervated by 5-HT projections and contain high to moderate densities of postsynaptic 5-HT1Areceptors (Hamon, 1997). Furthermore, these regions are considered to be critical sites for the antidepressant actions of SSRIs (Horovitz et al., 1966). However, at the very site of the DRN, NK1 and 5-HT1A receptors are not expressed by the same cells because we demonstrated previously that 5-HT1A receptors are located exclusively on the somas and dendrites of serotoninergic neurons (Sotelo et al., 1990), whereas we presently found that the latter neurons do not express NK1 receptors. Indeed, NK1 receptor-like immunoreactivity within the DRN was found mainly in the neuropil, surrounding the 5-HT-labeled neurons. This localization suggests that NK1 receptors might be involved in synaptic processing at the level of the somatodendritic domain of serotoninergic neurons. Interestingly, it has been shown recently that NK1 receptors could participate in local glutamatergic excitatory inputs on serotoninergic cells in the DRN (Liu and Aghajanian, 2000). In addition, facilitation of locus coeruleus noradrenergic system activity by NK1 receptor blockade (Millan et al., 2001) might indirectly affect the activity of DRN serotoninergic neurons through the tonic α1-adrenergic-mediated control originating in this nucleus (VanderMaelen and Aghajanian, 1983). Whether alterations in glutamatergic, noradrenergic, and other inputs contribute to 5-HT1A autoreceptor desensitization/downregulation in NK1−/− mice should warrant further investigations. In addition, special attention has to be paid to the early developmental period because such 5-HT1Aautoreceptor changes might have resulted from alterations in some NK1 receptor-mediated neurotrophic action of substance P during brain maturation (Barker, 1991).
In any case, the generation of mice lacking the NK1 receptor for substance P allowed the demonstration that this neuropeptide plays a key role in the control of behavior, notably in the adaptive responses to stress (De Felipe et al., 1998; Rupniak et al., 2000). In particular, NK1−/− mice have been shown to be less aggressive and less anxious than wild-type controls (De Felipe et al., 1998). Furthermore, spontaneous ultrasound calls after maternal separation were markedly reduced in NK1−/− compared with paired wild-type pups (Rupniak et al., 2000). In both guinea pig and mouse pups, specific NK1 receptor antagonists decreased ultrasonic vocalizations in a manner resembling that found after treatment with antidepressant or anxiolytic drugs (Rupniak et al., 2000). NK1 receptor antagonists (and NK1 receptor gene knock-out) thus may possibly act by reducing the effects of psychological stress. Using various pharmacological and behavioral models, 5-HT1A autoreceptor desensitization has previously been proposed to reflect an adaptation to stress (Lanfumey et al., 1999, 2000) through complex reciprocal interactions between the 5-HT system and the hypothalamo–pituitary–adrenal axis. Whether this axis also contributes to the adaptive changes in 5-HT1A autoreceptors reported herein in NK1−/− mice is another relevant question to be addressed in future investigations.
Footnotes
This research was supported by the Institut National de la Santéet de la Recherche Médicale (France), the Bristol-Myers Squibb Foundation (Unrestricted Biomedical Research Grant Program), and the Ministerio de Educacion y Ciencia (SAF00-0122) (Spain). N.F. was a recipient of a fellowship from the Ministère de l'Education Nationale et de la Recherche (France) during performance of this work. R.M. was supported by the Fundacion La Caixa, and I.A. was supported by the Communidad de Madrid (Spain). We are grateful to pharmaceutical companies (SmithKline Beecham, Troponwerke-Bayer, and Wyeth-Ayerst) for generous gifts of drugs. We thank Janine Webb and Susan Boyce for performing the hypothermia studies.
Correspondence should be addressed to Nicolas Froger, Institut National de la Santé et de la Recherche Médicale U288, Neuropsychopharmacologie Moléculaire, Cellulaire et Fonctionnelle, Faculté de Médecine Pitié-Salpêtrière, 91 Boulevard de l'Hôpital, 75634 Paris Cedex 13, France. E-mail:nifroger@ext.jussieu.fr.
REFERENCES
- 1.Albert PR, Zhou QY, Van Tol HH, Bunzow JR, Civelli O. Cloning, functional expression and mRNA tissue distribution of the rat 5-HT1A receptor gene. J Biol Chem. 1990;265:5825–5832. [PubMed] [Google Scholar]
- 2.Artigas F, Romero L, De Montigny C, Blier P. Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci. 1996;19:378–383. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- 3.Asberg M, Thoren P, Träksman L. Serotonin depression in a biochemical subgroup within the affective disorders. Life Sci. 1976;191:478–480. doi: 10.1126/science.1246632. [DOI] [PubMed] [Google Scholar]
- 4.Barker R. Substance P and neurodegenerative disorders. A speculative review. Neuropeptides. 1991;20:73–78. doi: 10.1016/0143-4179(91)90054-m. [DOI] [PubMed] [Google Scholar]
- 5.Bel N, Artigas F. Chronic treatment with fluvoxamine increases extracellular serotonin in frontal cortex but not in raphe nuclei. Synapse. 1993;15:243–245. doi: 10.1002/syn.890150310. [DOI] [PubMed] [Google Scholar]
- 6.Bengel D, Murphy DL, Andrews AM, Wichems CH, Feltner D, Heils A, Mossner R, Westphal H, Lesch KP. Altered brain serotonin homeostasis and locomotor insensitivity to 3,4-methylenedioxymethamphetamine (“ecstasy”) in serotonin transporter-deficient mice. Mol Pharmacol. 1998;53:649–655. doi: 10.1124/mol.53.4.649. [DOI] [PubMed] [Google Scholar]
- 7.Bill DJ, Knight M, Forster EA, Fletcher A. Direct evidence for an important species difference in the mechanism of 8-OH-DPAT-induced hypothermia. Br J Pharmacol. 1991;103:1857–1864. doi: 10.1111/j.1476-5381.1991.tb12342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blier P, De Montigny C. Electrophysiological investigations on the effect of repeated zimelidine administration on serotoninergic neurotransmission in the rat. J Neurosci. 1983;3:1270–1278. doi: 10.1523/JNEUROSCI.03-06-01270.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaput Y, De Montigny C, Blier P. Effects of a selective 5-HT reuptake blocker, citalopram, on the sensitivity of 5-HT autoreceptors: electrophysiological studies in the rat brain. Naunyn Schmiedebergs Arch Pharmacol. 1986;333:342–348. doi: 10.1007/BF00500007. [DOI] [PubMed] [Google Scholar]
- 10.Dechant KL, Clissold SP. Paroxetine: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in depressive illness. Drugs. 1991;41:225–253. doi: 10.2165/00003495-199141020-00007. [DOI] [PubMed] [Google Scholar]
- 11.De Felipe C, Herrero JF, O'Brien JA, Palmer JA, Doyle CA, Smith AJH, Laird JMA, Belmonte C, Cervero F, Hunt SP. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature. 1998;392:394–397. doi: 10.1038/32904. [DOI] [PubMed] [Google Scholar]
- 12.Delgado PL, Charney DS, Price LH, Aghajanian GK, Landis H, Heninger GR. Serotonin function and the mechanism of antidepressant action: reversal of antidepressant induced remission by rapid depletion of plasma tryptophan. Arch Gen Psychiatry. 1990;47:411–418. doi: 10.1001/archpsyc.1990.01810170011002. [DOI] [PubMed] [Google Scholar]
- 13.Descarries L, Watkins KC, Garcia S, Beaudet A. The serotonin neurons in nucleus raphe dorsalis of adult rat: a light and electron microscope radioautographic study. J Comp Neurol. 1982;207:239–254. doi: 10.1002/cne.902070305. [DOI] [PubMed] [Google Scholar]
- 14.Fabre V, Beaufour C, Evrard A, Rioux A, Hanoun N, Lesch KP, Murphy DL, Lanfumey L, Hamon M, Martres MP. Altered expression and functions of serotonin 5-HT1A and 5-HT1B receptors in knock-out mice lacking the 5-HT transporter. Eur J Neurosci. 2000;12:2299–2310. doi: 10.1046/j.1460-9568.2000.00126.x. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher A, Forster EA, Bill DJ, Brown G, Cliffe IA, Hartley JE, Jones DE, McLenachan A, Stanhope KJ, Critchley DJP, Childs KJ, Middlefell VC, Lanfumey L, Corradetti R, Laporte AM, Gozlan H, Hamon M, Dourish CT. Electrophysiological, biochemical, neurohormonal and behavioural studies with WAY-100635, a potent, selective, and silent 5-HT1A receptor antagonist. Behav Brain Res. 1996;73:337–353. doi: 10.1016/0166-4328(96)00118-0. [DOI] [PubMed] [Google Scholar]
- 16.Franklin KB, Paxinos G. Mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 17.Fuller RW, Perry KN, Molloy B. Effect of 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine on the depletion of brain serotonin by 4-chloroamphetamine. J Pharmacol Exp Ther. 1975;193:796–803. [PubMed] [Google Scholar]
- 18.Gardier A, Malagié I, Trillat A, Jacquot C, Artigas F. Role of 5-HT1A autoreceptors in the mechanism of action of serotoninergic antidepressant drugs: recent findings from in vivo microdialysis studies. Fundam Clin Pharmacol. 1996;10:16–27. doi: 10.1111/j.1472-8206.1996.tb00145.x. [DOI] [PubMed] [Google Scholar]
- 19.Gozlan H, Thibault S, Laporte AM, Lima L, Hamon M. The selective 5-HT1A antagonist radioligand [3H]WAY 100635 labels both G-protein-coupled and free 5-HT1A receptors in rat brain membranes. Eur J Pharmacol Mol Pharmacol. 1995;288:173–186. doi: 10.1016/0922-4106(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 20.Haj-Dahmane S, Hamon M, Lanfumey L. K+ channel and 5-hydroxytryptamine1A autoreceptor interactions in the rat dorsal raphe nucleus: an in vitro electrophysiological study. Neuroscience. 1991;41:495–505. doi: 10.1016/0306-4522(91)90344-n. [DOI] [PubMed] [Google Scholar]
- 21.Hamon M. The main features of central 5-HT1A receptors. In: Baumgarten HG, Göthert M, editors. Serotoninergic neurons and 5-HT receptors in the CNS. Handbook of experimental pharmacology. Springer; Berlin: 1997. pp. 239–268. [Google Scholar]
- 22.Hensler J, Kovachich GB, Frazer A. A quantitative autoradiographic study of serotonin1A receptor regulation: effects of 5,7-dihydroxytryptamine and antidepressant treatments. Neuropsychopharmacology. 1991;4:131–144. [PubMed] [Google Scholar]
- 23.Horovitz ZP, Piala JJ, High JP, Burke JC, Leaf RC. Effects of drugs on the mouse-killing (muricide) test and its relationship to amygdaloid function. Int J Neuropharmacol. 1966;5:405–411. doi: 10.1016/0028-3908(66)90005-0. [DOI] [PubMed] [Google Scholar]
- 24.Jolas T, Haj-Dahmane S, Kidd EJ, Langlois X, Lanfumey L, Fattaccini CM, Vantalon V, Laporte AM, Adrien J, Gozlan H, Hamon M. Central pre- and postsynaptic 5-HT1A receptors in rats treated chronically with a novel antidepressant, cericlamine. J Pharmacol Exp Ther. 1994;268:1432–1443. [PubMed] [Google Scholar]
- 25.Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, Reines SA, Liu G, Snavely D, Wyatt-Knowles E, Hale JJ, Mills SG, MacCoss M, Swain CJ, Harrison T, Hill RG, Hefti F, Scolnick EM, Cascieri MA, Chicchi GG. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998;281:1640–1645. doi: 10.1126/science.281.5383.1640. [DOI] [PubMed] [Google Scholar]
- 26.Lanfumey L, Pardon MC, Laaris N, Joubert C, Hanoun N, Hamon M, Cohen-Salmon C. 5-HT1A autoreceptor desensitization by chronic ultramild stress in mice. NeuroReport. 1999;10:3369–3374. doi: 10.1097/00001756-199911080-00021. [DOI] [PubMed] [Google Scholar]
- 27.Lanfumey L, Mannoury la Cour C, Froger N, Hamon M. 5-HT-HPA interactions in two models of transgenic mice relevant to major depression. Neurochem Res. 2000;25:1199–1206. doi: 10.1023/a:1007683810230. [DOI] [PubMed] [Google Scholar]
- 28.Le Poul E, Laaris N, Doucet E, Laporte AM, Hamon M, Lanfumey L. Early desensitization of somato-dendritic 5-HT1A autoreceptors in rats treated with fluoxetine or paroxetine. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:141–148. doi: 10.1007/BF00176767. [DOI] [PubMed] [Google Scholar]
- 29.Le Poul E, Boni C, Hanoun N, Laporte AM, Laaris N, Chauveau J, Hamon M, Lanfumey L. Differential adaptation of brain 5-HT1A and 5-HT1B receptors and 5-HT transporter in rats treated chronically with fluoxetine. Neuropharmacology. 2000;39:110–122. doi: 10.1016/s0028-3908(99)00088-x. [DOI] [PubMed] [Google Scholar]
- 30.Liu RJ, Aghajanian GK. Neurokinin NK1 and NK3 receptors activate local glutamatergic inputs to serotonergic neurons of the dorsal raphe nucleus. Soc Neurosci Abstr. 2000;26:1927. doi: 10.1016/S0893-133X(02)00305-6. [DOI] [PubMed] [Google Scholar]
- 31.Lloyd KG, Farley IJ, Deck JHN, Hornykiewicz O. Serotonin and 5-hydroxyindoleacetic acid in discrete areas of brainstem of suicide victims and control patients. Adv Biochem Psychopharmacol. 1974;11:387–397. [PubMed] [Google Scholar]
- 32.Maes M, Meltzer HY. The serotonin hypothesis of major depression. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. Raven; New York: 1995. pp. 933–944. [Google Scholar]
- 33.Malagié I, Trillat AC, Douvier E, Anmella MC, Dessalles MC, Jacquot C, Gardier AM. Regional differences in the effect of the combined treatment of WAY 100635 and fluoxetine: an in vivo microdialysis study. Naunyn Schmiedebergs Arch Pharmacol. 1996;354:785–790. doi: 10.1007/BF00166906. [DOI] [PubMed] [Google Scholar]
- 34.Mannoury la Cour C, Boni C, Hanoun N, Lesch KP, Hamon M, Lanfumey L. Functional consequences of 5-HT transporter gene disruption on 5-HT1A receptor-mediated regulation of dorsal raphe and hippocampal cell activity. J Neurosci. 2001;21:2178–2185. doi: 10.1523/JNEUROSCI.21-06-02178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLean S, Ganong AH, Seeger TF, Bryce DK, Pratt KG, Reynolds LS, Siok CJ, Lowe JA, Heym J. Activity and distribution of binding sites in brain of a nonpeptide substance P (NK1) receptor antagonist. Science. 1991;251:437–439. doi: 10.1126/science.1703324. [DOI] [PubMed] [Google Scholar]
- 36.Meller E, Goldstein M, Bohmaker K. Receptor reserve for 5-hydroxytryptamine1A-mediated inhibition of serotonin synthesis: possible relationship to anxiolytic properties of 5-hydroxytryptamine1A agonists. Mol Pharmacol. 1990;37:231–237. [PubMed] [Google Scholar]
- 37.Millan MJ, Lejeune F, De Nanteuil G, Gobert A. Selective blockade of neurokinin (NK)1 receptors facilitates the activity of adrenergic pathways projecting to frontal cortex and dorsal hippocampus in rats. J Neurochem. 2001;76:1949–1954. doi: 10.1046/j.1471-4159.2001.00211.x. [DOI] [PubMed] [Google Scholar]
- 38.Moratalla R, Elibol B, Vallejo M, Graybiel AM. Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron. 1996;17:147–156. doi: 10.1016/s0896-6273(00)80288-3. [DOI] [PubMed] [Google Scholar]
- 39.Papp M, Vassout A, Gentsch C. The NK1-receptor antagonist NKP608 has an antidepressant-like effect in the chronic mild stress model of depression in rats. Behav Brain Res. 2000;115:19–23. doi: 10.1016/s0166-4328(00)00230-8. [DOI] [PubMed] [Google Scholar]
- 40.Pradhan S, Hanson G, Lovenberg W. Inverse relation of substance P-like immunoreactivity in dorsal raphe nucleus to serotonin levels in pons-medulla following administration of cocaine and 5-hydroxytryptophan. Biochem Pharmacol. 1981;30:1071–1076. doi: 10.1016/0006-2952(81)90444-5. [DOI] [PubMed] [Google Scholar]
- 41.Rupniak NMJ, Carlson EJ, Harrison T, Oates B, Seward E, Owen S, de Felipe C, Hunt S, Wheeldon A. Pharmacological blockade or genetic deletion of substance P (NK1) receptors attenuates neonatal vocalisation in guinea-pigs and mice. Neuropharmacology. 2000;39:1413–1421. doi: 10.1016/s0028-3908(00)00052-6. [DOI] [PubMed] [Google Scholar]
- 42.Santarelli L, Gobbi G, Debs PC, Sibille EL, Blier P, Hen R, Heath MJ. Genetic and pharmacological disruption of neurokinin 1 receptor function decreases anxiety-related behaviors and increases serotonergic function. Proc Natl Acad Sci USA. 2001;98:1912–1917. doi: 10.1073/pnas.041596398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirayama Y, Mitsushio H, Takashima M, Ichikawa H, Takahashi K. Reduction of substance P after chronic antidepressants treatment in the striatum, substantia nigra and amygdala of the rat. Brain Res. 1996;739:70–78. doi: 10.1016/s0006-8993(96)00812-8. [DOI] [PubMed] [Google Scholar]
- 44.Siebert PD, Larrick JW. Competitive PCR. Nature. 1992;359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- 45.Sotelo C, Cholley B, El Mestikawy S, Gozlan H, Hamon M. Direct immunohistochemical evidence of the existence of 5-HT1A autoreceptors on serotonergic neurons in the midbrain raphe nuclei. Eur J Neurosci. 1990;2:1144–1154. doi: 10.1111/j.1460-9568.1990.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 46.VanderMaelen CP, Aghajanian GK. Electrophysiological and pharmacological characterization of serotoninergic dorsal raphe neurons recorded extracellularly and intracellularly in rat brain slices. Brain Res. 1983;289:109–119. doi: 10.1016/0006-8993(83)90011-2. [DOI] [PubMed] [Google Scholar]
- 47.Walker PD, Riley LA, Hart RP, Jonakait GM. Serotonin regulation of tachykinin biosynthesis in the rat neostriatum. Brain Res. 1991;546:33–39. doi: 10.1016/0006-8993(91)91155-t. [DOI] [PubMed] [Google Scholar]