

Abstract
Hypoxic encephalopathy is the most common cause of neonatal seizures and can lead to chronic epilepsy. In rats at postnatal days 10–12 (P10–12), global hypoxia induces spontaneous seizures and chronically decreases seizure threshold, thus mimicking clinical aspects of neonatal hypoxia. We have shown previously that the acute and chronic epileptogenic effects of hypoxia are age-dependent and require AMPA receptor activation. In this study, we aimed to determine whether hypoxia-induced seizures and epileptogenesis are associated with maturational and seizure-induced changes in AMPA receptor composition and function. Northern and Western blots indicated that glutamate receptor 2 (GluR2) mRNA and protein expression were significantly lower in neocortex and hippocampus at P10–12 compared with adult. After hypoxia-induced seizures at P10, GluR2 mRNA was significantly decreased within 48 hr, and GluR2 protein was significantly decreased within 96 hr. AMPA-induced Co2+ uptake by neurons in hippocampal slices indicated higher expression of Ca2+-permeable AMPA receptors in immature pyramidal neurons compared with adult. In slices obtained 96 hr after hypoxia-induced seizures, AMPA-induced Co2+ uptake was significantly increased compared with age-matched controls, and field recordings revealed increased tetanus-induced afterdischarges that could be kindled in the absence of NMDA receptor activation. In situ end labeling showed no acute or delayed cell death after hypoxia-induced seizures. Our results indicate that susceptibility to hypoxia-induced seizures occurs during a developmental stage in which the expression of Ca2+-permeable AMPA receptors is relatively high. Furthermore, perinatal hypoxia-induced seizures induce increased expression of Ca2+-permeable AMPA receptors and an increased capacity for AMPA receptor-mediated epileptogenesis without inducing cell death.
Keywords: epilepsy, AMPA receptor, glutamate, neonatal, calcium, hippocampus, cobalt
Hypoxic encephalopathy is the most common cause of neonatal seizures and is associated with an increased risk of epilepsy in later life (Volpe, 2000). Our laboratory demonstrated previously that neonatal rats exposed to transient global hypoxia on postnatal days 10–12 (P10–12) exhibit spontaneous electrographic and behavioral seizures that result in decreased seizure thresholds throughout adulthood, thus mimicking clinical aspects of neonatal hypoxic encephalopathy (Jensen et al., 1991, 1992). Additionally, hippocampal slices obtained from animals that experienced hypoxia-induced seizures exhibit long-term enhancement of synaptic plasticity and ictal-like population discharges within area CA1 (Jensen et al., 1998). The precise mechanisms that underlie these epileptogenic effects of hypoxia in the neonatal brain have not been fully characterized. However, the AMPA subtype of glutamate receptor (GluR) appears to play a critical role, because both the acute and chronic effects of hypoxia can be blocked by pretreatment with an AMPA receptor antagonist but not by NMDA receptor antagonists or by GABA receptor agonists (Jensen et al., 1995).
AMPA receptors are hetero-oligomers assembled from four molecular subunits termed GluR1–4 (alternatively GluRA–D; for review, seeDingledine et al., 1999). In the adult brain, most native AMPA receptors contain a GluR2 subunit, but in the immature brain, evidence suggests that the expression of GluR2 is lower relative to other subunits (Pellegrini-Giampietro et al., 1992; Durand and Zukin, 1993;Monyer et al., 1994). Functionally, recombinant AMPA receptors that lack a GluR2 subunit exhibit increased permeability to Ca2+ and other divalent cations compared with those that contain at least one GluR2 subunit (Burnashev et al., 1992; Hollman and Heinemann, 1994). These observations suggest that the proportion of AMPA receptors that are permeable to Ca2+ is higher in the immature brain compared with the adult. However, maturational differences in AMPA receptor permeation properties have yet to be demonstrated.
Ca2+-permeable AMPA receptors have been proposed to play a role in certain neurodegenerative disorders by allowing excessive glutamate-stimulated Ca2+ entry into vulnerable neurons (Pellegrini-Giampietro et al., 1997; Weiss and Sensi, 2000). In particular, decreases in neuronal GluR2 expression after a variety of neurological insults, including prolonged seizures (Pollard et al., 1993; Friedman et al., 1994), are thought to result in pathologically increased and toxic Ca2+ entry through AMPA receptors. In immature rats, transient knockdown of GluR2 expression by intrahippocampal infusion of antisense GluR2 mRNA was shown to elicit seizure-like behaviors and neurodegeneration in hippocampal area CA3 (Friedman and Koudinov, 1999). These data suggested that the regulation of GluR2 expression can have critical consequences for epileptogenesis and neuronal injury in early development.
The epileptogenic effects of hypoxia are only observed in the immature brain (Jensen et al., 1991) and are highly dependent on AMPA receptor activation (Jensen et al., 1995). Given a possible relationship between relative GluR2 expression and seizures, we hypothesized that the lower GluR2 expression in the immature brain contributes to the age-dependent susceptibility to hypoxia-induced seizures. In the present study, we aimed to test the hypotheses that the susceptibility to hypoxia-induced seizures occurs during an age window when GluR2 expression is relatively low, and that increased hippocampal excitability after perinatal hypoxia-induced seizures is associated with further decreases in GluR2 expression. Additionally, we aimed to determine whether changes in hippocampal GluR2 expression are associated with changes in the numbers of Ca2+-permeable AMPA receptors expressed by principal neurons. Our findings demonstrate that the dynamic regulation of GluR2 expression during development and by hypoxia-induced seizures determines the relative abundance of Ca2+-permeable AMPA receptors and support a role for these receptors in perinatal hypoxia-induced epileptogenesis. Notably, in contrast to other paradigms that result in decreased GluR2 expression, our results also indicate that perinatal hypoxia-induced seizures do not cause acute cell death, suggesting that GluR2 downregulation can be associated with epileptogenesis in the absence of cell loss.
MATERIALS AND METHODS
Animals. Male Long–Evans Hooded rats (Charles River Laboratories, Wilmington, MA) were used in this study. Rats were housed in the animal care facility in a 12-hr light/dark cycle. All procedures were approved and in accordance with guidelines set by the Institutional Animal Care and Use Committee.
Hypoxia-induced seizures. At P10, animals were removed from the litter and placed in an airtight chamber on a heating pad to maintain temperature at 32–34°C. The O2concentration measured with an oxygen meter placed inside the chamber was lowered to 5–7% by infusion of N2 gas into the chamber. Seizure activity produced by this model consists of a period of myoclonic jerks followed by tonic–clonic activity of the trunk and neck, and the numbers of both myoclonic jerks and tonic–clonic seizures were counted by an observer. After 12 min, the O2 concentration was lowered by ∼1%/min until the onset of apnea. The total duration of hypoxia ranged between 14 and 17 min. Only rats exhibiting at least one tonic–clonic seizure during hypoxia were used in this study.
Northern blotting and quantification of GluR2 subunit mRNA.Rats were decapitated, and the hippocampus and frontoparietal neocortex were isolated and placed in liquid nitrogen. Liver RNA was used as a negative control. RNA was isolated from tissue blocks as described previously (Tolentino et al., 1995), electrophoresed in denaturing gels, and then transferred and fixed to a charged membrane using a modification of the basic Northern blot method that results in increased sensitivity (Tolentino et al., 1995). RNA was hybridized to32P-labeled DNA. Probes were prepared from GluR2 cDNA (a gift from Dr. James Boulter, University of California Los Angeles Medical School, Los Angeles, CA). After hybridization, blots were exposed in PhosphorImager cassettes and imaged using a Molecular Dynamics (Sunnyvale, CA) 445 SI PhosphorImager system. Digital images were analyzed with the IP Lab Gel densitometry and gel analysis software package (Signal Analytics Corporation, Vienna, VA). Quantitative data were compared between the hypoxic samples and littermate controls killed at the same age. Blots were stripped and reprobed with another subunit probe. To control for variability in loading and sample handling, we rehybridized all blots with a probe for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which does not undergo significant tissue-specific changes with hypoxia or development. Relative values for experimental and control signals from the same blot were compared using Student's t test.
In situ hybridization for localizing GluR2 receptor subunit RNA. Rats were killed at 48–96 hr after hypoxia and perfused with 4% paraformaldehyde. Coronal brain sections (20 μm thickness) were prepared and stored at −20°C until prehybridization.35S-[α-35S]UTP (>1000 Ci/mmol; NEN, Boston, MA)-labeled antisense riboprobes for GluR2 were generated by runoff transcription using linearized plasmid DNA as a template for T7 RNA polymerase. Transcripts were hydrolyzed to an average length of 200 bp using alkaline hydrolysis to ensure efficient penetration of the probe into the tissue sections (Tolentino et al., 1995). Hybridization was performed at 52°C for 18–24 hr in 50% deionized formamide, 0.3 m sodium chloride, 20 mm Tris-HCl, pH 7.4, 5 mm EDTA, 10 mmNa2PO4-H2O, pH 8, 10% dextran sulfate, 1× Denhardt's solution, and 500 μg/ml total yeast RNA with 3.5 × 104cpm/μl 35S-labeled RNA probe under parafilm. After hybridization, slides were washed in 5× SSC and 10 mm dithiothreitol at 50°C and in 50% formamide, 2× SSC, and 10 mm dithiothreitol at 65°C. Slides were rinsed in washing buffer (two times for 10 min each), in washing buffer and RNase A (20 μg/ml; Sigma, St. Louis, MO; 15 min), and then in washing buffer without RNase and then washed at 37°C for 15 min in 2× SSC and then for 15 min in 0.1× SSC. Sections were dehydrated rapidly and apposed to high-performance autoradiography film (Hyperfilm; Amersham Pharmacia Biotech, Arlington Heights, IL) for 55 hr. For quantification, six consecutive sections spanning the dorsal hippocampus were selected from each brain (3 control and 4 hypoxia, a total of 84 sections through 14 hippocampi), images were acquired on a personal computer, and the relative optical signal density was measured using a Northern Light Precision Illuminator and Micro Computer Imaging Device software (Imaging Research, Inc.). The circular measuring frame was placed over the region of interest to include the pyramidal cell layer, and six consecutive measurements were taken. The region of interest was defined as area CA1 proper medial to the point of transection by a straight line drawn through upper and lower blades of the dentate gyrus.
Western blotting. Membrane protein preparations were made according to the protocol described by Wenthold et al. (1992). Protein concentrations were determined for each homogenized sample using a Bradford assay (Bio-Rad, Hercules, CA). Homogenization buffer was added to bring each sample to the same concentration, and then a Bradford assay again was performed on the diluted samples to ensure comparable protein concentrations. Equal volumes of each sample were then loaded onto gel. The protein was then transferred to a membrane (polyvinylidene difluoride), and a Coomassie blue stain (Sigma) was performed on the gel to check again for consistent protein loading across lanes. Membranes were incubated with polyclonal IgG GluR1- and GluR2-specific antibodies [initial antibodies were a gift from Dr. R. J. Wenthold (Wenthold et al., 1992), and additional antibodies were obtained from Chemicon, Temecula, CA], incubated with a secondary anti-IgG antibody conjugated to 125I, and exposed in PhosphorImager cassettes. Digital images were quantified using the IP Lab Gel software, and subunit protein was compared between control and hypoxic rats using Student's t test.
Detection of DNA fragmentation by in situ end labeling. Animals were perfused transcardially with 20 ml of PBS followed by 40 ml of ice-cold 4% paraformaldehyde in 0.1 mPBS, pH 7.4. Brains were removed, post-fixed for 24 hr, and then cryoprotected in 20% sucrose overnight. Fifty micrometer sections were cut on a freezing microtome and processed for in situ nick translation using a modification of the protocol developed by Wijsman et al. (1993) and described in detail previously (Weiss et al., 1996). Briefly, free-floating sections were incubated in 1[times] SSC (300 mm NaCl and 30 mmNa-citrate, pH 7) at 80°C for 20 min and then treated for 10 min with pronase (1 μg/ml; Roche Molecular Biochemicals, Indianapolis, IN). Digestion was stopped in 2% glycine, and the sections were briefly rinsed in water and incubated for 1 hr at room temperature with 50 μg/ml DNA polymerase I (Promega, Madison, WI) and 10 μm each of biotin-21-dUTP (Clontech), dCTP, dATP, and dGTP dissolved in buffer A (50 mmTris-HCl, pH 7.5, 5 mmMgCl2, 10 mmβ-mercaptoethanol, and 0.005% BSA). Biotin end-labeled DNA fragments were detected using avidin-biotin-peroxidase (Vectastain Elite kit; Vector Laboratories, Burlingame, CA) with nickel (II) sulfate intensification. Sections were mounted onto gelatin-coated slides, dehydrated, cleared, and coverslipped in Permount.
Cobalt staining method to assess divalent cation permeability. Rat pups were decapitated, the brains were removed, and hippocampus was dissected rapidly and immersed in ice-cold uptake buffer (in mm: 57.5 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 12 glucose, 139 sucrose, and 10 HEPES, pH 7.4, bubbled with 100% O2). Four hundred micrometer slices of hippocampus were cut in a plane perpendicular to the septotemporal axis using a manual tissue chopper (Stoelting). AMPA-stimulated Co2+ uptake was performed using a modification of procedures described previously (Pruss et al., 1991). Slices were collected in a Ca2+-free HEPES-buffered Krebs' saline (in mm: 118 NaCl, 4.6 KCl, 1.2 MgSO4, 10 glucose, and 25 Na-HEPES, pH 7.4) and rinsed two times for 5 min in uptake buffer. Slices were then stimulated for 20 min with 100 μm AMPA (Sigma) in uptake buffer containing 5 mm CoCl2 and the NMDA receptor antagonist APV (100 μm; Sigma) at room temperature in an airtight chamber continuously infused with 100% O2. AMPA stimulation was performed with or without the specific AMPA receptor antagonist 2,3-dihydro-6-nitro-7-sulfamoyl-benz (F) quinoxaline (NBQX) (100 μm; Research Biochemicals, Natick, MA) or a selective blocker of Ca2+-permeable AMPA receptors, 1-napthyl-acetyl-spermine (100 μm; RBI). Slices were then rinsed quickly in Krebs' saline and then three times for 5 min each in Krebs' saline with 2 mm EDTA to remove extracellular Co2+, and intracellular Co2+ was precipitated by incubation in 0.2% (NH4)S for 5 min. Slices were then fixed by submersion in 4% paraformaldehyde for 1–2 hr, rinsed, and stored in PBS until silver enhancement. For silver enhancement, slices were transferred to enhancement solution without silver (in mm: 292 sucrose, 15.5 hydroquinone, and 42 citric acid), warmed to 50°C, and then incubated for 45 min at 50°C in enhancement solution containing 0.1% AgNO3, added fresh every 15 min. Slices were then rinsed twice in double-distilled water, dehydrated in methanol:ethyleneglycol monomethyl ether (1:1) for 4–12 hr, and transferred to a warm mixture of 1% cetyl alcohol in polyethylene glycol distearate to be infiltrated overnight at 37°C. Slices were embedded in a mold containing the same mixture at room temperature. Twenty micrometer sections were cut on a microtome (RM2135; Leica, Nussloch, Germany), mounted wet, air-dried, cleared in acetone, hydrated, counterstained in methylene green, dehydrated, cleared in xylene, and coverslipped in Permount.
For the quantification of Co2+ staining, six consecutive sections were selected from the middle third of each slice to avoid edge artifacts and to minimize data from injured cells near the slice surfaces. A 250 μm line segment was drawn through the pyramidal cell layer in the CA1/CA2 region, and the numbers of Co2+-positive cell bodies that intersected this line segment were counted. This was done to minimize the numbers of interneurons counted. Counts were averaged across sections, and the average counts were compared between the control versus hypoxia-treated groups by Student's t test.
Slice preparation and electrophysiological recordings.Hippocampal slices were prepared from hypoxia-treated and age-matched control pups 4–9 d after hypoxia treatment, and electrophysiological recordings were obtained using previously published methods (Sanchez et al., 2000). Briefly, after decapitation, the brains were rapidly dissected from the skull and placed in ice-cold artificial CSF (ACSF; in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1.3 MgSO4, 10d-glucose, and 26 NaHCO3, bubbled with 95%O2 and 5%CO2). For extracellular recordings, one hippocampus was removed, and 400 μm slices were cut using a manual tissue chopper (Stoelting) and transferred to a flow-through interface chamber perfused with oxygenated ACSF at 40–50 ml/hr at 33.5°C. For whole-cell recordings, 300 μm slices were cut using a vibratome and incubated in oxygenated ACSF at room temperature. All slices were incubated for 60–90 min before recording. Field recordings were obtained at 33.5°C, and whole-cell recordings were obtained at room temperature.
The intracellular solution for whole-cell recordings consisted of (in mm): 123 Cs-gluconate, 4 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 2 Na-ATP, 0.3 GTP, and 0.1 spermine, pH 7.2. Spermine was added fresh to the patch solution immediately before use. Borosilicate pipettes (4–6 MΩ) were used to form gigaohm seals on CA1 pyramidal neurons under visual guidance. Voltage-clamp recordings were obtained using an Axopatch 200A amplifier (Axon Instruments, Union City, CA), and data were digitized and collected onto a computer using the PClamp6 acquisition system (Axon Instruments). Whole-cell AMPA receptor-mediated currents were evoked by bath application of kainate (Sigma), and I–V relationships were determined by subtracting responses to continuous voltage ramps (−60 to +50 mV in 1.25 sec) before kainate application from responses to the same voltage protocols during kainate application. Kainate-evoked currents were recorded with 1 μmtetrodotoxin (Calbiochem, La Jolla, CA) and 20 μm bicuculline (Sigma) in the bath to block voltage-gated Na+ channels and GABAA receptors, respectively. Slopes of the plotted I–V curves were determined by fitting straight lines over the voltage ranges of −60 to −40 mV and +10 to +30 mV, and the I–V rectification ratio was defined as the ratio of the slope measured at positive potentials to that measured at negative potentials. Rectification ratios were averaged across cells and compared between control and hypoxia-treated groups using Student'st test.
For experiments to study in vitro kindling-like epileptogenesis, field population spikes were recorded from area CA1 stratum pyramidale through glass microelectrodes filled with ACSF (1–2MΩ) using an A-M Systems 1800 AC amplifier. Schaffer collateral axons were electrically stimulated through a bipolar tungsten stimulating electrode (Fred Haer) placed in area CA3 stratum radiatum. Input–output curves were obtained by applying single pulses (0–450 μA, 0.1 msec duration) every 30 sec. High-frequency stimulation (100 Hz for 2 sec) was then applied once every 10 min using a stimulus intensity twice that which evoked the largest population spike (typically 700–800 μA). Evoked population spikes were acquired and analyzed on a computer using SCOPE (Neuropro; RC Electronics), and continuous data after tetanic stimulation were recorded using Fetchex (pCLAMP6; Axon) and analyzed using Clampfit 8.0 (Axon Instruments) and Igor Pro (Wavemetrics).
Quantification of electrically evoked afterdischarges. The numbers of population spikes evoked by each tetanic stimulus were counted by eye and compared between hypoxic and control slices using a two-way repeated measures ANOVA. For analysis of NMDA receptor-independent in vitro kindling-like epileptogenesis, the numbers of afterdischarge spikes were normalized by the number of spikes in the initial afterdischarge (response to first tetanus), and the maximum percent increase was determined. Slices were defined as showing kindling-like epileptogenesis if they exhibited an afterdischarge that was at least a 150% of the initial afterdischarge (i.e., at least a 50% increase), and Fisher's exact test was used to compare the proportion of slices from each group (control versus hypoxia treatment) that exhibited kindling-like epileptogenesis.Post hoc analyses using different criteria for defining kindling-like epileptogenesis (ranging from a 20 to 100% increase in afterdischarge) altered the proportions of slices defined as exhibiting epileptogenesis but did not alter the statistical significance.
RESULTS
Maturational and seizure-induced regulation of GluR2 mRNA expression
We first used Northern blot analysis to examine maturational and perinatal seizure-induced changes in GluR2 mRNA expression. As indicated by the representative blot in Figure1, GluR2 mRNA levels were clearly less in both hippocampus and neocortex of rats aged P10–12 compared with mature rats aged P60 (n = 5 per group), confirming previously published findings (Pellegrini-Giampietro et al., 1991;Durand and Zukin, 1993). We further analyzed neocortical and hippocampal tissue obtained from hypoxia-treated and age-matched control animals killed at 10 min and 2, 6, 24, and 48 hr after hypoxia treatment. In the hippocampi of hypoxia-treated animals, GluR2 mRNA showed a progressive, significant decline with time, to a maximum decrease to 50% of control at 48 hr after hypoxia (Fig.2; p < 0.05, ANOVA;n = 5 per group per time point). Control experiments showed no significant change in GluR1 mRNA (n = 5 per group; data not shown).
Fig. 1.

GluR2 mRNA expression was found to be relatively low during the age window of susceptibility to hypoxia-induced seizures. Shown is a representative Northern blot comparing GluR2 mRNA levels in P10–12 rat brain with those of the adult. Note the lower expression of GluR2 mRNA in both the hippocampus and neocortex of rat at ages P10 and P12 compared with adult (P60). GAPDH was used as a control and was not significantly different across lanes.
Fig. 2.
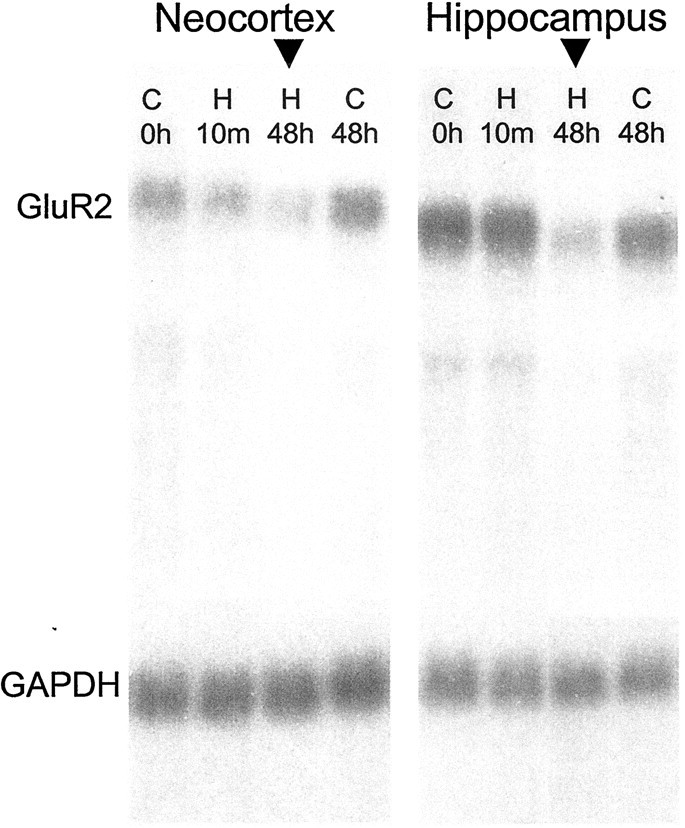
GluR2 mRNA was significantly decreased within 48 hr after hypoxia-induced seizures at P10. Shown is a representative Northern blot comparing GluR2 mRNA levels in P14 rat brain from control animals and animals that experience hypoxia-induced seizures at P10. In hippocampus, GluR2 mRNA showed a progressive, significant decline with time, to a maximum decrease to 50% of control at 48 hr after hypoxia (p < 0.05, ANOVA).
We next used in situ hybridization to localize the maturational and seizure-induced changes in GluR2 mRNA. In situ hybridization revealed clear age-dependent differences in mRNA expression in principal neurons of the hippocampus, with hippocampal pyramidal neurons showing lower GluR2 mRNA expression at P12 compared with P60 (data not shown). Semiquantitative analysis using densitometry measurements (see Materials and Methods) of sections from animals 48–96 hr after hypoxia-induced seizures showed that the seizure-induced decreases in GluR2 mRNA were most evident in principal neurons throughout the hippocampus, particularly in the CA1/CA2 pyramidal cell layer (Fig. 3). Densitometry revealed a significant decrease in GluR2 mRNA in sections from hypoxia-treated animals compared with controls (mean optical density, 79 ± 3.8% of control; p < 0.03;n = 3 control animals and 4 hypoxia-treated animals).
Fig. 3.

GluR2 mRNA was significantly decreased in the hippocampal pyramidal cell layers after perinatal hypoxia-induced seizures. In situ hybridization showed that GluR2 mRNA was significantly decreased in the pyramidal cell layers within 48 hr after hypoxia-induced seizures at P10. The most significant decrease was observed in CA1/CA2 pyramidal cells (arrows).
Maturational and seizure-induced changes in GluR2 protein expression
To determine whether the maturational and seizure-induced changes in GluR2 mRNA were associated with changes in the expression of GluR2 protein, we next used Western blot analysis. Consistent with the Northern blot data, Western blots revealed apparently less GluR2 protein expression in both hippocampus and neocortex at P10 compared with P60 (Fig. 4). Additionally, GluR2 expression was significantly decreased in both hippocampus and neocortex in hypoxia-treated animals at 96 hr after hypoxia compared with age-matched controls (Fig. 5). Compared with tissue from littermate controls, GluR2 protein was decreased by 25.4 ± 13.69% in hippocampus (n = 15; p < 0.01) and was decreased by 28 ± 8% in neocortex (n = 5; p < 0.03) at 96 hr after hypoxia-induced seizures. No significant changes were observed in GluR1 protein expression in neocortex or hippocampus.
Fig. 4.

GluR2 protein expression was found to be relatively low at an age when susceptibility to hypoxia-induced seizures is highest. Shown is a representative Western blot comparing hippocampal GluR2 protein expression in P10 rat with that in the adult (P60). GluR2 expression was clearly less in immature rat hippocampus compared with adult.
Fig. 5.

GluR2 protein expression was significantly decreased within 96 hr after hypoxia-induced seizures. Shown are Western blots comparing GluR1 and GluR2 expression. GluR2 protein expression was significantly decreased in hippocampus within 96 hr after hypoxia treatment. In contrast, GluR1 expression did not change significantly after hypoxia-induced seizures.
Decreased GluR2 expression is associated with an increase in Ca2+-permeable AMPA receptors
To determine whether lower GluR2 expression in immature rat hippocampus was indicative of a higher proportion of Ca2+-permeable AMPA receptors, we used the cobalt-staining technique (Pruss et al., 1991; see Materials and Methods). This histological technique specifically identifies neurons that express divalent-permeable AMPA receptors by visualizing AMPA-stimulated Co2+ uptake in the presence of NMDA receptor antagonists. As shown in Figure6, exposure of hippocampal slices to AMPA (100 μm) in the presence of the NMDA receptor antagonist APV (400 μm) in 2 mmCo2+-containing medium resulted in many Co2+-stained pyramidal neurons in P10 slices, with no staining of pyramidal neurons in adult slices. Additionally, Co2+ accumulation in immature slices was completely blocked by the AMPA receptor antagonist CNQX (200 μm; data not shown), indicating that the Co2+ accumulation had been dependent on the activation of AMPA receptors. These results suggested that the lower GluR2 expression in P10–12 rat hippocampus is associated with a greater number of native Ca2+-permeable AMPA receptors compared with adult.
Fig. 6.

Co2+ staining showed that the lower GluR2 expression in the immature hippocampal pyramidal cell layers was associated with increased numbers of divalent-permeable AMPA receptors. A, High-power micrograph of the CA1/CA2 pyramidal cell layer in a section of Co2+-stained P10 hippocampus; B, same for a P60 animal. Clearly, a large number of pyramidal neurons in the P10 hippocampus exhibited AMPA-induced Co2+ uptake, whereas no uptake was observed in pyramidal cells of the adult hippocampus. Scale bar, 20 μm.
To determine whether the decreased GluR2 expression after hypoxia-induced seizures resulted in increased numbers of Ca2+-permeable AMPA receptors, we performed Co2+ staining on slices obtained from same-age control and hypoxia-treated animals 96 hr after hypoxia treatment. As shown in Figure 7, AMPA-induced Co2+ uptake was observed in a greater number of neurons in area CA1 in the hippocampi of animals that experienced hypoxia-induced seizures at P10 compared with those of age-matched controls. The average number of Co2+-positive pyramidal neurons per 250 μm (see Materials and Methods) were 4.3 ± 0.8 for the control group (n = 5 animals) and 12.5 ± 2.3 for hypoxia-treated group (n = 5; p < 0.02) (Fig. 7). AMPA-induced Co2+ uptake was inhibited by NBQX (100 μm; data not shown), indicating that Co2+ uptake required activation of AMPA receptors. Furthermore, AMPA-induced Co2+ uptake was inhibited by 1-napthyl-acetyl-spermine (100 μm), a selective blocker of divalent-permeable AMPA receptor channels (Fig.7C). This indicated that the increased Co2+ accumulation was likely attributable to increased entry through Ca2+-permeable AMPA receptors. These results indicated that the decreased GluR2 expression 96 hr after hypoxia-induced seizures at P10 resulted in increased numbers of Ca2+-permeable AMPA receptors in hippocampal pyramidal neurons, particularly in area CA1.
Fig. 7.

Co2+ staining revealed increased AMPA-induced Co2+ uptake by pyramidal neurons in P14 hippocampal slices from animals that experienced hypoxia-induced seizures at P10 (B) compared with age-matched controls (A). The numbers of Co2+-positive neurons intersecting a 250 μm straight line segment drawn through the CA1 pyramidal cell layer of each section were counted and averaged (6 sections per animal). The average numbers of Co2+-positive neurons counted in this manner were 4.3 ± 0.8 for the control group (n = 5 animals) and 12.5 ± 2.3 for the hypoxia-treated group (n = 5; p< 0.02). Additionally, coapplication of the selective antagonist 1-napthyl-acetyl-spermine (100 μm) clearly inhibited AMPA-induced Co2+ uptake (C), indicating that the Co2+ entry was at least in part through divalent-permeable AMPA receptors. Scale bar, 20 μm.
Altered AMPA receptor current–voltage relationships after hypoxia-induced seizures
Recombinant AMPA receptors that lack a GluR2 subunit not only exhibit high permeability to divalent cations but also display inwardly rectifying I–V relationships (Hollman et al., 1991;Verdoorn et al., 1991). For native AMPA receptors, these properties also are correlated with decreased GluR2 mRNA expression (Bochet et al., 1994; Jonas et al., 1994; Geiger et al., 1995). Because our data thus far indicated decreased GluR2 expression and increased expression of divalent-permeable AMPA receptors in CA1 pyramidal neurons, it was expected that AMPA receptor-mediated currents in these cells also would exhibit increased inward rectification in their I–Vrelationships. To test this hypothesis, we obtained whole-cell voltage-clamp recordings from CA1 pyramidal neurons in hippocampal slices and examined the I–V relationships for kainate-evoked currents. Continuous I–V curves were generated by plotting the subtracted current responses to voltage ramps before and during bath application of 200 μmkainate as a function of the command potential (Fig.8A,B). Rectification ratios were then calculated by dividing the fitted slope of theI–V curve from +10 to +30 mV by the slope at −60 to −40 mV. Some inward rectification in the I–V curves (ratio <1) was observed in neurons from the control group, but the rectification ratios were significantly less in the hypoxia-treated group, indicating increased inward rectification in the I–V curves. The mean ± SD rectification ratios were 0.93 ± 0.14 for neurons from the control group (n = 5) and 0.72 ± 0.11 for the hypoxia-treated group (n = 8) and were significantly different (p < 0.02). Notably, the degree of rectification for both groups appeared to be intermediate to that observed for hippocampal neurons that exhibit a type I (linear or outwardly rectifying) or type II (inwardly rectifying)I–V relationship for kainate-evoked currents (Iino et al., 1990; Bochet et al., 1994).
Fig. 8.

I–V relationships for kainate-evoked currents showed increased inward rectification in CA1 pyramidal neurons from animals that experienced hypoxia-induced seizures. I–V curves were generated by subtracting the current responses to continuous voltage ramps without kainate from those with kainate in the bath. Representative subtracted currents are shown for one neuron from the control group (A) and one from the hypoxia-treated group (B). The magnitude of rectification in the I–V curves was determined by taking the ratio of the slopes fitted over the region of +20 to +40 mV to that over the region of −60 to −40 mV.C, The bar graphs indicate the mean rectification ratios determined in this manner and illustrate the increase in the magnitude of inward rectification forI–V curves obtained from the hypoxia-treated group.
Enhanced AMPA receptor-dependent epileptogenesis after hypoxia-induced seizures
In immature rat hippocampal slices, repeated high-intensity, high-frequency (tetanic) electrical stimulation of CA3 Schaffer collaterals results in progressively longer ictal-like population afterdischarges that can be recorded from area CA3 or area CA1 (Stasheff et al., 1985). This form of in vitro“kindling-like” epileptogenesis is thought to depend on Ca2+ influx through NMDA receptors (Stasheff et al., 1989), because NMDA receptor blockade prevents the progressive increase but not the expression of tetanus-induced afterdischarges. Given the above evidence for increased permeability of AMPA receptors to Ca2+, we asked whether the pathologically increased numbers of Ca2+-permeable AMPA receptors in hippocampal pyramidal neurons after hypoxia-induced seizures could mediate this form of in vitro epileptogenesis in the absence of NMDA receptor activation. To answer this question, we examined population afterdischarges in area CA1 evoked by this type of stimulation with NMDA receptors blocked in hippocampal slices obtained from animals at 4–9 d after hypoxia-induced seizures and from age-matched control animals. In all slices, tetanic stimulation applied in the presence of the NMDA receptor antagonist APV (100 μm) induced afterdischarges (Fig.9). As expected, APV prevented the increase in the number of population spikes in the afterdischarges evoked by repeated tetanic stimulation in the majority of control slices, and kindling-like epileptogenesis (defined as a 50% increase beyond the initial afterdischarge; see Materials and Methods) was observed in only 1 of 10 control slices. In contrast, increased afterdischarges were observed in the majority of slices from hypoxia-treated animals in the presence of APV, with 7 of 10 slices exhibiting NMDA receptor-independent kindling-like epileptogenesis (p < 0.05, Fisher's exact test). This finding indicated that the GluR2 downregulation and increased number of divalent-permeable AMPA receptors were correlated with an increased capacity for AMPA receptor-mediated epileptogenesis.
Fig. 9.

Repeated tetanic stimulation in the presence of the NMDA receptor antagonist APV (100 μm) revealed that hippocampal slices from rat pups that experienced hypoxia-induced seizures 4–9 d earlier (at P10) had the capacity for purely AMPA receptor-mediated epileptogenesis, whereas control slices did not. Shown are representative field recordings from a control slice and a slice from a same-age hypoxia-treated animal. Tetanus-induced afterdischarges were always observed in the presence of APV but increased with repeated stimulation only in the majority of slices (7 of 10) from hypoxia-treated animals. This type of increase was observed in only 1 of 10 control slices.
Hypoxia-induced seizures do not cause acute neuronal death in perinatal brain
Transient GluR2 knockdown in immature hippocampus was shown previously to result in both seizure-like events and hippocampal neuronal degeneration (Friedman and Koudinov, 1999). To determine whether the GluR2 decrease that resulted from hypoxia-induced seizures was associated with neuronal death, we used in situend-labeling (ISEL) to identify injured or dying cells in hippocampus after hypoxia-induced seizures. ISEL at 12, 24, 48, 72, and 96 hr and 7 d after hypoxia-induced seizures at P10 revealed no increase in the numbers of ISEL-positive cells compared with control littermates (Fig. 10; n = 3 per group per time point). Thus, perinatal hypoxia-induced seizures result in altered AMPA receptor subunit expression and function in the complete absence of cell loss.
Fig. 10.

ISEL revealed no cell injury at any point within 1 week after perinatal hypoxia-induced seizures at P10. High-power micrographs of the hippocampal CA1 region show no ISEL-positive cells at 24 hr (A), 48 hr (B), and 7 d (C) after hypoxia-induced seizures.D, Positive control hippocampal CA1 region from a rat that experienced kainate-induced status epilepticus at P45, 3 d earlier.
DISCUSSION
Altered GluR2 expression, AMPA receptor function, and hippocampal excitability after perinatal hypoxia-induced seizures
Our data indicate that perinatal hypoxia-induced seizures result in delayed changes in the molecular composition and function of AMPA receptors expressed by pyramidal neurons in the hippocampus. Specifically, we observed that hypoxia-induced seizures in the P10 rat resulted in significantly decreased GluR2 mRNA expression within 48 hr and significantly decreased GluR2 protein expression within 96 hr after hypoxia treatment. In situ hybridization showed that the decrease in GluR2 mRNA in hippocampus occurred primarily in pyramidal neurons, especially those in area CA1. GluR2-lacking AMPA receptor channels are relatively permeable to divalent cations (Jonas and Burnashev, 1995); therefore, these observations implied that perinatal hypoxia-induced seizures resulted in increased expression of divalent-permeable AMPA receptors. Staining in hippocampal slices for AMPA receptor-mediated Co2+ uptake confirmed this and further showed that increased divalent-permeable AMPA receptors were expressed in the same cell populations that exhibited decreased GluR2 mRNA. Additionally, whole-cell currents evoked by the nondesensitizing AMPA receptor agonist kainate exhibited inwardly rectifying current–voltage relationships, as are typical of AMPA receptors with high divalent permeability (Hollman et al., 1991;Verdoorn et al., 1991). Taken together, these data indicate that perinatal hypoxia-induced seizures result in delayed persistent changes in AMPA receptor composition and function that are consistent with pathologically increased AMPA receptor-mediated Ca2+ influx in principal neurons of the hippocampus.
Our data further indicate that increased AMPA receptor-mediated Ca2+ influx may contribute to the chronic epileptogenic effects of perinatal hypoxia-induced seizures. We demonstrated previously that hyperexcitability persists in hippocampal slices from adult animals after perinatal hypoxia-induced seizures (Jensen et al., 1998). Here we showed at 4–9 d after hypoxia-induced seizures that hippocampal slices exhibited purely AMPA receptor-mediated (NMDA receptor-independent) epileptogenesis in response to repeated tetanic stimulation. The prevention of thisin vitro kindling-like epileptogenesis by NMDA receptor blockade only in control slices suggested that increased Ca2+ influx through AMPA receptors in slices from hypoxia-treated animals was sufficient to trigger mechanisms of plasticity that are normally activated by Ca2+ influx through NMDA receptors. It also is possible that changes in AMPA receptor function other than increased Ca2+ permeability (see below) could have contributed to the enhancement of AMPA receptor-mediated plasticity, because the precise mechanisms that link AMPA receptor activation to epileptogenesis in this model have yet to be identified. Nonetheless, our data indicate that seizure-induced changes in AMPA receptor permeation properties are associated with increased hippocampal excitability and may contribute to the long-term epileptogenic effects of perinatal hypoxia (Jensen et al., 1991,1992).
Maturational regulation of GluR2 expression, AMPA receptor function, and susceptibility to hypoxia-induced seizures
Our previous work demonstrated that the susceptibility to hypoxia-induced seizures is only observed in rats during a narrow age window of P10–12 (Jensen et al., 1991). Additionally, we previously observed that hypoxia-induced seizures at this age are resistant to antiepileptic compounds that augment GABA receptor function or inhibit NMDA receptor function but are blocked by systemically tolerated doses of the AMPA receptor antagonist NBQX (Jensen et al., 1995). Notably, AMPA receptor density as revealed by binding studies in rat peaks at approximately P10 before gradually declining to adult levels (Insel et al., 1990). These findings suggested a key role for the maturational state of AMPA receptor expression and function in the generation of hypoxia-induced seizures.
Our present data indicate that GluR2 expression is developmentally regulated such that expression is relatively low at approximately P10 compared with adulthood in Long–Evans rats, consistent with previous reports in other rat strains (Pellegrini-Giampietro et al., 1992;Durand and Zukin, 1993; Monyer et al., 1994). These observations implied that higher proportions of neurons in the immature brain express Ca2+-permeable AMPA receptors compared with the adult brain, and we now have confirmed this by demonstrating increased numbers of neurons labeled positively for AMPA-induced Co2+ uptake in P10 hippocampal slices compared with adult slices. The physiological significance of this developmental difference is not yet known. Ca2+ entry via AMPA receptors has been shown to mediate various forms of synaptic plasticity in spinal cord dorsal horn neurons (Gu et al., 1996), cerebellar stellate cells (Liu and Cull-Candy, 2000), and hippocampal interneurons (Laezza et al., 1999). Thus, the higher numbers of Ca2+-permeable AMPA receptors in immature hippocampal pyramidal neurons suggest that AMPA receptor-mediated plasticity in principal hippocampal networks may have an important yet unknown physiological role in early brain development. Given that the susceptibility to hypoxia-induced seizures occurs specifically at a developmental stage in which the numbers of Ca2+-permeable AMPA receptors are relatively high and AMPA receptors are expressed at high levels (Insel et al., 1990), a consequence of increased AMPA receptor-mediated plasticity may be to increase susceptibility to hypoxia-induced seizures and epileptogenesis.
Role of altered AMPA receptor subunit composition in epileptogenesis
Our data indicate that the decrease in GluR2 expression by maturation or hypoxia-induced seizures is associated with an increase in the permeability of AMPA receptors to divalent cations. Additionally, our data indicate that the seizure-induced switch in AMPA receptor composition and function is associated with an increased capacity for AMPA receptor-mediated epileptogenesis in hippocampus, because in vitro kindling-like epileptogenesis was not blocked by an NMDA receptor antagonist in slices from hypoxia-treated animals. The dependence of this form of in vitroepileptogenesis on NMDA receptor activation in control slices in this and previous studies (Stasheff et al., 1989; Stasheff et al., 1993) suggested that the plasticity that underlies this form of epileptogenesis is mediated by glutamate-stimulated Ca2+ entry, as is generally the case for NMDA receptor-mediated plasticity (Malenka and Nicoll, 1993). Kindling-like epileptogenesis of slices from hypoxia-treated animals in the absence of NMDA receptor activation therefore suggested that the increased AMPA receptor-mediated Ca2+entry was sufficient to trigger mechanisms of plasticity and epileptogenesis that are normally triggered by NMDA receptor-mediated Ca2+ entry in this model.
It is important to note, however, that AMPA receptors that lack the GluR2 subunit may differ from GluR2-containing AMPA receptors in ways other than just their permeability characteristics. For example, GluR2 subunits show specific interactions with certain postsynaptic density elements such asN-ethylmaleimide-sensitive fusion protein (Osten et al., 1998; Song et al., 1998; Luthi et al., 1999), glutamate receptor-interacting protein (Li et al., 1999), and AMPA receptor-binding protein (Srivastava et al., 1998). These subunit-specific interactions may generate additional differences in the localization, function, and modulation of GluR2-containing versus GluR2-lacking AMPA receptors. Furthermore, studies of recombinant heteromeric AMPA receptors suggest that GluR2-containing AMPA receptors are likely to exhibit significantly lower channel conductances compared with those lacking a GluR2 subunit (Swanson et al., 1997). Thus, the alteration in AMPA receptor function after decreased GluR2 expression that critically contributes to enhancing epileptogenesis may yet be independent of a change in permeation properties or may depend on a combination of functional alterations.
Additionally, although our previous work indicated that animals that experienced hypoxia-induced seizures at P10 exhibit decreased seizure thresholds into adulthood (Jensen et al., 1992), we do not yet know the complete time course of changes in hippocampal GluR2 expression. Preliminary long-term experiments suggest that the seizure-induced GluR2 downregulation may not be long-lasting. During development, the susceptibility to hypoxia-induced seizures is highest at a time of heightened synaptic plasticity (Swann et al., 1999). It is possible that GluR2 downregulation at this time could prolong this period of heightened plasticity and enable the establishment of long-lasting aberrant synaptic connections. Thus, long-term epileptogenic changes in hippocampal synaptic function could conceivably arise from even a temporary decrease in GluR2 expression during early maturation.
Lack of cell death associated with decreased GluR2 after perinatal hypoxia-induced seizures
Pathological decreases in GluR2 expression have been proposed to underlie neuronal injury and death in a variety of situations (Weiss and Sensi, 2000). Our present data are the first to demonstrate seizure-induced GluR2 downregulation and subsequent enhanced hippocampal excitability in the complete absence of neuronal death or injury. This lack of injury may be attributable to maturational factors, because the immature brain is relatively resistant to seizure-induced injury compared with the adult (Sperber, 1996; Holmes, 1997). Significant neuronal degeneration in immature hippocampus was induced by antisense knockdown of GluR2 (Friedman and Koudinov, 1999), but the magnitude of the decrease in GluR2 expression induced in this manner appeared to be much greater than that observed after perinatal hypoxia-induced seizures. This lack of injury may also have been attributable to the lesser degree of seizure severity in our model. Other seizure models in which GluR2 downregulation has been demonstrated, such as after kainate-induced status epilepticus (Pollard et al., 1993; Friedman et al., 1994), involve much more prolonged and repeated seizures compared with those induced by perinatal hypoxia. Regardless of mechanism, the long-term pro-epileptic changes in brain function that result from perinatal hypoxia-induced seizures represent a form of pure epileptogenesis without compensatory responses to cell loss; therefore, the associated decrease in GluR2 expression and alterations in AMPA receptor function strongly argue for their potential contribution to epileptogenesis in the immature brain. The mechanism of injury after GluR2 downregulation has been postulated to be an overload of AMPA receptor-mediated Ca2+ entry into neurons that do not have adequate Ca2+-buffering mechanisms (Weiss and Sensi, 2000; Pellegrini-Giampietro et al., 1997).
Summary
Our findings indicate that the dynamic expression of the AMPA receptor subunit GluR2 is inversely correlated with the expression of Ca2+-permeable AMPA receptors in hippocampus. Our data further confirm that GluR2 expression is regulated by maturation such that the susceptibility to hypoxia-induced seizures is highest when the total expression of AMPA receptors is high and the expression of GluR2 is relatively low. Furthermore, perinatal hypoxia-induced seizures result in further decreased GluR2 expression that is associated with altered AMPA receptor properties and increased divalent permeability. The changes in AMPA receptor function are further associated with enhanced NMDA receptor-independent epileptogenesis in immature hippocampus. Because perinatal hypoxia-induced seizures do not result in hippocampal neurodegeneration, this decreased GluR2 expression and enhanced epileptogenesis cannot be compensatory responses to cell loss; therefore, GluR2 downregulation is associated only with hippocampal epileptogenesis in this setting. Although increased AMPA receptor-mediated Ca2+ influx is the most likely mechanism contributing to enhanced epileptogenesis after GluR2 downregulation, other functional properties of GluR2-deficient AMPA receptors may contribute as well. Identification of the mechanisms by which GluR2 expression is downregulated after perinatal seizures and the specific functional changes that contribute to epileptogenesis in the developing brain may lead to more specific strategies to treat neonatal hypoxic encephalopathy to minimize the risk of epilepsy.
Footnotes
This research was supported by National Research Service Award AG00222 from the National Institute on Aging (R.M.S.), by an Epilepsy Foundation of America research training fellowship (R.M.S.), by National Institutes of Health Grants K08 NS02068 (S.K.) and R01 NS31718 (F.E.J.) from the National Institute on Neurological Disorders and Stroke, by grants from the Klingenstein Foundation (G.C.) and the EJLB Foundation (G.C.), and by Children's Hospital Mental Retardation Research Center Grant P30 HD18655 from the National Institute of Child Health and Human Development. We thank Dr. Robert J. Wenthold for generously providing glutamate receptor antibodies. We also thank Matthew Corbett, David Hershberger, Elijah Owens, and Jorge Benzecry for contributing to this work.
R.M.S. and S.K. contributed equally to this work.
Correspondence should be addressed to Dr. Frances E. Jensen, Division of Neuroscience, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115. E-mail: jensen@a1.tch.harvard.edu.
REFERENCES
- 1.Bochet P, Audinat E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 2.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 3.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 4.Durand GM, Zukin RS. Developmental regulation of RNAs encoding rat brain kainate/AMPA receptors: a Northern analysis study. J Neurochem. 1993;61:2239–2246. doi: 10.1111/j.1471-4159.1993.tb07465.x. [DOI] [PubMed] [Google Scholar]
- 5.Friedman LK, Koudinov AR. Unilateral GluR2(B) hippocampal knockdown: a novel partial seizure model in the developing rat. J Neurosci. 1999;19:9412–9425. doi: 10.1523/JNEUROSCI.19-21-09412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennet MVL, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 8.Gu JG, Albuquerque CJ, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- 9.Hollman M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 10.Hollman M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 11.Holmes GL. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia. 1997;38:12–30. doi: 10.1111/j.1528-1157.1997.tb01074.x. [DOI] [PubMed] [Google Scholar]
- 12.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. J Physiol (Lond) 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Insel TR, Miller LP, Gelhard RE. The ontogeny of excitatory amino acid receptors in the rat forebrain I: N-methyl-d-aspartate and quisqualate receptors. Neuroscience. 1990;35:31–43. doi: 10.1016/0306-4522(90)90117-m. [DOI] [PubMed] [Google Scholar]
- 14.Jensen FE, Applegate CD, Holtzman D, Belin T, Burchfiel J. Epileptogenic effects of hypoxia on immature rodent brain. Ann Neurol. 1991;29:629–637. doi: 10.1002/ana.410290610. [DOI] [PubMed] [Google Scholar]
- 15.Jensen FE, Holmes GH, Lombroso CT, Blume H, Firkusny I. Age dependent long term changes in seizure susceptibility and neurobehavior following hypoxia in the rat. Epilepsia. 1992;33:971–980. doi: 10.1111/j.1528-1157.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- 16.Jensen FE, Alvarado S, Firkusny IR, Geary C. NBQX blocks the acute and late epileptogenic effects of perinatal hypoxia. Epilepsia. 1995;36:966–972. doi: 10.1111/j.1528-1157.1995.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 17.Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC. Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol. 1998;79:73–81. doi: 10.1152/jn.1998.79.1.73. [DOI] [PubMed] [Google Scholar]
- 18.Jonas P, Burnashev N. Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron. 1995;15:987–990. doi: 10.1016/0896-6273(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 19.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 20.Laezza F, Doherty JJ, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- 21.Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- 22.Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- 23.Luthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24:389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 24.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 25.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 26.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 27.Pellegrini-Giampietro DE, Bennett MVL, Zukin RS. Differential expression of three glutamate receptor genes in developing rat brain: an in situ hybridization study. Proc Natl Acad Sci USA. 1991;88:4157–4161. doi: 10.1073/pnas.88.10.4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellegrini-Giampietro DE, Bennett MVL, Zukin RS. Are Ca2+-permeable kainate/AMPA receptors more abundant in immature brain? Neurosci Lett. 1992;144:65–69. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- 29.Pellegrini-Giampietro DE, Gorter JA, Bennett MVL, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- 30.Pollard H, Heron A, Moreau J, Ben-Ari Y, Khrestchatisky M. Alteration of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 31.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation permeable kainate receptors on neurons and glia. Neuron. 1991;7:509–518. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez RM, Wang C, Gardner G, Orlando L, Tauck DL, Rosenberg PA, Aizenman E, Jensen FE. Novel role for the NMDA receptor redox modulatory site in the pathophysiology of seizures. J Neurosci. 2000;20:2409–2417. doi: 10.1523/JNEUROSCI.20-06-02409.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 34.Sperber EF. The relationship between seizures and damage in the maturing brain. Epilepsy Res. 1996;S12:365–376. [PubMed] [Google Scholar]
- 35.Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21:581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- 36.Stasheff SF, Bragdon AC, Wilson WA. Induction of epileptiform activity in hippocampal slices by trains of electrical stimuli. Brain Res. 1985;344:296–302. doi: 10.1016/0006-8993(85)90807-8. [DOI] [PubMed] [Google Scholar]
- 37.Stasheff SF, Anderson WW, Clark S, Wilson WA. NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science. 1989;245:648–651. doi: 10.1126/science.2569762. [DOI] [PubMed] [Google Scholar]
- 38.Stasheff SF, Mott D, Wilson WA. Axon terminal hyperexcitability associated with epileptogenesis in vitro. II. Pharmacological regulation by NMDA and GABAA receptors. J Neurophysiol. 1993;70:976–984. doi: 10.1152/jn.1993.70.3.976. [DOI] [PubMed] [Google Scholar]
- 39.Swann JW, Pierson MG, Smith KL, Lee CL. Developmental neuroplasticity: roles in early life seizures and chronic epilepsy. Adv Neurol. 1999;79:203–216. [PubMed] [Google Scholar]
- 40.Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tolentino PJ, Dikkes P, Tsuruda L, Ebert K, Fink JS, Villa-Komaroff L, Lamperti ED. Quantitative analysis of the expression of a VIP transgene. Brain Res Mol Brain Res. 1995;33:47–60. doi: 10.1016/0169-328x(95)00105-2. [DOI] [PubMed] [Google Scholar]
- 42.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 43.Volpe JJ. Neurology of the newborn. Saunders; Philadelphia: 2000. Neonatal seizures. pp. 178–214. [Google Scholar]
- 44.Weiss JH, Sensi SL. Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci. 2000;23:365–371. doi: 10.1016/s0166-2236(00)01610-6. [DOI] [PubMed] [Google Scholar]
- 45.Weiss S, Cataltepe O, Cole AJ. Anatomical studies of DNA fragmentation in rat brain after systemic kainate administration. Neuroscience. 1996;74:541–551. doi: 10.1016/0306-4522(96)00148-0. [DOI] [PubMed] [Google Scholar]
- 46.Wenthold RJ, Yokotani N, Doi K, Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies. J Biol Chem. 1992;267:501–507. [PubMed] [Google Scholar]
- 47.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH. A new method to detect apoptosis in paraffin section: in situ end-labeling of fragmented DNA. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]