

Abstract
We generated mitochondrial late-onset neurodegeneration (MILON) mice with postnatal disruption of oxidative phosphorylation in forebrain neurons. They develop normally and display no overt behavioral disturbances or histological changes during the first 5 months of life. The MILON mice display reduced levels of mitochondrial DNA and mitochondrial RNA from 2 and 4 months of age, respectively, and severely respiratory chain-deficient neurons from 4 months of age. Surprisingly, these respiratory chain-deficient neurons are viable for at least 1 month without showing signs of neurodegeneration or major induction of defenses against oxidative stress. Prolonged neuronal respiratory chain deficiency is thus required for the induction of neurodegeneration. Before developing neurological symptoms, MILON mice show increased vulnerability to excitotoxic stress. We observed a markedly enhanced sensitivity to excitotoxic challenge, manifest as an abundance of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reactive cells after kainic acid injection, in 4-month-old MILON mice, showing that respiratory chain-deficient neurons are more vulnerable to stress. At ∼5–5.5 months of age, MILON mice start to show signs of disease, followed by death shortly thereafter. The debut of overt disease in MILON mice coincides with onset of rapidly progressive neurodegeneration and massive cell death in hippocampus and neocortex. This profound neurodegenerative process is manifested as axonal degeneration, gliosis, and abundant TUNEL-positive nuclei. The MILON mouse model provides a novel and powerful tool for additional studies of the role for respiratory chain deficiency in neurodegeneration and aging.
Keywords: mitochondria, respiratory chain, reactive oxygen species, ROS, neurodegeneration, oxidative stress, apoptosis, oxidative phosphorylation, OXPHOS, cre-loxP, Tfam, mitochondrial transcription factor A, MILON, neocortex, hippocampus
Deficient function of the mitochondrial respiratory chain is increasingly recognized as an important cause of neurodegeneration (Larsson and Clayton, 1995;Larsson and Luft, 1999; Wallace, 1999). The respiratory chain is located in the mitochondrial inner membrane, and it generates most of the cellular ATP through the process of oxidative phosphorylation. The respiratory chain consists of ∼100 different polypeptides distributed in five enzyme complexes, which are encoded by either nuclear or mitochondrial genes. The mitochondrial DNA (mtDNA) encodes 13 subunits of respiratory chain complexes I, III, IV, and V, whereas nuclear DNA encodes all of the remaining respiratory chain proteins and all proteins needed for maintenance and expression of mtDNA. More than 50 point mutations and hundreds of rearrangements of mtDNA, as well as a variety of nuclear gene mutations, have been found in patients with mitochondrial disease (Larsson and Clayton, 1995; Larsson and Luft, 1999; Wallace, 1999). Mitochondrial dysfunction has also been implicated in the pathogenesis of age-associated neurodegenerative diseases, such as Parkinson's, Huntington's, and Alzheimer's diseases, as well as in naturally occurring aging (Wallace, 1992). Somatic mtDNA mutagenesis has been proposed to contribute to the aging process by creating a vicious circle, in which such mutations impair the synthesis of respiratory chain subunits and thus reduce respiratory chain function, leading to increased production of reactive oxygen species (ROS), which, in turn, additionally damages mtDNA.
We developed a system for tissue-specific inactivation of mtDNA expression in the mouse by conditional knock-out of the nuclear mitochondrial transcription factor A gene (Tfam), which encodes a mitochondrial protein necessary for transcription and replication of mtDNA (Larsson et al., 1998). Previous reports from our laboratory have established that animals homozygous for aloxP-flanked Tfam allele (TfamloxP/TfamloxP), which also harbor a transgene directing tissue-specific expression of bacteriophage P1 recombinase (cre), will develop a profound tissue-specific mtDNA depletion and respiratory chain deficiency (Wang et al., 1999; Li et al., 2000; Silva et al., 2000). We demonstrated recently that decreased mtDNA expression in vivo is associated with induction of antioxidant defenses and increased apoptosis in Tfam knock-out mouse hearts (Wang et al., 2001). There is a connection between respiratory chain deficiency, ROS production, and apoptosis induction (Geromel et al., 2001), but the molecular pathways are poorly understood. Correlative data suggest that these processes are linked by unknown pathways and are involved in the pathophysiology of various types of age-associated neurodegeneration (Wallace, 1992). We now investigated this issue further by postnatal disruption of Tfam in neurons of hippocampus and neocortex to create mitochondrial late-onset neurodegeneration (MILON) mice.
MATERIALS AND METHODS
Matings and genotyping of transgenic animals. TfamloxP/TfamloxPmice of mixed genetic background (Larsson et al., 1998; Wang et al., 1999) were mated to +/CaMKII-cre mice (Xu et al., 2000). Compound heterozygotes (+/TfamloxP, +/CaMKII-cre) were identified and backcrossed to theTfamloxP/TfamloxPstrain to generate MILON mice with the genotypeTfamloxP/TfamloxP,+/CaMKII-cre. This mating gave rise to normal litter sizes (mean litter size, 10.3 pups) with genotypes of the expected Mendelian distribution (of 542 animals, the distribution was as follows:TfamloxP/TfamloxP, 25.5%; +/TfamloxP, +/CaMKII-cre, 26.6%; MILON, 23.2%; and +/TfamloxP, 24.7%). The Tfamgenotype and the cre recombinase gene were identified at weaning by PCR analyses (Larsson et al., 1998; Wang et al., 1999). Animal studies were approved by the animal welfare ethics committee and performed in compliance with Swedish law.
Southern, Northern, and Western blot analyses. Southern, Northern, and Western blot analyses were performed as described previously (Larsson et al., 1996). Phosphoimaging (BAS 1500; Fujifilm, Toyko, Japan), and appropriate software (Image Gauge v3.41; Fujifilm) were used to measure relative levels of mtDNA and mitochondrial RNA (mtRNA) in brain samples of neocortex and cerebellum fromTfamloxP/TfamloxP; +/TfamloxP, +/CaMKII-cre and MILON animals of ages 1, 2, 4, and 6 months (n = 4–6). The mtDNA levels were normalized to the nuclear 18S rRNA gene, whereas the complete smear of mtRNA was normalized to 18S rRNA transcript levels. Southern blots were also probed with Tfam cDNA to calculate the relative Tfam gene recombination frequency, as described previously (Wang et al., 1999). For gene expression studies, RNA from neocortex was extracted and Northern blots were performed as described previously (Wang et al., 2001). One-sided, unpairedt tests were used to assess statistical significance. Western blot analyses of Tfam protein levels were performed on total protein extracts from neocortex of MILON mice andTfamloxP/TfamloxPlittermate controls at the age of 2, 3, 4, and 5 months as described previously (Larsson et al., 1996).
In situ hybridization. Probes against the mitochondrial transcripts cytochrome c oxidase subunit I (5′-TGGGT CCCCT CCTCC AGCGG GATCA AAGAA AGTTG TGTTT AGGTT GCGG-3′) and nicotineamide adenine dinucleotide (NADH) dehydrogenase subunit 4 (5′-CCATT TGAAG TCCTC GGGCC ATGAT TATAG TACGG CTGTG GATCC GTTCG-3′) were 3′-end labeled with35S and used to detect mtRNA in 14 μm cryostat sections from fresh frozen brains by in situhybridization (Dagerlind et al., 1992). A random oligonucleotide probe was used as a negative control. Hybridizations were performed at 42°C for 16–18 hr, and sections were rinsed five times in 1× SSC, dehydrated, and exposed to photographic emulsion (Kodak NTB2; Eastman Kodak, Rochester, NY) for 4 d.
Biochemistry and enzyme histochemistry. The activities of the respiratory chain complexes were measured in fresh frozen samples of neocortical and hippocampal tissue from MILON andTfamloxP/TfamloxPcontrol mice at 2, 4, and 5 months of age (n = 4) as described previously (Rustin et al., 1994). Glutathione peroxidase (Gpx) and mitochondrial superoxide dismutase (Sod2) activities were determined as described previously (Wang et al., 2001). Enzyme histochemical analyses of succinate dehydrogenase (SDH) and cytochrome c oxidase (COX) activities were performed on 14 μm cryostat sections of fresh frozen brains of MILON and control mice as described previously (Wang et al., 1999).
Histochemistry and immunohistochemistry. MILON andTfamloxP/TfamloxPlittermate controls were perfused with Ca2+-free Tyrode's solution, followed by 4% paraformaldehyde with 0.4% picric acid in 0.16m phosphate buffer. The brains were dissected out, post-fixed overnight, and equilibrated to 10% sucrose containing 0.1% sodium azide. Primary antibodies used for indirect immunohistochemistry (Hökfelt et al., 1973; Zetterström et al., 1994) included polyclonal antibodies against neurofilament-10 (NF-10) (1:100; Sigma, St. Louis, MO), glial fibrillary acidic protein 19 (GFAP-19) (1:500; Sigma), von Willebrand factor (1:300; Dako, Glostrup, Denmark), cleaved caspase 3 (1:100; Cell Signaling Technology, Beverly, MA), cleaved caspase 7 (1:100; Cell Signaling Technology), and nitrotyrosine (1:50; Cell Signaling Technology). Cryostat sections (14 μm) were incubated with primary antibodies overnight at 4°C, rinsed, and incubated with appropriate FITC-labeled secondary antibodies. Sections were analyzed by fluorescence microscopy. Proliferating cell nuclear antigen (PCNA) expression was studied with a monoclonal antibody (1:50; Dako). Brain sections were permeabilized with ice-cold ethanol/acetic acid (1:2) and blocked (M.O.M Immunodetection kit; Vector Laboratories, Burlingame, CA) before application of specific antibodies. Peroxidase activity was detected with 3,3′-diaminobenzidine (DAB substrate kit for peroxidase; Vector Laboratories), and the sections were counterstained with methyl green (Dako). Controls included omitting the primary antibody.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling and DNA ladder gel electrophoresis. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) (Apoptag in situ Apoptosis Detection kit; Intergen, Purchase, NY) was performed according to the instructions of the manufacturer. DNA from neocortex and hippocampus was extracted, and the DNA ladder assay performed as described previously (Wang et al., 2001). We also used a kit (ApoAlert LM-PCR Ladder Assay; Clontech, Palo Alto, CA) for detection of DNA ladders in samples containing small fractions of apoptotic cells following the instructions of the manufacturer.
Kainic acid induction of seizures. Kainic acid was dissolved in Ringer's isotonic saline (pH 6) and administered intraperitoneally at a dose of 20 or 30 mg/kg body weight in MILON and control mice at the age of 4 months. Mice were monitored continuously for 2–3 hr after injection to determine the onset and level of seizures. Seizure levels were rated (Sperk et al., 1985): level 1, immobility; level 2, forelimb and/or tail extension, rigid posture; level 3, repetitive movements, head bobbing; level 4, rearing; and level 5, rearing and falling. Mice injected with saline were included as negative controls. Brains were collected 24 hr after seizure onset.
RESULTS
Forebrain-specific disruption of the Tfam gene
We generated mice with disruption of Tfam in forebrain neurons by matingTfamloxP/TfamloxPmice (Wang et al., 1999) to mice heterozygous for a transgene expressing cre recombinase from the calcium-dependent calmodulin kinase II promoter, +/CaMKII-cre (Xu et al., 2000). The resulting MILON mice (genotypeTfamloxP/TfamloxP,+/CaMKII-cre) displayed a highly tissue-specific pattern ofTfam knock-out as determined by PCR (Fig.1a) and Southern blot analyses (data not shown). The knock-out (Tfam−) allele was present in neocortex but, with the exception of testis, not in other tissues, including cerebellum (Fig. 1a). TheCaMKII-cre transgenic mouse used in this study has been shown previously to express cre recombinase from postnatal day 14 (P14), and maximal recombination of loxP-flanked alleles was observed at P29 in neocortex, hippocampus, and some other regions (Xu et al., 2000). The CaMKII-cre transgene causes recombination in ∼50% of neocortical and hippocampal neurons harboring a loxP-flanked allele (Xu et al., 2000). Consistent with these observations, we found that the levels ofTfam− alleles were equal in 1-month-old (relative level, 24 ± 3%) and 5-month-old (25 ± 2%) presymptomatic MILON mice, demonstrating that maximal Tfamrecombination was obtained by 1 month after birth. Tfam protein levels were clearly reduced in neocortex of MILON mice from 2 months of age and throughout life as shown by Western blot analysis (Fig.1b). The MILON mice appeared normal and showed no overt behavioral abnormalities until the age of 5–6 months, when they started to display signs of deteriorating physical condition progressing rapidly until death within 1–2 weeks. There was thus a surprisingly long time span of ∼4–5 months between the completedTfam recombination and obvious signs of disease in the MILON mice.
Fig. 1.

Characterization of Tfamrecombination, Tfam protein expression, mtDNA copy number, and mtRNA levels in MILON mice and controls.a, PCR analysis to determine the tissue-specific recombination pattern in a 2-month-old MILON mouse. TheTfam knock-out allele (Tfam−) is only present in neocortex and testis. b, Western blot analyses of cortical protein extracts from MILON (M;TfamloxP/TfamloxP,+/CaMKII-cre) and control (C;TfamloxP/TfamloxP) animals. Tfam protein is detected with a polyclonal anti-mouse Tfam antibody, whereas a monoclonal anti-actin antibody is used as a loading control. The Tfam protein levels are notably decreased in the MILON mice from 2 months of age. c, Results from phosphoimager quantification of Southern blots to determine mtDNA copy number in neocortex of control (C;black bars), heterozygous knock-out (H; +/TfamloxP,+/CaMKII-cre; gray bars), and MILON (M; white bars) mice. All values are normalized to the nuclear 18S rRNA gene and depicted as the percentage of the mean value of age-matched controls (n = 4–6 pairs of MILON and control mice). *p < 0.05; **p < 0.01. The levels of mtDNA are significantly decreased in MILON mice from 2 months of age. d, Results from phosphoimager quantification of Northern blots to determine mtRNA levels in neocortex of control (C; black bars), heterozygous knock-out (H; gray bars), and MILON (M; white bars) mice. All values are normalized to 18S rRNA and depicted as the percentage of the mean value of age-matched controls (n = 4–6 pairs of MILON and control mice). **p < 0.01; ***p < 0.001. The levels of mtRNA are significantly decreased in MILON mice from 4 months of age.
Reduced mtDNA expression in neocortex and hippocampus of MILON mice
There was an ∼40% reduction of mtDNA copy number and mtRNA levels in neocortex at 2 and 4 months of age, respectively, and onward (Fig. 1c,d). Furthermore, in situhybridizations showed a drastic decrease in mtRNA levels in neocortex and hippocampus, especially in the CA1 and CA3 regions (Franklin and Paxinos, 1997) at age 4 months (Fig.2b). The CA2 region of hippocampus and the polymorphic layer of the dentate gyrus displayed no evident reduction of mtRNA levels. Analyses of respiratory chain enzyme activities in neocortical samples demonstrated decreased activities of NADH dehydrogenase (complex I) and COX (complex IV), which both contain critical mtDNA-encoded subunits, in 4- and 5-month-old MILON mice (Fig.2a). The activity of SDH (complex II), which is exclusively nucleus encoded, remained unchanged in MILON mice (Fig. 2a). Severe respiratory chain deficiency in individual cells was demonstrated by enzyme histochemistry to determine COX and SDH activities on brain tissue sections. Hippocampus, piriform cortex, and amygdala in 4-month-old MILON mice contained many COX-deficient cells. In hippocampus, the CA3 region was most affected with massive amounts of COX-deficient cells, but also CA1 and the dentate gyrus contained numerous COX-deficient cells (Fig. 2b). A large number of COX-deficient cells were also seen throughout neocortex and were particularly abundant in cingulum cortex of 4-month-old MILON mice (data not shown).
Fig. 2.
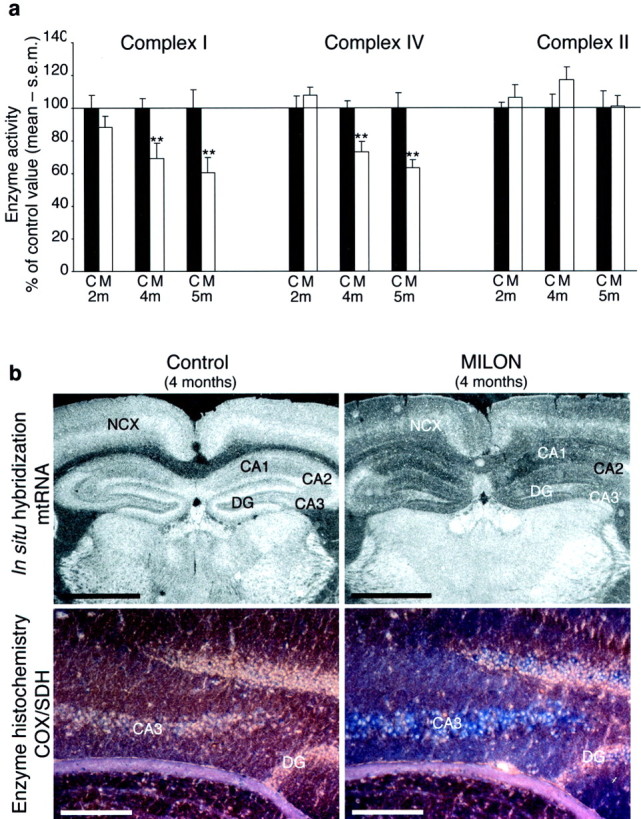
Biochemical and histochemical assessment of respiratory chain function and in situ hybridization to detect mtRNA. a, Enzyme activities of respiratory chain complexes I, II, and IV in neocortex from 2-, 4-, and 5-month-old control (C; black bars) and MILON (M; white bars) mice. Values are depicted as the percentage of the mean value of age-matched controls. **p < 0.01. The activity of complexes I and IV are significantly decreased in MILON mice from 4 months of age.b, Top row, In situhybridization to detect the mtDNA-encoded COXI transcript (mtRNA) in brain from a 4-month-old presymptomatic MILON mouse and a littermate control. Scale bars, 1 mm. NCX, Neocortex;DG, dentate gyrus. The levels of mtRNA are decreased in neocortex and hippocampus in MILON mice. Bottom row, Enzyme histochemical double staining for COX and SDH activities. Cells with COX and SDH activity appear brown, whereas cells with deficient COX activity appear blue. Scale bars, 0.2 mm.
It should be emphasized that the CaMKII-cre transgene causes recombination of loxP-flanked alleles in neurons but not in the other cell types of neocortex and hippocampus (Xu et al., 2000). It is thus likely that the moderate reduction of respiratory chain function (Fig. 2a) in tissue extracts of neocortex, containing many different cell types, is explained by the profound COX deficiency present in individual Tfam knock-out neurons (Fig. 2b).
Corticohippocampal nerve cell loss and gliosis in MILON mice
Next, we investigated the neuropathological consequences of respiratory chain deficiency in forebrain neurons. Early-stage (age 5–5.5 months) and end-stage (age 5.5–6 months) symptomatic MILON mice displayed a progressive marked nerve cell loss in neocortex and hippocampus (Fig. 3). There was substantial degeneration in neocortex and a severe disruption of cortical organization in end-stage MILON mice as determined by cresyl violet staining (Fig. 3). Early-stage symptomatic MILON mice displayed a substantial nerve cell loss, as well as a cellular infiltration indicative of an inflammatory response in the CA1 region of hippocampus (Fig. 3). This was further supported by the observation of macrophages, including characteristic gitter cells. Other hippocampal areas appeared normal in these animals. In end-stage animals, the pyramidal cell layer of the medial part of CA1 was completely absent, CA2 was intact, and CA3 only slightly affected. Although the granule cell layer of the dentate gyrus was present, many of the nuclei appeared condensed. Such pyknotic nuclei, which could be consistent with apoptosis, were found in all affected areas of the hippocampal formation.
Fig. 3.
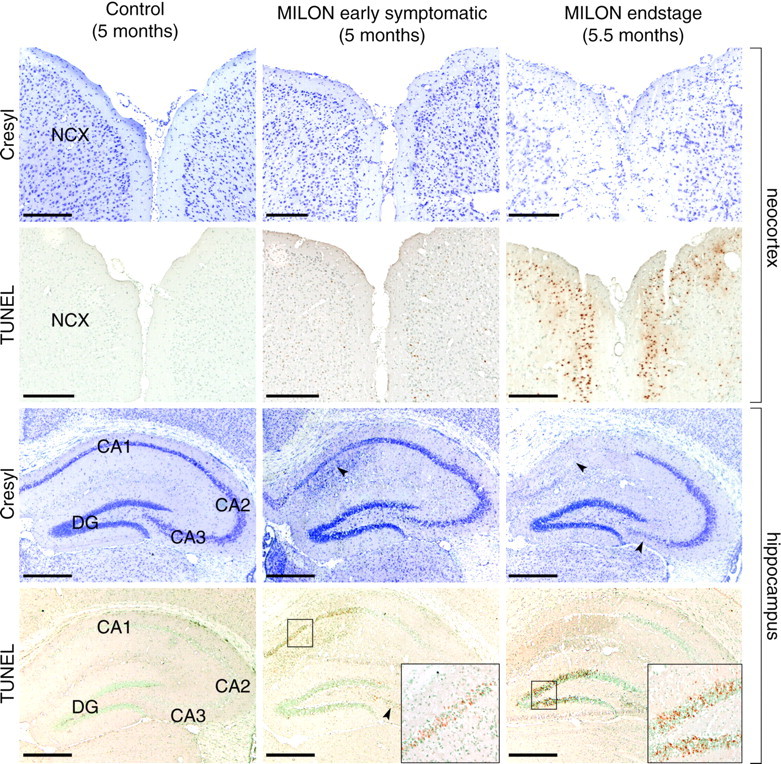
Characterization of cell loss in neocortex and hippocampus of a 5-month-old control mouse, a 5-month-old early symptomatic MILON mouse, and a 5.5-month-old end-stage MILON mouse stained with cresyl violet or TUNEL. NCX, Neocortex;DG, dentate gyrus The arrowheads indicate cell loss in the CA1 and CA3 regions. The boxes are close-ups of indicated areas. Scale bars, 0.5 mm. There is cell loss and cell infiltration in the CA1 area of hippocampus of early symptomatic MILON mice. There is extensive cell loss and degenerative changes in neocortex and in the CA1, CA3, and dentate gyrus areas of the hippocampal formation of end-stage MILON mice.
We used the cell cycle S-phase marker PCNA to detect dividing cells. Numerous PCNA-positive cells were found in hippocampus, especially in early-stage symptomatic MILON animals, and in neocortex (data not shown). The majority of these dividing cells in hippocampus were glia or inflammatory response cells, as judged from morphological appearance. Some labeled cells were found clustered in the subgranular zone of the dentate gyrus, indicative of increased neurogenesis.
Immunohistochemistry further supported the presence of neurodegeneration and an inflammatory response in end-stage MILON mice (Fig. 4). Neurofilament (NF-10) immunohistochemistry showed extensive axonal degeneration, as evidenced by increased axonal beading and fragmentation in both early- and end-stage symptomatic MILON mice. Axonal degeneration was unevenly distributed in neocortex, being more pronounced anteriorly and also increasing with the progression of disease. Early-stage animals showed signs of axonal degeneration in hippocampus, as well as neocortex. In end-stage animals having lost the medial pyramidal cell layer of CA1, axonal degeneration was no longer noted in this area of hippocampus, but there was neurofilament accumulation in some granular cell somata of the dentate gyrus. This was probably the result of a loss of axonal connections to CA3.
Fig. 4.
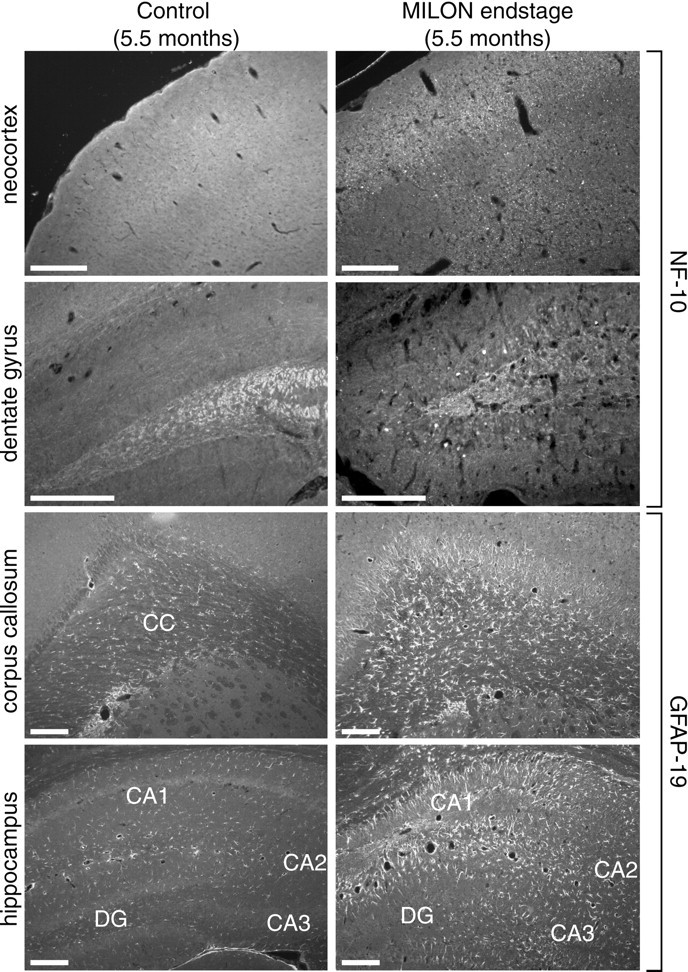
Characterization of neurodegenerative changes and gliosis in a MILON and a control mouse at the age of 5.5 months. Neurofilaments were detected with a polyclonal antibody against NF-10, and astrocytes were detected with a polyclonal antibody against GFAP. Antibody reactivity was visualized with immunofluorescence.CC, Corpus callosum; DG, dentate gyrus. Scale bars, 0.25 mm. NF-10 immunofluorescence shows extensive axonal beading in neocortex and accumulation of neurofilaments in granular cell somata of dentate gyrus in MILON mice. GFAP immunofluorescence shows extensive gliosis in corpus callosum and hippocampus.
We demonstrated the presence of gliosis by using GFAP antibodies to detect reactive astrocytes (Fig. 4). Neocortex did not contain any significant amount of reactive astrocytes, but there was a large increase of GFAP-immunoreactive cells in corpus callosum, with many astrocytes having processes directed into the degenerating neocortex. In all hippocampal regions, we observed massive gliosis, corresponding fully to the increased cellularity noted with cresyl violet staining. There was no difference in the distribution of reactive astrocytes between early symptomatic and end-stage MILON mice. However, the amount of gliosis was clearly larger in end-stage MILON mice. Pathological vascularization, as indicated by large abnormal vessels, particularly in neocortical areas, could also be seen using antibodies against the von Willebrand factor (data not shown). None of the different neuropathological changes described above were present in 2-, 3-, or 4-month-old MILON mice, in heterozygous knock-out animals, or in controls.
To assess the magnitude of cell death at different time points, we performed TUNEL. No TUNEL-positive nuclei could be detected in 2-, 3-, or 4-month-old presymptomatic MILON mice, further supporting thatTfam knock-out neurons are viable for several months. In early- and end-stage symptomatic animals (age 5–6 months), TUNEL-positive nuclei were seen in all areas suffering from cell loss, as demonstrated by cresyl violet staining (Fig. 3). Moreover, TUNEL reactivity was often observed before any cell loss was evident using a cresyl violet stain. TUNEL in neocortex was limited to the outer layers, corresponding well to the areas most depleted of mtRNA, as seen with in situ hybridization. The first areas of hippocampus to display TUNEL-positive cells were CA1 and to some extent also CA3. Later, when medial CA1 was completely lost and CA3 was slightly affected, most TUNEL-positive cells were seen in the granule cell layer of the dentate gyrus. Because TUNEL may detect necrotic as well as apoptotic cell death, we used complementary methods to detect apoptosis. DNA gel electrophoresis of cortical and hippocampal samples from symptomatic 5- to 6-month-old MILON mice yielded a smear, and a DNA ladder was faintly visible. By using a sensitive PCR assay, we detected DNA ladders in cortical and hippocampal samples of MILON mice at the age of 5–6 months but not in MILON mice at the age of 4 months or in control mice (Fig. 5d). Northern Blot analysis did not show any increase in transcripts encoding the proapoptotic Bax or anti-apoptotic Bcl-xL proteins in 2-, 4-, and 5-month-old mutants (Fig. 5a,b). We could not detect activated caspase 3 or 7 by immunohistochemistry of brain sections from MILON mice at different ages (data not shown). Northern blot analyses showed normal levels of transcripts for glyceraldehyde-3-phosphate dehydrogenase (Gapdh) (Fig. 5a), suggesting that upregulation of glycolysis does not occur in MILON mice.
Fig. 5.

Gene expression profiles, enzyme activities, and DNA ladder gel analysis in MILON and control mice of different ages.a, Results from phosphoimager quantification of Northern blots to determine mitochondrial Sod2, Gpx, Bax, Bcl-xL, Gapdh, and mtRNA transcript levels in neocortex of control (C;black bars) and MILON (M; white bars) mice at the age of 2, 4, and 5 months. Transcript levels were normalized to 18S rRNA and are presented as the percentage of the mean value of age-matched controls. **p < 0.01; ***p < 0.001. b, Northern blot analysis of transcript levels in neocortex from controls (C) and MILON (M) mice at different ages. The same blot was reprobed to detect different transcripts as indicated in the panel. c, Sod2 and Gpx enzyme activities in neocortex from controls (C; black bars) and MILON (M; white bars) mice at different ages. Values are presented as the percentage of the mean enzyme activity of age-matched controls. d, PCR DNA ladder gel assay of DNA prepared from neocortex of MILON (M) and control (C) mice at different ages. DNA fragmentation is present in neocortex of 5- to 6-month-old symptomatic MILON mice, as indicated by arrowheads. A staurosporine-treated human osteosarcoma cell line 143B was used as a positive control.
MILON mice display a low induction of antioxidant defenses
We next evaluated whether respiratory chain deficiency in neurons affect ROS production and ROS defense mechanisms. Northern blot analyses of RNA samples from neocortex of MILON mice showed moderately increased Gpx transcript levels at 5 months of age and a tendency toward increased Sod2 transcript levels at 2, 4, and 5 months of age (Fig. 5a,b). We also measured the Sod2 and Gpx enzyme activities at different ages and found no significant differences between MILON and control mice (Fig. 5c).
Western blot analysis and immunohistochemistry to detect protein nitrosylation, another marker for oxidative stress induced by the conversion of superoxide and nitric oxide to peroxynitrite, did not show any difference between 4-to 6-month-old mutant and control animals (data not shown). The activity of the nucleus-encoded respiratory chain complex II, an iron–sulfur group-containing enzyme readily impaired by oxidative stress, was unchanged in the mutants (Fig. 2). These results suggest unexpectedly low levels of ROS production in the MILON mice.
Excitotoxic stress induces marked neuronal cell death in MILON mice
Surprisingly, there were no signs of neurodegeneration, as determined by cresyl violet, TUNEL, GFAP, NF-10, and PCNA labeling of brain sections (Figs. 3, 4), in 4-month-old MILON mice with widespread and profound respiratory chain deficiency in forebrain neurons (Fig.2). We therefore challenged MILON mice with kainic acid injections to determine whether they would be more susceptible to stress-induced neuronal death.
We injected 16 mice (MILON, n = 8; controls,n = 8) with 20 mg/kg kainic acid at the age of 4 months and observed that three MILON and three control mice developed level 4 or 5 seizures, whereas the remaining mice developed level 2 seizures. Next, we injected 13 mice (MILON, n = 6; controls,n = 7) with 30 mg/kg kainic acid at the age of 4 months and observed that six MILON and six control mice developed level 3–5 seizures, whereas no seizures were induced in one of the control animals. There was thus no significant difference in the probability to develop high-level (grade 3–5) seizures after kainic acid injections between MILON and control mice at the age of 4 months in response to 20 or 30 mg/kg kainic acid.
Next, we investigated the neuropathological consequences of seizures by harvesting brains 24 hr after kainic acid injection and performing TUNEL staining. There was no difference in the low amount of TUNEL-positive cells that could be seen in hippocampus of MILON (n = 2) and control (n = 2) mice with level 2 seizures. MILON mice (n = 2) injected with saline displayed no TUNEL-positive cells in hippocampus. However, MILON mice (n = 5) with grade 3–5 seizures displayed larger numbers of TUNEL-positive cells in the CA3 region of hippocampus than did controls (n = 5) (Fig.6). There was no overlap between the five MILON specimens with abundant TUNEL-reactive cells in the CA3 area of hippocampus and the five control specimens with much less TUNEL reactivity, when the amount of TUNEL-reactive cell profiles in hippocampus was rated by two independent observers using a semiquantitative scale on coded slides.
Fig. 6.

TUNEL analysis of neuronal cell death 24 hr after injection of kainic acid in MILON and control mice at the age of 4 months. Scale bars, 0.2 mm. MILON mice consistently display more TUNEL-labeled cells in the CA3 area compared with controls with similar seizure activity.
DISCUSSION
Neurodegenerative diseases in man typically become manifest late in life. There are few animal models that replicate the late-onset and progressive features of this major group of human CNS diseases. Here, we characterize the MILON mice, who are apparently healthy well into adulthood, at which time they develop progressive lethal corticohippocampal neurodegeneration. Before the development of overt neurological symptoms, the MILON mice are considerably more vulnerable to excitotoxic challenge. We also characterized some of the mechanisms leading to nerve cell death in the MILON mice and found that neurons are able to survive for at least 1 month after shutting off oxidative phosphorylation and that nerve cell death is preceded by only minimal activation of defense mechanisms against reactive oxygen species.
Human mitochondrial disorders have an overall incidence (∼1:10,000) comparable with other common genetic forms of neurodegeneration (Chinnery et al., 2000; Darin et al., 2001). Point mutations or deletions affecting tRNA genes of human mtDNA will impair mitochondrial translation and result in the same type of respiratory chain deficiency affecting multiple complexes as in the MILON mice. Affected patients mainly have encephalomyopathic syndromes and display mosaic tissue-specific patterns of respiratory chain deficiency that, at least partly, are determined by the distribution of mutated mtDNA in different cell types (Larsson and Clayton, 1995). We reported previously that cell type-specific disruption of Tfam in mouse heart (Wang et al., 1999; Li et al., 2000) and pancreatic β-cells (Silva et al., 2000) faithfully mimics phenotypes found in humans with syndromes caused by mtDNA deletions (▵mtDNA). Similar to the MILON mouse, neuropathological findings in patients with mitochondrial disorders include nerve cell loss, gliosis, and white matter changes (Leigh, 1951; Oldfors et al., 1990; Oldfors et al., 1995). There is typically a time lag between the occurrence of respiratory chain deficiency and onset of mitochondrial neurodegeneration in human patients. In some cases, this can be attributed to heteroplasmy, i.e., a mixture of normal and mutated mtDNA, with increasing levels of mutated mtDNA with time in affected organs (Larsson et al., 1990). However, this time lag is also observed in mitochondrial neurodegeneration syndromes attributable to nuclear mutations, such as Friedreich's ataxia (Rotig et al., 1997), in which temporal differences in gene dosage is not an issue. Similar to humans, prolonged respiratory chain deficiency is also required for the induction of neurodegeneration in MILON mice and therefore probably reflects an inherent property of neurons to withstand long periods of respiratory chain deficiency. Interestingly, MILON mice exhibited marked neuronal cell death in response to exogenous stress; similarly, patients with Leigh syndrome and other mitochondrial encephalomyopathies may exhibit profound clinical deterioration in response to moderate stress, e.g., viral infections.
Aging is associated with increased levels of ▵mtDNA, attributable to somatic mutagenesis, and some studies have shown a decline of respiratory chain function with increasing age (Wallace, 1992). There are always multiple forms of ▵mtDNA in old subjects with high levels in postmitotic tissues, such as brain and skeletal muscle. An uneven distribution of ▵mtDNA with clonal expansions within single cells has been reported in several studies (Oldfors et al., 1993; Brierley et al., 1998; Cottrell et al., 2000). A recent enzyme histochemical study of brains from old subjects demonstrated an age-related increase of respiratory chain-deficient pyramidal neurons in hippocampus (Cottrell et al., 2001), further supporting the concept of uneven distribution of age-associated mtDNA mutations in postmitotic cells. It is likely that presumptive functional impairment and neuropathological changes caused by ▵mtDNA will be determined by the fraction of respiratory chain-deficient neurons. It is also possible that affected neurons may impair the function of many respiratory-competent neurons through transneuronal degeneration mechanisms. The generation of chimeras between MILON and control mice should enable us to establish threshold fractions of respiratory chain-deficient cortical neurons needed to induce severe neurodegeneration and thus shed light on the role of the increasing numbers of COX-deficient neurons during aging.
We reported previously that disruption of Tfam in mouse cardiomyocytes causes dilated cardiomyopathy with heart conduction blocks (Wang et al., 1999; Li et al., 2000). Interestingly, these severely respiratory chain-deficient hearts display signs of apoptosis, as manifested by moderate increase of TUNEL-reactive cells, expression of activated caspase 3 and 7, increased levels of transcripts for Bax and Bcl-xL, and DNA fragmentation on gels (Wang et al., 2001). We observed much more abundant TUNEL-reactive cells in the MILON mice than in the mitochondrial cardiomyopathy mice, but, surprisingly, activated caspase 3 or 7 was not detected by immunohistochemical assays, the levels of transcripts for Bax and Bcl-xL were not changed, and DNA fragmentation was only observed by using a sensitive PCR assay in symptomatic MILON mice. These findings suggest that the pathways leading to cell death may be different in respiratory chain-deficient cardiomyocytes and neurons. It is known that differences in intracellular ATP levels are of importance for the execution of apoptosis (Leist et al., 1997). Consistent with this hypothesis, we found massive upregulation of transcripts encoding the glycolytic enzyme Gapdh in respiratory chain-deficient cardiomyocytes (Wang et al., 2001) but not in MILON mice neurons. Future studies of MILON mice, including pharmacological treatments as well as breeding to other transgenic mouse strains, should help elucidate molecular pathways leading to neuronal cell death. The MILON mice should thus help unravel the molecular mechanisms leading to death of nerve cells with defective mitochondrial function and model CNS aspects of mitochondrial diseases.
Footnotes
N.-G.L. was supported by Swedish Medical Research Council Grants 13X-12197 and 13P-12204 and funds from the Karolinska Institutet, Torsten and Ragnar Söderbergs Stiftelse, the Human Frontiers Science Program, and the Swedish Foundation for Strategic Research. L.S. was supported by a stipend from the Swedish Foundation for Strategic Research. P.R. was supported by the Association Françaises contre les Myopathies (AMF). L.O. was supported by grants from the Swedish Medical Research Council, United States Public Health Service, the AMF, Hedlunds Stiftelse, the Human Frontiers Science Program, and funds from the Karolinska Institutet. B.X was supported by the Howard Hughes Medical Institute and National Institute of Neurological Disorders and Stroke Grant P01-16033.
L.S. and M.E. have contributed equally to this work.
Correspondence should be addressed to Nils-Göran Larsson, Department of Medical Nutrition, Karolinska Institutet, Novum, Huddinge Hospital, S-141 86 Huddinge, Sweden. E-mail:nils-goran.larsson@mednut.ki.se.
REFERENCES
- 1.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–223. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 2.Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- 3.Cottrell DA, Ince PG, Blakely EL, Johnson MA, Chinnery PF, Hanna M, Turnbull DM. Neuropathological and histochemical changes in a multiple mitochondrial DNA deletion disorder. J Neuropathol Exp Neurol. 2000;59:621–627. doi: 10.1093/jnen/59.7.621. [DOI] [PubMed] [Google Scholar]
- 4.Cottrell DA, Blakely EL, Johnson MA, Ince PG, Borthwick GM, Turnbull DM. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol Aging. 2001;22:265–272. doi: 10.1016/s0197-4580(00)00234-7. [DOI] [PubMed] [Google Scholar]
- 5.Dagerlind A, Friberg K, Bean AJ, Hökfelt T. Sensitive mRNA detection using unfixed tissue: combined radioactive and non-radioactive in situ hybridization histochemistry. Histochemistry. 1992;98:39–49. doi: 10.1007/BF00716936. [DOI] [PubMed] [Google Scholar]
- 6.Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49:377–383. [PubMed] [Google Scholar]
- 7.Franklin KBJ, Paxinos G. The mouse brain. Academic; San Diego: 1997. [Google Scholar]
- 8.Geromel V, Kadhom N, Cebalos-Picot I, Ouari O, Polidori A, Munnich A, Rötig A, Rustin P. Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the neurogenic ataxia retinitis pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum Mol Genet. 2001;10:1221–1228. doi: 10.1093/hmg/10.11.1221. [DOI] [PubMed] [Google Scholar]
- 9.Hökfelt T, Fuxe K, Goldstein M, Joh TH. Immunohistochemical localization of three catecholamine synthesizing enzymes: aspects on methodology. Histochemie. 1973;33:231–254. doi: 10.1007/BF00274236. [DOI] [PubMed] [Google Scholar]
- 10.Larsson NG, Clayton DA. Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet. 1995;29:151–178. doi: 10.1146/annurev.ge.29.120195.001055. [DOI] [PubMed] [Google Scholar]
- 11.Larsson NG, Luft R. Revolution in mitochondrial medicine. FEBS Lett. 1999;455:199–202. doi: 10.1016/s0014-5793(99)00854-6. [DOI] [PubMed] [Google Scholar]
- 12.Larsson NG, Holme E, Kristiansson B, Oldfors A, Tulinius M. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res. 1990;28:131–136. doi: 10.1203/00006450-199008000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Larsson NG, Garman JD, Oldfors A, Barsh GS, Clayton DA. A single mouse gene encodes the mitochondrial transcription factor A and a testis-specific nuclear HMG-box protein. Nat Genet. 1996;13:296–302. doi: 10.1038/ng0796-296. [DOI] [PubMed] [Google Scholar]
- 14.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 15.Leigh D. Subacute necrotizing encephalomyopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci USA. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oldfors A, Fyhr I-M, Holme E, Larsson N-G, Tulinius M. Neuropathology in Kearns-Sayre syndrome. Acta Neuropathol. 1990;80:541–546. doi: 10.1007/BF00294616. [DOI] [PubMed] [Google Scholar]
- 19.Oldfors A, Larsson N-G, Lindberg C, Holme E. Mitochondrial DNA deletions in inclusion body myositis. Brain. 1993;116:325–336. doi: 10.1093/brain/116.2.325. [DOI] [PubMed] [Google Scholar]
- 20.Oldfors A, Holme E, Tulinius M, Larsson NG. Tissue distribution and disease manifestations of the tRNA(Lys) A->G(8344) mitochondrial DNA mutation in a case of myoclonus epilepsy and ragged red fibres. Acta Neuropathol. 1995;90:328–333. doi: 10.1007/BF00296519. [DOI] [PubMed] [Google Scholar]
- 21.Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 22.Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 23.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 24.Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338:289–295. doi: 10.1016/0006-8993(85)90159-3. [DOI] [PubMed] [Google Scholar]
- 25.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 26.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Brüning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci USA. 2001;98:4038–4043. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 30.Zetterström RH, Simon A, Giacobini MM, Eriksson U, Olson L. Localization of cellular retinoid-binding proteins suggests specific roles for retinoids in the adult central nervous system. Neuroscience. 1994;62:899–918. doi: 10.1016/0306-4522(94)90482-0. [DOI] [PubMed] [Google Scholar]