

Abstract
GABAergic IPSCs have a relatively slow decay (deactivation) that appears to result from GABAA receptor channel openings that occur well beyond the predicted duration of free GABA at central synapses. Open and desensitized states have been suggested to prevent dissociation of agonist from the receptor, thus prolonging deactivation. However, simultaneous assessment of GABA binding and channel gating has not been possible. We developed a functional assay for occupancy of the GABA binding site or sites to test the GABA “trapping” hypothesis. Deactivation currents were compared in the absence and presence of bicuculline, a competitive antagonist that also allosterically inhibits GABAA receptors. This provided a model-independent, functional test of the hypothesis that GABA is trapped on the receptor during gating: bicuculline could only inhibit the channel if it was open but unbound by GABA. Although bicuculline inhibited spontaneous and neurosteroid-activated GABAAreceptor currents, it failed to alter the deactivation time course of GABA-activated GABAA receptor currents. Protection of deactivation current from bicuculline block indicated that GABA remained bound to the receptors while the channel was open, thus suggesting that all open states, as well as all closed and desensitized states from which channel opening can occur, must be GABA liganded states. Trapping may be specific to agonists, because the positive allosteric modulator diazepam unbound from GABAA receptors independent of GABA binding and channel activity.
Keywords: GABAA receptor, deactivation, inverse agonist, GABA binding, concentration jump, diazepam
At rest, ligand-gated ion channels generally exist in stable closed conformations. Agonist binding triggers conformational changes that culminate in transitions of the channel among open, closed, and desensitized states. Much theoretical and experimental work has focused on the relationship between agonist binding and channel gating, yet their coupling remains poorly understood. Early models of ligand-gated ion channels (see Scheme 1) indicated that, in the simplest case, an agonist-binding step precedes isomerization to the open state (Del Castillo and Katz, 1957). Implicit in such a scheme is that while bound channels are visiting the open state, agonist dissociation cannot occur, and thus, the agonist is “trapped” on the receptor when the channel is open. In other words, the equilibrium concept of “affinity” has no meaning (because it is infinite) for open channels. For more complex kinetic models, multiple open, pre-open, and desensitized states may represent infinite affinity states (Haas and Macdonald, 1999). Cyclic schemes that allow agonist association and dissociation from all states imply a similar phenomenon, represented as higher (although not infinite) affinity for “active” states. In any case, agonist binding induces a conformational change in the receptor complex that favors gating transitions. These transitions are thought to reciprocally influence the binding site.
Although it has been difficult to explore experimentally the interaction between binding and gating (Colquhoun, 1998), two studies have addressed the concept of agonist trapping by GABAA receptors. At central inhibitory synapses, the postsynaptic response to vesicular release of GABA lasts longer than predicted by the low affinity of the channels for GABA and the relatively short burst duration of single-channel currents. Jones and Westbrook (1995, 1996) proposed that desensitized state or states transiently maintain synaptic GABAA receptors in a bound state, thus prolonging the postsynaptic response to GABA release. More recently, Chang and Weiss (1999a) combined binding and electrophysiological analysis to provide evidence that channel opening prevents dissociation of GABA from GABACreceptors. However, any suggestion that agonist remains bound during occupancy of specific states (e.g., open or desensitized) requires a kinetic model, and therefore, interpretation is limited by the accuracy of the model (Colquhoun, 1999).
This series of experiments was designed to investigate the question of agonist trapping in a model-independent manner. A “double-jump” protocol allowed drug application specifically during channel deactivation. To probe the relationship between open and bound channels, we reasoned that application of an antagonist that bound to the GABA binding site (a competitive antagonist) and also allosterically inhibited open channels in the absence of GABA (via the same site) would provide a functional assay for occupancy of the GABAA receptor binding site or sites during deactivation. Bicuculline is a competitive antagonist of GABAA receptors (Macdonald and Olsen, 1994) that also allosterically inhibits channel activity in the absence of GABA (Barker et al., 1989; Ueno et al., 1997; Neelands et al., 1999). In the presence of bicuculline during deactivation, any unliganded receptors contributing to the deactivation current would be inhibited by bicuculline, disproving the GABA-trapping hypothesis. If, however, no acceleration of deactivation is produced by bicuculline, the GABA-trapping hypothesis would be confirmed.
MATERIALS AND METHODS
Expression of recombinant GABAAreceptors. The cDNAs encoding rat α1, β3, and γ2L, GABAAR subunit subtypes were individually subcloned into the plasmid expression vector pCMVNeo. The L245S point mutation in the γ2L subunit was made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by sequencing. Oligonucleotide primers were synthesized by the University of Michigan DNA synthesis core facility (Ann Arbor, MI). Human embryonic kidney cells (HEK293T; a gift from P. Connely, COR Therapeutics, San Francisco, CA) were maintained in DMEM, supplemented with 10% fetal bovine serum, at 37°C in 5% CO2 and 95% air. This cell line is a derivative of HEK293 cells that constitutively express the SV-40 T antigen to increase plasmid replication (DuBridge et al., 1987). Cells were transfected with 4 μg of each subunit plasmid along with 1–2 μg of pHOOK (Invitrogen, Carlsbad, CA) for immunomagnetic bead separation (Greenfield et al., 1997), using a modified calcium phosphate coprecipitation technique, as previously described (Angelotti et al., 1993). The next day, cells were replated, and recordings were made 18–30 hr later.
Electrophysiology. Patch-clamp recordings were performed on transfected fibroblasts bathed in an external solution consisting of (in mm): NaCl 142; KCl 8; MgCl2 6; CaCl2 1; HEPES 10; and glucose 10, pH 7.4, 325 mOsm. Electrodes were formed from thin-walled (whole-cell) or thick-walled (excised patch) borosilicate glass (World Precision Instruments, Pittsburgh, PA) with a Flaming Brown electrode puller (Sutter Instruments, San Rafael, CA), fire-polished to resistances of 0.8–1.5 MΩ (whole cell), or 4–12 MΩ (excised patch) when filled with an internal solution consisting of (in mm): KCl 153; MgCl21; MgATP 2; HEPES 10; and EGTA 5, pH 7.3, 300 mOsm. This combination of internal and external solutions produced a chloride equilibrium potential near 0 mV. Although large-conductance changes were observed in some cells during this study, no evidence for chloride shifts was detected in control experiments. I–V relations derived from peak current and current after 10 sec of GABA (1 mm) application revealed similar reversal potentials (data not shown). Unless otherwise stated, cells and patches were voltage-clamped at −10 to −60 mV using an Axon 200A amplifier (Axon Instruments, Foster City, CA). No voltage-dependent effects were observed between −10 and −60 mV. Unless otherwise stated, cells were gently lifted from the recording dish soon after establishing the whole-cell patch clamp configuration. Drugs were applied (via gravity) to whole cells using a rapid perfusion system consisting of three-barrel square glass connected to a Warner Perfusion Fast-Step (Warner Instruments, Hamden, CT). The glass was pulled to a final barrel size of ∼250 μm. The solution exchange time was estimated routinely by stepping a dilute external solution across the open electrode tip to measure a liquid junction current. The 10–90% rise times for solution exchange were consistently ≤1–2 msec. The exchange around lifted cells is likely to be slower than the open tip measurements. Although we did not quantify this, we inferred from the current rise times that solution exchange occurred within 10 msec, which is sufficiently fast for these experiments. For concentration jumps with excised patches, a theta tube attachment was used, with 10–90% exchange times of ≤400 μsec.
Analysis of currents. Currents were low-pass filtered at 2–5 kHz, digitized at 10 kHz, and analyzed using the pClamp8 software suite (Axon Instruments). The deactivation time courses of GABAA receptor currents were fit using the Levenberg–Marquardt least squares method with one or two component exponential functions of the form Σanτn, wheren is the best number of exponential components, ais the relative amplitude of the component, and τ is the time constant. A second component was accepted only if it significantly improved the fit compared with a single exponential function, as determined by an F test on the sum of squared residuals. Three component fits were not considered. The correlation coefficients of fitted curves were usually >90%. For comparison of deactivation time courses, a weighted summation of the fast and slow decay components (af * τf + as * τs) was used. For comparisons of the rate of block onset, the fast exponential component was used (see Fig. 5 for example). The bicuculline block of spontaneous currents could be described with two exponential functions; however, because the relative contribution was dominated by the fast component (>90%), we only used its time constant for comparison. Note that because of limitations associated with solution exchange around cells, the rate of block onset represents an upper estimate of the macroscopic blocking rate (the actual rate might be faster) (see Fig. 5C, for example). Numerical data were expressed as mean ± SEM. Statistical significance, using Student's t test (paired or unpaired as appropriate) was taken as p < 0.05.
Fig. 5.

Onset of bicuculline block is limited by GABA unbinding. A, Deactivation currents of α1β3γ2L (L245S) GABAA receptors after a concentration jump into 1 mm GABA (solid bar) were partially blocked by bicuculline (hatched bar). Cells were voltage clamped at 0 to −5 mV. The onset of block was time-dependent (the separation of deactivation currents during control wash and bicuculline wash increased with time). B, To illustrate the time course of bicuculline inhibition, the traces were subtracted (middle) for comparison with the direct effect of bicuculline (left) in the same cell. The traces are normalized and overlaid to demonstrate the slow onset rate when bicuculline was applied during deactivation (right). C, Summary chart showing the onset rate of bicuculline inhibition for various conditions. Although the time course of block was often best fit by a two exponential function, only the faster time constant is shown, which accounted for >90% of the decay. The number of cells is shown for each condition. Note the logarithmic ordinate.
RESULTS
In a typical lifted cell transiently expressing α1β3γ2L GABAA receptors, current persists for hundreds of milliseconds (slow deactivation) after removal of free GABA (Fig.1A). In an outside-out patch containing a few GABAA receptor channels, a brief pulse (5 msec) of 1 mm GABA elicited a rapid inward current as channels synchronously opened. Repetitive openings could be seen hundreds of milliseconds after the GABA pulse was terminated (Fig. 1B).
Fig. 1.

GABAA receptor channel openings outlast the duration of agonist exposure. A, Macroscopic current response of a lifted HEK293T cell expressing α1β3γ2L GABAA receptors exposed to 1 mm GABA for 400 msec (solid bar). Despite precise control of the solution bathing the cell by the concentration jump technique, the current relaxation after agonist removal requires many hundreds of milliseconds. The cell was voltage clamped at −15 mV.B, Individual openings are observed for hundreds of milliseconds after a 5 msec (arrow) pulse of 1 mm GABA delivered to an excised patch containing a few α1β3γ2L GABAA receptors. The patch was voltage clamped at −70 mV.
The duration of deactivation currents is longer than predicted by the low whole-cell GABA EC50 of these channels (∼10 μm), and the short burst durations (∼15 msec) and is the basis for suggestions that conformations associated with channel gating and desensitization may represent high-affinity states. Previous work has suggested that desensitized states (Jones and Westbrook, 1995;Dominguez-Perrot et al., 1997; Haas and Macdonald, 1999) as well as open states (Chang and Weiss, 1999a) may serve as transient high-affinity conformations, effectively slowing GABA unbinding and thus prolonging deactivation. Thus, one hypothesis for this persistent current is that GABA remains bound to or trapped by the open and desensitized channels.
To test the hypothesis that GABA is trapped on the receptors during deactivation, without making any assumptions about the number or connectivity of kinetic states, we used bicuculline as a functional assay for the occupancy of the GABA binding site or sites. We began by characterizing bicuculline inhibition of α1β3γ2L GABAA receptor channels that are open in the absence of GABA (Fig. 2). α1β3γ2L GABAA receptors opened spontaneously with low probability. Bicuculline (100 μm) rapidly and reversibly inhibited the spontaneous GABAA receptor current in voltage-clamped lifted cells transfected with that isoform (n = 4) (Fig. 2A). α1β3γ2L(L254S) GABAA receptor channels had frequent spontaneous openings and, consistent with a previous report (Chang and Weiss, 1999b), these spontaneous openings were also rapidly and reversibly inhibited by bicuculline (100 μm) in an excised patch (n = 4) (Fig. 2B). (Although we consistently observed an overshoot in the holding current after bicuculline washout for both constructs, the basis for this phenomenon remains unclear.) The rate of block onset reflects solution exchange time, the binding rate of bicuculline, and the rate at which bound bicuculline inhibits the channel. It has also been demonstrated that GABAAreceptor channels can be opened in the absence of GABA by neurosteroids and that the neurosteroid-activated currents are bicuculline-sensitive (Barker et al., 1989; Ueno et al., 1997). A lifted cell expressing α1β3γ2L GABAA receptors was first jumped from control solution into 10 μm alphaxalone, then back into control (Fig. 2C). The rebound current after termination of alphaxalone application likely indicated an additional low-affinity site for open channel block. The same cell was jumped a second time from control solution into alphaxalone, and then into 100 μm bicuculline alone, which strongly inhibited the deactivation current as indicated by the accelerated decay rate (Table 1). Similar inhibition was observed if 1 μm alphaxalone was used as the agonist, a concentration that did not result in a rebound current (Fig.2D). Thus, bicuculline clearly inhibited GABAA receptor channels in the absence of GABA. We currently have no explanation for the slower rate of block onset after activation by neurosteroid compared with block of spontaneous currents, although this could indicate a state-specific interaction of bicuculline with the channel.
Fig. 2.

Bicuculline inhibited GABAAreceptor currents in the absence of GABA. A, α1β3γ2L channels open spontaneously with low probability. A 100 mm concentration of bicuculline (hatched bar) rapidly and reversibly blocked the spontaneous activity in a lifted cell expressing α1β3γ2L GABAA receptors voltage clamped at −30 mV. A transient “overshoot” current was observed after bicuculline washout (dotted line inA and B). B, α1β3γ2L GABAA receptors containing the L245S mutation in the γ2L subunit have a higher spontaneous opening probability. A 100 μm concentration of bicuculline (hatched bar) rapidly and reversibly blocked the spontaneous activity in an excised patch containing the mutant GABAA receptors voltage clamped at −30 mV. C, GABAAreceptor currents activated by direct application of alphaxalone (solid bar) were blocked by 100 μmbicuculline (hatched bar). The cell was jumped first from control solution into 10 μm alphaxalone and then back to control solution (control wash), then the same cell was jumped from control into 10 μm alphaxalone and then into 100 μm bicuculline (bicuc wash). The dotted line (in C andD) shows the smaller holding current in the presence of bicuculline. D, Inhibition of the deactivation current was also observed when 1 μm alphaxalone was used, a concentration that did not produce a rebound current.
Table 1.
Deactivation time constants
| Drug | Wash | τ (msec) | n | Drug | Wash | τ (msec) | n |
|---|---|---|---|---|---|---|---|
| 1 μmAlph | Control | 310.2 ± 48.7 | 4 | 1 mmGABA | Control | 376.3 ± 56.9 | 5 |
| 1 μm Alph | 100 μmBic | 132.7 ± 32.4 | 4 | 1 mm GABA | 100 μm PTX | 61.0 ± 15.6 | 5 |
| 10 μmAlph | Control | 665.0 ± 47.2 | 3 | 3 μmGABA | Control | 283.4 ± 22.0 | 5 |
| 10 μmAlph | 100 μm Bic | 60.8 ± 5.7 | 3 | 3 μm GABA | 1 μm DZP | 362.9 ± 21.3 | 5 |
| 1 mm GABA | Control | 328.3 ± 29.8 | 7 | 1 mm GABA | Control | 318.7 ± 43.7 | 10 |
| 1 mm GABA | 100 μmBic | 320.1 ± 31.5 | 7 | 1 mm GABA | 1 μm DZP | 435.4 ± 75.4 | 10 |
| 3 μmGABA | Control | 244.3 ± 32.3 | 6 | 1 mmGABA | Control | 328.7 ± 47.6 | 4 |
| 3 μmGABA | 100 μm Bic | 218.4 ± 27.7 | 6 | 1 mm GABA | 600 nm DMCM | 276.5 ± 42.0 | 4 |
| 100 μmBic1-a | n/a | 8.7 ± 1.6* | 4 | 100 μmBic1-b | n/a | 6.3 ± 0.6* | 7 |
Effect on spontaneous activity in α1β3γ2L GABAA receptors.
Effect on spontaneous activity of α1β3γ2L (L245S) GABAA receptors. τ (msec) is the weighted time constant of deactivation, except for
which indicates the time constant of inhibition onset.
Alph, Alphaxalone; Bic, bicuculline.
Next we used the double-jump protocol to activate the GABAA receptors with GABA and then to apply bicuculline only during current deactivation (in the absence of applied GABA). We reasoned that if GABA remained bound to the channels that were open during deactivation and if, once GABA dissociated from the receptor, no rebinding of GABA occurred, then the current during deactivation should not be inhibited by bicuculline. However, if GABA could unbind from open channels or if unliganded receptors could reopen during the deactivation current, then bicuculline could bind and allosterically block opening. This would result in accelerated deactivation, because the bicuculline inhibition occurs faster (as shown in Fig. 2, Table 1) than the deactivation rate after GABA application. A 400 msec pulse of 1 mm GABA applied to a lifted cell elicited a rapidly activating (<10 msec) current that desensitized biphasically and deactivated slowly (deactivation τ, ∼300 msec) (Fig. 3A, Table1). The deactivation time course after a 1 mmpulse of GABA was not significantly altered when the cell was washed into 100 μm bicuculline (n = 7) (Fig. 3A,E). Even when the activating concentration of GABA was decreased to only 3 μm (near the GABA EC50) (Fig. 3B), only a minor (9%;p < 0.05) acceleration of the deactivation time constant was observed when cells were washed into 100–200 μm bicuculline (n = 6) (Fig.3B,E, Table 1). This minimal effect might be attributed to block of spontaneous openings, which would be relatively more prevalent at lower current amplitudes for a given amount of spontaneous activity. In fact, a smaller holding current was observed during bicuculline wash (as well as picrotoxin wash), indicating block of spontaneous openings. Also, when we used a very low concentration of GABA (100 nm), greater acceleration of deactivation was observed during bicuculline wash (21.5%; n = 9; data not shown). Alternatively, partially liganded channels might allow a small degree of bicuculline inhibition. Monoliganded openings might occur with higher probability at the lower GABA concentration, and if bicuculline could act dominantly on the channel via the remaining site, a small degree of inhibition would be observed. Although Ueno et al. (1997) also suggested (based on the Hill coefficient of bicuculline block) that binding of a single molecule of bicuculline was sufficient to inhibit GABAA receptor currents, further work is required to test this possibility. Binding of bicuculline to the receptor channel should have resulted in channel closure and acceleration of deactivation after GABA application, because the onset of bicuculline block in the absence of GABA was extremely fast (Fig.2A,B, Table 1). The protection from bicuculline block indicated that bicuculline could not bind to channels contributing to the deactivation current, and thus we infer that such channels were still GABA-bound.
Fig. 3.

Bicuculline failed to inhibit deactivation currents after GABA application. A, Deactivation currents after a concentration jump into 1 mm GABA (solid bar) were identical whether the cell was washed into control solution (open bar) or into 100 μm bicuculline (hatched bar).B, A 100 μm concentration of bicuculline (hatched bar) produced a small degree of inhibition in the deactivation current after activation by a lower concentration of GABA (3 μm; EC50 ∼5 μm). Thedotted line shows the smaller holding current during bicuculline wash. A portion of the trace (in the circle) is expanded to show the small effect of bicuculline wash on the deactivation current. C, The noncompetitive antagonist picrotoxin (100 μm; hatched bar) significantly inhibited deactivation currents after activation by 1 mm GABA. Note the smaller holding current during picrotoxin wash. D, The benzodiazepine diazepam (1 μm; shaded bar) potentiated deactivation currents after activation with 1 mm GABA. The dotted line shows the larger holding current in the presence of diazepam. E, Deactivation current pharmacology is summarized as the percentage of change in the weighted time constant of deactivation. The number of data points is indicated next to eachbar. Asterisks indicate significant differences compared with control deactivation rate for each condition.
Trapped GABA should not, however, afford any protection from inhibition by a noncompetitive antagonist applied during deactivation. Accordingly, when cells were jumped into 100 μmpicrotoxin, the deactivation current was substantially accelerated (n = 5) (Fig. 3C,E, Table 1). Note the lower holding current during the picrotoxin wash, indicating block of baseline spontaneous openings.
If GABA was trapped on the channels during deactivation, drugs acting at a separate modulator site such as the benzodiazepine site should alter the deactivation time course. Benzodiazepines enhance γ subunit-containing GABAA receptor currents by decreasing the EC50 for GABA, without affecting efficacy (Rogers et al., 1994). Consistent with this proposed mechanism, deactivation was slower when cells were washed into 1 μm diazepam after activation by GABA (Fig.3D,E, Table 1). Similar enhancement was obtained whether 1 mm or 3 μm GABA was used to activate the channels, and the effect was completely abolished by coapplication of the benzodiazepine receptor antagonist flumazenil (10 μm) (n = 4; data not shown). This slowed deactivation was in fact caused by delayed unbinding of GABA (and the resulting increase in “late” openings) because 100 μm bicuculline failed to block significantly diazepam-enhanced deactivation currents (n = 3; data not shown). Additionally, deactivation currents were accelerated during wash into DMCM (600 nm) (n= 4) (Fig. 3E, Table 1), an inverse agonist at the benzodiazepine site that reduces the affinity for GABA.
Under conditions where binding and rebinding of GABA could occur, bicuculline was able to block GABAA receptor currents (Fig. 4). During a prolonged (12 sec) application of GABA at a low concentration (3 μm, near the EC50), bicuculline and GABA coapplication blocked >90% of the current (n = 5) (Fig. 4A). Inhibition onset was slow and similar to the rate of deactivation during control wash, consistent with GABA unbinding preceding bicuculline binding. A slow phase of desensitization was observed during the 3 μmGABA applications. The GABA current after removal of bicuculline was larger than predicted by a control pulse in the same cell (Fig.4A, gray trace), consistent with bicuculline occupying the GABA binding site and preventing GABA-induced desensitization during that time. Similar current profiles were obtained when two GABA pulses were separated by control wash instead of bicuculline coapplication (data not shown).
Fig. 4.
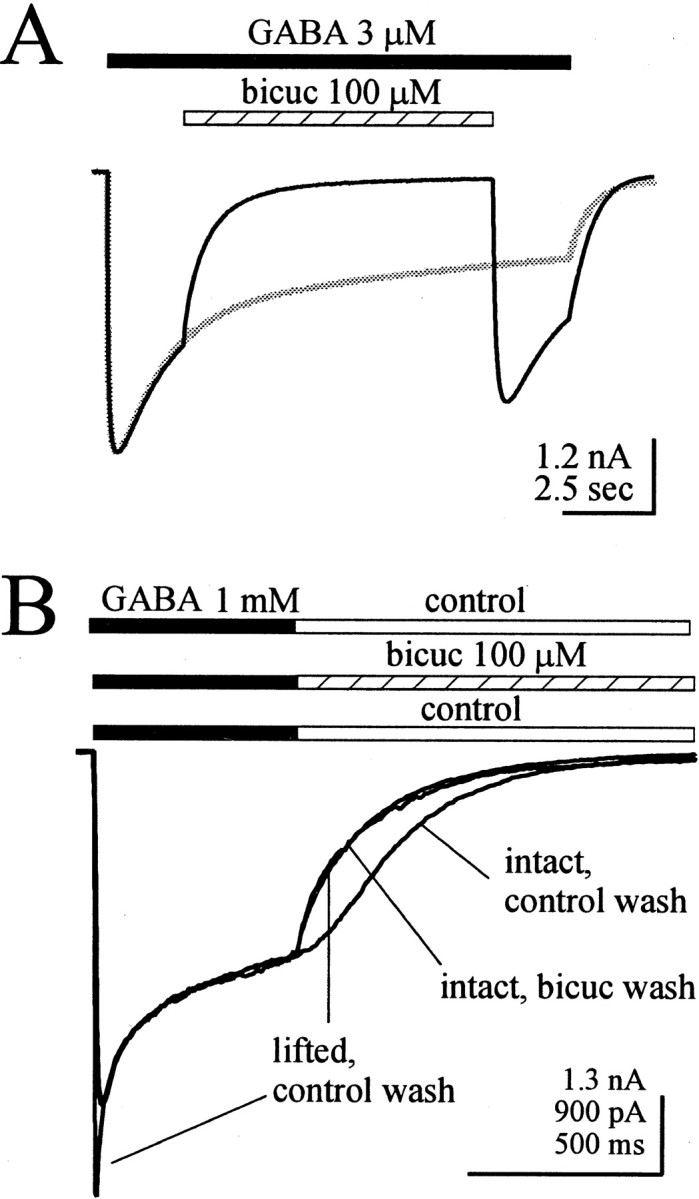
Bicuculline inhibited GABAA receptor currents under conditions in which unbinding and rebinding of GABA occurs. A, A 100 μm concentration of bicuculline (hatched bar) blocked the current during a long application of 3 μm GABA (solid bar) to a lifted cell expressing α1β3γ2L GABAA receptors. The response to 3 μm GABA alone from the same cell is superimposed in gray. Note the larger current after removal of bicuculline. B, Current responses to 400 msec jumps into 1 mm GABA in the same cell before and after lifting the cell from the recording dish. Deactivation currents were inhibited during bicuculline wash (hatched bar) before the cell was lifted, and this deactivation current overlapped with that observed in the same cell after it was lifted. Currents were normalized to the amplitude at the offset of the GABA pulse. The larger vertical scale value applies to the intact cell current response.
A more striking example of GABA binding and rebinding came from a comparison of double jump experiments in the same cells before and after lifting them from the recording dish (Fig. 4B). Under conditions of imprecise solution exchange, such as might occur around a cell adhering to the recording dish, some rebinding of GABA might occur during the washout period. When concentration jumps using 1 mm GABA were applied to cells adherent to the culture dish, current rise times were slower (>10 msec) than those observed in lifted cells and patches, and the fast component of desensitization (τ < 15 msec) was difficult to resolve, consistent with relatively poor rates of solution exchange. Bicuculline partially inhibited the deactivation current (Fig.4B) in double-jump experiments performed on the intact cells. The half-time of decay was decreased by 24.6 ± 6.6% (n = 5; p < 0.01). However, when the same cell was lifted off of the culture dish, thus ensuring more precise solution exchanges (accompanied by faster current rise times and increased resolution of fast desensitization), the bicuculline wash no longer affected the current (n = 4). Moreover, the control (no bicuculline) deactivation current in the lifted cells overlapped the time course of deactivation current when the cell was jumped into bicuculline before lifting.
Because α1β3γ2L(L245S) mutant GABAAreceptors had increased spontaneous openings, they allowed an additional test of GABA trapping (Fig.5). A 400 msec pulse of 1 mmGABA presumably saturated all of the GABA binding sites. At the instant cells were subsequently jumped into bicuculline, there were no unliganded channels available for bicuculline binding, and thus there was no reduction in current. Because only the spontaneous openings would be blocked, GABA unbinding would slow the onset of inhibition by bicuculline (which was very rapid in the absence of GABA) (Fig. 2, Table 1). Indeed, the extent of block increased with time (Fig.5A), suggesting that fully liganded receptors were protected, but after eventual unbinding of GABA, they resumed their bicuculline-sensitive spontaneous openings. To illustrate this slow onset of bicuculline block, the control traces were subtracted from the bicuculline wash traces. Direct application of bicuculline blocked spontaneous channel activity very rapidly (Fig. 5B, left, C), although the initial rate of block onset (τ fast) may have been limited by solution exchange time around lifted cells (therefore it represents an upper limit estimate for the rate of block onset). In contrast, the block proceeded at a much slower rate during the washout period (Fig. 5B, middle, subtracted trace, C). The traces are overlaid to demonstrate the striking difference in onset of block (Fig. 5B, right, C). The subtracted rate was similar to the deactivation rate during control wash; further evidence that deactivation of GABA-bound receptors was unaltered by the bicuculline wash.
These results demonstrated that GABA was trapped by open states and thus raised a related question. Are allosteric modulators of the GABAA receptor such as diazepam also trapped? To investigate potential “activity dependence” or “trapping” of diazepam binding, we first compared deactivation currents obtained with the following double-jump protocols: (1) 3 μm GABA, control wash; (2) 3 μm GABA, 1 μm diazepam wash; (3) 3 μm GABA + 1 μm diazepam coapplication, control wash; (4) 3 μm GABA + 1 μm diazepam coapplication, 1 μm diazepam wash (Fig. 6). When GABA and diazepam were coapplied, deactivation currents were slower than control deactivation currents (Fig. 6) (3 μm GABA, control wash). The relatively slow onset of enhancement in the diazepam wash (also see Fig. 3D) was probably because diazepam had a slow macroscopic on-rate (see below). When coapplication of 3 μm GABA and 1 μmdiazepam was followed by a 1 μm diazepam wash, no additional effect on deactivation was observed. This suggested that diazepam remained bound at least as long as GABA remained bound to the receptor, that is, at least the duration of the deactivation current.
Fig. 6.

Diazepam unbinding is slower than GABA unbinding. Diazepam unbinds at least as slowly as GABA from α1β3γ2L GABAA receptors. Deactivation currents are shown for applications of 3 μm GABA (solid bar) with control wash (open bar), 3 μm GABA with 1 μm diazepam wash (shaded bar), 3 μm GABA + 1 μm diazepam coapplication with control wash, and 3 μm GABA + 1 μm diazepam coapplication with 1 μm diazepam wash. Currents were normalized to the amplitude at the offset of the GABA pulse.
These results prompted us to determine specifically whether either GABA binding or channel gating could influence the macroscopic unbinding of diazepam. Therefore, we compared deactivation of diazepam-modulated currents in the presence and absence of GABA, and in GABAA receptors with short- and long-duration mean open times. As stated earlier, α1β3γ2L channels exhibited low-probability spontaneous openings that were blocked by selective GABAA receptor antagonists bicuculline and picrotoxin (Figs. 2A, 3D). Direct application of 1 μm diazepam increased the bicuculline-sensitive spontaneous channel activity, reaching peak enhancement within 1 sec (10–90% rise time; ∼700 msec) (n = 7) (Fig.7A1) (bicuculline sensitivity not shown). The amplitude of this enhancement was generally <2% of the maximal GABA-evoked response. After diazepam washout, this increase in holding current relaxed with a time constant of ∼4 sec (Fig.7C). This deactivation reflected unbinding of diazepam, because test pulses of GABA delivered at intervals after the removal of free diazepam were enhanced within a similar time window (time constant of 2.1 sec) (Fig. 7D). When a 1 sec pulse of 1 μm diazepam was coapplied during the current evoked by 300 nm GABA (also with a 10–90% rise time, ∼700 msec), the subsequent current relaxation had a time constant of ∼4 sec (n = 4) (Fig. 7A2,C). Similar results were obtained when a higher GABA concentration (2 μm) was used (data not shown). Next we tested the interaction of diazepam with α1β3γ2L (L245S) GABAA receptors, because they exhibited longer spontaneous (and GABA-activated) mean open times (our unpublished data). These channels deactivated significantly slower than wild-type α1β3γ2L channels when brief (<5 msec) pulses of 1 mm GABA were applied to excised patches (Fig.7B1), consistent with the significantly increased open times of the mutated channels compared with wild type. Direct application of diazepam increased the spontaneous activity of α1β3γ2L (L245S) GABAA receptor channels. This effect was consistent with a previous report involving spontaneous gating caused by an L to S mutation in the β3 subunit (Thompson et al., 1999), although the basis for benzodiazepine modulation of spontaneous GABAA receptor currents remains unclear. However, the deactivation after a 1 sec pulse of 1 μmdiazepam was still ∼4 sec (n = 4) (Fig.7B2,C), similar to that observed in wild-type α1β3γ2L GABAA receptors in the absence or presence of GABA. Thus, diazepam unbinding, as indicated by deactivation rate, was not sensitive to differences in channel gating that strongly affect GABA unbinding.
Fig. 7.
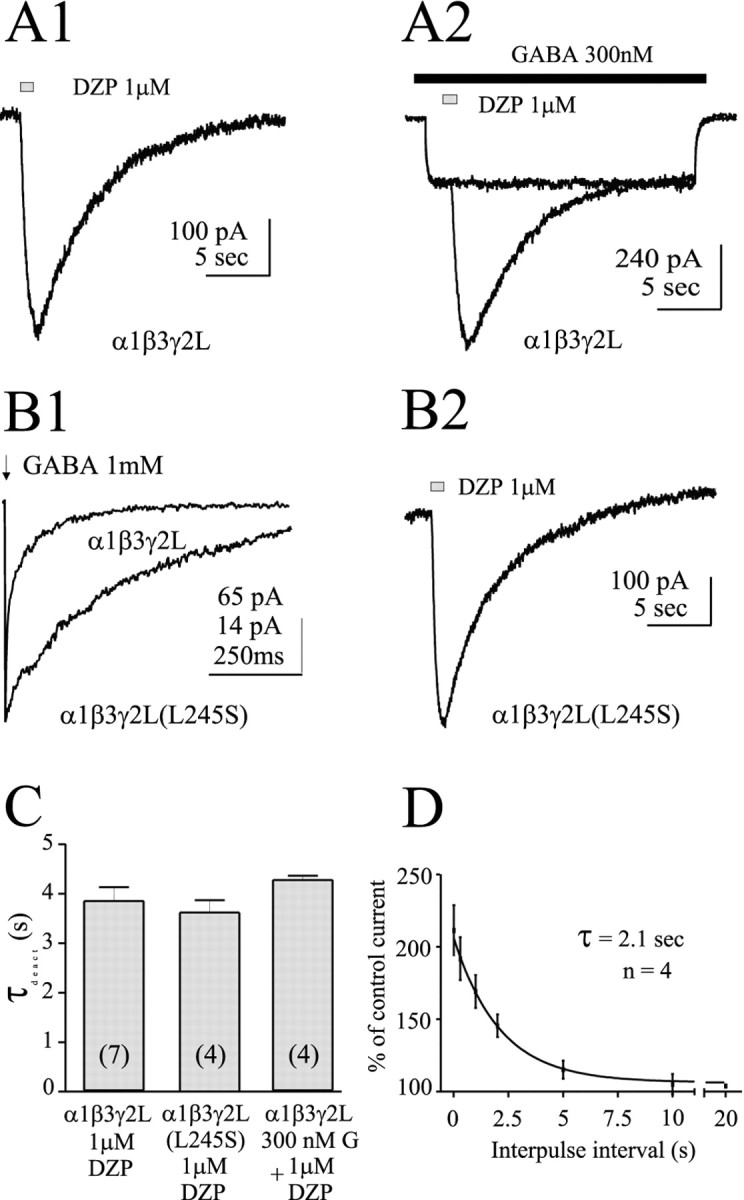
Diazepam unbinding is independent of channel gating. A1, In the absence of GABA, diazepam (1 μm) enhanced spontaneous activity in lifted cells expressing α1β3γ2L GABAA receptors.A2, Diazepam enhanced the response to a steady-state application of 300 nm GABA (different cell than inB1). B1, Deactivation of α1β3γ2L and α1β3γ2L(L245S) GABAA receptors after a 5 msec pulse of 1 mm GABA (arrow). Longer mean open times caused by this mutation slowed deactivation. B2, In the absence of GABA, 1 μm diazepam enhanced spontaneous activity in α1β3γ2L(L245S) GABAAreceptors. C, Summary chart showing the rate of deactivation after a 1 sec application of 1 μm diazepam was indistinguishable under different GABA-binding and intrinsic gating conditions. The number of cells is indicated inparentheses. D, A 1 sec application of 1 μm diazepam applied in the absence of GABA enhances subsequent responses to 1 μm GABA in lifted cells expressing α1β3γ2L GABAA receptors. Cells were washed with control solution for the interpulse interval.
DISCUSSION
This study strongly suggests that GABA is trapped on GABAA receptor channels by open states and closed or desensitized (preopen) states from which reopening can occur. Gating transitions are thought to delay unbinding of GABA independent of the microscopic affinity for GABA, resulting in a prolonged IPSC current caused by reopening before dissociation eventually occurs. Several studies have inferred a role for gating and desensitization in synaptic responses (Jones and Westbrook, 1995; Dominguez-Perrot et al., 1997;Haas and Macdonald, 1999; Chang and Weiss, 1999a), and we have identified a critical portion of TM1 involved in the functional coupling between desensitization and slow deactivation (Bianchi et al., 2001). However, it has been difficult to simultaneously assess the GABA binding site and channel gating. This study provides evidence for a constraint on the relationship between agonist binding and channel gating: GABA unbinding cannot occur from open and preopen states. Two common ways to study ion channel function are through binding assays and electrophysiological recording. Specific information about binding has been inferred through interpretation of currents in the context of a kinetic model. In many cases such models are empirical descriptions of macroscopic data and are therefore of unknown relevance to the behavior of individual channels. When binding is measured, nothing is known about the function of detected receptors. This can be problematic if conformational changes induced by binding affect dissociation [resulting in errors of affinity estimates (Colquhoun, 1998)], or if an unknown subset of receptors with intact binding sites are not functional (Chang and Weiss, 1999a; Colquhoun, 1999). Furthermore, the temporal resolution of conventional binding assays is orders of magnitude slower than the time scale of channel transitions, limiting the relevance of such measurements to steady-state behavior.
We developed a functional assay for occupancy of GABA binding sites. Selective application of bicuculline during GABAAreceptor deactivation was used to probe GABA binding sites during channel gating without making any assumptions about the nature of the gating process. Bicuculline blocked spontaneous channel activity in the absence of GABA as well as openings triggered by neurosteroid binding to a site distinct from that of GABA. However, after channel activation by GABA, bicuculline had virtually no effect on GABAA receptor deactivation currents. Bound GABA effectively “protected” the channels from bicuculline inhibition because bicuculline could not access its binding site. Although a relatively high concentration of bicuculline was used [more than two orders of magnitude higher than the IC50 found byUeno et al. (1997) in the presence of 3 μm GABA], we cannot rule out the possibility that higher concentrations of bicuculline would affect deactivation. We believe this is unlikely, because only minimal effects on deactivation were observed despite varying the GABA concentration by >300-fold. Interestingly, previous studies could not distinguish between a shared (or overlapping) binding site for GABA and bicuculline, versus a separate binding site for bicuculline that rendered the GABA binding site lower affinity. Our data argues against the idea of a second site, because such a site would be available for bicuculline to bind during deactivation and would accelerate deactivation by favoring unbinding. However, we cannot completely rule out the possibility of a very strong negative cooperativity between structurally distinct GABA and bicuculline binding sites.
The increased spontaneous activity of α1β3γ2L (L245S) mutated receptors offered an additional tool to test the idea that bound GABA protected channels from bicuculline inhibition. In the absence of GABA, the onset of bicuculline block was extremely fast (<10 msec). However, when α1β3γ2L (L245S) receptors were washed into bicuculline after a pulse of saturating GABA, the onset of block was substantially delayed (>500 msec; n = 5) (Fig. 5). This was expected if the spontaneous openings of unliganded channels were the only ones available for bicuculline block during the washout period. Thus, the onset of bicuculline inhibition was limited by GABA unbinding, further evidence that GABA remains trapped on the receptors.
In situations where GABA binding and unbinding were occurring, bicuculline inhibition of GABAA receptor currents was observed. In one case, channels activated by 3 μmGABA (below the GABA EC50) were clearly sensitive to bicuculline coapplication. During the coapplication of bicuculline, the blocked channels no longer entered desensitized states, consistent with the mutual exclusivity of GABA and bicuculline binding. This was apparent in the strong rebound current after removal of bicuculline (Fig. 4A). In the second case, we compared deactivation and its sensitivity to bicuculline between single cells in the intact and lifted configurations. Bicuculline washes partially blocked the deactivation currents of cells adherent to the culture dish. Lifting the same cells substantially improved the solution exchange, as evidenced by faster rise times and increased resolution of fast desensitization. Moreover, deactivation currents were no longer sensitive to bicuculline. Therefore, when the probability of rebinding of GABA was reduced, either by jumping an intact cell into excess bicuculline, or by lifting the cell to achieve better solution exchange, the deactivation time courses were the same.
It has been proposed that GABA binding induces some structural rearrangement around the binding site (Wagner and Czajkowski, 2001). Whether such a rearrangement is a local consequence of GABA binding or a result of channel gating is unknown. If channel gating prevents dissociation of bound GABA via a “Venus fly trap or clam shell” mechanism, then is it also true that channel gating in the absence of GABA limits access to the GABA binding site? If so, the rate of bicuculline block of open channels should be limited by channel closure. However, spontaneous openings in lifted cells expressing α1β3γ2L and α1β3γ2L(L245S) receptors were blocked with a fast time constant. Because the spontaneous open duration of wild type and the mutated channels differed by at least fivefold (∼300 μsec and ∼2 msec, respectively; our unpublished data), it was possible that the bicuculline block was not limited by channel closure. However, the solution exchange time around lifted cells might be too slow to distinguish subtle differences in block rate. Also, the block of neurosteroid-activated currents was slower than the block of spontaneous whole-cell currents. We do not understand this difference, but it may suggest that only certain states are available for block by bicuculline in the absence of GABA. Block of spontaneous α1β3γ2L(L245S) receptor currents in excised patches occurred with a very fast time constant (∼2 msec), but no comparison could be made with block of spontaneous wild-type receptors in patches because the spontaneous currents were undetectable or too small to measure accurately. This time constant was in the range we observed for spontaneous open durations, raising the possibility that channel closure is required for block. Similar arguments could be made for the interaction of GABA with spontaneously gating channels. We found that current rise time was fast (∼600 μsec; our unpublished data) when patches containing α1β3γ2L(L245S) GABAAreceptors were jumped into 1 mm GABA, similar to the rise times observed in wild-type receptors (Haas and Macdonald, 1999). Furthermore, all of the receptors became liganded during the GABA application (Fig. 5). These two observations suggested that channels could bind GABA even when they were open. However, these arguments assume that spontaneous and GABA-gated channel openings are associated with similar receptor conformations, which is not known except to the extent that the single-channel conductance is the same (data not shown). If the GABA binding site was accessible whether or not spontaneous openings were occurring, then bicuculline might be able to actively close open channels as well as prevent the reopening of closed channels. Single-channel recordings of spontaneous activity can distinguish among these possibilities, because the active closure of open channels by bicuculline predicts shorter mean open durations.
The selective delivery of modulators during the deactivation current presents a pharmacological tool that would be useful in several contexts, such as for screening potential competitive antagonists, where there is controversy between competitive and noncompetitive actions, or where there is a proposed “mixed” mechanism. Moreover, deactivation current modulation eliminates the potentially confounding factor of agonist rebinding inherent in steady-state experiments. For positive modulators such as benzodiazepines that have proposed mechanisms associated only with GABA affinity, effects onkoff can be specifically investigated. Although it is thought that diazepam does not affect GABAA receptor gating (as indicated by single-channel analysis), there has been some debate on whether diazepam increased GABA sensitivity through an effect onkon orkoff (Rogers et al., 1994; Lavoie and Twyman, 1996). Our results indicated an effect onkoff, although we cannot exclude an additional increase in kon. Note that diazepam increased the spontaneous activity of α1β3γ2L GABAA receptors. Although this may indicate an additional mechanism of action, the relatively small currents elicited by direct application of diazepam cannot quantitatively account for the enhancement of deactivation observed in the diazepam wash experiment (Fig. 3D). Similar reasoning suggested that the benzodiazepine inverse agonist DMCM decreases GABA sensitivity at least through an effect on koff. Although the structural correlate of changingkoff is not known, it is possible that the changes were not limited to the GABA binding site. For example, diazepam altered the accessibility of engineered cysteine residues in the third transmembrane domain that were distant from the GABA binding sites (Williams and Akabas, 2000). Despite the possibility that diazepam binding induces a distinct conformation of the receptor, its unbinding (as indicated by deactivation rate) was unaffected by either GABA binding or by channel opening. Interestingly, many of the residues important for benzodiazepine binding (at the α/γ interface) are homologous to those important for GABA binding (at the α/β interface) (Sigel and Buhr, 1997), and subunit interface structures have been suggested to represent a general ligand-binding motif. However, our data suggests a fundamental difference in the coupling of gating-related conformations (such as the open state) and the recognition sites for GABA and diazepam. Although the mechanism by which channel gating detains GABA at its binding site remains poorly understood, it appears that the process does not effectively generalize to the benzodiazepine binding site to detain diazepam.
It remains to be seen how the binding of other modulators such as barbiturates or neurosteroids may be affected by gating. It is also unknown whether agonist trapping generalizes to other types of ligand-gated channels. Certain competitive antagonists block spontaneous channel activity in mutant neuronal acetylcholine (ACh)-gated receptors (Bertrand et al., 1997), and thus the double jump paradigm could be used on ACh receptor channels. More generally, it would be relevant to test classical competitive antagonists for different ligand-gated channels to determine if bicuculline-like allosteric inhibition (inverse agonism) is observed. Not only will this allow phenomena such as agonist trapping to be investigated for other channel families, but it will also compel a re-evaluation of competitive antagonists in general as simply “sitting” in the agonist binding site without any additional effect on the receptor. This information is of practical interest because competitive antagonists are often used in ligand-binding studies to assay binding site availability and affinity without the bias of efficacy intrinsic to the agonists themselves.
Footnotes
This work was supported National Institutes of Health Grant R01-NS33300 (R.L.M.) and National Institute on Drug Abuse Training Fellowship T32-DA07281-03 (M.T.B.).
Correspondence should be addressed to Dr. Robert L. Macdonald, Department of Neurology, Vanderbilt University, 2100 Pierce Avenue, Nashville, TN 37212. E-mail: Robert.Macdonald@mcmail.vanderbilt.edu.
REFERENCES
- 1.Angelotti TP, Uhler MD, Macdonald RL. Assembly of GABAA receptor subunits: analysis of transient single-cell expression utilizing a fluorescent substrate/marker gene technique. J Neurosci. 1993;13:1418–1428. doi: 10.1523/JNEUROSCI.13-04-01418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker JL, Harrison NL, Lange GD, Owen DG. Potentiation of γ-aminobutyric-acid-activated chloride conductance by a steroid anesthetic in cultured rat spinal neurones. J Physiol (Lond) 1989;386:485–501. doi: 10.1113/jphysiol.1987.sp016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertrand S, Devillers-Thiery A, Palma E, Buisson B, Edelstein SJ, Corringer P-J, Changeux J-P, Bertrand D. Paradoxical allosteric effects of competitive inhibitors on neuronal α7 nicotinic receptor mutants. NeuroReport. 1997;8:3591–3596. doi: 10.1097/00001756-199711100-00034. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi MT, Haas KF, Macdonald RL. Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAA receptors. J Neurosci. 2001;21:1127–1136. doi: 10.1523/JNEUROSCI.21-04-01127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999a;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 6.Chang Y, Weiss DS. Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999b;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colquhoun D. Binding, gating, affinity and efficacy: The interpretation of structure-activity relationships for agonists and the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colquhoun D. GABA and the single oocyte: relating binding to gating. Nat Neurosci. 1999;2:201–202. doi: 10.1038/6298. [DOI] [PubMed] [Google Scholar]
- 9.Del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc Roy Soc B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 10.Dominguez-Perrot C, Feltz P, Poulter MO. Recombinant GABAA receptor desensitization: the role of the gamma2 subunit and its physiological significance. J Physiol (Lond) 1997;497:145–159. doi: 10.1113/jphysiol.1996.sp021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuBridge RB, Tang P, Hsai HC, Leong P-M, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenfield LJ, Sun F, Jr, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL. Expression of functional GABAA receptors in transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- 13.Haas K, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol (Lond) 1999;514.1:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 15.Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 16.Lavoie AM, Twyman RE. Direct evidence for diazepam modulation of GABAA receptor microscopic affinity. Neuropharmacology. 1996;35:1383–1392. doi: 10.1016/s0028-3908(96)00077-9. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 18.Neelands TR, Fisher J, Bianchi M, Macdonald RL. Spontaneous and γ-aminobutyric acid (GABA)-activated GABAa receptor channels formed by ε subunit-containing isoforms. Mol Pharmacol. 1999;55:168–178. doi: 10.1124/mol.55.1.168. [DOI] [PubMed] [Google Scholar]
- 19.Rogers CJ, Twyman RE, Macdonald RL. Benzodiazepine and beta-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J Physiol (Lond) 1994;475:69–82. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. TIPS. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- 21.Thompson S-A, Smith MZ, Wingrove PB, Whiting PJ, Wafford KA. Mutation at the putative GABAA ion channel gate reveals changes in allosteric modulation. Br J Pharmacol. 1999;127:1349–1358. doi: 10.1038/sj.bjp.0702687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci. 1997;17:625–634. doi: 10.1523/JNEUROSCI.17-02-00625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner DA, Czajkowski C. Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci. 2001;21:67–74. doi: 10.1523/JNEUROSCI.21-01-00067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DB, Akabas MH. Benzodiazepines induce a conformational change in the region of the γ-aminobutyric acid type A receptor α1-subunit M3 membrane-spanning segment. Mol Pharmacol. 2000;58:1129–1136. doi: 10.1124/mol.58.5.1129. [DOI] [PubMed] [Google Scholar]