

Abstract
Although accumulating evidence indicates that cAMP response element-binding protein (CREB) phosphorylation mediates not only synaptic plasticity but also survival of certain neurons, it remains uncertain whether CREB phosphorylation induced after metabolic insult leads to CRE-mediated gene transcription and is involved in cell survival or not. In the present study, we clarified that (1) CREB phosphorylation and ischemic tolerance induced after preconditioning ischemia in the hippocampal neurons was abolished by MK801 administration in gerbil global ischemia model, (2) CREB phosphorylation induced after exposure to glutamate in cultured neurons was inhibited by removal of extracellular calcium, by MK801 and by an inhibitor of calcium–calmodulin-dependent protein kinase (CaMK) II and IV, (3) inhibitor of CaMK II–IV or CRE-decoy oligonucleotide suppressed upregulation of BCL-2 expression and accelerated neuronal damage after exposure to glutamate, and (4) CREB phosphorylation induced in the hippocampal neurons after ischemia and in cultured neurons after exposure to glutamate was followed by CRE-mediated gene transcription in transgenic mice with a CRE–LacZ reporter. Our results suggest that CREB phosphorylation in neurons after ischemia and exposure to glutamate is induced by NMDA receptor-gated calcium influx and subsequent activation of CaMK II–IV and that CREB phosphorylation after metabolic stress might show a neuroprotective response through CRE-mediated gene induction.
Keywords: CREB, ischemia, BCL-2, β-galactosidase, glutamate, CRE-decoy oligonucleotide
Ischemic stress has been known to induce expression of various genes in neurons (Lipton, 1999), and some of the gene products are believed to play key roles in cellular adaptive responses, such as ischemic tolerance (Kitagawa et al., 1990;Kirino et al., 1991) and cell death mechanisms such as apoptosis (Ninatori et al., 1995). Therefore, elucidation of the molecular mechanism for gene expression in neurons after ischemic insult is important in developing novel strategies against stroke. It is widely known that heat shock protein (HSP) genes are induced after ischemic insult (Yagita et al., 1999), in which activation and binding of heat shock factor to heat shock element in the promotor region of each HSP gene (Morimoto, 1993) is a key step in induction of HSP expression (Higashi et al., 1995). Several other genes induced after ischemia, including c-Fos (Kiessling et al., 1993) and brain-derived neurotrophic factor (BDNF) (Lindvall et al., 1992), have a consensus cAMP response element (CRE, TGACGTCA) in the promoter region (Sheng et al., 1990; Shieh et al., 1998), suggesting the activation of CRE-binding protein (CREB) after ischemia. CREB is abundant in the brain and particularly in neurons. Phosphorylation of the Ser-133 residue is necessary but not sufficient for activation of CRE-mediated gene transcription (Hu et al., 1999). Diverse extracellular stimuli cause phosphorylation of CREB by protein kinase A (PKA), extracellular signal-related protein kinase (ERK), and calcium–calmodulin-dependent protein kinase (CaMK) (Finkbeiner, 2000), and CREB phosphorylation has recently been found to be important in both activity-dependent neuronal plasticity and neurotrophin-mediated neuronal survival (Ghosh and Greenberg, 1995; Bonni et al., 1999; Riccio et al., 1999). It was also suggested that CREB phosphorylation in response to brain insults (Walton and Dragunow, 2000) or activity deprivation by deafferentation (Zirpel et al., 2000) might be involved in nerve cell survival. Although CREB phosphorylation in the cultured neurons after glutamate (Hardingham et al., 1999) and NMDA (Sala et al., 2000) treatment was examined, the signal pathway leading to CREB phosphorylation and its functional role after ischemic stress or exposure to glutamate are not fully elucidated. In the present study, we aimed at clarifying the relation between the severity of insults and CREB phosphorylation, and the relation between CREB phosphorylation and CRE-mediated gene transcription, and finding the upstream pathway leading to CREB phosphorylation and the functional role of CREB phosphorylation in neuronal injury.
MATERIALS AND METHODS
Animals. Animals used in the present study were fed standard laboratory chow and given ad libitum access to water before surgery. All experimental procedures were approved by the Institutional Animal Center Use Committee of the Osaka University Graduate School of Medicine. CRE–LacZ transgenic mice were obtained from a colony at the University of Washington (Seattle, WA) (Imprey et al., 1996) and backcrossed at least six generations to wild-type C57BL/6 mice (Charles River, Yokohama, Japan). The transgene was maintained exclusively in heterozygotes. Mice were genotyped by PCR as described (Imprey et al., 1996).
Chemicals and reagents. The following drugs were used: (+)-MK801 (Sigma, St. Louis, MO), CNQX (Sigma), nifedipine (Sigma), EGTA (Sigma), Rp-8-Cl-cAMPs (Biolog, Hayward, CA), KT5720 (Calbiochem, San Diego, CA), chelerythrine (Calbiochem), Rp-8-Br-cGMPs (Biolog), KN93 (Seikagaku, Tokyo, Japan; Alexis, San Diego, CA), PD98059 (Calbiochem), genistein (Sigma), wortmannin (Sigma), and staurosporine (Alexis). All polyclonal antibodies, anti-CREB (Upstate Biotechnology, Lake Placid, NY), anti-phosphoCREB (Upstate Biotechnology), and anti-microtubule-associated proteins (MAPs) (Sigma) had been raised in rabbits. Anti-NeuN (Chemicon, Temecula, CA), anti-α-tubulin (Sigma), anti-microtubule-associated protein 2 (MAP2) (Sigma), anti-BCL-2 (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-β-galactosidase (Santa Cruz Biotechnology) were monoclonal antibodies.
Transient global ischemia in gerbils. Adult Mongolian gerbils of both sexes, weighing 60–80 gm, were used. Transient forebrain ischemia was produced by bilateral occlusion of the common carotid artery with aneurysm clips for 1, 2, or 5 min under anesthesia with 2% halothane. During surgical procedures, rectal temperature was maintained at 37 ± 0.5°C with a heat lamp. The clips were removed after the occlusion procedure. For histologic analysis, 30 gerbils were used. Six sham-operated animals were used as controls. For induction of tolerance, 24 gerbils were subjected to 2 min of ischemia (n = 12) or sham operation (n = 12), and 4 d later they were subjected to 5 min of ischemia as described previously (Kitagawa et al., 1990). MK801 (3 mg/kg) or vehicle was administered intraperitoneally 60 min before 2 min of preconditioning ischemia or sham operation. Seven days after the last ischemia, gerbils were decapitated, and their brains were promptly removed, divided into coronal sections (5 mm in thickness), and fixed in 5% acetic acid in ethanol at 4°C for 4 hr and embedded in paraffin. Coronal 5 μm sections corresponding to the stereotactic planes 1.4–1.6 mm caudal to the bregma were stained with hematoxylin and eosin. Intact neurons in the hippocampal CA1 sector were counted in a blind manner, and neuronal density per millimeter was calculated. For Western blot analysis, control gerbils (n = 7) and ischemic gerbils at 0, 2, 5, 15, 30, 60, and 180 min (n= 7 for each reperfusion period) after 5 min ischemia and at 15 min after 1 min and 2 min ischemia (n = 7 for each ischemic period) were decapitated under deep ether anesthesia, and the hippocampi were rapidly dissected out in iced saline and frozen in liquid nitrogen. Additional control gerbils (n = 3) and ischemic gerbils were perfusion-fixed with Zamboni's solution (2% paraformaldehyde and 0.2% picric acid) at 5, 15, 30, and 60 min (n = 3 for each reperfusion period) after 5 min ischemia under deep pentobarbital anesthesia, and brains were post-fixed in the same fixative overnight at 4°C. After three washes in PBS containing 20% sucrose, the brains were frozen in powdered dry ice and cut into 4-μm-thick coronal sections with a freezing microtome for immunohistochemical examination.
Transient global ischemia in CRE–LacZ transgenic mice. We also used male transgenic mice, age 12–16 weeks, carrying a CRE–LacZ reporter to confirm that CREB phosphorylation after ischemia induces CRE-mediated transcription. Transient forebrain ischemia was produced in CRE–LacZ transgenic mice as previously described (Kitagawa et al., 1998). In brief, a polyacrylamide column for measurement of cortical microperfusion by a laser Doppler flowmetry (Unique Medical, Osaka, Japan) was attached to the intact skull under halothane anesthesia. Both common carotid arteries were then exposed at the neck, occluded with microanerysmal clips for 15 min, and then reperfused. Only mice (n = 6) that showed <10% of baseline cortical microperfusion were used. After reperfusion for 60 min, brains were processed for immunohistochemistry as described before. Four sham-operated transgenic mice were used as controls.
Neuronal cell culture. Primary cultures of the rat hippocampal neurons were obtained as described previously (Mattson and Kater, 1988). Briefly, neuronal cultures were prepared from the hippocampus of embryonic day 18 (E18)∼19 rat embryos. The cells were dissociated with papain (papain dissociation system; Worthington, Lakewood, NJ) and plated onto six-well plates (Falcon, Becton Dickinson, and Company, Franklin Lakes, NJ) or two-chamber glass slides (Falcon) coated with polyethylenimine. Cells at a final concentration of 5.0∼7.0×105 cells/ml were cultured in high-glucose DMEM (Sigma) containing 10% fetal calf serum (FCS) (Sigma), 100 IU of penicillin per milliliter and 100 μg of streptomycin sulfate per milliliter. At 24 hr after seeding, the medium was changed to Neurobasal medium (Life Technologies, Rockville, MD) supplemented with B-27 (Life Technologies). Cells were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 and were used after 6∼7 d in vitro when the majority of cells showed a neuronal phenotype. We also used transgenic mice with a CRE–LacZ reporter gene to investigate whether CREB phosphorylation after exposure to glutamate induced CRE-mediated transcription. Primary cultures were prepared from the hippocampi of E17∼E18 mouse embryos after mating of heterozygotes as described before.
Cells were treated with glutamate (20, 50, or 100 μm) for 15 min after 6 or 7 d in vitro. Glutamate and all other chemicals were added directly to the medium. Before exposure to glutamate, neurons were pretreated for 30 min with one of the following chemicals; non-NMDA receptor antagonist CNQX (10 μm), NMDA receptor antagonist MK-801 (10 μm), L-type Ca2+channel blocker nifedipine (10 μm), Ca2+-free medium with Ca-chelator EGTA (1 mm), PKA inhibitor Rp-8-cAMPs (100 μm), KT5720 (2 μm), PKC inhibitor chelerythrine (10 μm), PKG inhibitor Rp-8-Br-cGMPs (50 μm), CaMK inhibitor KN93 (30 μm), mitogen-activated protein kinase kinase (MEK) inhibitor PD98059 (50 μm), tyrosine kinase inhibitor genistein (200 μm), phosphatidylinositol 3-kinase (PI3K) inhibitor wortmannin (400 nm), and the broad spectrum protein kinase inhibitor staurosporine (100 nm). Neurons were also coincubated with these drugs during exposure to glutamate for 15 min that followed pretreatment. The incubation medium was then changed completely.
Treatment of cells in culture with CRE-decoy oligonucleotide. Treatment with CRE-decoy oligonucleotide was performed as described recently with some modifications (Park et al., 1999, 2001). CRE-decoy and control oligonucleotides used in the present study were phosphorothioate oligonucleotides (OligoExpress; Amersham Pharmacia Biotech, Tokyo, Japan). Their sequences were as follows: 24 mer CRE-decoy, 5′-TGACGTCATGACGTCATGACGTCA-3′ and 24 mer nonsense–sequence control, 5′-CTAGCTAGCTAGCTAGCTAGCTAG-3′. Because the CRE cis-element, TGACGTCA, is palindromic, it was shown that CRE-decoy oligonucleotide self-hybridized to form a duplex–hairpin and compete with CRE enhancers for binding transcription factors, and specifically interfere with CRE-directed transcription in vivo (Park et al., 1999). To increase the delivery of oligonucleotide into the cell, cationic lipidN-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP; Boehringer Mannheim, Mannheim, Germany) was used in the oligonucleotide treatment. The CRE-decoy and control oligonucleotides were added (1 d before exposure to glutamate) to the cells at 150 nm in the presence of DOTAP. At 24 hr of incubation, glutamate (100 μm) were added directly to the medium as described before.
For cellular localization of CRE-decoy oligonucleotide, cells were incubated with 150 nm of fluorescein isothiocyanate (FITC)-labeled CRE-decoy oligonucleotide (5′-end labeled; Oligoexpress; Amersham Pharmacia Biotech, Arlington Heights, IL) in the presence of DOTAP. Six hours after incubation, the medium was removed, and cells were washed twice with PBS and were cultured in fresh growth medium. The distribution of FITC-labeled oligonucleotides was analyzed by fluorescence inverted microscope.
Western blot analysis. Cultured neurons were extracted, and the adult frozen hippocampi were homogenized in 2% SDS solution and boiled for 5 min. Protein concentrations were determined with a DC Protein Assay (Bio-Rad, Hercules, CA), and samples were mixed with 2× Laemmli sample buffer. An equal amount of protein for each sample was separated by 12.5% SDS-PAGE and transferred onto a polyvinylidine difluoride sheet (Immobilon P; Millipore, Bedford, MA). Membranes were then incubated in a blocking buffer (PBS and 0.1% Tween 20) containing 5% skimmed milk powder for 1 hr at room temperature. Blots were then incubated with polyclonal anti-CREB (1:400), anti-phosphorylated CREB (anti-pCREB; 1:200), anti-α-tubulin (1:1000), or anti-BCL-2 (1: 100) at 4°C overnight. The membranes were then washed three times in the blocking buffer and incubated for 1 hr with anti-rabbit IgG or anti-mouse IgG conjugated to HRP (1:1000; Amersham Pharmacia Biotech). The final reaction products were visualized using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Buckinghamshire, UK), and the membranes were exposed to x-ray film. Densitometric analysis of CREB, pCREB, and other proteins was performed with a microcomputer imaging device (MCID/M2, version 3.0 Image Analysis; Imaging Research Inc., St. Catharines, Ontario, Canada).
Immunohistochemistry and immunocytochemistry. After treatment with 2% H2O2 for 10 min to eliminate endogenous peroxidase activity, brain sections were incubated with anti-CREB antibody (1:400), anti-pCREB antibody (1:200), or anti-NeuN antibody (1:200) overnight at 4°C. Sections were then incubated with secondary antibody: biotinylated anti-rabbit IgG (1:120), rhodamine-conjugated anti-rabbit IgG (Calbiochem; 1:200), or FITC-conjugated anti-mouse IgG (Calbiochem; 1:200). Sections reacted with biotinylated anti-rabbit IgG were incubated further with avidin-biotinylated horseradish peroxidase complex (ABC) (1:50 in 50 mm Tris-NaCl; ABC Elite kit; Vector Laboratories, Burlingame, CA) for 45 min. Reaction products were visualized by treatment for 1–5 min with 0.05% 3,3-diaminobenzidine tetrahydrochloride (DAB) solution in 50 mm Tris-NaCl containing 0.005% hydrogen peroxide or with 3-amino-9-ethylcarbazole (AEC) solution (Vector Laboratories). To evaluate the specificity of the anti-pCREB antibody, an absorption test was performed with the antibody pre-absorbed with the synthetic peptide (KRREILSRRPpSYRK) specific to the recognition site. Brain sections reacted with immunofluorescent secondary antibodies were examined under laser confocal-scanning microscopy (ZEISS LSM 410). X-gal staining was performed as follows: frozen sections after perfusion fixation with Zamboni's solution were washed three times with PBS while on ice and then incubated overnight at 37°C in the reaction solution consisting of 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mm MgCl2, and 1 mg/ml 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside (X-gal) (Sigma) in PBS. After reaction with X-gal, the sections were immunostained with anti-pCREB antibody as described above and finally visualized with the AEC solution.
For immunocytochemical studies, neurons were cultured in two-chamber glass slides and incubated as described above. After exposure to glutamate, the neurons were fixed immediately in 4% paraformaldehyde (PFA) for 15 min and permeabilized with 0.01% Triton X-100. Cells were then incubated with anti-CREB (1:400), anti-pCREB (1:400), anti-MAPs (1:200), monoclonal anti-MAP2 (1:100), anti-β-galactosidase (1:100), or anti-BCL2 antibody (1:100) for 1 hr at room temperature. The sections were then washed in three changes of PBS, incubated for 1 hr in a 1:200 dilution of rhodamine- or FITC-labeled secondary antibody, and evaluated using a confocal microscope.
Cell viability assay. Quantitative assessments of neuronal injury were accomplished by measuring the lactate dehydrogenase (LDH) activity in the media 24 hr after exposure to glutamate (20, 50, or 100 μm for 15 min) with the cytotoxicity detection kit (Boehringer Mannheim) and by counting the viable cells in the photographs taken under phase-contrast microscopy.
Statistical analysis. Statistical analysis was performed by one-way ANOVA followed by Scheffe's test. p < 0.05 was considered statistically significant.
RESULTS
Phosphorylation status of CREB after transient global ischemia in gerbils
We performed Western blot analyses to examine the temporal profile of the phosphorylation status of CREB in the post-ischemic hippocampus after 5 min of ischemia. As shown in Figure1A, total CREB levels were unchanged in the non-ischemic (control) animals and during ischemia and reperfusion. In contrast, pCREB levels decreased during ischemia, but increased during reperfusion peaking at 15 min, and then gradually declined.
Fig. 1.

CREB phosphorylation at Ser133in gerbil hippocampus after transient forebrain ischemia.A, Western blot of proteins isolated from post-ischemic hippocampal tissues at time 0–180 min after transient ischemia for 5 min. In the top panel, total CREB levels were not changed. However, pCREB levels were below the detection limit at the end of ischemia (0 min) but began to increase relative to controls and remained phosphorylated for at least 3 hr after reperfusion. The data in the bottom panel (bar graph) are presented as fold increase in pCREB, as determined by the optical density analysis of phosphorylated and total CREB levels. CREB phosphorylation was increased significantly from 5 to 30 min after reperfusion. Each column and bar represents mean ± SD (n = 7 for each recirculation period); *p < 0.05 versus control.B, Immunostaining for pCREB in the gerbil hippocampus after reperfusion for 15 min. pCREB was detected primarily in the nuclei of pyramidal cells, dentate granule cells, and neocortical neurons from 15 to 60 min after reperfusion. Left panel, Lower magnification image of the hippocampus. Scale bar, 500 μm.Middle panel, Higher magnification image of CA1 pyramidal neurons. Scale bar, 50 μm. Right panel, Absorption test to confirm the specificity of the anti-pCREB antibody.
To study pCREB levels in neurons, we also performed an immunohistochemical study using ABC–DAB and immunofluorescent staining of brain sections from animals after recirculation for 15 min. pCREB was detected in the nuclei of hippocampal pyramidal cells, dentate granule cells, and neocortical neurons during recirculation for 15 min (Fig. 1B) to 60 min (data not shown). The immunoreaction was effectively blocked by preabsorption of the anti-pCREB antibody with the synthetic peptide (KRREILSRRPpSYRK) (Fig.1B).
We investigated the presence of pCREB in CA1 pyramidal neurons by double immunostaining for pCREB and NeuN, a neuron-specific marker, to confirm that pCREB was expressed primarily in neurons. Although the level of staining and the distribution of NeuN were not affected by ischemia (Fig. 2, top), the number of ischemia-induced pCREB-positive cells was increased markedly after reperfusion for 15 min (Fig. 2, middle). In the hippocampus, most pCREB-positive cells were also NeuN-positive (Fig. 2,bottom).
Fig. 2.
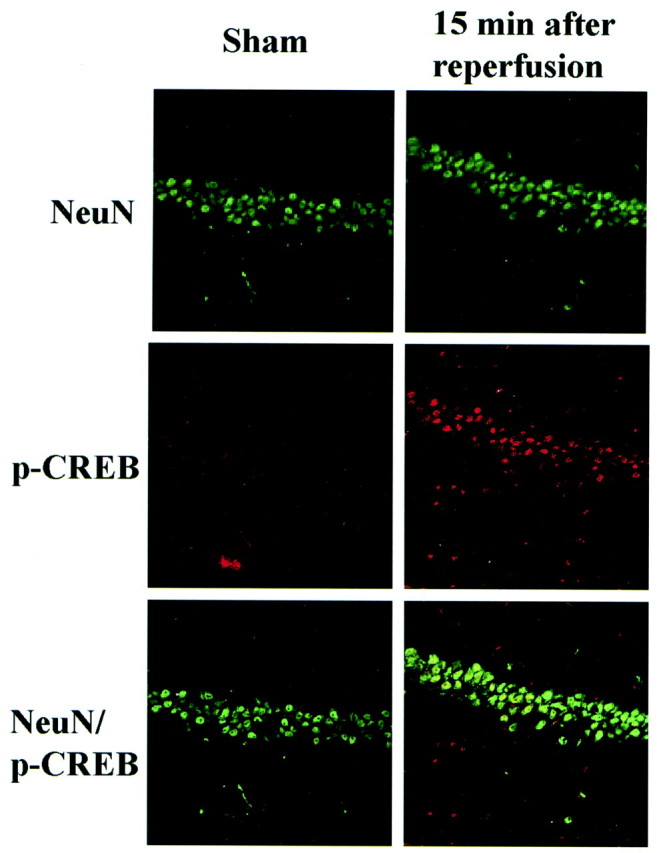
Expression of pCREB in hippocampal neurons after ischemia. High-magnification confocal images of double immunostaining of NeuN (green) and pCREB (red) in CA1 pyramidal neurons 15 min after reperfusion (right column) compared with sham-operated animals (left column). Top, The number of pyramidal neurons in the CA1 region was unchanged with an antibody specific for NeuN (green). Middle, The number of pCREB-positive cells (red) was increased markedly after reperfusion for 15 min. Bottom, Double immunofluorescence with both antibodies (yellow), showing colocalization in many neurons after reperfusion.
We also investigated the effects of milder ischemia (i.e., 1 or 2 min) on CREB phosphorylation. There was no enhancement of CREB phosphorylation in the gerbil hippocampus after reperfusion for 15 min after global ischemia for 1 min, however, after ischemia for 2 min, CREB phosphorylation was significantly enhanced compared with that in control animals, although it was still less than that after ischemia for 5 min (Fig. 3A). We also investigated whether the NMDA receptor played a role in CREB phosphorylation and induction of tolerance after forebrain ischemia. We found that enhancement of CREB phosphorylation after transient forebrain ischemia was prevented by intraperitoneal injection of 3 mg/kg of an NMDA receptor antagonist MK801, 60 min before ischemia (Fig. 3B). Two minutes of transient bilateral carotid occlusion induced tolerance in the CA1 neurons against subsequent lethal (5 min) ischemic insult (Fig. 3C), as described previously (Kitagawa et al., 1990). Administration of MK801 before preconditioning ischemia for 2 min also diminished induction of tolerance in CA1 neurons (Fig. 3C), as described previously (Kato et al., 1992).
Fig. 3.
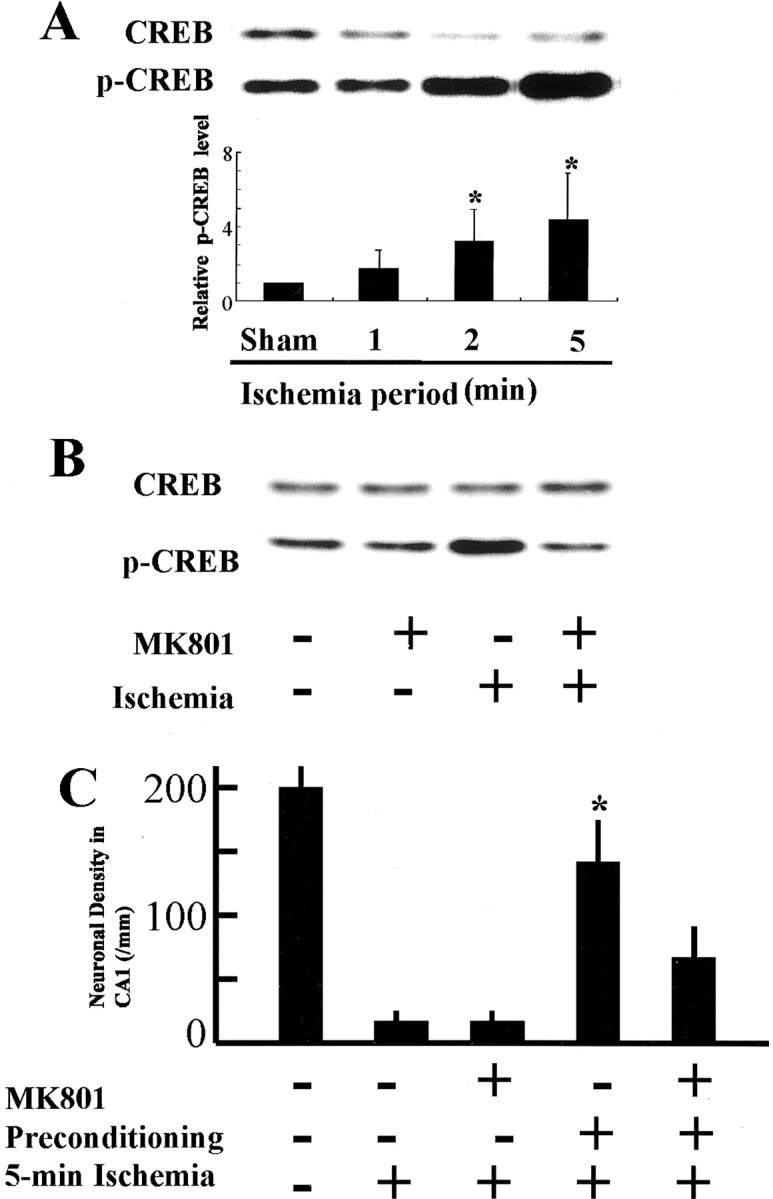
CREB phosphorylation and ischemic tolerance induced after sublethal stress were inhibited by MK801. Effects of the length of ischemia and NMDA receptor antagonist MK801 on CREB phosphorylation after reperfusion for 15 min (A, B).A, Western blot analyses of proteins extracted from the gerbil hippocampus after reperfusion for 15 min after transient forebrain ischemia for 1, 2, or 5 min (top panel). The bar graph (bottom panel) is presented as fold increase in pCREB levels after global ischemia. There was no enhancement of CREB phosphorylation after 1 min of ischemia, whereas both 2 and 5 min of ischemia induced significant increase in pCREB levels. Each column and bar represents the mean ± SD (n = 7 for each ischemic period); *p < 0.05 versus sham-operated animals.B, Intraperitoneal administration of MK801 60 min before transient forebrain ischemia prevented CREB phosphorylation observed after ischemia-reperfusion (n = 3 each).C, Intraperitoneal administration of MK801 before 2 min of preconditioning ischemia abolished neuronal protection. Ischemic tolerance was induced by 2 min of ischemia 4 d before 5 min of ischemia. Each column and bar represents the mean ± SD of neuronal density in the CA1 (n = 6 for each experiment). *p < 0.05 versus all other groups with 5 min of ischemia.
CREB phosphorylation after exposure to glutamate in cultured neurons
Glutamate (100 μm) induced phosphorylation of Ser133 in CREB in primary cultures from rat hippocampus. CREB phosphorylation peaked at 10 min and returned to the baseline level by 180 min after exposure to glutamate (Fig.4). The pattern with rapid and transient enhancement of CREB phosphorylation was similar to that observed in gerbil hippocampus after global ischemia (Fig. 1).
Fig. 4.

CREB phosphorylation at Ser133induced by exposure to glutamate in cultured neurons. Top panel, Immunoblots of primary cultures from rat hippocampus after exposure to glutamate (100 μm) for the indicated periods. Levels of CREB proteins were unchanged, but those of pCREB peaked at 10 min after exposure and returned to the basal level 180 min later. The bar graph is presented as fold increase in pCREB levels with the optical density analysis of pCREB and total CREB. Each column and bar represents the mean ± SD (n = 6 for each exposure period); *p < 0.05 versus control.
To confirm the notion that the observed transient enhancement of CREB phosphorylation reflected the neuronal response to glutamate excitotoxicity, a double immunocytochemical study was performed for pCREB and NeuN. The number of pCREB-positive cells increased markedly compared with the number in untreated cells, whereas the number of NeuN-positive neurons was unchanged at 10 min after exposure to glutamate (Fig. 5). The increase in the number of double-positive neurons indicated that pCREB expression was enhanced in glutamate-stimulated neurons.
Fig. 5.
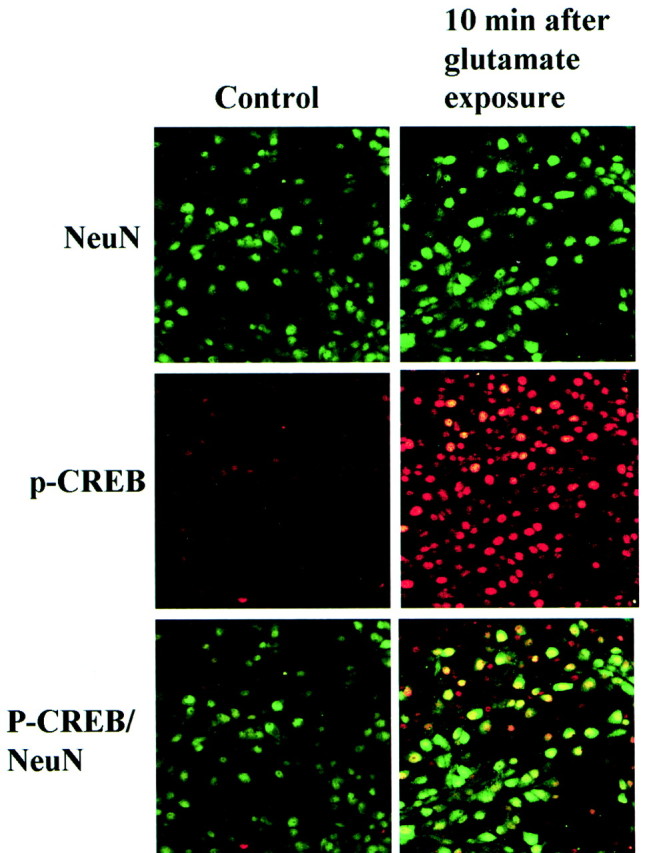
pCREB in cultured neurons after exposure to glutamate. NeuN (green) and pCREB (red) immunofluorescence in cultured hippocampal neurons 10 min after exposure to glutamate (right column) compared with controls (left column).Top, The number of neurons stained with an anti-NeuN antibody (green) was unchanged.Middle, The number of pCREB-positive cells (red) increased markedly 10 min after exposure to glutamate. Bottom, NeuN-positive–pCREB-positive cells (yellow). CREB phosphorylation was enhanced in most neurons after exposure to glutamate.
We also investigated CREB phosphorylation after milder exposure to glutamate in cultured neurons. LDH assay revealed that the concentration of glutamate ranging from 50 to 100 μM caused death in 30–50% of cultured neurons, whereas neurons exposed to 20 μm of glutamate were undamaged (Fig.6A). CREB phosphorylation status then studied after 10 min of exposure at each glutamate concentration. There was a significant increase in pCREB at 20, 50, and 100 μm of glutamate (Fig.6B). These results suggest that CREB phosphorylation may also be induced by sublethal stimuli and that CREB phosphorylation may play an important role in the neuronal response to metabolic stress.
Fig. 6.

Glutamate-induced CREB phosphorylation after sublethal stress. A, Evaluation of neuronal death by the measurement of LDH released into culture media or by the counting of damaged cells. Open columns indicate the percentage of cell death (LDH release) 24 hr after treatment with the indicated concentrations of glutamate, and shaded columns indicate the percentage of loss of cells (cell counting) 24 hr after exposure to 100 μm glutamate. B, Effects of different concentrations of glutamate (20, 50, and 100 μm) on CREB phosphorylation 10 min after an addition of glutamate, as determined by Western blot analysis. Although the total CREB level in each lane was similar, CREB phosphorylation was enhanced by 20, 50, and 100 μm glutamate. Fold increase in CREB phosphorylation is quantified in a bar graph. Each column and bar represents mean ± SD (n = 6 for each glutamate concentration); *,†p < 0.05 versus controls.
Signal transduction pathway inducing CREB phosphorylation after exposure to glutamate in cultured neurons
To examine the involvement of Ca2+influx and protein kinases in CREB phosphorylation, cultures were pretreated with various antagonists and inhibitors for 30 min before exposure to glutamate (Fig. 7). Glutamate-mediated CREB phosphorylation was dependent on extracellular Ca2+. Removal of Ca2+ from the medium prevented CREB phosphorylation. MK801 blocked glutamate-induced CREB phosphorylation, but CNQX and nifedipine did not. The increased pCREB expression after exposure to glutamate was effectively blocked by KN93 (a specific inhibitor of CaMK), and by staurosporine (a broad spectrum protein kinase inhibitor). However, glutamate-induced CREB phosphorylation was not blocked in the presence of PKA inhibitors (Rp-8-Cl-cAMPs and KT5720), PKC inhibitor (chelerythrine), PKG inhibitor (RP-8-Br-cGMPs), MEK inhibitor (PD98059), tyrosine kinase inhibitor (genestein), or PI3K inhibitor (wortmannin) (Fig. 7).
Fig. 7.

Effects of receptor antagonists, Ca2+ channel blocker, Ca2+depletion, and protein kinase inhibitors on CREB phosphorylation in cultured hippocampal neurons after exposure to glutamate. As indicated, reagents used in this study were MK-801 (10 μm), nifedipine (10 μm), Ca-free medium with CA2+-chelator EGTA (1 mm), Rp-8-cAMPs (100 μm), KT5720 (2 μm), chelerythrine (10 μm), Rp-8-cGMPs (100 μm), KN93 (30 μm), PD98059 (50 μm), genistein (200 μm), wortmannin (400 nm), and staurosporine (100 nm). Cultured neurons were treated for 30 min before the addition of and during the 10 min exposure to 100 μmglutamate. Enhanced CREB phosphorylation induced by exposure to glutamate was prevented in the Ca2+-free condition and in the presence of MK-801, KN93, and staurosporine as indicated in the graphical quantification. Data are mean ± SD (n = 6 for each treatment); *p < 0.05 versus control.
CREB phosphorylation mediates neuronal survival through BCL-2 production after exposure to glutamate
The data shown in Figure 7 suggests that CaMK may participate in the signaling pathway that mediates the neuronal response to glutamate. To examine the role of CREB phosphorylation by CaMK on neuronal survival, BCL-2 expression by cultured neurons was investigated after exposure to glutamate (100 μm) because the BCL-2 gene contains a CRE in the promoter region (Riccio et al., 1999) and shows protection against cell death (Martinou et al., 1994). Immunocytochemical analyses revealed that, after exposure to glutamate, BCL-2 was identified to localize in those cells that showed immunoreactivity to a specific neuronal marker, MAPs (Fig.8A). Western blot analysis showed that BCL-2 expression was upregulated 6 hr after exposure to glutamate. The glutamate-induced increase in BCL-2 production was inhibited by pretreatment with KN93 (Fig.8B). We also investigated the effect of KN93 on neuronal survival. Pretreatment with KN93 (30 μm) for 30 min before exposure to glutamate significantly increased the level of neuronal damage compared with that in controls (Fig. 8C), suggesting that CaMK-mediated CREB phosphorylation may contribute to neuronal survival.
Fig. 8.

Pretreatment with KN93 inhibits glutamate-induced BCL-2 production and exacerbates the neuronal cell damage observed after exposure to glutamate. A, High-magnification confocal images of MAPs (green) and BCL-2 (red) immunostaining in cultured hippocampal neurons 6 hr after exposure to glutamate (100 μm). The double-immunofluorescence image suggests that the MAP-positive cells are all BCL-2-positive (yellow).B, Western blot analyses showing upregulation of BCL-2 6 hr after exposure to glutamate; this increase was prevented by pretreatment with KN93 (top panel). The bar graph is presented as fold increase of BCL-2 expression. The densitometric values were normalized to β-tubulin levels. Data are mean ± SD (n = 6 for each treatment); *p < 0.05 versus controls;†p < 0.05 versus glutamate treatment without KN93. C, Neuronal cell death 24 hr after exposure to glutamate was increased significantly when CREB phosphorylation was prevented by pretreatment with KN93 (30 μm). Data are mean ± SD (n = 6 for each treatment); *p < 0.05 versus control;†p < 0.05 versus glutamate treatment without KN93.
To determine the role of CREB in neuroprotection and survival in this model, we examined the effect of CRE-decoy oligonucleotide on CREB phosphorylation, BCL-2 production and cell viability after glutamate exposure. After 6 hr incubation at 150 nm, most neurons on the plate showed uptake of FITC-conjugated CRE-decoy oligonucleotide (Fig. 9A). CREB phosphorylation at 10 min after exposure to glutamate was not inhibited by CRE-decoy or control oligonucleotide (Fig. 9B), however, BCL-2 production at 6 hr after exposure to glutamate for 15 min was suppressed by CRE-decoy oligonucleotide (Fig. 9B). Pretreatment with CRE-decoy oligonucleotide also significantly increased the level of neuronal damage compared with that in controls (Fig. 9C). Pretreatment with control oligonucleotide did not suppress BCL-2 production or increase the level of cell damage compared with that in controls (Fig. 9B,C). Immunocytochemistry for MAP2 showed neuronal cytoplasma and dendrites in cultured neurons (Fig.9A). The number of MAP2-positive neurons decreased 24 hr after glutamate exposure for 15 min. Again, pretreatment with CRE-decoy oligonucleotide accelerated the decrease of the number of MAP2-positive neurons after glutamate exposure (Fig. 9A).
Fig. 9.
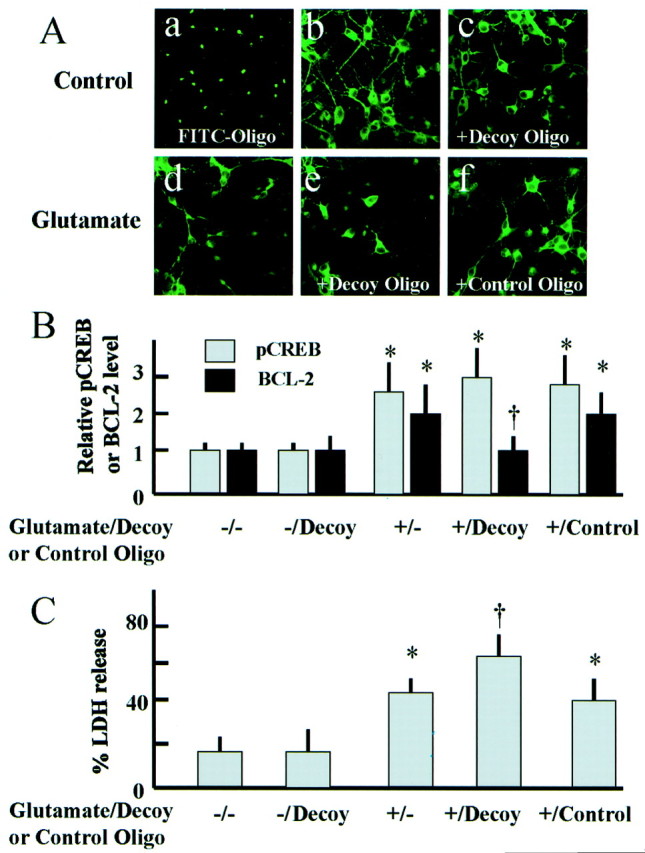
Pretreatment with CRE-decoy oligonucleotide inhibits glutamate-induced BCL-2 production and exacerbates the neuronal cell damage observed after exposure to glutamate.Aa, Cellular uptake of FITC-conjugated oligonucleotide (FITC-Oligo) after incubation for 6 hr.b–f, MAP2 immunostaining of control neurons (b, c) and of neurons 24 hr after exposure to glutamate (100 μm) (d–f). Pretreatment with CRE-decoy or control oligonucleotide was indicated with +Decoy Oligo and +Control Oligo in the figures, respectively. Most neurons on the plate showed uptake of FITC-conjugated oligonucleotide in the presence of DOTAP (a). In control neurons, immunocytochemistry for MAP2 showed neuronal cytoplasma and dendrites (b, c). The number of MAP2-positive neurons decreased 24 hr after glutamate exposure for 15 min (d). Pretreatment with CRE-decoy oligonucleotide (e), not with control oligonucleotide (f), accelerated the decrease of the number of MAP2-positive neurons after glutamate exposure.B, Effect of pretreatment with CRE-decoy or control oligonucleotide on CREB phosphorylation and BCL-2 production after exposure to glutamate (100 μm). The data in the bar graph is presented as fold increase in pCREB and BCL-2 levels. The densitometric values were normalized to CREB and β-tubulin levels, respectively. Data are mean ± SD (n = 6 for each treatment); *p < 0.05 versus controls;†p < 0.05 versus glutamate treatment without oligonucleotide. C, Effect of pretreatment with CRE-decoy or control oligonucleotide on neuronal cell death after exposure to glutamate (100 μm). Neuronal cell death 24 hr after exposure to glutamate (100 μm) increased significantly by pretreatment with CRE-decoy oligonucleotide. Data are mean ± SD (n = 6 for each treatment); *p < 0.05 versus control;†p < 0.05 versus glutamate treatment without oligonucleotide.
CREB phosphorylation and CRE-mediated gene transcription after ischemia and glutamate exposure in CRE–LacZ transgenic mice
In cultured neurons prepared from control transgenic mice, there were only few pCREB- or β-galactosidase-positive cells (Fig.10). However, the number of pCREB- and β-galactosidase- positive cells increased markedly 30 min after exposure to glutamate (100 μm) (Fig.10A,B). A subset of pCREB-positive neurons were also β-galactosidase-positive. Pretreatment with KN93, CaMK inhibitor, decreased the number of pCREB- and β-galactosidase-positive cells (Fig. 10).
Fig. 10.
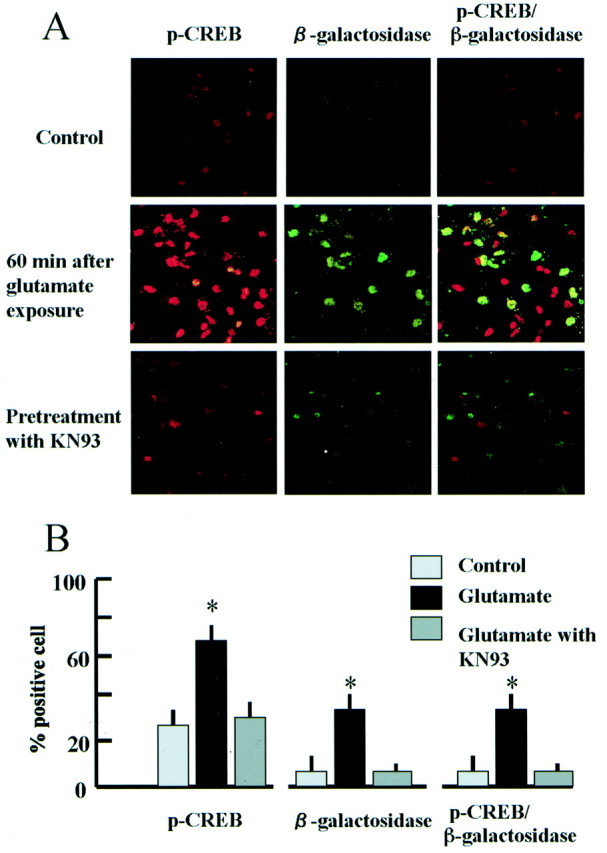
CRE-mediated LacZ transcription in cultured neurons was upregulated by exposure to glutamate. A,High-magnification confocal images of immunostaining for pCREB (red), β-galactosidase (green), and both proteins (yellow) in cultured hippocampal neurons from CRE–LacZ transgenic mice. Top, The number of pCREB-positive cells was small, and β-galactosidase expression was weak without stimulation. Middle, The number of pCREB-positive cells (red) increased markedly 60 min after exposure to glutamate (100 μm), and β-galactosidase expression was also enhanced in a subset of pCREB-positive cells (yellow).Bottom, Both CREB phosphorylation and β-galactosidase expression were inhibited by pretreatment with KN93. B, Frequency of pCREB-, β-galactosidase, or both-positive cells in control and glutamate exposure with or without KN93 (n = 4 for each group); *p < 0.05 versus control.
In brain sections from sham-operated CRE–LacZ transgenic mice, there were only few pCREB-positive or X-gal-positive cells (Fig.11A). After reperfusion for 60 min, most pyramidal neurons in the hippocampus were pCREB-positive (red cells in Fig. 11B,D). X-gal-positive cells (blue cells in Fig.11B) were also observed, but the number was smaller than that of pCREB-positive cells. Most X-gal-positive cells were also pCREB-positive (Fig. 11C,D).
Fig. 11.

CRE-mediated LacZ transcription was increased in pCREB-positive hippocampal neurons by ischemic stress. pCREB-positive cells were primarily neurons (red cells), and most X-gal-positive cells (blue cells) were pCREB-positive neurons (red cells) in CRE–LacZ transgenic mice after transient forebrain ischemia. A, Nonischemic control hippocampal CA1 sector. Neither pCREB nor X-gal reactivity was observed. Scale bar, 50 μm. B, Hippocampal CA1 pyramidal neurons in CRE–LacZ transgenic mice after transient ischemia. Scale bar, 50 μm. C, Higher magnification image of the rectangle in B. X-gal-positive cells (arrows) corresponded to red cells immunoreactive for pCREB. Scale bar, 10 μm. D,Frequency of pCREB-, X-gal-, or both-positive cells in control and ischemic hippocampus (n = 4 for each group); *p < 0.05 versus control.
DISCUSSION
The present study demonstrated that transient CREB phosphorylation and subsequent CRE-mediated gene transcription occurred after exposure to glutamate in cultured neurons and after ischemic insult in adult gerbil hippocampal neurons. Transient exposure to glutamate mimics the increase of the extracellular glutamate concentration during and after transient global ischemia. Previous studies have demonstrated that stimulation with glutamate or NMDA caused transient CREB phosphorylation in cultured neurons and hippocampal slices (Bito et al., 1996; Hardingham et al., 1999; Hu et al., 1999; Rajadhyaksha et al., 1999; Vanhoutte et al., 1999; Sala et al., 2000).
Our first goal was to clarify the extracellular and intracellular signal pathways leading to CREB phosphorylation after exposure to glutamate. CREB phosphorylation can be induced by extracellular signals such as glutamate, growth factors, and membrane depolarization, and is mediated through several kinases including PKA, PKC, CaMK, mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2, and the pp90 ribosomal S6 kinase family (Rsks) (Imprey et al., 1998;Finkbeiner, 2000). Recently, neurotrophin-mediated survival of cerebellar granule cells was found to be at least in part because of CREB phosphorylation by MAPK–Rsks (Bonni et al., 1999). Our pharmacological studies with several kinase inhibitors showed that CREB phosphorylation after exposure to glutamate was dependent on calcium influx through NMDA-type receptors and on CaMK II–IV, but not mediated through PKA, PKC, cGMP-dependent protein kinase, PI3 kinase, or the MAPK cascade. Our results were consistent with those observed after electrical stimulation of hippocampal neurons (Bito et al., 1996). Therefore, it is likely that the same intracellular pathway that mediates the activity-dependent neuronal plasticity is also involved in CREB phosphorylation after cytotoxic exposure to glutamate, although neurons in different brain regions, such as the striatum, may respond to glutamate and phosphorylate CREB through the MAPK cascade (Perkinton et al., 1999; Rajadhyaksha et al., 1999; Vanhoutte et al., 1999). The decline of CREB phosphorylation observed 1 hr after exposure to glutamate in our study would be ascribed to activation of protein phosphatases linked to NMDA receptors and calcium influx (Bito et al., 1996; Sala et al., 2000).
What is the role of CREB phosphorylation after exposure to glutamate?
In our study, several treatments including calcium removal and preincubation with MK801, staurosporine, and KN93 inhibited CREB phosphorylation after exposure to glutamate. We used KN93 to inhibit CaMKII–IV, because CaMKII–IV can phosphorylate CREB directly, and CaMKIV overexpression was shown to inhibit apoptosis induced by potassium deprivation in cerebellar granule neurons (See et al., 2001). Pretreatment with KN93 inhibited CREB phosphorylation and increased the degree of neuronal injury after exposure to glutamate, suggesting that CREB phosphorylation after exposure to glutamate may be important for cell survival. The experiment with CRE-decoy oligonucleotide also supported the neuroprotective role of CRE-mediated gene expression that follows CREB phosphorylation in exposure to glutamate. CREB overexpression in PC12 and Neuro2A cells was also shown to inhibit apoptosis induced by okadaic acid (Walton et al., 1999). Recently, the BCL-2 gene was found to have a CRE in the 5′ promoter region, and cell survival mediated by neurotrophin-induced CREB phosphorylation in sympathetic and cortical neurons was associated with increased BCL-2 expression (Riccio et al., 1999). Accumulating evidence indicates that overexpression of BCL-2 provides protection against apoptosis (Martinou et al., 1994) and ischemic neuronal death (Lawrence et al., 1996;Kitagawa et al., 1998). Thus, the protective effect of CREB phosphorylation against glutamate- or ischemia-induced neuronal degeneration may be attributable to increased expression of BCL-2. In the present study, we observed increased BCL-2 expression 6 hr after exposure to glutamate, which was inhibited by pretreatment of cultured neurons with KN93 or CRE-decoy oligonucleotide, suggesting that BCL-2 expression was induced by CaMK-mediated CREB activation and involved in the neuroprotective role of CREB after exposure to glutamate.
CREB phosphorylation has been examined in several experimental stroke models. At first, Walton et al. (1996) showed that ischemia-resistant granule cells produced increase in pCREB immunoreactivity, peaking 6 and 48 hr after a unilateral hypoxic–ischemic injury in the 21-d-old rat. Later, Hu et al. (1999) demonstrated that CREB phosphorylation was induced in the adult rat hippocampus, mainly in the resistant dentate granule cells, several hours after transient global ischemia for 15 min. It was also shown that in the focal-ischemia model, CREB phosphorylation was marked in the peri-infarct area (Tanaka et al., 1999; Irving et al., 2000). CREB phosphorylation in the surviving neurons was also shown in the chick cochlear nucleus after activity deprivation by cochlea removals (Zirpel et al., 2000). However, the temporal profile of CREB phosphorylation immediately after ischemia has not been fully examined, although early phosphorylation of CREB can be expected from the expression pattern of immediate early genes such as c-Fos after ischemia (Kiessling et al., 1993), exposure to glutamate (Hardingham et al., 1999), and electrical stimulation (Bito et al., 1996; Sgambato et al., 1998). Hata et al. (1998) also reported involvement of CREB activation in c-fos mRNA expression after middle cerebral artery occlusion in CREB knock-out mice. Our study demonstrated that CREB was phosphorylated in hippocampal neurons immediately after a brief period (5 min) of ischemia and returned to control levels by 60 min after reperfusion. The molecular pathway leading to ischemia-induced early CREB phosphorylation in vivo is primarily unknown, but our experiments with MK801 suggested that the activation of NMDA-type receptor was important. This mechanism has been known for CREB phosphorylation in response to high-frequency stimulation causing long-term potentiation (Schulz et al., 1999).
Both in vivo and in vitro findings in the present study suggested that common pathways for CREB phosphorylation were activated in response to electrical stimulation and cytotoxic exposure to glutamate including Ca2+ influx, NMDA-type receptor activation, and CaM kinase. CREB phosphorylation observed after exposure to glutamate in vitro and ischemiain vivo may represent the cellular protective response against metabolic stresses. This notion is supported by the finding that both lethal and nonlethal stresses with glutamate in cultured neurons and brief ischemia for 2 min in gerbil hippocampus induced CREB phosphorylation. The latter finding suggests that CREB phosphorylation and subsequent gene expression may play an important role in the acquisition of ischemic tolerance, where nonlethal ischemic stress makes neurons resistant to subsequent severe ischemic insult (Kitagawa et al., 1990; Kirino et al., 1991).
Footnotes
This work was supported by a Grant-in-aid for Scientific Research on Priority Areas (A). We thank Y. Nishizawa and R. Morimoto for secretarial assistance.
Correspondence should be addressed to Dr. Kazuo Kitagawa, Division of Strokology, Department of Internal Medicine and Therapeutics (A8), Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita City, Osaka 565-0871, Japan. E-mail:kitagawa@medone.med.osaka-u.ac.jp.
REFERENCES
- 1.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 2.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and –independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 3.Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh A, Greenberg ME. Calcium signaling in neurons: Molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 5.Hardingham GE, Chawia S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- 6.Hata R, Gass P, Mies G, Wiessner C, Hossmann KA. Attenuated c-fos mRNA induction after middle cerebral artery occlusion in CREB knockout mice does not modulate focal ischemic injury. J Cereb Blood Flow Metab. 1998;18:1325–1335. doi: 10.1097/00004647-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Higashi T, Nakai A, Uemura Y, Kikuchi H, Nagata K. Activation of heat shock factor 1 in rat brain during cerebral ischemia or after heat shock. Mol Brain Res. 1995;34:262–270. doi: 10.1016/0169-328x(95)00163-m. [DOI] [PubMed] [Google Scholar]
- 8.Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH. Persistent phosphorylation of cyclic AMP responsive element-binding protein and activating transcription factor-2 transcription factors following transient cerebral ischemia in rat brain. Neuroscience. 1999;89:437–452. doi: 10.1016/s0306-4522(98)00352-2. [DOI] [PubMed] [Google Scholar]
- 9.Hu SC, Chrivia J, Ghosh A. Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron. 1999;22:799–808. doi: 10.1016/s0896-6273(00)80738-2. [DOI] [PubMed] [Google Scholar]
- 10.Imprey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 11.Imprey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 12.Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurons and glia following focal cerebral ischemia in the rat. Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- 13.Kato H, Liu Y, Araki T, Kogure K. MK-801,but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- 14.Kiessling M, Stumn G, Xie Y, Herdegen T, Aguzzi A, Bravo R, Gass P. Differential transcription and translation of immediate early genes in the gerbil hippocampus after transient global ischemia. J Cereb Blood Flow Metab. 1993;13:914–924. doi: 10.1038/jcbfm.1993.114. [DOI] [PubMed] [Google Scholar]
- 15.Kirino T, Tsujita Y, Tamura A. Induced tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood Flow Metab. 1991;11:299–307. doi: 10.1038/jcbfm.1991.62. [DOI] [PubMed] [Google Scholar]
- 16.Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou JC, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- 17.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, Kamada T. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 18.Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: Evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab. 1998;18:570–579. doi: 10.1097/00004647-199805000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM. Overexpression of BCL-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo. J Neurosci. 1996;16:486–496. doi: 10.1523/JNEUROSCI.16-02-00486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia. Proc Natl Acad Sci USA. 1992;89:648–652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 22.Mattson MP, Kater SB. Isolated hippocampal neurons in cryopreserved long-term cultures: development of neuroarchitecture and sensitivity to NMDA. Int J Dev Neurosci. 1988;6:439–452. doi: 10.1016/0736-5748(88)90050-0. [DOI] [PubMed] [Google Scholar]
- 23.Martinou JC, Dubois-Dauphins M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto RI. Cells in stress: Transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 25.Ninatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1010. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park YG, Nesterova M, Agrawal S, Cho-Chung YS. Dual blockade of cyclic AMP response element- (CRE) and AP-1-directed transcription by CRE-transcription factor decoy oligonucleotide. J Biol Chem. 1999;274:1573–1580. doi: 10.1074/jbc.274.3.1573. [DOI] [PubMed] [Google Scholar]
- 27.Park YG, Park S, Lim SO, Lee KS, Ryu CK, Kim I, Cho-Chung YS. Reduction in cyclin D1/Cdk4/retinoblastoma protein signaling by CRE-decoy oligonucleotide. Biochem Biophys Res Commun. 2001;281:1213–1219. doi: 10.1006/bbrc.2001.4521. [DOI] [PubMed] [Google Scholar]
- 28.Perkinton MS, Sihra TS, Williams RJ. Ca2+-permeable AMPA receptors induce phosphorylation of camp response element-binding protein through a phosphatidylinositol 3-kinase dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci. 1999;19:5861–5874. doi: 10.1523/JNEUROSCI.19-14-05861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajadhyaksha A, Barczak A, Macias W, Leveque JC, Lewis SE, Konradi C. L-type Ca2+ channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci. 1999;19:6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 31.Sala C, Rudolph-Correia S, Sheng M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci. 2000;20:3529–3536. doi: 10.1523/JNEUROSCI.20-10-03529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz S, Siemer H, Krug M, Holt V. Direct evidence for biphasic cAMP responsive element-binding protein phosphorylation during long-term potentiation in the rat dentate gyrus in vivo. J Neurosci. 1999;19:5683–5692. doi: 10.1523/JNEUROSCI.19-13-05683.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.See V, Boutillier AL, Bito H, Loeffler JP. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 2001;15:134–144. doi: 10.1096/fj.00-0106com. [DOI] [PubMed] [Google Scholar]
- 34.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 36.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka K, Nogawa S, Nagata E, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Temporal profile of CREB phosphorylation after focal ischemia in rat brain. NeuroReport. 1999;10:2245–2250. doi: 10.1097/00001756-199908020-00004. [DOI] [PubMed] [Google Scholar]
- 38.Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M. The role of the cyclic AMP-responsive element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Mol Brain Res. 1996;43:21–29. doi: 10.1016/s0169-328x(96)00144-1. [DOI] [PubMed] [Google Scholar]
- 40.Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- 41.Walton MR, Dragunow M. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- 42.Yagita Y, Kitagawa K, Taguchi A, Ohtsuki T, Kuwabara K, Mabuchi T, Matsumoto M, Yanagihara T, Hori M. Molecular cloning of a novel member of the HSP110 family of genes, ischemia-responsive protein 94 kDa (irp94), expressed in rat brain after transient forebrain ischemia. J Neurochem. 1999;72:1544–1551. doi: 10.1046/j.1471-4159.1999.721544.x. [DOI] [PubMed] [Google Scholar]
- 43.Zirpel L, Janowiak MA, Veltri CA, Parks TN. AMPA receptor-mediated, calcium-dependent CREB phosphorylation in a subpopulation of auditory neurons surviving activity deprivation. J Neurosci. 2000;20:6267–6275. doi: 10.1523/JNEUROSCI.20-16-06267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]