

Abstract
Background:
Olanzapine, regarded as one of the most efficacious antipsychotic medications for the treatment of schizophrenia, is associated with a high risk of weight gain and metabolic dysfunction. ALKS 3831, a clinical candidate for treatment of schizophrenia, is a combination of olanzapine and samidorphan, an opioid receptor antagonist. The addition of samidorphan is intended to mitigate weight gain and the metabolic dysregulation associated with the use of olanzapine.
Methods:
Non-clinical studies were conducted to assess the metabolic effects of olanzapine and samidorphan alone and in combination at clinically relevant exposure levels.
Results:
Chronic olanzapine administration in male and female rats shifted body composition by increasing adipose mass, which was accompanied by an increase in the rate of weight gain in female rats. Co-administration of samidorphan normalized body composition in both sexes and attenuated weight gain in female rats. In hyperinsulinemic euglycemic clamp experiments conducted prior to measurable changes in weight and/or body composition, olanzapine decreased hepatic insulin sensitivity and glucose uptake in muscle while increasing uptake in adipose tissue. Samidorphan appeared to normalize glucose utilization in both tissues, but did not restore hepatic insulin sensitivity. In subsequent studies, samidorphan normalized olanzapine-induced decreases in whole-body glucose clearance following bolus insulin administration. Results from experiments in female monkeys paralleled the effects in rats.
Conclusions:
Olanzapine administration increased weight gain and adiposity, both of which were attenuated by samidorphan. Furthermore, the combination of olanzapine and samidorphan prevented olanzapine-induced insulin insensitivity. Collectively, these data indicate that samidorphan mitigates several metabolic abnormalities associated with olanzapine in both the presence and the absence of weight gain.
Keywords: Olanzapine, atypical antipsychotic, opioid, metabolic dysfunction, weight gain
Introduction
Schizophrenia is a psychiatric disorder that affects more than 21 million people, and is among the top 10 causes of disability worldwide (World Health Organization, 2018). Second generation atypical antipsychotic medications, which have decreased risk of extrapyramidal effects associated with first generation antipsychotics, are commonly used to treat schizophrenia (Freedman, 2003). However, nearly all antipsychotic medications, including atypical agents, are associated with significant weight gain, metabolic dysfunction and increased risk of type 2 diabetes mellitus (T2DM) (Bak et al., 2014; De Hert et al., 2011b; Leucht et al., 2012). Olanzapine (OLZ) is considered to be one of the more efficacious atypical antipsychotics (Leucht et al., 2013; Lieberman et al., 2005), but its use is limited by frequent observations of weight gain and deleterious metabolic sequelae (De Hert et al., 2011a; Lieberman et al., 2005). Moreover, acute OLZ administration produces weight-independent insulin resistance in patients (Vidarsdottir et al., 2010). These data indicate that OLZ-induced metabolic adverse side effects may develop in the absence of changes in body weight and fat mass. As a result, pharmacotherapeutic options to treat schizophrenia with favorable metabolic profiles are warranted.
Preclinical and clinical studies have provided evidence for a critical role of the opioid system in mediating food reward, feeding behavior and metabolism. In genetic studies, single nucleotide polymorphisms within the OPRM1 gene (μ-opioid receptor) have been associated with T2DM susceptibility (Gallagher et al., 2006). Furthermore, in carriers of the OPRM1 locus (rs2281617) there is a decreased preference for fat intake (Haghighi et al., 2014). A 14-base pair deletion within the pro-opiomelanocortin (precursor for β-endorphin) gene was associated with weight gain and increased food motivation in dogs (Raffan et al., 2016). In contrast, a decrease in weight gain has been reported in μ-, κ-, and δ-opioid receptor knockout (KO) mice despite no differences in caloric intake in μ- and κ-opioid receptor KOs (Czyzyk et al., 2010, 2012; Tabarin et al., 2005). Notably, decreased weight gain in δ-opioid receptor KO mice was associated with a decrease in adipose accretion and an increase in thermogenic activity in brown fat (Czyzyk et al., 2012).
Depending on the physiological context, pharmacologic modulation of opioid signaling is associated with a variety of effects on feeding and metabolism. For example, a μ-selective opioid receptor inverse agonist, GSK1521498, and a pan opioid receptor antagonist, LY255582, reduced weight gain and adipose accretion in diet-induced obesity in rats (Ignar et al., 2011; Statnick et al., 2003). Furthermore, a fixed-dose combination of the norepinephrine and dopamine reuptake inhibitor, bupropion, and the μ-opioid receptor antagonist, naltrexone, decreased food intake with a commensurate decrease in weight in obese mice (Greenway et al., 2009; Sinnayah, 2007). In clinical studies, however, only the combination was effective as a weight loss agent (Plodkowski et al., 2009). Zhang et al. (2006) suggested that μ-opioid receptor antagonism was primarily responsible for decreased weight gain in diet-induced obese mice; however, equivocal results have been published on weight gain and insulin resistance in Syrian hamsters and obese Zucker rats (Jones and Corp, 2003). Nevertheless, these data indicate that the effects of opioid receptor modulation on metabolism need to be studied in a disease-relevant context, particularly when combined with another agent.
Studies in rodents suggest that naltrexone (NTX) may decrease OLZ-induced body weight gain through a food intake-dependent mechanism (Kurbanov et al., 2012). In addition, a small pilot study reported that NTX may attenuate OLZ-induced fat mass gain in patients with schizophrenia and schizoaffective disorder in the absence of OLZ-induced weight gain (Taveira et al., 2014). To date, however, neither NTX nor any opioid based mechanism is approved for OLZ-induced metabolic dysfunction, including weight gain. ALKS 3831, an oral, fixed-dose combination of OLZ and samidorphan, a new molecular entity (SAM; 3-carboxyamido-4-hydroxy naltrexone) structurally related to naltrexone, is currently under development for the treatment of schizophrenia. The addition of SAM is intended to mitigate the deleterious metabolic side effects, including weight gain, associated with OLZ administration while maintaining its clinical efficacy. In vitro, SAM binds with high affinity to μ-, κ-, and δ-opioid receptors and is an μ-opioid receptor antagonist with partial agonist activity at κ- and δ-opioid receptors (Bidlack et al., 2018; Wentland et al., 2005, 2009). Notably, when compared with NTX, SAM binds with higher affinity to μ-, κ, and δ-opioid receptors and functions as a more potent μ-opioid receptor antagonist (Bidlack et al., 2018; Raynor et al., 1994). The potential for SAM to affect targets in addition to µ-, κ-, and δ-opioid receptors was evaluated at 10 µM in an in vitro CEREP panel of 104 in vitro receptor, transporter and enzyme binding/inhibition assays1. Importantly, no additional stimulation/inhibition of any receptor, transporter or enzyme was detected for SAM (data not shown). In a phase 2 clinical trial, ALKS 3831 significantly mitigated weight gain relative to OLZ alone and retained similar antipsychotic efficacy to OLZ in schizophrenia patients (Martin et al., 2018). To explore the mechanism of action of ALKS 3831 in greater detail, a series of non-clinical studies in rats was designed to determine whether SAM would mitigate: 1) weight gain and adiposity following chronic administration of OLZ and 2) OLZ-induced metabolic abnormalities prior to weight gain following subacute administration of OLZ. Furthermore, to assess whether our observations were species specific, an exploratory study was conducted in non-human primates (NHPs) over an eight week period. To best emulate the clinical pharmacology of OLZ and SAM, dosing regimens in rats and NHPs were designed to target clinically relevant plasma concentrations.
Materials and methods
Studies
Animals
Sprague Dawley rats approximately 12 weeks old (Charles River Laboratories (CRL), Kingston, New York, USA) were used for all studies conducted at Alkermes, Inc. Male (~400−425 g) and female (~250−275 g) rats were used for studies of weight gain, body composition and metabolic markers. Femoral artery catheterized female rats (CRL) were used for bolus insulin studies. All rats used in these studies were housed, managed, and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and experiments were approved by the Alkermes Institutional Animal Care and Use Committee. Rats were housed two per cage, except following surgery, and were maintained on a 12:12-h light:dark cycle (lights off at 18:00 h) in a temperature and humidity controlled environment (22 ± 2°C; 45 ± 10% relative humidity). Rats were fed standard chow (Rodent Diet 5001, Lab Diets, St Louis, Missouri, USA) and tap water and allowed to eat and drink ad libitum. Hyperinsulinemic euglycemic clamp (HIEC) studies were carried out in accordance with the European Communities’ Council Directive 2010/63/EU in 10-week-old female Sprague Dawley rats (Charles River, France; ~250 g) at Physiogenex (Labège, France) and were evaluated and approved by the Ministry of Higher Education and Research and ethics committee (N° et CEEA-122). Rats were housed two per cage or individually on a 12:12-h light:dark cycle (lights off at 20:00 h) in a temperature and humidity controlled environment (22 ± 2°C; 50 ± 10% relative humidity). Rats were fed standard chow (RM1 (E) 801492, SDS, Essex, UK) and tap water and allowed to eat and drink ad libitum.
Test compounds and dosing regimens
SAM was synthesized as a salt (l-malate) by Cambridge Major Laboratories (W130 N10497 Washington Drive, Germantown, Wisconsin, USA) and dissolved in sterile saline for injection (Fresenius Kabi, Lake Zurich, Illinois, USA). A long-acting injectable formulation of olanzapine pamoate (OLZ) was prepared by Alkermes, Inc. and dissolved in 5 mM phosphate buffer containing 2% carboxymethylcellulose (CMC) and 0.2% tween (PS20). Vehicle-treated rats were administered the 5 mM phosphate buffer or sterile saline as appropriate. To establish clinically relevant concentrations of OLZ and SAM in rodents, a dosing regimen was designed to target Cmax steady state plasma levels of ALKS 3831 (20 mg OLZ/10 mg SAM when administered in a bilayer tablet for 14 days in humans diagnosed with schizophrenia (mean (SD); OLZ, 64.6 (28.9) ng/mL and SAM, 46 (15.1) ng/mL) (Sun et al., 2018). Pilot pharmacokinetic studies were conducted to determine the target concentrations of OLZ for subcutaneous (s.c.) injection and SAM for osmotic mini pump delivery in male and female rats (Supplementary Material Figure 1(a) and (b) online). The dose of OLZ was chosen based on previous reports to deliver clinically relevant and stable target plasma concentrations in female rats (Skrede et al., 2014). The concentration of SAM used in osmotic mini pumps was extrapolated from the antecedent pilot pharmacokinetic study and was tailored for pump flow rates and duration of experiments. Furthermore, SAM pump concentrations were tailored to account for the differences in SAM metabolism between male and female rats (Supplementary Figure 1(b)).
Based on the pilot studies above, OLZ (100 mg/kg) was injected s.c. in combination with SAM administered via Alzet® osmotic mini pump (models 2001 or 2ML4; Alzet, Cupertino, California, USA). Pumps for male rats were filled with SAM (50 or 125 mg/mL) dissolved in sterile H2O and delivered at 2.5 or 1 μL/h respectively; whereas pumps for female rats were filled with SAM (16 or 40 mg/mL) delivered at 2.5 or 1 μL/h respectively. Pumps were primed overnight according to manufacturer instructions in sterile saline at 37°C. The following day pumps were inserted via a mid-scapular s.c. surgical implantation performed under isoflurane anesthesia (1.5– 2%). Incisions were closed with wound clips. Sublingual bleeds were taken on days 6 and 29 to determine average OLZ and SAM plasma concentrations during the dosing regimen. Under these conditions the average values (± SEM) for females were: OLZ alone, 86.7 ± 42.4 ng/mL; SAM alone, 51.84 ± 4.24 ng/mL; OLZ + SAM, 76.6 ± 44.1 ng/mL and 48.3 ± 21.8 ng/mL, respectively. Similar exposure levels were achieved in male rats, average values (±SEM) were: OLZ alone, 76.85 ± 51.7 ng/mL, SAM alone, 45.85 ± 10.78 ng/mL; OLZ + SAM, 74.5 ± 44.5 ng/mL and 47 ± 12.3 ng/mL, respectively.
Metabolic effects of chronic OLZ, SAM and their combination
Body weight was monitored throughout the study and body composition measures of fat mass were assessed via Echo MRI (Echo MRI, LLC; Houston, Texas, USA) on days 0, 7, 14, 21, and 28. Prior to surgical pump implantation and OLZ dosing, body fat composition and weight were measured and rats were assigned to groups by total fat composition using the matched distribution randomization procedure in StudyLog (StudyLog Systems, Inc.; San Francisco, California, USA). Osmotic pumps containing SAM were then implanted and OLZ was dosed once weekly for 28 days (i.e. injection on study days 1, 8, 15, and 22). Food consumption (g) was measured once daily on days 2–14 at approximately 08:00 h prior to any dosing or animal handling.
HIEC studies of subacute OLZ, SAM, and the combination
A pilot study in female rats was conducted to determine: 1) the degree of insulin resistance induced by OLZ and 2) the lowest dose of insulin that enabled clamping without completely blunting hepatic glucose production (expected range: 0.2 to 0.8 U/kg/h). Prior to HIEC, female rats were allowed to recover for two days following femoral vein catheterization, vehicle osmotic mini pump implantation and OLZ (100 mg/kg; s.c.) administration prior to HIEC. A simplified HIEC without 3H-glucose tracer was performed after fasting the rats for 6 h. A 0.5 U/kg/h insulin infusion rate was chosen from pilot studies. Rats were catheterized and treated with OLZ (100 mg/kg), SAM (40 mg/mL; delivered via osmotic pump at 1 μL/h) or the combination. Rats recovered for 48 h and were fasted at 07:30 h the morning of the clamp experiment. Prior to clamp, blood samples (approximately 100 µL; Mini Collect K3EDTA tubes) were collected from the tail tip to measure plasma insulin and OLZ and SAM concentrations. Plasma was separated via centrifugation (14,000 × g for 2 min at 4°C; Eppendorf Centrifuge 5430R, A-45-30-11 rotor), transferred to 0.5 mL sample tubes and stored at −80°C. At 13:30 h, rats were perfused with 3H-glucose (4 µCi/kg per min at 13 µL/min) and insulin (0.5 U/kg per h) for 180 min. Blood glucose was measured every 10 min to maintain euglycemia (~100 mg/dL) by adjusting cold glucose infusion rate (GIR). Blood (10 µL) was collected from the tip of the tail every 10 min during the steady state period (120–180 min) for 3H-radioactivity analysis to determine whole-body glucose turnover and hepatic glucose production, as well as whole-body glycolysis and glycogen synthesis rates. 3H-glucose enrichments were determined from total blood after deproteinization by a Zn(OH)2 precipitate and the supernatant was used to measure 3H-radioactivity (“wet”) or evaporated to dryness prior to measuring 3H-radioactivity (“dry”). 3H-glucose specific activity was calculated by dividing 3H-radioactivity “dry” by blood glucose concentration. Whole-body glucose turnover rate was calculated by dividing the rate of 3H-glucose infusion by 3H-glucose plasma specific activity. At steady state, whole-body glucose turnover was equal to glucose infusion rate plus hepatic glucose production. Hepatic glucose production was then determined by subtracting glucose infusion rate from whole-body glucose turnover. The whole-body glycolysis rate was measured by dividing the amount of 3H-water (determined from the “wet” minus “dry” 3H-radioactivity) by the 3H-glucose specific activity. The whole-body glycogen synthesis rate was then calculated by the difference between the whole-body glucose turnover and the whole-body glycolysis rate.
14C-2-deoxyglucose (2-DG; 100 µCi) was also injected at time 120 min to measure tissue glucose uptake and blood was collected to measure 14C-tracer blood disappearance until time 180 min. At study termination, rats were euthanized by cervical dislocation after an intravenous (i.v.) injection of pentobarbital. Several tissue samples (inguinal (iWAT) and retroperitoneal (rWAT) adipose tissues, soleus, extensor digitorum longus (EDL) and gastrocnemius muscles, and liver) were harvested, weighed and flash frozen in liquid nitrogen and stored at −80°C prior to 14C-radioactivity measurements to determine glucose utilization. 14C-tracer disappearance was determined by measuring 14C-radioactivity from blood samples during the last hour of the hyperinsulinemic clamp after deproteinization by a Zn(OH)2 precipitate and an area under the curve was calculated. Tissues were dissolved with NaOH, neutralized with HCl and then 14C-2-deoxyglucose 6-phosphate and 14C-2-deoxyglucose radioactivity were assessed after differential precipitation using Zn hydroxide and perchloride acid solutions. Glucose utilization was then determined from 14C-2-deoxyglucose 6-phosphate radioactivity divided 14C-tracer disappearance area under the curve.
Subacute effects of SAM, OLZ, and the combination on glucose clearance following bolus insulin injection
A subset of femoral artery catheterized rats was used to assess whole-body glucose clearance following bolus insulin injection. Pilot studies in female rats were conducted using 0.375 U/kg and 0.75 U/kg, intraperitoneally of insulin (Sigma Aldrich, #I9278). The higher dose of insulin, 0.75 U/kg, commonly reported in the literature, produced signs of hypoglycemia (lethargy and immobility). As a result, rats were dosed with 0.375 U/kg of insulin for all studies. Rats were treated with OLZ, SAM or the combination 48 h prior to insulin sensitivity testing. Rats were placed in an automated blood sampling system (CULEX; BASi, West Lafayette, Indiana, USA) and fasted overnight for 16–18 h prior to test. The following morning, baseline glucose concentrations were measured in whole blood using a glucometer (Nova Biomedical StatStrip® Xpress; DSI, St. Paul, Minnesota, USA). Glucose concentrations were subsequently monitored for 120 min following insulin injection.
NHP studies
Experiments in NHPs were conducted at Battelle (Columbus, Ohio, USA). Fifteen female cynomolgus macaque monkeys, approximately 3.6 to 4.2 years of age and weighing 2.7 ± 0.07 kg (mean ± SEM) upon study start, were used for all studies. Monkeys were individually housed in stainless steel cages in accordance with the Guide for Care and Use of Laboratory Animals (National Research Council, 1996), and requirements as stated by the U.S. Department of Agriculture through the Animal Welfare Act regulations, as amended. Beginning two weeks prior to study start (to allow monkeys to acclimatize to ad libitum access) all monkeys were offered ad libitum access to high fat Harlan Teklad Custom Primate Diet TD.10600 (42% Fat Kcal, (21)), except during specified fasting periods. Fresh fruits, fresh vegetables, and/or supplements were offered as appropriate. Fresh water from the municipal water supply was provided ad libitum in home cages via an automatic watering system.
Dosing and assessment of weight and food consumption
Body weights were stable for one month prior to the initiation of the ad libitum feeding (2.9 ± 0.07 kg). On the day prior to study start, body weights were slightly increased with more variability (3.1 ± 0.09 kg) and thus monkeys were random block assigned to treatment groups by body weight. OLZ (Midas Pharmaceuticals, Inc. Parsippany, New Jersey, USA) was prepared in 1% CMC in deionized water and administered via oral gavage twice per day, once in the morning and once approximately 6 h later, at a volume of 2 mL/kg per day followed by 5 mL water flush of the gavage tube. Monkeys received 0.5 mg/kg per dose on days 1–3, 1 mg/kg per dose on days 4–6, 2 mg/kg per dose on days 7–9. The maximum dose of 3 mg/kg per dose (6 mg/kg per day) was administered on days 10–58. SAM (0.4 mg/kg per day in 0.125 mL sterile saline) was administered by intramuscular injection into the quadricep musculature (to minimize the number of daily oral gavages and handling stress) once per day following the morning dose of OLZ. The final target dose of OLZ was designed to target clinically relevant concentrations of OLZ (10–25 ng/mL) (Dorph-Petersen et al., 2005; Kapur et al., 1998, 1999). Blood samples were monitored through day 28 to verify target dose plasma concentrations of OLZ. Under these conditions, mean OLZ plasma concentrations measured on day 28 were 13.1 ± 4.1 (mean ± SEM) ng/mL. The SAM dose was chosen based on pharmacokinetic modeling to reach human therapeutically equivalent dose. Dosages were adjusted weekly (on days 1, 8, 15, 22, 29, 35, 42, 49, and 56) based on the most recent body weight. The treatment groups (n = 5 per group) were: 1) vehicle (VEH) (1.0% CMC/saline); 2) OLZ (OLZ alone on days 1–34, OLZ + SAM on days 35–58); and 3) OLZ + SAM.
Body weights were recorded prior to group assignment on day 1, and prior to dose administration every three days beginning on day 4. Food consumption was qualitatively evaluated once daily throughout the study period. Estimates of food consumption were visually assessed according to the following scale: 0 = no observable food consumed (none); 1 = up to and including one-quarter consumed; 2 = up to and including one-half consumed; 3 = up to and including three-quarters consumed; 4 = up to and including the entire amount consumed.
Computed tomography scans
Whole-body computed tomography (CT) images were conducted once pre-test and on days 29 (all groups) and 59 (OLZ group switched to OLZ + SAM only). CT imaging was performed using a 64 multi-slice system (Brilliance 64, Philips Healthcare, Cleveland, Ohio, USA). Iodinated contrast, Omnipaque 350 (6 mL) was used for the visualization of the vasculature using an automated injector (MEDRAD spectris dual) with a flow rate of 3 mL/s followed by 10 mL saline. Image analysis was performed using an extended brilliance multi-modality workstation (Philips Healthcare) and a Leonardo multi-modality workstation (Siemens Healthcare). For the purposes of this study, only CT images at the level of the lower pole of the kidney (abdominal adipose) and the sacral pelvic junction (subcutaneous adipose) were analyzed by personnel blinded to study identification numbers during the imaging process and initial reporting of the data.
Glucose tolerance test
Monkeys were fasted for approximately 12 h prior to blood sample collection on days 0, 28, and 58. Samples (2 mL whole blood) were collected immediately prior to dosing (pretest) and 5, 10, 20, 30, 40, 50, and 60 min after dosing. Each monkey was administered dextrose (600 mg/kg, i.v.) immediately following the pretest blood collection. Blood samples were then collected and 2–3 drops from each sample was used to measure glucose using an I-STAT handheld Blood Gas Analysis System that provides data in real time. The remaining samples were processed for separation into plasma and frozen at ~–70°C as described above. Plasma samples were subsequently thawed and analyzed for insulin concentrations on a Luminex 200TM with xMAP technology and xPONENTTM v 3.1 software (Austin, TX), using a Non-Human Primate Magnetic Hormone Panel (#NHPMHMAG-45k-02 kit; EMD Millipore Corp, Billerica, Massachusetts, USA) as per kit instructions.
Data analysis and statistics
Rat studies
Food consumption data were analyzed using a two-way ANOVA with repeated measures followed by a Dunnet’s test to identify significant differences from VEH-treated rats (5 mM phosphate buffer and saline). For weight gain, a slope analysis of baseline-corrected weight data was conducted for each group and analyzed via a one-way ANOVA followed by post hoc analysis (Tukey HSD). To assess fat mass changes, a baseline-corrected fat mass to total weight was calculated and analyzed with a two-way ANOVA with repeated measures followed by post hoc analysis (Tukey HSD). To assess changes in glucose utilization, treatment effects were analyzed using a one-way ANOVA followed by post hoc analysis (Fisher’s Least Significant Difference). To assess glucose clearance following bolus insulin injection the average change from baseline was analyzed using a mixed model repeated measurements with an unstructured variance–covariance matrix. Statistical values reaching p < 0.05 were considered significant. All data are expressed as mean ± standard error of the mean (SEM) and were analyzed by GraphPad Prism 6 or 7 (GraphPad software, La Jolla, California, USA).
Primate studies
Primate studies were conducted as an exploratory study and were underpowered to detect statistical differences in body weight and food consumption (at α < 0.05). Due to variability among all three treatment groups, a rolling average was generated where the mean for the preceding three measurements was plotted. For weight gain, a slope analysis was conducted for each treatment group. In addition, because these data have heterogeneity of variances, data for the CT and food consumption data are reported as group means ± SEM.
Results
Rat studies
Effects of OLZ and SAM on food consumption rates of weight gain and body composition
In female and male rats, using a two-way ANOVA, there was a significant effect of treatment on food consumption (F(3.36) = 10.04, p < 0.001, females; F(3.36) = 11.10; p < 0.001, males). A Dunnett’s post hoc analysis indicated that the changes in eating behavior did not persist throughout the 14 day assessment (Figure 1). When compared with VEH-treated rats, OLZ had no effect in females but produced a transient increase in food consumption in male rats for two days following drug administration. SAM produced a transient decrease in food consumption for 3–4 days in female rats and for one day in male rats following drug administration. An early decrease in weight was also noted during the first two days of SAM administration, which may have been driven by the initial effects of SAM on feeding. Importantly, no overt signs of sickness behavior were observed. In female rats, OLZ produced a robust increase in rate of weight gain compared with VEH controls (1.54 ±0.06 vs. 0.88 ± 0.08 g/day, respectively; F(3,56) = 37.21; p < 0.001; Figure 2(a)). This was not seen in male rats (2.93± 0.1 vs. 2.96 ± 0.08 g/day, respectively; F(3,56) = 11.29; p = NS; Figure 2(c)). In female rats, SAM decreased the rate of weight gain when compared with VEH-treated rats (0.53 ± 0.06 vs. 0.88 ± 0.08 g/day for the VEH group, F(3,56) = 37.21; p < 0.01; Figure 2(a)). Similarly, in male rats, SAM produced a decrease in the rate of weight gain versus VEH control (2.27 ± 0.09 vs. 2.96± 0.08 g/day for SAM and VEH group, respectively; F(3,56) = 11.29; p < 0.001; Figure 2(c)). In female rats, co-administration of SAM significantly attenuated the weight gain observed in the OLZ group (1.54 ± 0.06 vs. 1.26 ± 0.08 g/day, respectively; F(3,56) = 37.21; i < 0.05; Figure 2(a)). There was no significant difference in the rate of weight gain between rats treated with SAM alone or in combination with OLZ in male rats.
Figure 1.
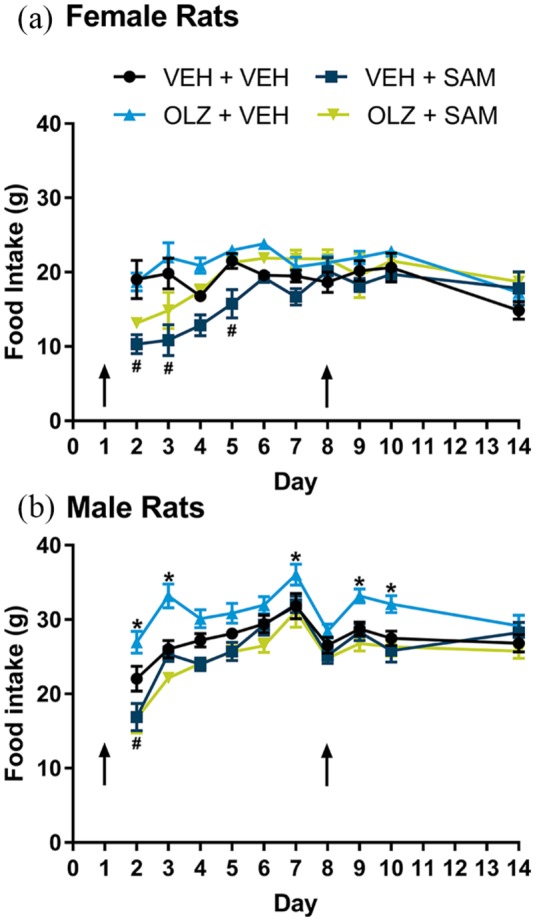
Food consumption in female and male rats. Food consumption was measured prior to animal handling at approximately 08:00 h one day after administration of OLZ, SAM or the combination (day 2) to day 14. (a) An acute transient decrease in food consumption was noted for 3–4 days following SAM administration female rats (#p < 0.05). (b) Similarly, an acute decrease in food consumption was noted one day following SAM administration in male rats (#p < 0.05). In male rats only, a transient increase in food consumption was noted for two days following each OLZ injection (*p < 0.05). Importantly, these effects did not persist throughout the 14 day assessment. Data are expressed as mean ± SEM. Arrows indicate time of OLZ injection.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Figure 2.

OLZ (100 mg/kg, subcutaneous (s.c.)) and SAM (female: 16 mg/mL or male: 50 mg/mL; delivered at 2.5 μL/h, s.c. infusion) alone or in combination were administered for 28 days. (a) Female rats; OLZ administration produced a statistically significant increase in the rate of weight gain versus control (*p < 0.001). SAM alone decreased the rate of weight gain versus vehicle (**p < 0.01). Co-administration of SAM decreased the effects of OLZ on weight (#p < 0.05). (b) Female rats. OLZ produced a significant (*p < 0.001) increase in adipose accretion versus vehicle control by day 28. SAM alone decreased adipose accretion (#p < 0.001) while co-administration of OLZ + SAM was similar to vehicle control by day 28. (c) Male rats; OLZ administration did not change the rate of weight gain significantly versus vehicle control. Addition of SAM decreased the rate of weight gain in both vehicle (*p < 0.001) and OLZ (#p < 0.01) treated rats. (d) Male rats; OLZ administration produced a significant increase in adipose accretion versus vehicle control by day 28 (*p < 0.01). Co-administration of OLZ + SAM was similar to vehicle control at day 28. Data are expressed as mean ± SEM. Arrows indicate time of OLZ injection.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Within the first 14 days of the study there was a significant increase in adiposity in both female and male rats treated with OLZ (Figure 2(b) and (d), respectively). The changes in body composition persisted throughout the 28 days’ duration of the study (treatment; F(3,34) = 56.81; p < 0.001: male; treatment; F(3,36) = 23.22; p < 0.001). SAM attenuated adipose accumulation in female rats (treatment × time interaction; F(12,136) = 10.17; p < 0.001) and prevented adipose accumulation in male rats (treatment × time interaction; F(12,144) = 14.7; p < 0.001) treated with OLZ by end of treatment (Figure 2(b) and (d), respectively).
HIEC study
Based on a pilot study (data not shown), an insulin infusion rate of 0.5 U/kg per h was selected to determine the effects of SAM on insulin sensitivity in liver, as well as muscle and adipose tissue. OLZ produced a 23% decrease in GIR compared with VEH control (Figure 3(a); F(3,44) = 9.37; p < 0.001). No changes were observed in glycogen synthesis, glycolysis, or whole-body glucose turnover (Figure 3(a)). The most pronounced effect of OLZ was a disinhibition of hepatic glucose production at the level of insulin infused, which is consistent with the induction of insulin resistance in the liver (Figure 3(b); F(3,44) = 11.82; p < 0.001). Glucose utilization rates were examined across liver, muscle and adipose tissue. To estimate the net effect of these changes on whole-body glucose metabolism, the effects across each tissue type were combined. The specific effect on each tissue assessment is provided in the Supplementary Material online (Supplementary Figure 2). OLZ had no effect on glucose utilization in the liver (Figure 3(a)). SAM co-administration did not affect whole-body GIR, either as a single agent, or when administered in combination with OLZ (Figure 3(a)). Consistent with this observation, SAM alone did not have an effect on hepatic glucose production, utilization, or glycogen synthesis and did not restore insulin sensitivity (Figure 3(a) and (b)).
Figure 3.
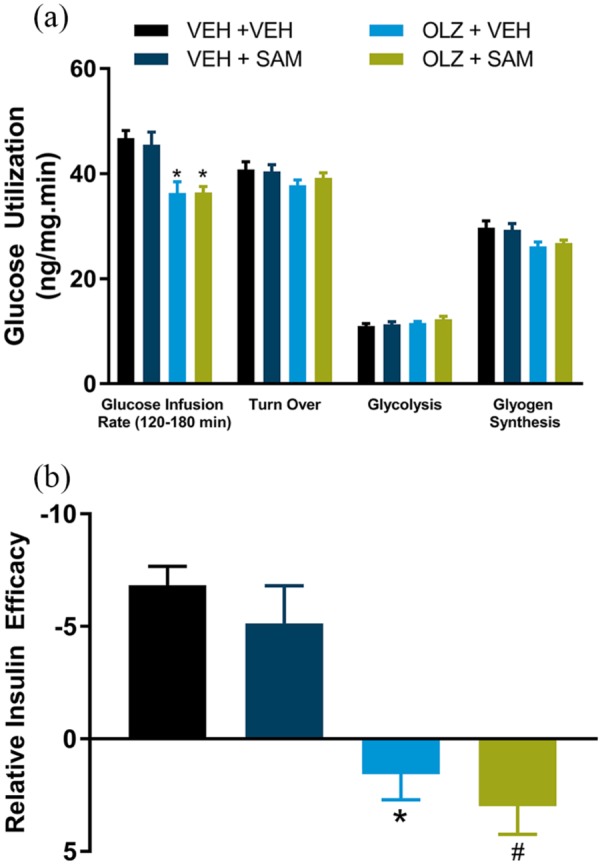
Hyperinsulinemic euglycemic clamp study in female rats. (a) At steady state (120–180 min), OLZ significantly decreased glucose infusion rate compared with vehicle control (*p < 0.001). No changes were observed in glycogen synthesis, glycolysis or whole-body glucose turnover. (b) OLZ significantly inhibited insulin-induced decreases in hepatic glucose production (*p < 0.01, OLZ + VEH; #p < 0.001, OLZ + SAM). Data are expressed as mean ± SEM.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
The net effect of OLZ across muscle tissues (gastrocnemius, EDL, and soleus muscle) was a statistically significant decrease in glucose utilization (Figure 4(a); F(3,137) = 6.22; p < 0.001). The opposite was observed in adipose tissue (iWAT and rWAT) where OLZ increased glucose utilization in adipose tissue when compared with VEH-treated rats (Figure 4(b); F(3,91) = 6.57; p < 0.05). SAM alone had no effect on glucose utilization in muscle compared with VEH treatment, but in combination appeared to attenuate OLZ-induced decreases in glucose utilization (Figure 4(a); F(3,137) = 6.22; p = 0.052). In adipose tissue, co-administration of SAM and OLZ prevented OLZ-induced increases in glucose utilization (Figure 4(b); F(3,91) = 6.57; p < 0.001).
Figure 4.
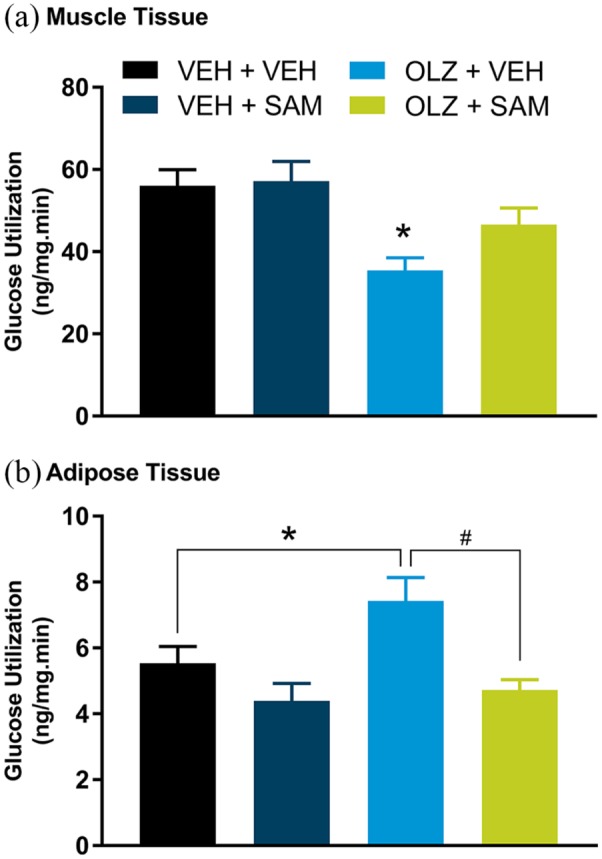
Tissue glucose utilization during hyperinsulinemic euglycemic clamp in female rats. (a) OLZ significantly decreased muscle glucose utilization (combined gastrocnemius, extensor digitorum longus and soleus muscle, *p < 0.05). Co-administration of SAM appeared to attenuate this effect. (b) OLZ significantly increased glucose utilization in adipose tissue (combined inguinal and retroperitoneal adipose tissue, *p < 0.05), which was blocked by co-administration with SAM (#p < 0.001). Data are expressed as mean ± SEM.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Effect of subacute SAM, OLZ, and the combination on glucose clearance after bolus injection of insulin
Consistent with results from the GIR study, glucose clearance in response to a bolus injection of insulin (0.375 U/kg) was blunted in female rats 48 h after OLZ treatment. Co-administration of SAM restored normal glucose clearance (Figure 5(a); treatment × time interaction; F(12, 120) = 2.95; p < 0.01). In male rats, OLZ significantly decreased glucose clearance (Figure 5(b); F(3,30) = 3.51; p < 0.05). Co-administration of SAM partially restored glucose clearance but this effect did not reach statistical significance (p = NS).
Figure 5.
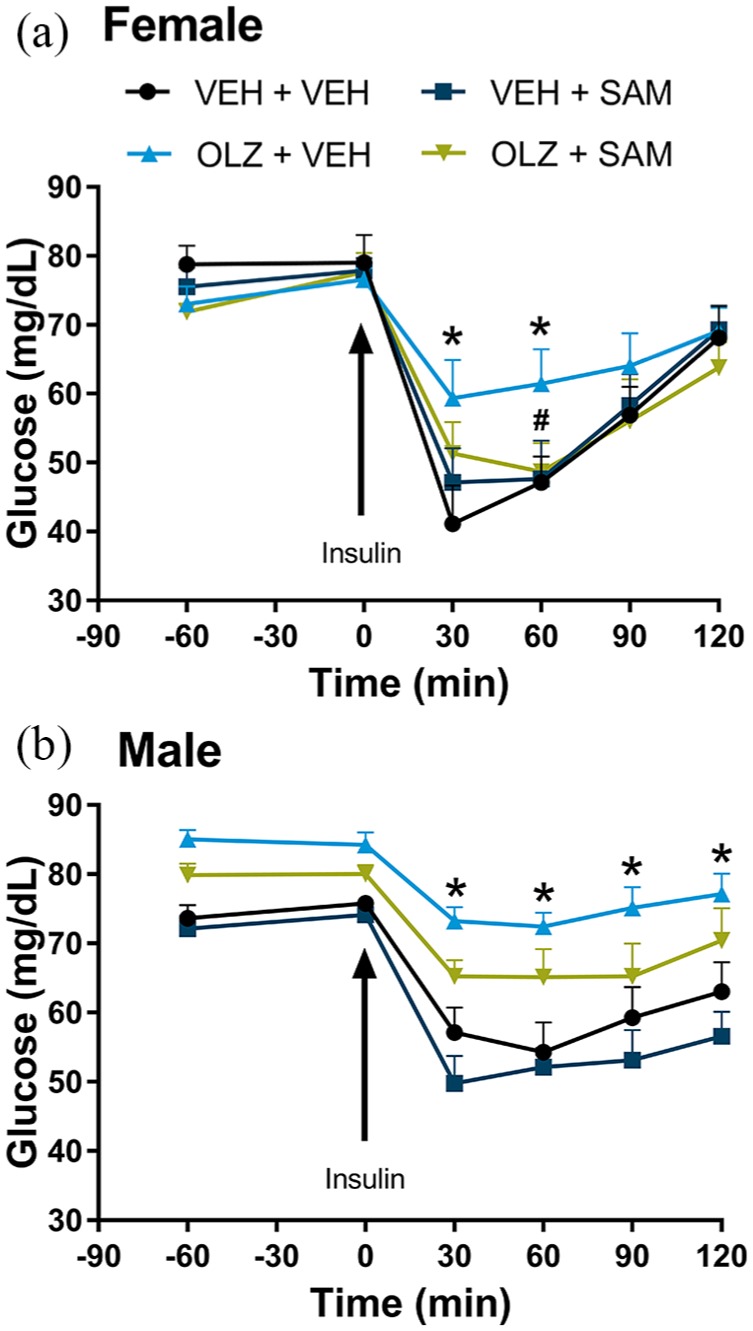
Glucose clearance following bolus insulin in female rats. (a) OLZ significantly decreased glucose clearance following bolus insulin administration in female rats, which was normalized by co-administration of SAM (VEH + VEH vs. OLZ + VEH, *p < 0.05; OLZ + VEH vs. OLZ + SAM, #p< 0.05). (b) OLZ significantly decreased glucose clearance following bolus insulin administration in male rats (VEH + VEH vs. OLZ + VEH, *p < 0.05), which was partially but not significantly restored by co-administration of SAM (p = NS). Arrows indicate time of insulin injection (0.375 U/kg). Data are expressed as mean ± SEM.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
NHP studies
Weight gain and food consumption
In Phase I of the study (days 1–34), control monkeys gained an average of 0.396 ± 0.173 kg. This was attributed to the ad libitum feeding of the palatable high-fat diet. OLZ-treated monkeys gained an average of 0.589 ± 0.461 kg over the same 34 day period. The increased average weight gain was ascribed to three of the five monkeys that gained between 16.0% and 40.6% of their initial body weight. The average body weight gain for monkeys treated with OLZ and SAM was 0.190 ± 0.263 kg over the first 34 days. In Phase II of the study (days 35–58), monkeys were followed for an additional 24 days. By day 58, control monkeys had gained an average of 0.610 ± 0.400 kg in comparison with their day 0 body weight. Monkeys treated with both OLZ and SAM for the full 58 days gained an average of 0.362 ± 0.207 kg compared with their day 0 body weight. The average weight gain in OLZ-treated monkeys that began receiving SAM on day 35 was 0.617 ± 0.400 kg on day 58, which was comparable to VEH, suggesting a decrease in the rate of weight gain (Figure 6).
Figure 6.

Rolling average of % weight change in non-human primates. A three-day rolling average of the cumulative % change in body weight for each treatment group was compared using a slope analysis. While all three treatment groups gained weight through the duration of the study, the monkeys receiving OLZ alone had the greatest rate of weight gain while those co-administered SAM had the smallest rate of weight gain. Furthermore, on day 35, monkeys that began receiving SAM no longer demonstrated a significant rate of weight gain.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Qualitative food consumption was tracked throughout the study (Figure 7). Monkeys treated with OLZ ate less than VEH-treated monkeys, suggesting their increased rate of weight gain could not be attributed to hyperphagia. Lower food consumption was observed through the duration of the study for monkeys receiving both OLZ and SAM, consistent with the reduced rate of weight gain observed versus the other two treatment groups. A transient decrease in food consumption was observed when SAM was co-administered on day 35 in monkeys previously treated with OLZ alone. This decrease was not seen one week after co-administration. Monkeys receiving OLZ + SAM through the duration of the study consistently ate less, and gained less weight.
Figure 7.

Rolling average of food consumption in non-human primates. A three-day rolling average of the qualitative food consumption for each treatment group was recorded. VEH-treated monkeys consumed the most food, likely due to the highly palatable high-fat diet. However, OLZ-treated monkeys did not eat as much compared with VEH-treated controls. Important to note is that this group gained more weight than the VEH-treated controls, suggesting a metabolic component associated with OLZ-induced weight gain. Food consumption was lowest in monkeys co-administered SAM. Furthermore, on day 35, monkeys that began receiving SAM ate less, but this effect was transient and food consumption returned to pre-change values approximately 12 days later. Data are expressed as mean ± SEM.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Effects of SAM on OLZ associated adipose accretion
Adipose accretion across all treatment groups was assessed by CT scan. In Phase I of the study, a difference in the percentage of fat volume change was identified in relation to anatomic position (Figure 8). Body fat increased between baseline measurements and day 29 in OLZ-treated monkeys. Abdominal fat level increased with a mean of 456% at the lower kidney pole whereas the pelvic regions increased at a mean of 251% (median 222%). In vehicle-treated monkeys, body fat increased less with a more variable distribution within the population. While body fat increased by a mean of 296%, the median was lower at 173% between baseline measurements and day 29. In addition, the difference between abdominal and pelvic fat gain was less pronounced. Monkeys treated with OLZ + SAM had the lowest body fat volume gain, with a mean of 270% (median of 167%) between baseline measurements and day 29. Similar to VEH-treated monkeys, the difference between abdominal and pelvic fat gain was less pronounced. Phase I differences at the level of the lower pole of the kidney between monkeys treated with OLZ and those treated with OLZ + SAM are shown in representative CT scan images (Figure 8). In Phase II, body fat increased by a mean of 407% (median of 295%) between baseline measurements and day 58 for the OLZ + SAM monkeys; a mean increase of 115% (median 112%) between day 29 and day 58, indicative of a slower rate of adipose accretion during Phase II.
Figure 8.

Volume of fat accretion in non-human primates. (a) All monkeys were scanned prior to study start and again on day 28. In addition, monkeys on OLZ alone that were transitioned to begin receiving SAM on day 35 were scanned again on day 57. Scans were targeted to assess abdominal adipose accretion and subcutaneous adipose accretion. As indicated in the top panel, monkeys administered OLZ alone gained appreciably more abdominal adipose accretion compared with monkeys treated with OLZ and SAM. Furthermore, when the OLZ alone group was switched to receive SAM, the rate of accretion was considerably lower compared with that of the first 28 days (bottom panel). Data are expressed as mean ± SEM. (b) Top: representative scans at the level of the lower pole of the kidney (abdominal adipose accretion) from an OLZ alone treated monkey taken on day 28 compared with its baseline scan. Bottom: A representative scan from an OLZ and SAM treated monkey taken on day 28 and compared with its baseline scan. The differences in adipose accretion are readily visible between the two treatment groups.
OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Effect of SAM on OLZ-induced insulin insensitivity
During the glucose tolerance tests (days 0, 28, and 58), plasma concentrations of glucose did not differ from pre-study baseline values among treatment groups or testing day. In the vehicle group, insulin concentrations did not differ from baseline values when examined on day 28 (Figure 9(b)); however, on day 58 insulin values were increased, suggesting development of insulin insensitivity (Figure 9(c)). In the OLZ only group, on day 28 (Figure 9(d)) the pattern of insulin concentrations in response to the glucose tolerance test was similar to that of the VEH group on day 58, suggesting a trend toward an increase in the rate of development of insulin insensitivity. In the OLZ-treated monkeys that began receiving SAM on day 35 (Phase II), the insulin response to the glucose challenge was comparable to baseline levels, suggesting a reversal of insulin insensitivity (Figure 9(e)). In monkeys treated with OLZ + SAM, the concentrations of insulin were comparable to baseline levels on both day 28 and day 58 (Figure 9(f) and (g)).
Figure 9.

Glucose tolerance test and insulin concentrations in non-human primates. A decrease in insulin sensitivity (purple lines) in VEH-treated monkeys exposed to high-fat diet was noted on day 58 (c) but not day 28 (b). OLZ treatment produced a more rapid decrease in insulin sensitivity (purple lines) as compared with baseline (a) and VEH-treated monkeys on day 28 (d). However, when this group was measured again after having been switched to receive SAM, this response was no longer observed (e). Co-administration of OLZ with SAM maintained insulin sensitivity throughout the study (f and g).
NHP: non-human primate; OLZ: olanzapine; SAM: samidorphan; VEH: vehicle
Discussion
The rat and primate studies conducted herein were designed to determine whether SAM would mitigate weight gain and metabolic side effects associated with OLZ treatment. In non-clinical species, different pharmacokinetic profiles often limit direct comparison of drug effects and efficacy in humans. This is also true for atypical antipsychotics that have shorter half lives in rodents and bind to multiple receptor targets with affinities varying by several orders of magnitude (Bymaster et al., 1996; Horska et al., 2016; Kapur et al., 2003). As a result, relatively high suprapharmacologic doses have traditionally been administered once or twice daily in rodent studies. The use of suprapharmacologic and frequent dosing strategies, however, is confounded by OLZ-induced sedation, animal stress and off target receptor binding effects not seen at therapeutic exposure levels in humans (van der Zwaal et al., 2014). Therefore, to best emulate the human pharmacology of ALKS 3831, a long-acting injectable formulation of OLZ was administered s.c. in combination with SAM delivered via an osmotic minipump in the rodent studies. Using this dosing strategy, plasma levels of OLZ and SAM were comparable to clinical exposure of ALKS 3831 (20 mg OLZ/10 mg SAM (Sun et al., 2018)).
Our finding that chronic administration of OLZ produced weight and metabolic changes in rats is consistent with literature reports of increased weight gain in female rats and increased adiposity in male and female rats (Chintoh et al., 2008; Cope et al., 2005; Horska et al., 2016; Minet-Ringuet et al., 2006; Smith et al., 2011) and male dogs (Ader et al., 2005). Importantly, the weight gain and body composition changes produced by OLZ in these studies occurred in the absence of increased food consumption. This was also the case in our studies, as OLZ administration had no long-term effect on food intake but produced significant changes in weight and body composition in female rats. Although a transient increase in food consumption was noted after OLZ administration in male rats, no changes in weight were observed despite a protracted increase in adiposity. These data suggest that the changes in weight and body composition in our studies were not caused by changes in eating behavior. In contrast, others have reported hyperphagia in female rats following oral and long-acting injectable formulations of OLZ (Davoodi et al., 2009; Skrede et al., 2014; Weston-Green et al., 2011), which demonstrates inconsistent effects of OLZ on food consumption. Consistent with previous literature reports, the weight effects of OLZ were more pronounced in female rats. The reasons for sex differences in OLZ-induced weight gain are not fully understood, but similar effects have been observed following antipsychotic use in humans (Jain et al., 2006; Seeman, 2004), though observations have been equivocal (Ascher-Svanum et al., 2005).
In assessing the effects of SAM on weight, a transient decrease in food consumption was noted in both male and female rats although no overt signs of sickness behavior were observed. In both female and male rats, SAM alone decreased the rate of weight gain when compared with VEH-treated rats. In the absence of a long-term change in food consumption, our data suggest that the effects of SAM were not caused by an overall decrease in eating behavior. Although, the effects of a transient decrease in consumption cannot be completely ruled out, the change in the rate of weight gain indicates that SAM has a long-term, food independent effect on weight. Importantly, SAM attenuated chronic OLZ-induced increases in the rate of weight gain in female rats. Similar findings were reported by Kurbanov et al. (2012), where administration of extended release naltrexone, a μ-opioid receptor antagonist, prevented OLZ-induced weight gain in female Wistar rats. In contrast to our findings, OLZ produced hyperphagia that was also attenuated by naltrexone. This suggests that the ability of naltrexone to attenuate OLZ-induced weight gain is dependent upon decreased food consumption. Although differences in rat strain and OLZ dosing regimen may explain discrepancies in food intake, our findings suggest that SAM can mitigate OLZ-induced weight gain in the absence of hyperphagia.
Although weight gain has been associated as a risk factor in the progression of T2DM, a gain in visceral adiposity and loss of muscle mass are also independent risk factors (Han et al., 2017; Kissebah and Peiris, 1989; Lebovitz and Banerji, 2005). In the current studies, OLZ significantly increased adiposity in both male and female rats compared with VEH-treated controls. These findings support previous demonstrations of OLZ-induced changes in body composition in the absence of weight gain (Albaugh et al., 2011; Cooper et al., 2007). Co-administration of SAM attenuated OLZ-induced changes in fat mass in both male and female rats. Given the lack of weight gain in male rats, these data suggest that co-administration of OLZ + SAM offers weight-independent metabolic benefits. Interestingly, Statnick et al. (2003) reported that the pan opioid receptor antagonist LY255582 reduced fat mass accretion with no change in lean body mass in obese Long–Evans rats on a high-fat diet. This effect was attributed to a combination of reduced food intake and stimulation of lipid utilization. Although it is tempting to speculate that the effects of SAM observed herein involve increased utilization of lipids, future studies would be necessary to assess the effects of OLZ and SAM on energy expenditure.
To better understand the metabolic drivers of increased rates of weight gain and adiposity, a HIEC metabolic study was conducted in female rats. Experiments were conducted 48 h following OLZ and SAM administration to isolate metabolic effects prior to significant weight gain or adiposity. As expected, OLZ decreased whole-body GIR and shifted the pattern of glucose utilization and disposition in all tissues tested. Similar to previous reports in rats (Chintoh et al., 2008; Houseknecht et al., 2007; Kowalchuk et al., 2017), OLZ decreased hepatic insulin sensitivity that in turn increased hepatic glucose production. In contrast, OLZ did not affect hepatic glucose utilization, or glycogen synthesis. Therefore, the primary driver of decreased whole-body GIR is likely a consequence of OLZ-driven decreases in hepatic insulin sensitivity. Interestingly, in a recent pancreatic euglycemic clamp study in male rats, an acute clinically relevant dose of OLZ blocked intracerebroventricular insulin-induced suppression of glucose production but had no effect on glucose uptake (Kowalchuk et al., 2017). These data support the hypothesis that OLZ alters hepatic glucose metabolism, at least in part, by inducing central insulin resistance. The cellular and molecular mechanisms by which OLZ could disrupt central insulin actions, however, remain unknown (discussed in Kowalchuk et al., 2017).
SAM alone did not affect the rate of glucose clearance, nor did it reverse the deficit in GIR caused by OLZ. SAM also had no significant effect on hepatic glucose production, glucose utilization or glycogen synthesis alone or in combination with OLZ. These results were surprising given that SAM reversed OLZ-induced decreases in glucose clearance following bolus dose of insulin administration in female rats. This discrepancy is likely caused by higher insulin concentrations following bolus administration that suppress hepatic glucose production. Collectively, these data may indicate that the effects of SAM are limited to non-hepatic peripheral glucose clearance.
OLZ decreased 2-DG uptake in muscle and increased uptake in adipose tissue, providing a potential mechanism for adipose accretion. Houseknecht et al. (2007) observed a similar pattern of glucose utilization, where acute and chronic administration of clozapine and OLZ resulted in a trend toward decreased glucose utilization in muscle and increased adipose glucose utilization. Furthermore, acute administration of OLZ in male rats produced a shift toward lipogenesis in adipose tissue (Albaugh et al., 2011) and changes in muscle gene expression consistent with an increased potential for metabolic disease and obesity (Lynch et al., 2015). Interestingly, the muscle transcript data from this study further suggested a transition from slow muscle fibers to fast-glycolytic fibers that are more susceptible to atrophy. Accordingly, acute muscle toxicity has been reported following high-dose OLZ administration in humans (Keyal et al., 2017; Waring et al., 2006). These data highlight excessive adipose accretion and muscle impairment as important metabolic abnormalities associated with OLZ use, even in the absence of weight gain.
Our data suggest that a primary metabolic benefit of SAM is reversal of OLZ-induced changes in muscle and fat tissue. Co-administration of SAM prevented OLZ-induced increases in adipose glucose uptake to vehicle-treated levels and attenuated OLZ-induced decreases in skeletal muscle glucose uptake. Shifts toward lipid catabolism have also been observed with other opioid receptor antagonists. For example, the pan opioid receptor antagonist LY255582 decreased body weight in a diet-induced obesity model through decreased food intake but also increased fat utilization and decreased glucose consumption (Statnick et al., 2003). This led to a decrease in fat mass and an increase in lean mass when compared with VEH-treated rats. SAM may operate through a similar mechanism when co-administered with OLZ. SAM is a highly selective modulator of the opioid system, and likely functions as an antagonist of µ- and δ-opioid receptors at the doses used in the present studies (Bidlack et al., 2018; Wentland et al., 2009). Opioid receptor antagonism, in particular at δ-opioid receptors, is consistent with the observed decrease in adipose accumulation and a potential shift in metabolism from carbohydrate to lipid metabolism. Additionally, mice lacking δ-opioid receptors are resistant to diet-induced obesity (Czyzyk et al., 2012).
To ensure our findings extended beyond rodents, an exploratory study was conducted in NHPs. While this study was not powered to detect statistical differences, the results largely paralleled the effects observed in rats. Through the first 28 days of administration (Phase I), OLZ increased the rate of weight gain despite a decrease in food consumption. Co-administration of SAM decreased the rate of weight gain observed in both the vehicle-treated group and the OLZ-treated group. This decrease in weight gain also correlated with a significant decrease in food consumption. In the second phase of the study (days 35–58), the OLZ-treated monkeys were switched to receive both OLZ and SAM. The addition of SAM was accompanied by a transient decrease in food consumption and a decrease in the rate of weight gain when compared with the first phase of the study. OLZ administration also produced a significant increase in adiposity compared with the VEH group. This increase in adiposity was not observed when SAM was co-administered with OLZ. Furthermore, the addition of SAM to the OLZ-treated monkeys at day 35 decreased the rate of adipose accretion through the subsequent 28 days of the study. These data, in combination with our rat studies, provide a species-independent demonstration of the ability of SAM to mitigate OLZ-induced adiposity.
Insulin sensitivity was assessed following bolus glucose infusion on days 28 and 58. In all cases, there was no significant change in the rate of glucose clearance compared with baseline assessments. At day 28, both vehicle and OLZ significantly increased circulating insulin, suggesting a decrease in insulin sensitivity. In monkeys treated with both SAM and OLZ, insulin sensitivity was similar to baseline measurements. Interestingly, in OLZ-treated monkeys receiving OLZ + SAM during the second phase of the study, insulin sensitivity was restored to baseline, despite having similar weights to the VEH control monkeys that had decreased insulin sensitivity. These data suggest that SAM can both prevent and restore insulin sensitivity in NHPs treated with OLZ.
While the studies above provide compelling data on the ability of SAM to mitigate OLZ-induced metabolic abnormalities, a few caveats should be considered when interpreting the data. Despite the fact that the majority of literature reports using knockout animals and selective antagonists against μ, κ-, and δ-opioid receptors suggest a role for the opioid system in metabolism, the current studies did not determine which opioid receptor or combination thereof is responsible for the effects of SAM on OLZ-induced metabolic dysfunction. Additionally, our studies did not directly assess whether increased adipose glucose utilization develops as a consequence of OLZ-induced insulin resistance. It is also possible that the primary effect of OLZ is to enhance adipose glucose uptake and that SAM prevents this effect. Furthermore, given the complex regulation of metabolism involving central and peripheral processes in several organs, our data are largely empirical and do not allow for firm conclusions at cellular and molecular levels concerning the potentially beneficial effects of SAM on OLZ-associated metabolic dysfunction. Finally, behavioral observations in the NHP studies noted early signs of lethargy in monkeys treated with OLZ alone and in combination with SAM. These transient observations may have affected appetite thereby influencing food consumption and weight. Additional studies are clearly needed to further address these potential study limitations.
Conclusions
OLZ is an efficacious antipsychotic but is associated with an increased risk of deleterious metabolic side effects. In our studies, co-administration of the opioid receptor antagonist SAM mitigated OLZ-induced increases in the rate of weight gain in female rats and NHPs. The hallmark metabolic consequence of OLZ administration was a change in body composition with increased adiposity in male and female rats and female NHPs. The data in the current study support the hypothesis that this change is due in part to an OLZ-induced decrease in glucose availability in muscle and an increase in glucose availability in adipose tissue, leading to an abnormal accumulation of glucose storage in adipose tissue. The major benefit of SAM appears to be blockade of adipose glucose uptake and prevention of changes in body composition produced by OLZ. Data from all studies conducted suggest that OLZ causes a decrease in the rate of glucose clearance and/or whole-body insulin sensitivity. Based on the HIEC study, this effect can largely be attributed to an OLZ-induced decrease in hepatic insulin sensitivity. The ability of SAM to reverse these effects was mixed as there were no effects on GIR within the rodent clamp study but normalization of glucose clearance in rodents after a bolus insulin administration. Moreover, in glucose tolerance tests conducted in NHPs, the addition of SAM restored normal insulin sensitivity when co-administered with OLZ, and restored normal insulin sensitivity in monkeys previously treated with OLZ for 28 days. Collectively, these data indicate that SAM mitigates several metabolic abnormalities associated with OLZ in both the presence and the absence of weight gain.
Supplemental Material
Supplemental material, Supplemental_Data for Samidorphan mitigates olanzapine-induced weight gain and metabolic dysfunction in rats and non-human primates by Jacobi I Cunningham, David J Eyerman, Mark S Todtenkopf, Reginald L Dean, Daniel R Deaver, Connie Sanchez and Mark Namchuk in Journal of Psychopharmacology
Eurofins Pharma Discovery Services CEREP Bioprint panel, https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewitem/BioPrint%C2%AE-Panel/P22-p
Footnotes
Declaration of conflicting interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors were employed by Alkermes, Inc. when studies were conducted.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Alkermes, Inc.
ORCID iD: Jacobi I Cunningham  https://orcid.org/0000-0003-4217-1927
https://orcid.org/0000-0003-4217-1927
Supplemental material: Supplemental material for this article is available online.
References
- Ader M, Kim SP, Catalano KJet al. (2005) Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: A placebo-controlled study of olanzapine and risperidone in dogs. Diabetes 54: 862–871. [DOI] [PubMed] [Google Scholar]
- Albaugh VL, Judson JG, She Pet al. (2011) Olanzapine promotes fat accumulation in male rats by decreasing physical activity, repartitioning energy and increasing adipose tissue lipogenesis while impairing lipolysis. Mol Psychiatry 16: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher-Svanum H, Stensland M, Zhao Zet al. (2005) Acute weight gain, gender, and therapeutic response to antipsychotics in the treatment of patients with schizophrenia. BMC Psychiatry 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak M, Fransen A, Janssen Jet al. (2014) Almost all antipsychotics result in weight gain: A meta-analysis. PLoS One 9: e94112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlack JM, Knapp BI, Deaver DRet al. (2018) In Vitro Pharmacological Characterization of Buprenorphine, Samidorphan, and Combinations Being Developed as an Adjunctive Treatment of Major Depressive Disorder. J Pharmacol Exp Ther 367: 267–281. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Calligaro DO, Falcone JFet al. (1996) Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 14: 87–96. [DOI] [PubMed] [Google Scholar]
- Chintoh AF, Mann SW, Lam TKet al. (2008) Insulin resistance following continuous, chronic olanzapine treatment: An animal model. Schizophr Res 104: 23–30. [DOI] [PubMed] [Google Scholar]
- Cooper GD, Pickavance LC, Wilding JPet al. (2007) Effects of olanzapine in male rats: Enhanced adiposity in the absence of hyperphagia, weight gain or metabolic abnormalities. J Psychopharmacol 21: 405–413. [DOI] [PubMed] [Google Scholar]
- Cope MB, Nagy TR, Fernandez JRet al. (2005) Antipsychotic drug-induced weight gain: Development of an animal model. Int J Obes (Lond) 29: 607–614. [DOI] [PubMed] [Google Scholar]
- Czyzyk TA, Nogueiras R, Lockwood JFet al. (2010) kappa-Opioid receptors control the metabolic response to a high-energy diet in mice. FASEB J 24: 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzyk TA, Romero-Pico A, Pintar Jet al. (2012) Mice lacking delta-opioid receptors resist the development of diet-induced obesity. FASEB J 26: 3483–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoodi N, Kalinichev M, Korneev SAet al. (2009) Hyperphagia and increased meal size are responsible for weight gain in rats treated sub-chronically with olanzapine. Psychopharmacology (Berl) 203: 693–702. [DOI] [PubMed] [Google Scholar]
- De Hert M, Correll CU, Bobes Jet al. (2011. a) Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry 10: 52–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Hert M, Detraux J, van Winkel Ret al. (2011. b) Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol 8: 114–126. [DOI] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Pierri JN, Perel JMet al. (2005) The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: A comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology 30: 1649–1661. [DOI] [PubMed] [Google Scholar]
- Freedman R. (2003) Schizophrenia. N Engl J Med 349: 1738–1749. [DOI] [PubMed] [Google Scholar]
- Gallagher CJ, Gordon CJ, Langefeld CDet al. (2006) Association of the mu-opioid receptor gene with type 2 diabetes mellitus in an African American population. Mol Genet Metab 87: 54–60. [DOI] [PubMed] [Google Scholar]
- Greenway FL, Dunayevich E, Tollefson Get al. (2009) Comparison of combined bupropion and naltrexone therapy for obesity with monotherapy and placebo. J Clin Endocrinol Metab 94: 4898–4906. [DOI] [PubMed] [Google Scholar]
- Haghighi A, Melka MG, Bernard Met al. (2014) Opioid receptor mu 1 gene, fat intake and obesity in adolescence. Mol Psychiatry 19: 63–68. [DOI] [PubMed] [Google Scholar]
- Han C, Liu Y, Sun Xet al. (2017) Prediction of a new body shape index and body adiposity estimator for development of type 2 diabetes mellitus: The Rural Chinese Cohort Study. Br J Nutr 118: 771–776. [DOI] [PubMed] [Google Scholar]
- Horska K, Ruda-Kucerova J, Babinska Zet al. (2016) Olanzapine-depot administration induces time-dependent changes in adipose tissue endocrine function in rats. Psychoneuroendocrinology 73: 177–185. [DOI] [PubMed] [Google Scholar]
- Houseknecht KL, Robertson AS, Zavadoski Wet al. (2007) Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: Implications for adverse metabolic effects. Neuropsychopharmacology 32: 289–297. [DOI] [PubMed] [Google Scholar]
- Ignar DM, Goetz AS, Noble KNet al. (2011) Regulation of ingestive behaviors in the rat by GSK1521498, a novel micro-opioid receptor-selective inverse agonist. J Pharmacol Exp Ther 339: 24–34. [DOI] [PubMed] [Google Scholar]
- Jain S, Bhargava M, Gautam S. (2006) Weight gain with olanzapine: Drug, gender or age? Indian J Psychiatry 48: 39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JE, Corp ES. (2003) Effect of naltrexone on food intake and body weight in Syrian hamsters depends on metabolic status. Physiol Behav 78: 67–72. [DOI] [PubMed] [Google Scholar]
- Kapur S, VanderSpek SC, Brownlee BAet al. (2003) Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther 305: 625–631. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky RB, Remington G. (1999) Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry 156: 286–293. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky RB, Remington Get al. (1998) 5-HT2 and D2 receptor occupancy of olanzapine in schizophrenia: A PET investigation. Am J Psychiatry 155: 921–928. [DOI] [PubMed] [Google Scholar]
- Keyal N, Shrestha GS, Pradhan Set al. (2017) Olanzapine overdose presenting with acute muscle toxicity. Int J Crit Illn Inj Sci 7: 69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissebah AH, Peiris AN. (1989) Biology of regional body fat distribution: Relationship to non-insulin-dependent diabetes mellitus. Diabetes Metab Rev 5: 83–109. [DOI] [PubMed] [Google Scholar]
- Kowalchuk C, Teo C, Wilson Vet al. (2017) In male rats, the ability of central insulin to suppress glucose production is impaired by olanzapine, whereas glucose uptake is left intact. J Psychiatry Neurosci 42: 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurbanov DB, Currie PJ, Simonson DCet al. (2012) Effects of naltrexone on food intake and body weight gain in olanzapine-treated rats. J Psychopharmacol 26: 1244–1251. [DOI] [PubMed] [Google Scholar]
- Lebovitz HE, Banerji MA. (2005) Point: Visceral adiposity is causally related to insulin resistance. Diabetes Care 28: 2322–2325. [DOI] [PubMed] [Google Scholar]
- Leucht S, Cipriani A, Spineli Let al. (2013) Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 382: 951–962. [DOI] [PubMed] [Google Scholar]
- Leucht S, Tardy M, Komossa Ket al. (2012) Antipsychotic drugs versus placebo for relapse prevention in schizophrenia: A systematic review and meta-analysis. Lancet 379: 2063–2071. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JPet al. (2005) Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353: 1209–1223. [DOI] [PubMed] [Google Scholar]
- Lynch CJ, Xu Y, Hajnal Aet al. (2015) RNA sequencing reveals a slow to fast muscle fiber type transition after olanzapine infusion in rats. PLoS One 10: e0123966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WF, Correll CU, Weiden PJet al. (2018) Samidorphan, an Opioid Antagonist, Mitigates Olanzapine-Induced Weight Gain: A Phase 2 Randomized Double-Blind Study in Patients with Schizophrenia. Am J Psychiatry. In press. [DOI] [PubMed] [Google Scholar]
- Minet-Ringuet J, Even PC, Goubern Met al. (2006) Long term treatment with olanzapine mixed with the food in male rats induces body fat deposition with no increase in body weight and no thermogenic alteration. Appetite 46: 254–262. [DOI] [PubMed] [Google Scholar]
- National Research Council (1996) Guide for the Care and Use of Laboratory Animals. Washington, DC: The National Academies Press; DOI: 10.17226/5140. [DOI] [Google Scholar]
- National Research Council (2011) Guide for the Care and Use of Laboratory Animals 8. Washington, DC: The National Academies Press; DOI: 10.17226/12910. [DOI] [Google Scholar]
- Plodkowski RA, Nguyen Q, Sundaram Uet al. (2009) Bupropion and naltrexone: A review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother 10: 1069–1081. [DOI] [PubMed] [Google Scholar]
- Raffan E, Dennis RJ, O’Donovan CJet al. (2016) A Deletion in the Canine POMC Gene Is Associated with Weight and Appetite in Obesity-Prone Labrador Retriever Dogs. Cell Metab 23: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Kong H, Hines Jet al. (1994) Molecular mechanisms of agonist-induced desensitization of the cloned mouse kappa opioid receptor. J Pharmacol Exp Ther 270: 1381–1386. [PubMed] [Google Scholar]
- Seeman MV. (2004) Gender differences in the prescribing of antipsychotic drugs. Am J Psychiatry 161: 1324–1333. [DOI] [PubMed] [Google Scholar]
- Sinnayah PWN, Evans AE, Cowley MA. (2007) Bupropion and naltrexone interact synergistically to decrease food intake in mice. Obesity (Silver Spring) 15: A179. [Google Scholar]
- Skrede S, Martins L, Berge RKet al. (2014) Olanzapine depot formulation in rat: A step forward in modelling antipsychotic-induced metabolic adverse effects. Int J Neuropsychopharmacol 17: 91–104. [DOI] [PubMed] [Google Scholar]
- Smith GC, Vickers MH, Shepherd PR. (2011) Olanzapine effects on body composition, food preference, glucose metabolism and insulin sensitivity in the rat. Arch Physiol Biochem 117: 241–249. [DOI] [PubMed] [Google Scholar]
- Statnick MA, Tinsley FC, Eastwood BJet al. (2003) Peptides that regulate food intake: Antagonism of opioid receptors reduces body fat in obese rats by decreasing food intake and stimulating lipid utilization. Am J Physiol Regul Integr Comp Physiol 284: R1399–R1408. [DOI] [PubMed] [Google Scholar]
- Sun L, McDonnell D, von Moltke L. (2018) Pharmacokinetics and Short-term Safety of ALKS 3831, a Fixed-dose Combination of Olanzapine and Samidorphan, in Adult Subjects with Schizophrenia. Clin Ther 40: 1845–1854 e1842. [DOI] [PubMed] [Google Scholar]
- Tabarin A, Diz-Chaves Y, Carmona Mdel Cet al. (2005) Resistance to diet-induced obesity in mu-opioid receptor-deficient mice: evidence for a “thrifty gene”. Diabetes 54: 3510–3516. [DOI] [PubMed] [Google Scholar]
- Taveira TH, Wu WC, Tschibelu Eet al. (2014) The effect of naltrexone on body fat mass in olanzapine-treated schizophrenic or schizoaffective patients: a randomized double-blind placebo-controlled pilot study. J Psychopharmacol 28: 395–400. [DOI] [PubMed] [Google Scholar]
- Van der Zwaal EM, Janhunen SK, la Fleur SEet al. (2014) Modelling olanzapine-induced weight gain in rats. Int J Neuropsychopharmacol 17: 169–186. [DOI] [PubMed] [Google Scholar]
- Vidarsdottir S, de Leeuw van Weenen JE, Frolich Met al. (2010) Effects of olanzapine and haloperidol on the metabolic status of healthy men. J Clin Endocrinol Metab 95: 118–125. [DOI] [PubMed] [Google Scholar]
- Waring WS, Wrate J, Bateman DN. (2006) Olanzapine overdose is associated with acute muscle toxicity. Hum Exp Toxicol 25: 735–740. [DOI] [PubMed] [Google Scholar]
- Wentland MP, Lou R, Lu Qet al. (2009) Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett 19: 2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentland MP, Lu Q, Lou Ret al. (2005) Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone. Bioorg Med Chem Lett 15: 2107–2110. [DOI] [PubMed] [Google Scholar]
- Weston-Green K, Huang XF, Deng C. (2011) Olanzapine treatment and metabolic dysfunction: A dose response study in female Sprague Dawley rats. Behav Brain Res 217: 337–346. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2018) Schizophrenia (Fact sheet no. 397, updated April 2018). Available at: http://www.who.int/mediacentre/factsheets/fs397/en/.
- Zhang J, Frassetto A, Huang RRet al. (2006) The mu-opioid receptor subtype is required for the anorectic effect of an opioid receptor antagonist. Eur J Pharmacol 545: 147–152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplemental_Data for Samidorphan mitigates olanzapine-induced weight gain and metabolic dysfunction in rats and non-human primates by Jacobi I Cunningham, David J Eyerman, Mark S Todtenkopf, Reginald L Dean, Daniel R Deaver, Connie Sanchez and Mark Namchuk in Journal of Psychopharmacology