

Abstract
In nature, co-assembly of polypeptides, nucleic acids, and polysaccharides is used to create functional supramolecular structures. Here, we show that DNA nanostructures can be used to template interactions between peptides and to enable the quantification of multivalent interactions that would otherwise not be observable. Our functional building blocks are peptide–oligonucleotide conjugates comprising de novo designed dimeric coiled-coil peptides covalently linked to oligonucleotide tags. These conjugates are incorporated in megadalton DNA origami nanostructures and direct nanostructure association through peptide–peptide interactions. Free and bound nanostructures can be counted directly from electron micrographs, allowing estimation of the dissociation constants of the peptides linking them. Results for a single peptide–peptide interaction are consistent with the measured solution-phase free energy; DNA nanostructures displaying multiple peptides allow the effects of polyvalency to be probed. This use of DNA nanostructures as identifiers allows the binding strengths of homo- and heterodimeric peptide combinations to be measured in a single experiment and gives access to dissociation constants that are too low to be quantified by conventional techniques. The work also demonstrates that hybrid biomolecules can be programmed to achieve spatial organization of complex synthetic biomolecular assemblies.
Keywords: self-assembly, DNA nanostructure, peptide, heterodimeric coiled coil, polyvalent binding, peptide−oligonucleotide conjugate
Natural biological molecules—lipids, nucleic acids, peptides, proteins, and polysaccharides—fold, assemble, and co-assemble to form a stunning variety of functional three-dimensional structures and complexes. Understanding and harnessing these processes underpins emerging applications in synthetic biology and biotechnology.1−3 Broadly speaking, there are two approaches to the manipulation of biomolecular assembly. Biomolecular engineering allows the chemistries of natural biomolecules to be altered to produce variants of wild-type parents whose interactions are tailored to specific applications.4−6 The alternative, de novo design, is completely bottom-up: polypeptide or nucleic acid sequences are generated from scratch by the designer to fold and assemble in prescribed ways.2,3,7,8 This second route has the potential to access a much larger design space but requires clear and reliable sequence-to-structure design rules: this is particularly challenging in the case of polypeptides. To begin to approach the complexity of natural systems and to harness fully the potential of de novo biomolecular assembly, we must learn to control and combine the assembly of different biomolecular types to generate structured, functional co-assemblies.9,10
The specificity of Watson–Crick pairing between the four DNA bases has enabled the fabrication of a rapidly increasing range of DNA nanostructures, especially DNA origami nanostructures, whose assembled shapes can be specified with high precision and reliability through design of the oligonucleotide components.11−18 This process is facilitated by rapid access to synthetic oligonucleotides with ever-decreasing price,19 the application of computer-aided design,20,21 and, most recently, the scaling up of DNA production and assembly.22 However, although well adapted to programmed nanostructure assembly, DNA has minimal intrinsic functionality. In contrast, the chemical diversity of the natural amino acids underpins the folding of a wide variety of protein structures with highly evolved functions including architecture, signaling, active and passive transport, and catalysis. However, the relationship between the amino acid sequence of a polypeptide and its structure and function is complex, which makes de novo protein design extremely challenging.3,7
For certain classes of protein structure, enough is understood to allow rational and reliable de novo design. For example, for α-helical coiled coils, which are bundles of two or more α-helices,23,24 established sequence-to-structure relationships have enabled the design of homomeric dimers, trimers and tetramers and the assembly of obligate heterodimers and trimers.25−27 More complex and elaborate architectures—for instance, antiparallel arrangements of helices and homomeric and heteromeric assemblies above a tetramer—have proved more challenging to design and have been discovered serendipitously or designed computationally.7,24,27,28 One way to access more complex structures is to make use of the high spatial resolution and predictable architectures of DNA nanostructures to template peptide assemblies and thus force otherwise unobtainable configurations of α-helices. Others have described the assembly of trimeric coiled-coil DNA conjugates,29,30 layered nanostructures formed from collagen-mimetic peptides and oligonucleotides,31 and DNA-directed peptide pores;9,10 conversely, proteins have been used to direct the folding of DNA scaffolds.32
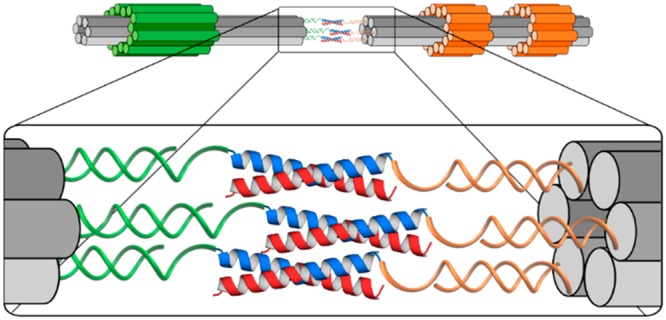
Here, we describe the formation of peptide–oligonucleotide conjugates that can be used to control the assembly of larger DNA–peptide structures and thereby to quantify multivalent peptide–peptide interactions. Single-stranded DNA (ssDNA) “handles”, extending from two distinct DNA origami nanostructures, hybridize to complementary oligonucleotide “tags” which are covalently linked to peptides. This places a prescribed number of peptides, each of which is half of a heterodimeric peptide coiled coil, on one end of each DNA origami. Dimerization of the composite nanostructures is driven by association of the peptides to form coiled coils (Scheme 1). Markers on the DNA origamis allow assembled species to be distinguished by transmission electron microscopy (TEM). Counting the distributions of all kinds of assembly in electron micrographs allows the dissociation constant (Kd) for each to be estimated and the effect of polyvalency on peptide–peptide interactions to be investigated.
Scheme 1. Assembly of Megadalton DNA Origami Structures Is Driven by Peptide–Oligonucleotide Conjugates through Formation of Peptide Coiled-Coil Heterodimers and Pairing of Oligonucleotide Tags with Complementary ssDNA Handles.

Peptide–oligonucleotide conjugates are formed via a Cu-free cycloaddition between the highlighted azide side chains of the peptides and the ring-strained dibenzylcyclooctyne groups of the oligonucleotide tags.
Results and Discussion
Peptides Compatible with Both Oligonucleotide Conjugation and Peptide Assembly Can Be Designed De Novo
Coiled-coil peptide sequences have heptad repeats, abcdefg, with hydrophobic residues predominating at the “a” and “d” sites.33,34 These form amphipathic α-helices whose assembly is driven by the formation of an a/d hydrophobic seam. Judicious placement of specific amino acids at these positions dictates an oligomeric state, with a = isoleucine (Ile) and d = leucine (Leu) directing dimers.24,26 The “e” and “g” sites which flank the hydrophobic core can be used to stabilize both homo- and heteromeric assemblies.35−37 Complementary patterns of charged residues at these sites have been used by many groups to create de novo heterodimers.35−40
The simplest previously reported coiled-coil heterodimers have glutamic acid (Glu) at all “e” and “g” sites in one peptide and lysine (Lys) at these sites in the complementary peptide.35−37,41 However, we confirmed that the basic peptide binds nonspecifically and tightly to DNA, consistent with strong electrostatic interactions, making it unsuitable for use in composite peptide–DNA nanostructures. We therefore designed a series of parallel heterodimeric coiled coils in which each peptide was, overall, close to or exactly charge-neutral at pH 7. Peptide sequences are given in Supporting Information (SI), Table S1. These peptides had blocks of both Glu- and Lys-based heptads, with Asn residues at some of the central “a” sites to add stability and increase specificity.42 They varied in length, having 3, 3.5, or 4 heptad repeats and, in the case of the 4-heptad pairs, either one or two asparagine residues at “a” sites to destabilize off-target homomers. The peptides were made by solid-phase peptide synthesis, purified by high-performance liquid chromatography (HPLC), and confirmed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (SI Figure S1). They were characterized in solution, both alone and in combination with their designed partners, using circular dichroism (CD) spectroscopy and analytical ultracentrifugation (AUC) (Figure 1 and SI Figures S2 and S3, Tables S2 and S3).
Figure 1.

Biophysical characterization of the coiled-coil heterodimer formed between charge-neutral peptides CC-Di-EK and CC-Di-KE. (a) CD spectra recorded at 5 °C and (b) thermal unfolding (filled symbols, solid line) and refolding (open symbols, dashed line) profiles, monitored through the CD signal at 222 nm, of the individual peptides CC-Di-EK (red, circles) and CC-Di-KE (blue, squares) and of an equimolar mixture of CC-Di-EK:KE (purple, triangles). The average of the spectra of the individual peptides is shown in black (no symbols). The spectrum of the mixture indicates the formation of α-helices. Representative data from a minimum of three replicates are shown. Conditions: each peptide 50 μM in PBS (pH 7.4), 1 mm path length quartz cuvette. (c) Sedimentation equilibrium data (top panel) from AUC of CC-Di-EK:KE at 55 μM of each peptide in PBS, pH 7.4 (circles). Fits assuming a single ideal species (black lines, residuals in bottom panel) return a mass of 6174 Da; i.e., 1.0 × dimer mass; r = radial distance from the center of the rotor and r0 = the reference radius; rotor speeds were 44 krpm (blue), 48 krpm (light blue), 52 krpm (lilac), 56 krpm (purple), and 60 krpm. (red). (d) Peptide sequences and optimized model of CC-Di-EK:KE with lysine-rich heptads shown in blue, glutamic acid-rich heptads in red, and asparagine residues at positions “a” highlighted in green. Full methods are given in the SI.
On the basis of CD and AUC characterization, the CC-Di-EK and CC-Di-KE pair (Figure 1), comprising four complete heptad repeats with asparagines at two “a” sites, was identified as a suitable coiled coil to link peptide–DNA nanostructures. CD spectra showed that unmixed CC-Di-EK and CC-Di-KE were either largely or fully unfolded at 5 °C, neutral pH, and 50 μM concentration, but that a 1:1 mixture of the peptides was fully α-helical43 and had a sigmoidal thermal unfolding transition with a midpoint (TM) of 43 °C (Figure 1a,b), consistent with the formation of a stable coiled-coil heterodimer. From the concentration dependence of the TM, the Kd for heterodimerization was estimated to be 102 ± 26 nM (SI Figure S4). Fits of AUC data assuming a single ideal species returned the mass of the CC-Di-EK:KE dimer (Figure 1c) and confirmed that the umixed peptides were monomers (SI Figure S3 and Table S3). Molecular models built and optimized in ISAMBARD44 indicate that these peptides form a stable coiled-coil dimer with a strong preference for the parallel helix orientation (Figure 1d and SI Figure S5). To confirm the preferred helix orientation, the peptides were synthesized with terminal Cys residues and linked as obligate heterodimers via disulfide bonds. With the peptides linked via the same ends—that is, both C-termini, consistent with parallel helix formation—the resulting conjugate was highly helical, thermally stable, and monomeric (SI Figures S2 and S3, Tables S2 and S3). In contrast, when linked via opposite ends, the behavior of the construct was more complex with larger oligomeric complexes indicated by AUC. These experiments support the conclusion that the unlinked CC-Di-EK and CC-Di-KE peptides form a stable, parallel, helical heterodimer. CD spectra showed no interaction between either CC-Di-EK or CC-Di-KE and the ssDNA tags (SI Figure S6a,b). This confirmed our design choice to use charge-neutral or acidic peptides for conjugation to DNA tags.
Other peptide pairs were not developed further. CD and AUC experiments (SI Figures S2 and S3, Tables S2 and S3) showed that the 4-heptad peptides with only a single asparagine displayed off-target homomeric interactions and that the 3-heptad pair was not fully folded. The 3.5-heptad pair CC-Di-EN3.5 was rejected because it had a +1 net charge.
Peptide–oligonucleotide conjugates were synthesized as follows. Azidonorleucine (Z) was added during peptide synthesis at the C-terminus of CC-Di-EK and at the N-terminus of CC-Di-KE to give CC-Di-EK-Z and Z-CC-Di-KE (SI Table S1). These were then conjugated to dibenzylcyclooctyne-functionalized oligonucleotide tags, α and β, respectively, by copper-free 1–3 Huisgen cycloaddition (Scheme 1 and SI Figure S6).45 The resulting conjugates, CC-Di-EK-α and β-CC-Di-KE, were isolated by reverse-phase HPLC and confirmed by LC-MS (SI Figures S6 and S7). Flexible single-stranded spacers (eight thymidines) were included in the oligonucleotide tags to separate the peptide attachment site and the duplex formed between tag and complementary handle (SI Figure S10 and Table S6). The CD spectra of the peptide–oligonucleotide conjugates when mixed were consistent with the formation of a fully folded α-helical species and had a thermal unfolding profile (TM = 34 °C) similar to that of the untagged peptides (SI Figure S8: the predicted TM for unconjugated peptides at this concentration is 36 °C).
Two Distinct DNA Nanostructures Whose Identities and Orientation Can Be Determined by Electron Microscopy
Two readily distinguishable, asymmetric DNA origami nanostructures, A and B, were designed with the assistance of caDNAno software20,21,46 (Figure 2a,b47 and SI Figure S9). Each had a central six-helix bundle with either one or two asymmetrically placed sleeves (bulges comprising a concentric layer of DNA helices) wrapped around it. The nanostructures were assembled by annealing a 2686 nucleotide single-stranded scaffold, derived from pUC19 plasmid,48 with 69 and 67 synthetic oligonucleotide staples, respectively (SI Table S6). The asymmetric positions of the sleeves allowed rapid determination of the identity and orientation of individual origamis in electron micrographs (Figure 2c,d). Each origami was functionalized at the end further from the sleeves with three single-stranded 23 nucleotide handles as extensions to selected staples (Figure 2a,b). Extensions were either complementary to the oligonucleotide tags α and β, referred to as A and B handles, respectively, or noncomplementary dummy sequences (SI Figure S10 and Table S6).
Figure 2.

DNA nanostructure design and characterization. (a,b) Representations of DNA nanostructures A (a) and B (b). The core of each structure is a 6-helix bundle (each double helix is represented by a gray cylinder). Staple extensions provide three single-stranded handle sequences at one end of the six-helix bundle (top left insets). The valency can be controlled by replacing one or more handles with a dummy sequence unrelated to that of the peptide-functionalized oligonucleotide tags. Asymmetrically positioned sleeves wrapped around the 6-helix bundle (green in (a) and orange in (b)) allow the two structures to be distinguished by TEM. Top right insets: atomic models of origamis A (a) and B (b) predicted by CanDo and visualized with QuteMol. (c,d) TEM images confirming the formation of origamis A (c) and B (d). Scale bars: 50 nm.
Peptide–Oligonucleotide Conjugates Bring Together DNA Nanostructures
Each DNA origami was decorated with one, two, or three copies of the corresponding half of the heterodimeric coiled-coil peptide pair, that is, A with CC-Di-EK-α and B with β-CC-Di-KE. Preassembled origamis displaying the specified number of complementary ssDNA handles were incubated at room temperature (approximately 20 °C) with a 3× stoichiometric excess of the corresponding oligonucleotide-tagged peptide. The resulting peptide-decorated origamis, named An and Bn, where n (n = 1, 2, 3) indicates the number of peptides attached to each DNA origami, were purified by gel electrophoresis (Figure 3a and SI Figure S11). Subsequent mixing of purified An and Bn resulted in the formation of heterodimeric complexes An:Bn, which, when imaged by TEM, were seen to be joined almost exclusively via their peptide-labeled ends (Figure 3b–d and SI Figure S12), consistent with dimerization driven by the formation of peptide coiled coils as designed.
Figure 3.

Directed assembly of DNA nanostructures through interactions of peptide–oligonucleotide conjugates. (a) DNA origamis A3 and B3 decorated with three peptides. (b–e) Representative TEM images of the assembly of peptide-decorated DNA origamis. The distribution between species depends on the number of peptides, n, displayed on each origami: (b) n = 3, (c) n = 2, (d) n = 1, and (e) n = 0. White numbers in (b) indicate representative heterodimers (1 and 2) and monomers (3 and 4). The initial concentrations of assemblies An and Bn were, respectively, 0.61 and 0.84 nM (b, n = 3), 0.84 and 1.09 nM (c, n = 2), 1.69 and 2.39 nM (d, n = 1), and 0.68 and 0.74 nM (e, n = 0). Scale bars: 50 nm. (f) Dissociation constants of heterodimers of origamis functionalized with different numbers of peptides were determined from linear fits (solid lines) to plots of [An] × [Bn] as a function of [An:Bn] over a range of total monomer concentrations from 0.25 to 2.4 nM. Data were weighted by estimated uncertainties (SI Tables S4, S5 and supporting discussion).
The use of two DNA origami structures that can be distinguished in electron micrographs allowed us to identify manually and count free assemblies An, Bn, and all complexes formed between them, that is, the intended heterodimer An:Bn, homodimers (An:An and Bn:Bn), and any larger multimers (Figure 3b–e and SI Figure S12 and Table S4). If it is assumed that the distribution between complexes on the TEM grid is representative of that present in solution, then, given the initial total concentrations of the two peptide-decorated DNA origamis, the equilibrium concentration of each species can be inferred. In each experiment, approximately 1000 origamis of each type were counted and classified as monomeric, homo- or heterodimeric, or as part of a larger multimer. Uncertainties in inferring equilibrium concentrations of these species from the observed distributions of particles in electron micrographs are discussed in the SI supporting discussion and quantified in SI Table S5.
As anticipated, in the absence of attached peptides (n = 0), very few complexes were counted (Figure 3e and SI Figure S12-1 and Table S4-1). This background count is likely to be due to chance observations of two origami structures in close proximity rather than to any interaction between the origamis: the data are consistent with a random distribution of origami nanostructures in each micrograph with a minimum detectable separation between the ends of adjacent origamis of 8 nm (SI supporting discussion). This minimum radius was used to calculate a background due to random coincidences which was subtracted from the observed numbers of dimers between nanostructures functionalized with one, two, or three peptides (SI Tables S4-2, S4-3, and S4-4).
For mono- and bifunctionalized origamis (n = 1, 2), the numbers of dimers observed were small, typically <2 and <4%, respectively, of the number of monomers counted, but for trifunctionalized origamis, the population of heterodimers reached 40% of the total number of particles. It was possible to obtain consistent estimates of the dissociation constants for all heterodimers and for n = 3 homodimers. Kd values were estimated from the fitted slopes of plots of [An] × [Bn] vs [An:Bn], [An]2vs [An:An], and [Bn]2vs [Bn:Bn] for a range of initial monomer concentrations (0.25–2.4 nM) (Figure 3f and SI Figure S13). Results are presented in Table 1.
Table 1. Dissociation Constants of Peptide-Mediated DNA Origami Dimers Inferred from the Relative Numbers of Complexes Observed in Electron Micrographs as Functions of Total Monomer Concentrations.
|
Kd (nM)a |
|||
|---|---|---|---|
| valency, n | complex An:Bn | complex An:An | complex Bn:Bn |
| 1 | 130 ± 20 | ||
| 2 | 25 ± 3 | ||
| 3 | 0.7 ± 0.1 | 24 ± 4 | 20 ± 3 |
See SI supporting discussion of experimental errors: there is an additional systematic error of approximately 20% in all Kd values corresponding to uncertainty in determining DNA origami concentrations.
Our estimated Kd of 130 ± 20 nM for the monofunctionalized heterodimer A1:B1 is close to the value of 102 ± 26 nM determined from the melting temperatures of free peptides in solution (SI Figure S4). This is consistent with our design hypothesis that interactions between peptide-functionalized origamis are dominated by specific peptide–peptide interactions and suggests that the preparation of samples for electron microscopy does not introduce significant sampling bias (SI supporting discussion). Increasing the number of displayed peptides increases the interaction strength, decreasing the dissociation constant by approximately 200× for the trifunctionalized heterodimer. Association between trifunctionalized homodimers A3:A3 and B3:B3 is clearly resolved and is 30× weaker than for the corresponding heterodimer, consistent with the designed association bias of the peptides.
Upon increasing the number of attached peptides from n = 1 to n = 2, the heterodimer Kd decreases only by a factor of approximately 5, which is orders of magnitude less than the factor that would be expected if two parallel peptide links could be formed independently.49,50 As shown in SI Figures S9 and S10, the ends of the origamis are not flat (the ends of the six bundled DNA helices are not aligned). Despite the presence of flexible T8 linkers between each peptide and the DNA duplex linking it to the origami, this may hinder the simultaneous formation of multiple coiled-coil links between pairs of origamis, providing a natural explanation for the unexpectedly small increase in affinity observed. The observed increase in affinity can be almost entirely attributed to the 4-fold increase in the number of ways of making a single coiled coil from two pairs of peptides, consistent with the hypothesis that a single peptide–peptide link dominates interactions between doubly functionalized origamis. If this combinatorial argument were extended to the n = 3 system, we would predict Kd ≈ 10 nM, as there are nine ways to make a single coiled-coil link between origamis displaying three peptides each. However, we observed an order of magnitude greater enhancement. This strongly suggests polyvalent binding:49,50 there is a significant probability that at least two of the three possible coiled-coil links are formed simultaneously.
Conclusions
In summary, we have shown that DNA origami nanostructures can be used to template and quantify the assembly of de novo designed peptides. To avoid nonspecific binding of the peptides to DNA, it was necessary to design charge-neutral, heterodimeric coiled-coil peptides which will prove useful in biomolecular construction and synthetic biology more generally. The use of distinguishable DNA origami conjugates allows the use of electron microscopy to estimate otherwise undetectable concentrations of free and bound peptides. The direct enumeration of interacting species provides an alternative way to determine Kd, which can be applied at concentrations much lower than would be practical for traditional methods such as CD spectroscopy or isothermal calorimetry. It also allows simultaneous measurement of dissociation constants for all observed species in a reaction mixture, in this case, homomeric and heteromeric complexes, something that would be impossible to deduce by measurement of a single average quantity such as circular dichroism. This semiquantitative technique also allows us to probe the effects of polyvalent interactions: the enhanced binding of trifunctionalized origami heterodimers indicates that the use of a DNA origami template to preposition multiple peptides is an effective method to control their interactions. This study shows the way to the development of more sophisticated DNA templates to control of the spatial orientation of peptide and protein assemblies and to the use of peptide–peptide interactions to direct DNA assembly.
Experimental Section
Peptide Synthesis
Peptides were synthesized via solid-phase peptide synthesis on a rink amide resin (Novabiochem, 0.1 mmol scale) using a microwave-assisted CEM Liberty Blue synthesizer (CEM Corporation) and standard deprotection and coupling chemistries. Peptide-grade dimethylformamide (DMF) was used throughout. Fmoc deprotections were performed with 20% morpholine in DMF (Merck-Millipore), followed by Cl-HOBt (AGTC Bioproducts, 0.5 M in DMF) and DIC (AGTC Bioproducts, 1 M in DMF) activated couplings of each amino acid (AGTC Bioproducts). N-Terminal acetylations were performed with pyridine (Fisher Scientific, 0.5 mL) and acetic anyhydride (BDH Laboratories, 0.25 mL) in DMF (15 min). Peptides were cleaved from the resin in trifluoroacetic acid (Acros Organics) with a triisopropylsilane scavenger (Sigma-Aldrich) and H2O at 90:5:5 vol % under agitation for 2 h. The cleavage mixtures were then filtered to remove the resin, and the volume was reduced to <5 mL by a flow of nitrogen before precipitation of the crude peptides by addition of cold diethyl ether. A solid peptide pellet was obtained by centrifugation followed by removal of the supernatant. This was then lyophilized from 1:1 H2O/MeCN to yield crude peptides as white powders.
DNA Origami Design
DNA origamis A and B were designed using caDNAno.3 Nicks separating staple strands were positioned manually, as shown in SI Figure S9. As shown in SI Figure S10, staple strands on helices 0, 2, and 4, at the end further from the marker bulges of each origami, were extended with 23 nucleotide handles to hybridize with corresponding peptide–oligonucleotide tags α or β. Some or all of these handles were substituted with dummy handles, 23 nucleotide extensions that are noncomplementary to the peptide–oligonucleotide conjugates, when origamis functionalized with 2, 1, or 0 peptides were produced.
A T8 spacer was included at the 5′ ends of the DNA tags to separate the peptides from the 23 nucleotide binding sequences complementary to the handles. Polythymidine extensions (in most cases T4) were added to staple strands at the ends of each origami, except at the handle positions, to minimize nonspecific helix-stacking interactions between origamis.
Equilibrium Concentration Calculations
The numbers (N) of monomers (An, Bn) and of dimers (An:Bn, An:An, Bn:Bn) and higher multimers, in which the proximity of the peptide-functionalized ends was consistent with the formation of a peptide-bound complex, were counted. A background corresponding to the random chance of observing two origami structures in close proximity (i.e., when their peptide-functionalized ends are within 8 nm of each other; see main text) was subtracted. The equilibrium concentration of each species was assumed to be proportional to the number counted and was estimated from counted numbers of each assembled complex and the total monomer concentrations (summed over all assemblies) (SI Table S4).
Kd Calculation and Fitting Model
Linear fits to graphs of [An] × [Bn] vs [An:Bn], [An]2vs [An:An], and [Bn]2vs [Bn:Bn] for six different initial concentrations [Bn]0 ≈ [An]0 from 0.25 to 2.4 nM were obtained using OriginPro (Figure 3f and SI Figure S13). Data were weighted by estimated uncertainties in both quantities (SI supporting discussion and Table S5). The dissociation constant was estimated as the gradient of a linear fit to these data which was constrained to pass through the origin, (0,0).
Acknowledgments
We thank Dr. R. Schreiber for discussions regarding DNA nanostructure design, ATDBio for DBCO–DNA synthesis and LCMS analyses, and the University of Bristol, School of Chemistry Mass Spectrometry Facility, for access to the EPSRC-funded Bruker Ultraflex MALDI TOF/TOF and Waters Synapt G2S instruments (EP/K03927X/1). This research was supported by the ERANET Scheme of the Seventh Framework Programme (FP7) of the European Commission, project title 14-ERASynBio BioOrigami, administered through the U.K. Biotechnology and Biological Sciences Research Council refs BB/M005615/1 and BB/M005739/1. A.J.T. and D.N.W. were supported by Royal Society–Wolfson Research Merit Awards.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b04251.
Additional methods; peptide interaction models; synthesis of peptide–oligonucleotide conjugates; biophysical characterization of peptides and peptide–oligonucleotide conjugates; design and characterization of DNA origami structures; TEM images of peptide-mediated DNA assemblies and determination of Kd values; error analysis (PDF)
Author Contributions
∥ J.J. and E.G.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Channon K.; Bromley E. H.; Woolfson D. N. Synthetic Biology through Biomolecular Design and Engineering. Curr. Opin. Struct. Biol. 2008, 18, 491–498. 10.1016/j.sbi.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Regan L.; Caballero D.; Hinrichsen M. R.; Virrueta A.; Williams D. M.; O’Hern C. S. Protein Design: Past, Present, and Future. Biopolymers 2015, 104, 334–350. 10.1002/bip.22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson D. N.; Bartlett G. J.; Burton A. J.; Heal J. W.; Niitsu A.; Thomson A. R.; Wood C. W. De Novo Protein Design: How Do We Expand into the Universe of Possible Protein Structures?. Curr. Opin. Struct. Biol. 2015, 33, 16–26. 10.1016/j.sbi.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St. Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunzel H. A.; Garrabou X.; Pott M.; Hilvert D. Speeding up Enzyme Discovery and Engineering with Ultrahigh-Throughput Methods. Curr. Opin. Struct. Biol. 2018, 48, 149–156. 10.1016/j.sbi.2017.12.010. [DOI] [PubMed] [Google Scholar]
- Chen K.; Huang X.; Kan S. B. J.; Zhang R. K.; Arnold F. H. Enzymatic Construction of Highly Strained Carbocycles. Science 2018, 360, 71–75. 10.1126/science.aar4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P. S.; Boyken S. E.; Baker D. The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Nangreave J.; Liu Y.; Yan H. Structural DNA Nanotechnology: State of the Art and Future Perspective. J. Am. Chem. Soc. 2014, 136, 11198–11211. 10.1021/ja505101a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt E.; Tusk S. E.; Bayley H. DNA Scaffolds Support Stable and Uniform Peptide Nanopores. Nat. Nanotechnol. 2018, 13, 739–746. 10.1038/s41565-018-0139-6. [DOI] [PubMed] [Google Scholar]
- Henning-Knechtel A.; Knechtel J.; Magzoub M. DNA-Assisted Oligomerization of Pore-Forming Toxin Monomers into Precisely-Controlled Protein Channels. Nucleic Acids Res. 2017, 45, 12057–12068. 10.1093/nar/gkx990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman N. C. DNA in a Material World. Nature 2003, 421, 427–431. 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Hogberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459, 414–418. 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veneziano R.; Ratanalert S.; Zhang K.; Zhang F.; Yan H.; Chiu W.; Bathe M. Designer Nanoscale DNA Assemblies Programmed from the Top Down. Science 2016, 352, 1534. 10.1126/science.aaf4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong L. L.; Hanikel N.; Yaghi O. K.; Grun C.; Strauss M. T.; Bron P.; Lai-Kee-Him J.; Schueder F.; Wang B.; Wang P. F.; Kishi J. Y.; Myhrvold C.; Zhu A.; Jungmann R.; Bellot G.; Ke Y. G.; Yin P. Programmable Self-Assembly of Three-Dimensional Nanostructures from 10,000 Unique Components. Nature 2017, 552, 72–77. 10.1038/nature24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Sigl C.; Dietz H. Gigadalton-Scale Shape-Programmable DNA Assemblies. Nature 2017, 552, 78–83. 10.1038/nature24651. [DOI] [PubMed] [Google Scholar]
- Pinheiro A. V.; Han D. R.; Shih W. M.; Yan H. Challenges and Opportunities for Structural DNA Nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torring T.; Voigt N. V.; Nangreave J.; Yan H.; Gothelf K. V. DNA Origami: A Quantum Leap for Self-Assembly of Complex Structures. Chem. Soc. Rev. 2011, 40, 5636–5646. 10.1039/c1cs15057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson R. The Changing Economics of DNA Synthesis. Nat. Biotechnol. 2009, 27, 1091–1094. 10.1038/nbt1209-1091. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3d DNA-Origami Shapes with Cadnano. Nucleic Acids Res. 2009, 37, 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.-N.; Kilchherr F.; Dietz H.; Bathe M. Quantitative Prediction of 3d Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012, 40, 2862–2868. 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius F.; Kick B.; Behler K. L.; Honemann M. N.; Weuster-Botz D.; Dietz H. Biotechnological Mass Production of DNA Origami. Nature 2017, 552, 84–87. 10.1038/nature24650. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Bassler J.; Dunin-Horkawicz S.. The Structure and Topology of α-Helical Coiled Coils. In Fibrous Proteins: Structures and Mechanisms; Parry D. A. D., Squire J. M., Eds.; Springer: Cham, 2017; pp 95–129. [Google Scholar]
- Woolfson D. N.Coiled-Coil Design: Updated and Upgraded. In Fibrous Proteins: Structures and Mechanisms; Parry D. A. D., Squire J. M., Eds.; Springer: Cham, 2017; pp 35–61. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Zhang T.; Kim P. S.; Alber T. A Switch between 2-Stranded, 3-Stranded and 4-Stranded Coiled Coils in Gcn4 Leucine-Zipper Mutants. Science 1993, 262, 1401–1407. 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Woolfson D. N. The Design of Coiled-Coil Structures and Assemblies. Adv. Protein Chem. 2005, 70, 79–112. 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- Fletcher J. M.; Boyle A. L.; Bruning M.; Bartlett G. J.; Vincent T. L.; Zaccai N. R.; Armstrong C. T.; Bromley E. H. C.; Booth P. J.; Brady R. L.; Thomson A. R.; Woolfson D. N. A Basis Set of De Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology. ACS Synth. Biol. 2012, 1, 240–250. 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- Thomson A. R.; Wood C. W.; Burton A. J.; Bartlett G. J.; Sessions R. B.; Brady R. L.; Woolfson D. N. Computational Design of Water-Soluble α-Helical Barrels. Science 2014, 346, 485–488. 10.1126/science.1257452. [DOI] [PubMed] [Google Scholar]
- Lou C.; Martos-Maldonado M. C.; Madsen C. S.; Thomsen R. P.; Midtgaard S. R.; Christensen N. J.; Kjems J.; Thulstrup P. W.; Wengel J.; Jensen K. J. Peptide-Oligonucleotide Conjugates as Nanoscale Building Blocks for Assembly of an Artificial Three-Helix Protein Mimic. Nat. Commun. 2016, 7, 12294. 10.1038/ncomms12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou C.; Christensen N. J.; Martos-Maldonado M. C.; Midtgaard S. R.; Ejlersen M.; Thulstrup P. W.; Sørensen K. K.; Jensen K. J.; Wengel J. Folding Topology of a Short Coiled-Coil Peptide Structure Templated by an Oligonucleotide Triplex. Chem. - Eur. J. 2017, 23, 9297–9305. 10.1002/chem.201700971. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Meyer T. A.; Modlin C.; Zuo X. B.; Conticello V. P.; Ke Y. G. Structurally Ordered Nanowire Formation from Co-Assembly of DNA Origami and Collagen-Mimetic Peptides. J. Am. Chem. Soc. 2017, 139, 14025–14028. 10.1021/jacs.7b08087. [DOI] [PubMed] [Google Scholar]
- Praetorius F.; Dietz H. Self-Assembly of Genetically Encoded DNA-Protein Hybrid Nanoscale Shapes. Science 2017, 355, eaam5488. 10.1126/science.aam5488. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Gruber M. The Structure of α-Helical Coiled Coils. Adv. Protein Chem. 2005, 70, 37–38. 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Bassler J. Coiled Coils – a Model System for the 21st Century. Trends Biochem. Sci. 2017, 42, 130–140. 10.1016/j.tibs.2016.10.007. [DOI] [PubMed] [Google Scholar]
- O’Shea E. K.; Lumb K. J.; Kim P. S. Peptide ‘Velcro’: Design of a Heterodimer Coiled Coil. Curr. Biol. 1993, 3, 658–667. 10.1016/0960-9822(93)90063-T. [DOI] [PubMed] [Google Scholar]
- Litowski J. R.; Hodges R. S. Designing Heterodimeric Two-Stranded α-Helical Coiled-Coils: The Effect of Chain Length on Protein Folding, Stability and Specificity. J. Pept. Res. 2001, 58, 477–492. 10.1034/j.1399-3011.2001.10972.x. [DOI] [PubMed] [Google Scholar]
- Thomas F.; Boyle A. L.; Burton A. J.; Woolfson D. N. A Set of De Novo Designed Parallel Heterodimeric Coiled Coils with Quantified Dissociation Constants in the Micromolar to Sub-Nanomolar Regime. J. Am. Chem. Soc. 2013, 135, 5161–5166. 10.1021/ja312310g. [DOI] [PubMed] [Google Scholar]
- Crooks R. O.; Lathbridge A.; Panek A. S.; Mason J. M. Computational Prediction and Design for Creating Iteratively Larger Heterospecific Coiled Coil Sets. Biochemistry 2017, 56, 1573–1584. 10.1021/acs.biochem.7b00047. [DOI] [PubMed] [Google Scholar]
- Gradišar H.; Jerala R. De Novo Design of Orthogonal Peptide Pairs Forming Parallel Coiled-Coil Heterodimers. J. Pept. Sci. 2011, 17, 100–106. 10.1002/psc.1331. [DOI] [PubMed] [Google Scholar]
- Negron C.; Keating A. E. A Set of Computationally Designed Orthogonal Antiparallel Homodimers That Expands the Synthetic Coiled-Coil Toolkit. J. Am. Chem. Soc. 2014, 136, 16544–16556. 10.1021/ja507847t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth M. C.; Rink W. M.; Meyer N. F.; Thomas F. Kinetic Studies on Strand Displacement in De Novo Designed Parallel Heterodimeric Coiled Coils. Chem. Sci. 2018, 9, 4308–4316. 10.1039/C7SC05342H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. M.; Bartlett G. J.; Boyle A. L.; Danon J. J.; Rush L. E.; Lupas A. N.; Woolfson D. N. N@a and N@D: Oligomer and Partner Specification by Asparagine in Coiled-Coil Interfaces. ACS Chem. Biol. 2017, 12, 528–538. 10.1021/acschembio.6b00935. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J.; Fasman G. D. Computed Circular Dichroism Spectra for the Evaluation of Protein Conformation. Biochemistry 1969, 8, 4108–4116. 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- Wood C. W.; Heal J. W.; Thomson A. R.; Bartlett G. J.; Ibarra A. Á.; Brady R. L.; Sessions R. B.; Woolfson D. N. Isambard: An Open-Source Computational Environment for Biomolecular Analysis, Modelling and Design. Bioinformatics 2017, 33, 3043–3050. 10.1093/bioinformatics/btx352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Song W. S.; Borges C.; Lindsay S.; Zhang P. M. Click Addition of a DNA Thread to the N-Termini of Peptides for Their Translocation through Solid-State Nanopores. ACS Nano 2015, 9, 9652–9664. 10.1021/acsnano.5b04984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D.-N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8, 221. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Tarini M.; Cignoni P.; Montani C. Ambient Occlusion and Edge Cueing for Enhancing Real Time Molecular Visualization. IEEE Trans. Vis. Comput. Graph. 2006, 12, 1237–1244. 10.1109/TVCG.2006.115. [DOI] [PubMed] [Google Scholar]
- Zhang P. H.; Too P. H. M.; Samuelson J. C.; Chan S. H.; Vincze T.; Doucette S.; Backstrom S.; Potamousis K. D.; Schramm T. M.; Forrest D.; Schwartz D. C.; Xu S. Y. Engineering Bspqi Nicking Enzymes and Application of N.Bspqi in DNA Labeling and Production of Single-Strand DNA. Protein Expression Purif. 2010, 69, 226–234. 10.1016/j.pep.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitov P. I.; Bundle D. R. On the Nature of the Multivalency Effect: A Thermodynamic Model. J. Am. Chem. Soc. 2003, 125, 16271–16284. 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- Jencks W. P. On the Attribution and Additivity of Binding Energies. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4046–4050. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.