

Abstract
The essential biological role of rare earth elements lay hidden until the discovery in 2011 that lanthanides are specifically incorporated into a bacterial methanol dehydrogenase. Only recently has this observation gone from a curiosity to a major research area, with the appreciation for the widespread nature of lanthanide-utilizing organisms in the environment and the discovery of other lanthanide-binding proteins and systems for selective uptake. While seemingly exotic at first glance, biological utilization of lanthanides is very logical from a chemical perspective. The early lanthanides (La, Ce, Pr, Nd) primarily used by biology are abundant in the environment, perform similar chemistry to other biologically useful metals and do so more efficiently due to higher Lewis acidity, and possess sufficiently distinct coordination chemistry to allow for selective uptake, trafficking, and incorporation into enzymes. Indeed, recent advances in the field illustrate clear analogies with the biological coordination chemistry of other metals, particularly CaII and FeIII, but with unique twists—including cooperative metal binding to magnify the effects of small ionic radius differences—enabling selectivity. This Outlook summarizes the recent developments in this young but rapidly expanding field and looks forward to potential future discoveries, emphasizing continuity with principles of bioinorganic chemistry established by studies of other metals. We also highlight how a more thorough understanding of the central chemical question—selective lanthanide recognition in biology—may impact the challenging problems of sensing, capture, recycling, and separations of rare earths.
Short abstract
We discuss how the emerging understanding of the principles underlying biological recognition and utilization of rare earths may facilitate capture and separations of these valuable elements.
Introduction: Expanding the Bioinorganic Periodic Table
Rare earth (RE) elements—the lanthanides, yttrium, and scandium—are critical components of numerous modern technologies, from permanent magnets in wind turbines and electric car batteries, to lasers and phosphors, to medical imaging agents.1,2 However, the combination of the insolubility3 and similarity in physical properties4 of REs presents myriad challenges in basic science, environmental sustainability,5,6 and economics7 for their separations in mining or recycling applications.8 The primary sources of REs are phosphate or carbonate minerals, such as bastnäsite, monazite, and xenotime,9,10 in which multiple REs often co-occur. REs are also present at significant concentrations in coal ash and acid mine drainage, although these lower-grade sources present further challenges for separation from more abundant metal ions, especially FeIII, MnII, CuII, and AlIII.11 These challenges have motivated investigations into diverse new approaches8—from small molecules,12−14 supramolecular assemblies such as metal–organic frameworks,15 novel extraction and chromatographic methods, and even bacterial cells16,17—to separate REs from other metals and from each other. The discovery in 201118−20 that methylotrophic bacteria specifically incorporate certain REs (the early lanthanides, La–Nd) into pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenases (ADHs) has opened new possibilities for more sustainable and efficient aqueous extraction and separations of these elements.8,21 The pace of discovery in this field has accelerated rapidly in the last year; here, we review these discoveries from a chemical perspective, highlighting potential, yet to be uncovered biological roles of lanthanides as well as broader applications of these findings to aid in RE mining, recycling, and separations.
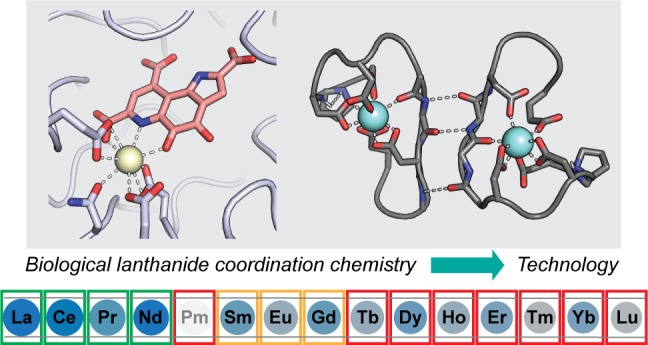
The aqueous chemistry of the REs is dominated by the +III oxidation state. Although other oxidation states are accessible, most notably CeIV and EuII, under normal physiological conditions it is generally believed that lanthanides would be limited to +III (LnIII).4,22−24 Important properties of LnIII ions have been extensively reviewed;4,25 the key characteristics relevant to lanthanide biochemistry (Figure 1) derive from their 4f electrons. The radial probability distribution of 4f orbitals is closer to the nucleus than for 6s and 5p; as a result, the 4f electrons poorly shield outer shells from increasing nuclear charge. Consequently, ionic radius decreases across the series (the lanthanide contraction): for a coordination number (CN) of 8, ionic radii span from 1.16 Å (LaIII) to 0.98 Å (LuIII).26,27 The ionic radius of PrIII is similar to that of CaII, 1.12 Å (for CN = 8). For this reason and the unique spectroscopic features of the 4f electrons, lanthanides have been used extensively in biochemistry as probes of calcium binding sites in proteins.4,28−30 Furthermore, because the 4f electrons have little impact on bonding, LnIII complexes are largely ionic in character—favoring hard, carboxylate ligands in biology—and flexible, sterically driven coordination geometries. These traits, combined with large ionic radii, allow LnIII (and especially the early lanthanides) to accommodate high CNs (8–12), rarely encountered with other common metal ions. The high charge of the LnIII ions makes them good Lewis acids and results in poor solubility of their hydroxide and phosphate salts, with Ksp’s of ∼10–30, translating to solubilities of ∼0.1–1 pM at pH 7.3
Figure 1.

Properties of the lanthanide series. The elements are scaled by ionic radius (LnIII, CN = 8);26 Lewis acidity increases from La to Lu. Elemental abundances in the crust31 range from blue (most abundant) to gray (least abundant). Pm has no stable isotopes and is not found in Nature. Boxes are colored according to currently known biological utilization.32 The other two REs, Y and Sc, are not shown. The ionic radius of Y is similar to that of Ho, and abundance is similar to that of La. Sc is not discussed in this Outlook.
Although once thought unlikely to be biologically relevant,33 there are good reasons why biology has utilized lanthanides. First, they are fairly abundant; crustal elemental abundances for La–Nd are ∼10–70 ppm, similar to other metals like Cu and Zn, whereas late lanthanides are 10–100 times rarer.31 Second, their higher charge-to-radius ratio versus other common biological Lewis acids such as CaII, MgII, and divalent first-row transition metal ions makes them equally or even more robust Lewis acid catalysts,34 and they would be more desirable when higher CNs, flexible coordination geometry, and/or redox-inertness are required for catalysis. Third, their functional and/or size similarities with CaII, FeIII, and MgII facilitate evolutionary connection to analogous pathways involving these metals, while their coordination chemistry is sufficiently distinct, facilitating selective recognition.
A central theme of this Outlook (relative to other recent reviews21,32,35−39) is that the above basic principles, in the context of existing knowledge about how biology handles other metal ions,40 anticipate the key aspects of lanthanide utilization. Results so far have revealed direct analogies between LnIII acquisition, transport, utilization, and storage to the extensively described bioinorganic chemistry of CaII and FeIII (Figure 2), and the analogies are likely to continue as more is learned. Cells must resolve the same challenge faced by the technological utilization of REs—the difficulty of separations. The differences in ionic radius and therefore ligand affinities between adjacent LnIII are small, and biology cannot afford dozens of chromatographic steps8,10 to filter one RE from another. At the same time, it is unable to utilize all REs indiscriminately. Instead, it has devised ways to accommodate a small swath of the periodic table roughly equally well, La–Nd, which are both the most abundant REs and those (due to their size) whose coordination chemistry is most distinct from that of other metals used by biology. This review examines how biology accomplishes this feat through its “lanthanome”41—the suite of proteins and other biomolecules involved in lanthanide utilization—and the potential broader applications thereof.
Figure 2.

Model for lanthanide uptake and utilization in M. extorquens based on the work of the Cotruvo,41−43 Vorholt,44 and Martinez-Gomez and Skovran45 laboratories. Unknown/postulated functions (the exact ligand for p1778, functions of p1779 and p1781, and LanM–MxcQ interaction) are indicated with parentheses and question marks.
Lanthanoenzymes
A specific biological role of lanthanides was first demonstrated in 2011 with genetic and biochemical characterization of XoxF as a lanthanide- and PQQ-dependent methanol dehydrogenase (Ln-MDH) in several methylotrophs, including Methylorubrum (formerly Methylobacterium) extorquens AM1 (Me).18−20 Methylotrophs are found in soil, water, and plants, where they play an important role in the carbon cycle because of their ability to utilize C1 compounds such as methanol or even methane (in the case of methanotrophs) as their sole carbon source.37,46,47 MDHs are critical enzymes for methylotrophs, catalyzing the oxidation of methanol to formaldehyde (Figure 3a).47,48 This reaction is a formal hydride transfer from the substrate to the metal-ligated C5 carbonyl of the PQQ cofactor, facilitated by the Lewis acidity of the metal ion.49,50 Single electrons are then transferred from the reduced PQQ cofactor to a c-type cytochrome (XoxG43,51,52 in the case of Ln-MDH). A Ca- and PQQ-dependent MDH (Ca-MDH, MxaFI) had been known for decades,53 but the function of XoxF had been cryptic.54,55 Whereas lanthanides were preferred but not required for growth of the methylotrophs in which XoxFs were first characterized, elegant work by Jetten, Op den Camp, and co-workers identified the first organism with an absolute requirement for lanthanides for growth—a novel thermoacidophilic methanotroph, Methylacidiphilum fumariolicum SolV (Mf), isolated from a volcanic mudpot.56 The mudpot water from which this organism was initially isolated had a pH of 1–2 and contained 2–3 μM REs, primarily La, Ce, and Nd.56 This result demonstrates both the high concentrations of soluble, bioavailable REs possible in acidic environments and the importance of REs for life in such an extreme niche. The fascinating environmental and ecological implications of this finding have been extensively reviewed;36,37,57 here, the discussion is restricted to chemical aspects. Structural characterization of the native XoxF lanthanoenzyme purified from Mf revealed an active site very similar to that of a Ca-MDH, with a CeIII ion coordinated to the PQQ and Asp, Glu, and Asn residues (Figure 3b). The only significant difference was an additional carboxylate ligand in XoxF, increasing CN from the 7 observed for CaII in Ca-MDH to the expected 9 for a LnIII ion.56,58
Figure 3.

(A) Reaction catalyzed by MDHs, with Em of the c-type cytochrome redox partners of Me Ca-MDH (MxaG) and Ln-MDH (XoxG) shown. The reactive C5 position of PQQ is denoted. Modified from ref (43). (B) Active site of Mf Ln-MDH (PDB code 4MAE), modeled with Ce.56 CeIII is a cream sphere; protein ligands are in gray sticks, and PQQ is in salmon. A ligand from the crystallization solution has been omitted.
XoxF-type ADHs are widespread in Nature, more so than the Ca-dependent MxaFI type, and they are proposed to have arisen prior to the Ca-MDHs.57,59,60 (Most XoxF-type ADHs are likely to be lanthanide-dependent enzymes; however, one XoxF has been found to copurify with Ca.61) Biological utilization of lanthanides, like that of iron, may have evolved in an environment in which lanthanides were more soluble and bioavailable than currently, and biology found them useful enough to retain them. Following the initial discovery of XoxF as a LnIII-dependent enzyme, several more lanthanide- and PQQ-dependent ADHs using methanol or other alcohols as substrates were found in methylotrophs51,58,62−65 and in a nonmethylotroph.66,67 These studies have shown that bacteria differ in lanthanide tolerances for growth, but all characterized to date prefer the early REs. For example, Mf grows most efficiently with La–Nd, but Sm–Gd also support growth;56 by contrast, Me will only use La–Nd, and Sm very poorly68 (Figure 1). These tolerances appear to be driven not only by RE bioavailability but also by coordination chemistry and reactivity considerations, as discussed below.
An intriguing and unique aspect of XoxF is that at least four different metals, La–Nd, are each able to confer activity when incorporated into the enzyme active site. Few enzymes, and particularly redox enzymes, are active with more than one or two different metal cofactors.69,70 Design of an enzyme that can tolerate several lanthanides in the same protein framework is a challenge because of differences in LnIII ionic radius and CN preference as well as interactions with substrate and intermediates, which may affect the methanol oxidation chemistry.24 Furthermore, in vivo, the electrons extracted from methanol to the LnIII-PQQ cofactor must then be transferred to XoxG,43,51,71 and the efficiency of this process would be expected to depend on the identity of the LnIII ion coordinated by PQQ.43
The chemical similarities of the lanthanide series make the Ln-MDH an excellent system to probe metal-dependent structure–function relationships in a single protein framework. Using Mf XoxF, which can be demetalated and reconstituted with all lanthanides in vitro, Daumann reported maximal activity with Pr, using a redox dye-based assay.24 Similar experiments cannot be carried out at present using Me XoxF because metal removal irreversibly inactivates the protein.43 Instead, the Me enzyme can be loaded with early REs in vivo, exhibiting highest activity with La.43,72 However, assays using the physiological electron acceptor, XoxG, tell a different story:43Me La-, Ce-, and Nd-XoxFs all exhibit the same maximum velocity, but the Km for XoxG increases from La to Nd, inversely proportional to ionic radius. We interpreted this result as reflecting an increasing reduction potential (Em) of the LnIII-PQQ cofactor with increasing LnIII Lewis acidity, suggesting that redox matching of XoxG and the LnIII-PQQ cofactor is an important constraint for lanthanide utilization in vivo. This proposal is supported by the observation that the Em of Mf XoxG52 is ∼80 mV higher than that of Me XoxG,43 as Mf is tolerant of heavier lanthanides, up to Gd, in vivo. Furthermore, Me grows more slowly with Nd than La,68 suggesting that later REs, even if they could be acquired, likely would not support efficient growth. Thus, lanthanide utilization in MDH seems to be a compromise between lanthanide availability, radius, Lewis acidity, and Em of its redox partner.24,43,64
Further studies of XoxFs24,43 and functional model complexes73 metalated with different REs, native or otherwise, will enable spectroscopic analysis of the MDH reaction mechanism, about which significant questions still exist (reviewed recently32). Such studies are challenging with the spectroscopically inert CaII. Substitution of different REs into XoxF also may be useful in trapping different intermediates by changing the rate constants for various steps in the reaction. These studies will be facilitated by a more complete understanding of the mechanism of activation of XoxF, which may involve XoxJ (speculated to bind XoxF to aid in its activation43) as well as potential chaperone proteins for the PQQ and LnIII cofactors41,45,74 (Figure 2). Understanding of this cofactor assembly pathway might allow for expression in a simpler system (e.g., Escherichia coli) to facilitate bioengineering applications such as a methanol biofuel cell.75 Because, unlike Ca-MDH, XoxF can efficiently oxidize not only methanol to formaldehyde but also (in vitro but likely not in vivo)56,72 formaldehyde to formate, it may simplify a methanol to CO2 cell to just two enzymes, XoxF and a formate dehydrogenase. Finally, the proposed effect of LnIII coordination on the Em of PQQ in XoxF43 suggests the feasibility of performing electrochemical separations of REs using RE-PQQ complexes, as has been accomplished recently with other redox-active ligands.13 The speciation of aqueous LnIII-PQQ complexes is heterogeneous,76,77 but such an approach may be possible with the help of PQQ-binding proteins.
Another open question is whether lanthanoenzymes that are not PQQ-dependent exist. The known lanthanoenzymes are all periplasmic, but discovery of lanthanide import to the cytosol41 suggests that lanthanoproteins may occur throughout the cell. However, studies so far have not identified putative enzymes other than ADHs that are obviously upregulated by lanthanides,44,72 so LnIII ions may substitute for other metals36 such as CaII, MgII, or FeIII in certain enzymes or cofactors. LnIII ions will likely bind tightly to any enzymes, facilitating their proteomic identification.
Lanthanide Recognition in Cells
From 2011 to 2018, the only lanthanide-binding proteins characterized were various PQQ-dependent ADHs.18−20,56,58,62,64,66 Biological utilization of other metal ions involves proteins responsible for selective import/export, trafficking, and regulation.78−80 Because these proteins are tasked with ensuring correct metalation of enzymes, their metal selectivity is especially critical. We reasoned that identification and characterization of lanthanoproteins with these functions may be more informative than the lanthanoenzymes about the central question of how biology selectively recognizes lanthanides.
The first insights into biological recognition of lanthanides were provided by discovery and characterization of lanmodulin (LanM), reported in late 2018.42,81 LanM was immediately intriguing to us as its amino acid sequence contained 4 EF-hand motifs (Figure 4a). EF hands are 12-residue CaII-binding motifs, widespread in biology and usually found in pairs for cooperative binding, such as in the CaII sensor, calmodulin.82,83 As a testament to the coordination similarities of CaII and LnIII ions, typical EF hands bind LnIII with slightly higher (∼100-fold) affinity than CaII, although LnIII coordination is not physiologically relevant in these cases.84−86 However, these similarities have also been exploited in the development of more selective “lanthanide-binding tags” with nanomolar lanthanide affinity, derived by engineering of natural EF hands.87,88 LanM exhibits several remarkable properties for an EF-hand protein.42 First, it undergoes a large, cooperative conformational change in response to binding of all trivalent REs. Second, the NMR solution structure of LanM in the presence of YIII (a diamagnetic RE used as a LnIII surrogate) revealed metal coordination by 5 carboxylates and a backbone carbonyl (CN = 8–9, depending on metal site), whereas CaII sites in typical EF hands are 7-coordinate (Figure 4b).81 Third, each EF hand possesses a Pro residue at the second position, a feature present in no previously characterized EF hands.83 The Pro N is engaged in an unusual Ni+1–H···Ni hydrogen bond with the backbone NH of the subsequent residue, a coordinating Asp. Finally, and perhaps most remarkably, LanM responds to picomolar free concentrations of REs, whereas millimolar CaII is required for a conformational change (108-fold selectivity). This selectivity is much greater than would be expected on the basis of charge and radius alone (∼104).4,86,89 Furthermore, LanM binds early REs with higher affinity than late REs, whereas chelators typically prefer the more Lewis-acidic, late REs.8 These intriguing and unusual properties have motivated us to study LanM as a model system for determining the rules of highly selective biological lanthanide coordination. While much work remains, our studies have suggested that the prolines contribute ∼100-fold to RE selectivity, apparently by uncoupling initial CaII binding from a conformational change.42
Figure 4.

Lanmodulin as a model system to study biological principles of selective RE recognition. (A) Solution structure of YIII–LanM (PDB code 6MI5).43 YIII ions are in cyan, and EF loops are shown in gray. Metal coordination by EF4 is weak and likely not physiologically relevant.42 (B) Detail of YIII coordination in LanM (EF3), with coordinating residues and the Ni+1–H···Ni hydrogen bond involving the proline residue shown. (C) The hydrogen bonding connectivity of the EF2/3 pair illustrates the importance of cooperativity in RE recognition. (D) Working model for selectivity for LnIII (and YIII) over CaII.
The proposed importance of the prolines and their Ni+1–H···Ni hydrogen bonds is 2-fold: (1) to constrain the structure of the EF loop such that metals that prefer fewer ligands (e.g., CaII) force the carboxylates into an arrangement incompetent to produce a conformational change until superphysiological concentrations (Figure 4d) and (2) to buttress against loop constriction as LnIII ionic radius decreases from La to Nd, thereby retaining communication with the adjacent metal site for optimal affinity and cooperativity (Figure 4c). Further structural and spectroscopic work is needed to draw more specific conclusions about metal coordination.
LanM’s large, highly RE-selective conformational change suggests a role in lanthanide sensing, perhaps as the substrate of a two-component system (Figure 2) that may be involved in switching between Ca- and RE-dependent physiologies (the “lanthanide switch”38,44,67,68,90). Although it is one of very few proteins upregulated in response to early REs in several organisms,44,72,91 LanM is not essential for lanthanide-dependent growth, pointing to probable redundancies in lanthanide-handling machinery. Furthermore, it is only found in certain methylotrophs, suggesting that other organisms encode unrelated proteins with a similar function.38
We also have identified a second RE-binding protein (META1p1781).41 It shares LanM’s unusual property of preferentially binding early over late REs, but it does not possess an EF hand. Although not yet as thoroughly characterized as LanM, this protein is dimeric and binds 2 LnIII ions per dimer, suggesting that cooperative metal binding may contribute to metal selectivity, as in LanM. It seems virtually guaranteed that more RE-binding proteins will be discovered, and their characterization will provide a compendium of biological lanthanide recognition strategies that may be useful for broader applications.
LanM’s high selectivity for REs suggests that it may be useful for green, aqueous separation of total REs from other metals. Large-scale separation methods generally involve repeated, acid- and organic solvent-intensive extraction steps using ligands with low selectivities.8,10 LanM may not be optimal as is for separations of adjacent REs, as Nature has engineered it to respond similarly to La–Nd. However, it may be a privileged scaffold for evolution of selective peptide-based ligands for REs and other metal ions. These applications may include metal harvesting and recycling, sensing (e.g., rapid detection of REs in the field using LaMP1, a LanM-derived fluorescent protein-based sensor we reported41), and metallotherapeutic and imaging agents.2 A particular advantage of LanM is the cooperative metal binding exhibited by its EF hand pairs, which could allow for greater selectivities for adjacent REs than is possible at a single metal-binding site (as in currently used ligands), a general principle highlighted by recent synthetic ligands for size-based separations.12,14,92
Lanthanide Uptake
While the solubilities of LnIII are picomolar at pH 7, they are still 8–9 orders of magnitude higher than that of FeIII, which bacteria acquire using secreted small molecules called siderophores.93−95 As a result, several investigators speculated that lanthanides might be acquired via a lanthanophore.32,37,39,42,57,96 In Gram-negative bacteria, FeIII–siderophore complexes are typically taken up using TonB-dependent systems comprising specific outer- and inner-membrane transporters, a periplasmic binding protein, and an enzyme for siderophore degradation for iron release.97−99
When we first reported LanM in Me AM1,42 we noted that the gene encoding it was adjacent to a putative TonB-dependent transporter, suggesting that the lanM gene cluster encodes a lanthanophore uptake system. Genetic studies by Vorholt44 in a closely related strain showed that the putative transporters of this cluster were essential for lanthanide-mediated growth. Contemporaneously, we used the LaMP1 sensor to reveal several details of lanthanide uptake. First, we showed for the first time that La–Nd are selectively taken up into the Me cytosol (Figure 5, confirming the importance of the transporters). Second, we showed that the periplasmic binding protein of the putative uptake system does not bind free LnIII ions (implying that it may bind a LnIII complex instead). Finally, we demonstrated an activity in the spent medium from cells grown without REs that is capable of selectively outcompeting LaMP1 for binding early REs (physical evidence for a lanthanophore).41 These data all but confirmed the secretion of a molecule required for selective uptake of early REs (Figure 2) and laid out an approach for its isolation and characterization.
Figure 5.
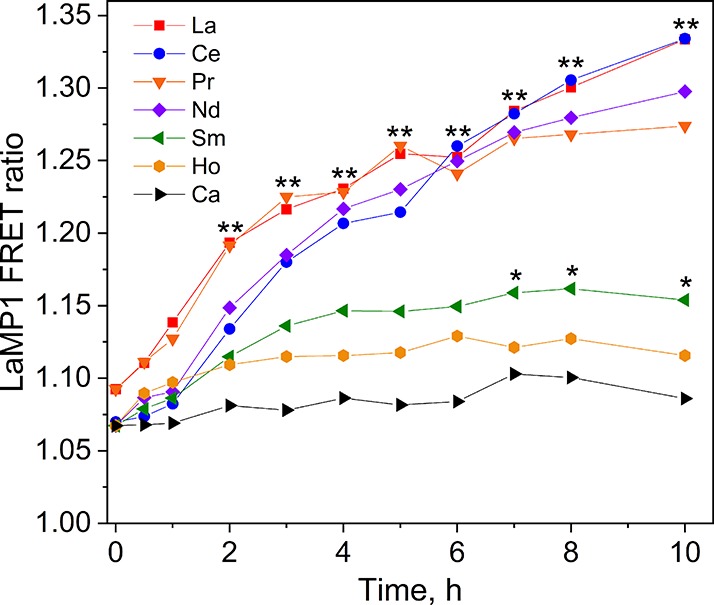
M. extorquens selectively uptakes early lanthanides into its cytosol. Cells expressing the LaMP1 sensor were grown without lanthanides, and at t = 0, 2 μM LnIII was added to each culture. Increased FRET ratio indicates increased sensor-bound LnIII ions. Error bars omitted for clarity. * p < 0.05 (Sm vs Ca), ** p < 0.01 (La–Nd vs Ca). Modified from ref (41).
These observations also provided key information about the mechanism by which the lanthanophore achieves its selectivity. The strong ionic radius dependence of lanthanide-dependent regulation68 and cytosolic lanthanide uptake41 argued that the lanthanophore does not simply rely on the rather small affinity differences between REs for a single site.8 Instead, it likely selects for early REs based on (1) differences in overall RE-complex shape, perhaps exploiting the ability of early REs to accommodate higher CNs, with only one conformation recognized by the importer, or (2) cooperative binding of two or more metal ions. Both potential approaches have been anticipated by elegant small-molecule work focused on RE separations, for example, that of Schelter using ionic radius-dependent dimerization equilibrium of an RE complex12,92 and that of Sun using supramolecular tetrahedral cages templated by metal binding,14 as well as by LanM’s RE recognition strategy. Determination of the lanthanophore’s structure will resolve these questions.
It is not known whether all lanthanide-utilizing organisms have lanthanophores, and whether the diversity of their architectures matches that of siderophores.93,94,100 For example, Mf may not require a dedicated lanthanophore because of higher RE solubility in its acidic environmental niche.32,43 Klebensberger has recently reported that iron interferes with lanthanide uptake in Pseudomonas putida,101 highlighting the complexity of selective metal uptake. Finally, it is possible that some previously characterized siderophores might instead (or in addition) be lanthanophores.
As with LanM, there are clear potential applications of lanthanophores for RE harvesting and separations. In fact, siderophores and simplified derivatives are already being explored in this regard,102,103 but the lanthanophore’s selectivity for early REs makes it an attractive alternative. However, the highest-value REs are the rarer, heavy ones, which the Me lanthanophore may not bind effectively; one can envision synthetic derivatives that are selective for different sets of REs. As more lanthanide-utilizing organisms are characterized, lanthanophores with different metal selectivities may also be found. One potential caveat is that, in biology, metal selectivity relies on both affinity differences and conformational changes necessary for specific recognition by a receptor. For example, LanM undergoes a conformational change with 108-fold selectivity for REs over CaII, but only 106 derives from affinity differences; the rest appears to derive from a conformational effect. By contrast, in RE harvesting and separation applications, selectivity likely must rely on affinity differences alone. Therefore, engineering of biological ligands may be required prior to their implementation as technologies for RE extraction and separations.
Lanthanide Storage
Given the selective cytosolic uptake of early REs by Me(41) and Me’s ability to deplete cultures of early REs,104 it was reasonable to suggest that bacteria would take advantage of the low solubility of RE (poly)phosphate complexes for lanthanide storage in “lanthanosomes,” similar to Ca, which is stored in organisms from bacteria to humans in organelles called acidocalcisomes.105 In fact, analogous mechanisms exist for storage of many metals: copper is stored in acidocalcisome-like structures in algae106 and zinc in similar zincosomes in C. elegans,107 and even iron storage in ferritin as an FeIII-oxyhydroxide with variable phosphate content108 is related. This lanthanide storage mechanism was recently demonstrated by Skovran and Martinez-Gomez,45 although details remain to be defined. The bioaccumulation of lanthanides presents possibilities for whole-cell lanthanide extraction methods, although the probable storage of only early REs, slow lanthanide accumulation by Me,104 and the need to carry out further separations from the rest of the bacterial constituents may hinder such an approach.
Lanthanides beyond Bacteria
Specific biological functions of lanthanides are best characterized in bacteria. However, REs have been used for decades as fertilizers, having been found to accumulate in plants and enhance growth, crop yields, and drought resistance (refs (109 and 110) and references therein). The precise mechanisms by which REs have these benefits have yet to be elucidated, but understanding them may enable more thoughtful approaches to improving agricultural yields. The targets may relate to plant photosynthesis or cell wall structure and/or to effects on the methylotrophs such as Me that colonize plants because of their substantial methanol production.36,111
Unlike bacteria studied to date, which strongly prefer early REs, plants seem to accumulate REs in proportion to their concentrations in the soil,44,112 suggesting that much of the accumulation occurs nonspecifically, perhaps through Ca or Fe uptake pathways, such as the broad-spectrum metallophore, nicotianamine.113,114 It is also conceivable that plants take advantage of the increased bioavailability of REs resulting from production of lanthanophores by bacteria residing in the phyllosphere. For example, much like the mammalian protein siderocalin binds FeIII–siderophore complexes as an immune defense mechanism to interfere with iron acquisition by invading pathogens,115 it is possible that plants produce proteins that can bind LnIII–lanthanophore complexes—either to limit colonization by plant pathogens or as part of a mutualistic association between plant and lanthanide-utilizing microbes to make REs more accessible to the plant. RE uptake may also be linked to the beneficial effects of microbe-derived PQQ for plant growth;116 the recent identification of an RE-regulated PQQ acquisition system in Me(74) supports the existence of RE and PQQ signaling networks in plant–microbe systems, the mechanisms and targets of which have yet to be fully elucidated.
The proportion of plant-accumulated REs present within cells versus outside is not known. Extracellular REs may be incorporated in place of CaII in the cell wall. Methanol is produced by de-esterification of pectins, which exposes carboxylates for metal ligation to facilitate cell–cell adhesion.117,118 This synergy between cell wall strength and structure, CaII or RE binding, and methanol production (which could be used by associated methylotrophs) may be relevant to the observed positive effects of REs on plants. Cellular RE uptake has been described to occur via endocytosis,119−121 but this mechanism may be a result of formation of insoluble RE aggregates under the in vitro assay conditions. However, in the plant, at least some of the accumulated REs are presumably present in a soluble, bioavailable form in cells, as CeIII has been reported to be incorporated into chlorophyll, particularly in Mg-limited soils.112,122 This substitution, presumably occurring via competition for binding to the Mg chelatase that inserts MgII into protoporphyrin IX during chlorophyll biosynthesis, was associated with enhanced photosynthetic rate of isolated chloroplasts, although it is not clear if this association is causal.112,122 (Relatedly, methylotrophs like Me are known to produce bacteriochlorophyll,123 so it bears investigation whether they can incorporate REs more specifically into porphyrin-derived or other cofactors.) CaII is also present in the Mn4Ca oxygen evolving complex (OEC) of photosystem II,124 and it is tempting to suggest that REs may be inserted into the OEC in place of Ca. However, model studies125,126 and in vitro substitution experiments127 suggest that the greater Lewis acidity of REs would make the cluster more difficult to oxidize, decreasing water oxidation activity. Most of the above studies have focused on the cheaper and more abundant early REs, but studies using other REs would help disambiguate effects on associated bacteria versus on the plant itself.
Finally, lanthanides may also affect the organisms that consume plants. Studies have suggested positive impacts of RE provision in animal husbandry, suggesting a role for REs in higher organisms.128 However, REs are not perfect mimics of Ca,4 as recent studies of calmodulin129,130 and cadherin131 have shown, suggesting that REs may not play a general role as Ca substitutes in Ca-dependent proteins. Instead, the growth-promoting effects of REs in higher organisms might plausibly be linked to the microbiome; for example, both beneficial (E. coli) and pathogenic (Pseudomonas aeruginosa) bacteria possess PQQ-dependent dehydrogenases, and these or other enzymes might benefit from or be inhibited by REs. However, at the moment, no specific mechanism for the effects of REs in organisms other than bacteria has been firmly established.
Conclusions and Outlook
It is fitting that the International Year of the Periodic Table (2019) would be the annus mirabilis of biological lanthanides, when a number of the key principles and mechanistic details of lanthanide recognition and utilization have come into view, having long laid hidden (Greek: lanthánein, the root of lanthanum). These new findings include discovery and characterization of lanmodulin (late 2018),42,81 discovery of the lanthanide uptake pathway41,42,44 and its reliance on a lanthanophore,41 discovery of cytosolic lanthanide uptake41 and storage,45 and (likely) characterization of the lanthanophore. The principles gleaned from these recent discoveries allow us to anticipate many of the key roles that lanthanides may play in biology, but the full scope and many details remain to be determined.
A curious aspect of biological lanthanide utilization, and one that has important implications for the translational impact of the biological ligands, is why the organisms characterized to date strongly prefer early REs, whereas any RE would be a suitable Lewis acid catalyst. We propose that this observation reflects a compromise between several factors. A single set of biological ligands can only retain specificity within a narrow window of ionic radius. The higher abundance of the early REs makes that window an attractive one, but abundance alone is not a sufficient explanation, as Y is similarly abundant but not used, as far as is currently known. The discovery of organisms that can selectively utilize late REs would be biologically and biotechnologically interesting. However, this possibility might be precluded by biology’s requirement for selective acquisition and trafficking mechanisms: a major advantage of the early REs is that their larger ionic radius allows for high CNs and thus more effective discrimination versus abundant metal ions that would compete for the polyanionic ligands most favorable for LnIII coordination. The data so far support LanM, META1p1781, and the lanthanophore being outworkings of the same fundamental principle in protein and small-molecule form: using cooperative metal binding to give exquisite metal selectivity by amplification of subtle differences in ionic radius and coordination preferences. There are clear, broader applications of this principle for binding of other REs and other metals by engineered protein, small-molecule, and cell-based approaches, for applications in RE harvesting and beyond. The interface of biology, biochemistry, coordination chemistry, and engineering will be a rich area of exploration as we learn more about the fundamentals and applications of biological lanthanide utilization.
Acknowledgments
J.A.C. thanks the whole Cotruvo lab for discussions, and E.R. Featherston and J.A. Mattocks for assistance in preparation of Figures 3–5. J.A.C. gratefully acknowledges the Penn State Department of Chemistry, the Huck Institutes for the Life Sciences, and a Louis Martarano Career Development Professorship for funding.
The author declares the following competing financial interest(s): J.A.C. is an inventor on a patent application submitted by the Pennsylvania State University related to some of the work described here.
References
- Haxel G. B.; Hedrick J. B.; Orris G. J. Rare earth elements - Critical resources for high technology. U.S.G.S. Fact Sheet 2002, 087–02. 10.3133/fs08702. [DOI] [Google Scholar]
- Kostelnik T. I.; Orvig C. Radioactive main group and rare earth metals for imaging and therapy. Chem. Rev. 2019, 119, 902–956. 10.1021/acs.chemrev.8b00294. [DOI] [PubMed] [Google Scholar]
- Firsching F. H.; Brune S. N. Solubility products of the trivalent rare-earth phosphates. J. Chem. Eng. Data 1991, 36, 93–95. 10.1021/je00001a028. [DOI] [Google Scholar]
- Evans C. H.Biochemistry of the Lanthanides; Plenum Press: New York, 1990. [Google Scholar]
- Yang X. J.; Lin A.; Li X.-L.; Wu Y.; Zhou W.; Chen Z. China’s ion-adsorption rare earth resources, mining consequences and preservation. Environ. Dev. 2013, 8, 131–136. 10.1016/j.envdev.2013.03.006. [DOI] [Google Scholar]
- Arshi P. S.; Vahidi E.; Zhao F. Behind the scenes of clean energy: The environmental footprint of rare earth products. ACS Sustainable Chem. Eng. 2018, 6, 3311–3320. 10.1021/acssuschemeng.7b03484. [DOI] [Google Scholar]
- Bomgardner M. M. The struggle to mine rare earths. Chem. Eng. News 2015, 93 (30), 36–39. [Google Scholar]
- Cheisson T.; Schelter E. J. Rare earth elements: Mendeleev’s bane, modern marvels. Science 2019, 363, 489–493. 10.1126/science.aau7628. [DOI] [PubMed] [Google Scholar]
- Long K. R.; Van Gosen B. S.; Foley N. K.; Cordier D.. The Principal Rare Earth Elements Deposits of the United States: A Summary of Domestic Deposits and a Global Perspective. In Non-Renewable Resource Issues. International Year of Planet Earth; Sinding-Larsen R., Wellmer F. W., Eds.; Springer: Dordrecht, 2012. [Google Scholar]
- Xie F.; Zhang T. A.; Dreisinger D.; Doyle F. A critical review on solvent extraction of rare earths from aqueous solutions. Miner. Eng. 2014, 56, 10–28. 10.1016/j.mineng.2013.10.021. [DOI] [Google Scholar]
- Erickson B. Rare-earth recovery: U.S. efforts to extract valuable elements from coal waste surge. Chem. Eng. News 2018, 96 (28), 29–33. [Google Scholar]
- Bogart J. A.; Cole B. E.; Boreen M. A.; Lippincott C. A.; Manor B. C.; Carroll P. J.; Schelter E. J. Accomplishing simple, solubility-based separations of rare earth elements with complexes bearing size-sensitive molecular apertures. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14887–14892. 10.1073/pnas.1612628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H.; Cole B. E.; Qiao Y.; Bogart J. A.; Cheisson T.; Manor B. C.; Carroll P. J.; Schelter E. J. Electro-kinetic separation of rare earth elements using a redox-active ligand. Angew. Chem., Int. Ed. 2017, 56, 13450–13454. 10.1002/anie.201706894. [DOI] [PubMed] [Google Scholar]
- Li X.-Z.; Zhou L.-P.; Yan L.-L.; Dong Y.-M.; Bai Z.-L.; Sun X.-Q.; Diwu J.; Wang S.; Bünzli J.-C.; Sun Q.-F. A supramolecular lanthanide separation approach based on multivalent cooperative enhancement of metal ion selectivity. Nat. Commun. 2018, 9, 547. 10.1038/s41467-018-02940-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. Y.; Peng W. L.; Meng P. P.; Feng X. F.; Li J. Q.; Wu H. Q.; Yan C. S.; Xiong Y. Y.; Luo F. Lanthanide separation using size-selective crystallization of Ln-MOFs. Chem. Commun. 2017, 53, 5737–5739. 10.1039/C7CC01898C. [DOI] [PubMed] [Google Scholar]
- Bonificio W. D.; Clarke D. R. Rare-earth separation using bacteria. Environ. Sci. Technol. Lett. 2016, 3, 180–184. 10.1021/acs.estlett.6b00064. [DOI] [Google Scholar]
- Park D. M.; Brewer A.; Reed D. W.; Lammers L. N.; Jiao Y. Recovery of rare earth elements from low-grade feedstock leachates using engineered bacteria. Environ. Sci. Technol. 2017, 51, 13471–13480. 10.1021/acs.est.7b02414. [DOI] [PubMed] [Google Scholar]
- Hibi Y.; Asai K.; Arafuka H.; Hamajima M.; Iwama T.; Kawai K. Molecular structure of La3+-induced methanol dehydrogenase-like protein in Methylobacterium radiotolerans. J. Biosci. Bioeng. 2011, 111, 547–549. 10.1016/j.jbiosc.2010.12.017. [DOI] [PubMed] [Google Scholar]
- Fitriyanto N. A.; Fushimi M.; Matsunaga M.; Pertiwiningrum A.; Iwama T.; Kawai K. Molecular structure and gene analysis of Ce3+-induced methanol dehydrogenase of Bradyrhizobium sp. MAFF211645. J. Biosci. Bioeng. 2011, 111, 613–617. 10.1016/j.jbiosc.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Nakagawa T.; Mitsui R.; Tani A.; Sasa K.; Tashiro S.; Iwama T.; Hayakawa T.; Kawai K. A catalytic role of XoxF1 as La3+-dependent methanol dehydrogenase in Methylobacterium extorquens strain AM1. PLoS One 2012, 7, e50480 10.1371/journal.pone.0050480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gomez N. C.; Vu H. N.; Skovran E. Lanthanide chemistry: From coordination in chemical complexes shaping our technology to coordination in enzymes shaping bacterial metabolism. Inorg. Chem. 2016, 55, 10083–10089. 10.1021/acs.inorgchem.6b00919. [DOI] [PubMed] [Google Scholar]
- Bogart J. A.; Lewis A. J.; Schelter E. J. DFT study of the active site of the XoxF-type natural, cerium-dependent methanol dehydrogenase enzyme. Chem. - Eur. J. 2015, 21, 1743–1748. 10.1002/chem.201405159. [DOI] [PubMed] [Google Scholar]
- Prejanò M.; Marino T.; Russo N. How can methanol dehydrogenase from Methylacidiphilum fumariolicum work with the alien CeIII ion in the active center? A theoretical study. Chem. - Eur. J. 2017, 23, 8652–8657. 10.1002/chem.201700381. [DOI] [PubMed] [Google Scholar]
- Lumpe H.; Pol A.; Op den Camp H. J. M.; Daumann L. J. Impact of the lanthanide contraction on the activity of a lanthanide-dependent methanol dehydrogenase - a kinetic and DFT study. Dalton Trans. 2018, 47, 10463–10472. 10.1039/C8DT01238E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton S. A.; Raithby P. R. Systematics and surprises in lanthanide coordination chemistry. Coord. Chem. Rev. 2017, 340, 220–231. 10.1016/j.ccr.2017.01.011. [DOI] [Google Scholar]
- Shannon R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Seitz M.; Oliver A. G.; Raymond K. N. The lanthanide contraction revisited. J. Am. Chem. Soc. 2007, 129, 11153–11160. 10.1021/ja072750f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrocks W. D.; Sudnick D. R. Lanthanide ion probes of structure in biology. Laser-induced luminescence decay constants provide a direct measure of the number of metal-coordinated water molecules. J. Am. Chem. Soc. 1979, 101, 334–340. 10.1021/ja00496a010. [DOI] [Google Scholar]
- Mulqueen P.; Tingey J. M.; Horrocks W. D. J. Characterization of lanthanide(III) ion binding to calmodulin using luminescence spectroscopy. Biochemistry 1985, 24, 6639–6645. 10.1021/bi00344a051. [DOI] [PubMed] [Google Scholar]
- Bentrop D.; Bertini I.; Cremonini M. A.; Forsen S.; Luchinat C.; Malmendal A. Solution structure of the paramagnetic complex of the N-terminal domain of calmodulin with two Ce3+ ions by 1H NMR. Biochemistry 1997, 36, 11605–11618. 10.1021/bi971022+. [DOI] [PubMed] [Google Scholar]
- Rumble J. R.CRC Handbook of Chemistry and Physics, 100th ed. (Internet Version 2019); CRC Press/Taylor & Francis: Boca Raton, FL, 2019. [Google Scholar]
- Daumann L.Essential and ubiquitous: The emergence of lanthanide metallobiochemistry. Angew. Chem. 2019, in press. 10.1002/ange.201904090. [DOI] [PubMed] [Google Scholar]
- Lim S.; Franklin S. J. Lanthanide-binding peptides and the enzymes that might have been. Cell. Mol. Life Sci. 2004, 61, 2184–2188. 10.1007/s00018-004-4156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin D. D.Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution, 2nd ed.; Pergamon Press: New York, 1982. [Google Scholar]
- Skovran E.; Martinez-Gomez N. C. Just add lanthanides. Science 2015, 348, 862–863. 10.1126/science.aaa9091. [DOI] [PubMed] [Google Scholar]
- Chistoserdova L. Lanthanides: New life metals?. World J. Microbiol. Biotechnol. 2016, 32, 138. 10.1007/s11274-016-2088-2. [DOI] [PubMed] [Google Scholar]
- Semrau J. D.; DiSpirito A. A.; Gu W.; Yoon S. Metals and methanotrophy. Appl. Environ. Microbiol. 2018, 84, e02289–17. 10.1128/AEM.02289-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistoserdova L. New pieces to the lanthanide puzzle. Mol. Microbiol. 2019, 111, 1127–1131. 10.1111/mmi.14210. [DOI] [PubMed] [Google Scholar]
- Picone N.; Op den Camp H. J. M. Role of rare earth elements in methanol oxidation. Curr. Opin. Chem. Biol. 2019, 49, 39–44. 10.1016/j.cbpa.2018.09.019. [DOI] [PubMed] [Google Scholar]
- Lippard S. J.; Berg J. M.. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, 1994. [Google Scholar]
- Mattocks J. A.; Ho J. V.; Cotruvo J. A. Jr. A selective, protein-based fluorescent sensor with picomolar affinity for rare earth elements. J. Am. Chem. Soc. 2019, 141, 2857–2861. 10.1021/jacs.8b12155. [DOI] [PubMed] [Google Scholar]
- Cotruvo J. A. Jr.; Featherston E. R.; Mattocks J. A.; Ho J. V.; Laremore T. N. Lanmodulin: A highly selective lanthanide-binding protein from a lanthanide-utilizing bacterium. J. Am. Chem. Soc. 2018, 140, 15056–15061. 10.1021/jacs.8b09842. [DOI] [PubMed] [Google Scholar]
- Featherston E. R.; Rose H. R.; McBride M. J.; Taylor E. M.; Boal A. K.; Cotruvo J. A. Jr.. Biochemical and structural characterization of XoxG and XoxJ and their roles in lanthanide-dependent methanol dehydrogenase activity. ChemBioChem 2019, in press. 10.1002/cbic.201900184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner A. M.; Hemmerle L.; Vonderach T.; Nüssli R.; Bortfeld-Miller M.; Hattendorf B.; Vorholt J. A. Use of rare earth elements in the phyllosphere colonizer Methylobacterium extorquens PA1. Mol. Microbiol. 2019, 111, 1152–1166. 10.1111/mmi.14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszczenko-Jasińska P.; Vu H. N.; Subuyuj G. A.; Crisostomo R. V.; Cai J.; Raghuraman C.; Ayala E. M.; Clippard E. J.; Lien N. F.; Ngo R. T.; Yarza F.; Hoeber C. A.; Martinez-Gomez N. C.; Skovran E.. Lanthanide transport, storage, and beyond: genes and processes contributing to XoxF function in Methylorubrum extorquens AM1. 2019, bioRxiv 647677. bioRxiv preprint server. https://www.biorxiv.org/content/10.1101/647677v1.
- Anthony C.The Biochemistry of Methylotrophs; Academic: London, 1982. [Google Scholar]
- Chistoserdova L.; Kalyuzhnaya M. G.; Lidstrom M. E. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. 10.1146/annurev.micro.091208.073600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony C., Methanol Dehydrogenase, a PQQ-Containing Quinoprotein Dehydrogenase. In Enzyme-Catalyzed Electron and Radical Transfer: Subcellular Biochemistry; Holzenburg A., Scrutton N. S., Eds.; Springer US: Boston, MA, 2000; pp 73–117. [DOI] [PubMed] [Google Scholar]
- Anthony C.; Williams P. The structure and mechanism of methanol dehydrogenase. Biochim. Biophys. Acta, Proteins Proteomics 2003, 1647, 18–23. 10.1016/S1570-9639(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Reddy S. Y.; Bruice T. C. Mechanism of methanol oxidation by quinoprotein methanol dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 745–749. 10.1073/pnas.0610126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Huang J.; Zhao F.; Chistoserdova L. Physiological effect of XoxG(4) on lanthanide-dependent methanotrophy. mBio 2018, 9, e02430-17 10.1128/mBio.02430-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versantvoort W.; Pol A.; Daumann L. J.; Larrabee J. A.; Strayer A. H.; Jetten M. S. M.; van Niftrik L.; Reimann J.; Op den Camp H. J. M. Characterization of a novel cytochrome cGJ as the electron acceptor of XoxF-MDH in the thermoacidophilic methanotroph Methylacidiphilum fumariolicum SolV. Biochim. Biophys. Acta, Proteins Proteomics 2019, 1867, 595–603. 10.1016/j.bbapap.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Anthony C.; Zatman L. J. The microbial oxidation of methanol. 2. The methanol-oxidizing enzyme of Pseudomonas sp. M 27. Biochem. J. 1964, 92, 614–621. 10.1042/bj0920614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovran E.; Palmer A. D.; Rountree A. M.; Good N. M.; Lidstrom M. E. XoxF is required for expression of methanol dehydrogenase in Methylobacterium extorquens AM1. J. Bacteriol. 2011, 193, 6032–6038. 10.1128/JB.05367-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S.; Christen P.; Kiefer P.; Vorholt J. A. Functional investigation of methanol dehydrogenase-like protein XoxF in Methylobacterium extorquens AM1. Microbiology 2010, 156, 2575–2586. 10.1099/mic.0.038570-0. [DOI] [PubMed] [Google Scholar]
- Pol A.; Barends T. R. M.; Dietl A.; Khadem A. F.; Eygensteyn J.; Jetten M. S. M.; Op den Camp H. J. M. Rare earth metals are essential for methanotrophic life in volcanic mudpots. Environ. Microbiol. 2014, 16, 255–264. 10.1111/1462-2920.12249. [DOI] [PubMed] [Google Scholar]
- Keltjens J. T.; Pol A.; Reimann J.; Op den Camp H. J. M. PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl. Microbiol. Biotechnol. 2014, 98, 6163–6183. 10.1007/s00253-014-5766-8. [DOI] [PubMed] [Google Scholar]
- Deng Y. W.; Ro S. Y.; Rosenzweig A. C. Structure and function of the lanthanide-dependent methanol dehydrogenase XoxF from the methanotroph Methylomicrobium buryatense 5GB1C. J. Biol. Inorg. Chem. 2018, 23, 1037–1047. 10.1007/s00775-018-1604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhnaya M. G.; Lapidus A.; Ivanova N.; Copeland A. C.; McHardy A. C.; Szeto E.; Salamov A.; Grigoriev I. V.; Suciu D.; Levine S. R.; Markowitz V. M.; Rigoutsos I.; Tringe S. G.; Bruce D. C.; Richardson P. M.; Lidstrom M. E.; Chistoserdova L. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 2008, 26, 1029–1034. 10.1038/nbt.1488. [DOI] [PubMed] [Google Scholar]
- Huang J.; Yu Z.; Groom J.; Cheng J. F.; Tarver A.; Yoshikuni Y.; Chistoserdova L. Rare earth element alcohol dehydrogenases widely occur among globally distributed, numerically abundant and environmentally important microbes. ISME J. 2019, 13, 2005–2017. 10.1038/s41396-019-0414-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. L.; Wessels H. J. C. T.; Pol A.; Op den Camp H. J. M.; Jetten M. S. M.; van Niftrik L.; Keltjens J. T. XoxF-type methanol dehydrogenase from the anaerobic methanotroph ″Candidatus Methylomirabilis oxyfera″. Appl. Environ. Microbiol. 2015, 81, 1442–1451. 10.1128/AEM.03292-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good N. M.; Vu H. N.; Suriano C. J.; Subuyuj G. A.; Skovran E.; Martinez-Gomez N. C. Pyrroloquinoline quinone ethanol dehydrogenase in Methylobacterium extorquens AM1 extends lanthanide-dependent metabolism to multicarbon substrates. J. Bacteriol. 2016, 198, 3109–3118. 10.1128/JB.00478-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F.; Lidstrom M. E. XoxF acts as the predominant methanol dehydrogenase in the type I methanotroph Methylomicrobium buryatense. J. Bacteriol. 2016, 198, 1317–1325. 10.1128/JB.00959-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Yu Z.; Chistoserdova L. Lanthanide-dependent methanol dehydrogenases of XoxF4 and XoxF5 clades are differentially distributed among methylotrophic bacteria and they reveal different biochemical properties. Front. Microbiol. 2018, 9, 1366. 10.3389/fmicb.2018.01366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause S. M. B.; Johnson T.; Samadhi Karunaratne Y.; Fu Y.; Beck D. A. C.; Chistoserdova L.; Lidstrom M. E. Lanthanide-dependent cross-feeding of methane-derived carbon is linked by microbial community interactions. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 358–363. 10.1073/pnas.1619871114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrmann M.; Billard P.; Martin-Meriadec A.; Zegeye A.; Klebensberger J. Functional role of lanthanides in enzymatic activity and transcriptional regulation of pyrroloquinoline quinone-dependent alcohol dehydrogenases in Pseudomonas putida KT2440. mBio 2017, 8, e00570-17 10.1128/mBio.00570-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrmann M.; Berthelot C.; Billard P.; Klebensberger J. The PedS2/PedR2 two-component system is crucial for the rare earth element switch in Pseudomonas putida KT2440. mSphere 2018, 3, e00376-18 10.1128/mSphere.00376-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu H. N.; Subuyuj G. A.; Vijayakumar S.; Good N. M.; Martinez-Gomez N. C.; Skovran E. Lanthanide-dependent regulation of methanol oxidation systems in Methylobacterium extorquens AM1 and their contribution to methanol growth. J. Bacteriol. 2016, 198, 1250–1259. 10.1128/JB.00937-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobota J. M.; Imlay J. A. Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5402–5407. 10.1073/pnas.1100410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotruvo J. A. Jr.; Stubbe J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036. 10.1039/c2mt20142a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalimuthu P.; Daumann L. J.; Pol A.; Op den Camp H. J. M.; Bernhardt P. V. Electrocatalysis of a europium-dependent bacterial methanol dehydrogenase with its physiological electron acceptor cytochrome cGJ. Chem. - Eur. J. 2019, 25, 8760–8768. 10.1002/chem.201900525. [DOI] [PubMed] [Google Scholar]
- Good N. M.; Moore R. S.; Suriano C. J.; Martinez-Gomez N. C. Contrasting in vitro and in vivo methanol oxidation activities of lanthanide-dependent alcohol dehydrogenases XoxF1 and ExaF from Methylobacterium extorquens AM1. Sci. Rep. 2019, 9, 4248. 10.1038/s41598-019-41043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSkimming A.; Cheisson T.; Carroll P. J.; Schelter E. J. Functional synthetic model for the lanthanide-dependent quinoid alcohol dehydrogenase active site. J. Am. Chem. Soc. 2018, 140, 1223–1226. 10.1021/jacs.7b12318. [DOI] [PubMed] [Google Scholar]
- Ho J. V.; Cotruvo J. A. Jr. A periplasmic binding protein for pyrroloquinoline quinone. Biochemistry 2019, 58, 2665–2669. 10.1021/acs.biochem.9b00358. [DOI] [PubMed] [Google Scholar]
- Kim Y. H.; Campbell E.; Yu J.; Minteer S. D.; Banta S. Complete oxidation of methanol in biobattery devices using a hydrogel created from three modified dehydrogenases. Angew. Chem., Int. Ed. 2013, 52, 1437–1440. 10.1002/anie.201207423. [DOI] [PubMed] [Google Scholar]
- Dekker R. H.; Duine J. A.; Frank J.; Verweil P. E. J.; Westerling J. Covalent addition of H2O, enzyme substrates and activators to pyrrolo-quinoline quinone, the coenzyme of quinoproteins. Eur. J. Biochem. 1982, 125, 69–73. 10.1111/j.1432-1033.1982.tb06652.x. [DOI] [PubMed] [Google Scholar]
- Lumpe H.; Daumann L. J. Studies of redox cofactor pyrroloquinoline quinone and its interaction with lanthanides(III) and calcium(II). Inorg. Chem. 2019, 58, 8432–8441. 10.1021/acs.inorgchem.9b00568. [DOI] [PubMed] [Google Scholar]
- Finney L. A.; O’Halloran T. V. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936. 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Jacobsen F. E.; Giedroc D. P. Coordination chemistry of bacterial metal transport and sensing. Chem. Rev. 2009, 109, 4644–4681. 10.1021/cr900077w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron K. J.; Robinson N. J. How do bacterial cells ensure that metalloproteins get the correct metal?. Nat. Rev. Microbiol. 2009, 7, 25–35. 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- Cook E. C.; Featherston E. R.; Showalter S. A.; Cotruvo J. A. Jr. Structural basis for rare earth element recognition by Methylobacterium extorquens lanmodulin. Biochemistry 2019, 58, 120–125. 10.1021/acs.biochem.8b01019. [DOI] [PubMed] [Google Scholar]
- Gifford J. L.; Walsh M. P.; Vogel H. J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Halling D. B.; Liebeskind B. J.; Hall A. W.; Aldrich R. W. Conserved properties of individual Ca2+-binding sites in calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E1216–E1225. 10.1073/pnas.1600385113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.-L.; Aquaron R. R.; Leavis P. C.; Gergely J. Metal-binding properties of calmodulin. Eur. J. Biochem. 1982, 124, 7–12. 10.1111/j.1432-1033.1982.tb05900.x. [DOI] [PubMed] [Google Scholar]
- Gilli R.; Lafitte D.; Lopez C.; Kilhoffer M.-C.; Makarov A.; Briand C.; Haiech J. Thermodynamic analysis of calcium and magnesium binding to calmodulin. Biochemistry 1998, 37, 5450–5456. 10.1021/bi972083a. [DOI] [PubMed] [Google Scholar]
- Snyder E. E.; Buoscio B. W.; Falke J. J. Calcium(II) site specificity: Effect of size and charge on metal ion binding to an EF-hand-like site. Biochemistry 1990, 29, 3937–3943. 10.1021/bi00468a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz M.; Sherawat M.; Franz K. J.; Peisach E.; Allen K. N.; Imperiali B. Structural origin of the high affinity of a chemically evolved lanthanide-binding peptide. Angew. Chem., Int. Ed. 2004, 43, 3682–3685. 10.1002/anie.200460028. [DOI] [PubMed] [Google Scholar]
- Allen K. N.; Imperiali B. Lanthanide-tagged proteins - an illuminating partnership. Curr. Opin. Chem. Biol. 2010, 14, 247–254. 10.1016/j.cbpa.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Horrocks W. D. J.Lanthanide ion probes of biomolecular structure. In Adv. Inorg. Biochem.; Eichhorn G. L., Marzilli L. G., Eds.; Elsevier: New York, 1982; Vol. 4, pp 201–261. [Google Scholar]
- Chu F.; Beck D. A. C.; Lidstrom M. E. MxaY regulates the lanthanide-mediated methanol dehydrogenase switch in Methylomicrobium buryatense. PeerJ 2016, 4, e2435 10.7717/peerj.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S.; Suzuki Y.; Fujitani Y.; Mitsui R.; Nakagawa T.; Shintani M.; Tani A. Lanthanide-dependent regulation of methylotrophy in Methylobacterium aquaticum strain 22A. mSphere 2018, 3, e00462-17 10.1128/mSphere.00462-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole B. E.; Falcones I. B.; Cheisson T.; Manor B. C.; Carroll P. J.; Schelter E. J. A molecular basis to rare earth separations for recycling: tuning the TriNOx ligand properties for improved performance. Chem. Commun. 2018, 54, 10276–10279. 10.1039/C8CC04409K. [DOI] [PubMed] [Google Scholar]
- Sandy M.; Butler A. Microbial iron acquisition: marine and terrestrial siderophores. Chem. Rev. 2009, 109, 4580–4595. 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhalfa H.; Crumbliss A. L. Chemical aspects of siderophore mediated iron transport. BioMetals 2002, 15, 325–339. 10.1023/A:1020218608266. [DOI] [PubMed] [Google Scholar]
- Raymond K. N.; Dertz E. A.; Kim S. S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3584–3588. 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W.; Haque M. F. U.; Dispirito A. A.; Semrau J. D. Uptake and effect of rare earth elements on gene expression in Methylosinus trichosporium OB3b. FEMS Microbiol. Lett. 2016, 363, fnw129. 10.1093/femsle/fnw129. [DOI] [PubMed] [Google Scholar]
- Moeck G. S.; Coulton J. W. TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol. Microbiol. 1998, 28, 675–681. 10.1046/j.1365-2958.1998.00817.x. [DOI] [PubMed] [Google Scholar]
- Noinaj N.; Guillier M.; Barnard T. J.; Buchanan S. K. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 2010, 64, 43–60. 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney G. E.; Rosenzweig A. C. Methanobactins: maintaining copper homeostasis in methanotrophs and beyond. J. Biol. Chem. 2018, 293, 4606–4615. 10.1074/jbc.TM117.000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom J. D.; Ford S. M.; Pesesky M. W.; Lidstrom M. E. A mutagenic screen identifies a TonB-dependent receptor required for the lanthanide metal switch in the Type I methanotroph ″Methylotuvimicrobium buryatense″ 5GB1C. J. Bacteriol. 2019, 201, e00120–19. 10.1128/JB.00120-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrmann M.; Berthelot C.; Billard P.; Klebensberger J.. Rare earth element (REE)-dependent growth of Pseudomonas putida KT2440 depends on the ABC-transporter PedA1A2BC and is influenced by iron availability. 2019, bioRxiv 670216. bioRxiv preprint server. https://www.biorxiv.org/content/10.1101/670216v1. [DOI] [PMC free article] [PubMed]
- Deblonde G. J.-P.; Sturzbecher-Hoehne M.; Rupert P. B.; An D. D.; Illy M.-C.; Ralston C. Y.; Brabec J.; de Jong W. A.; Strong R. K.; Abergel R. J. Chelation and stablization of berkelium in oxidation state +IV. Nat. Chem. 2017, 9, 843–849. 10.1038/nchem.2759. [DOI] [PubMed] [Google Scholar]
- Allred B. E.; Rupert P. B.; Gauny S. S.; An D. D.; Ralston C. Y.; Sturzbecher-Hoehne M.; Strong R. K.; Abergel R. J. Siderocalin-mediated recognition, sensitization, and cellular uptake of actinides. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10342–10347. 10.1073/pnas.1508902112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogendoorn C.; Roszczenko-Jasińska P.; Martinez-Gomez N. C.; de Graaff J.; Grassl P.; Pol A.; Op den Camp H. J. M.; Daumann L. J. Facile arsenazo III-based assay for monitoring rare earth element depletion from cultivation media for methanotrophic and methylotrophic bacteria. Appl. Environ. Microbiol. 2018, 84, e02887–17. 10.1128/AEM.02887-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docampo R.; de Souza W.; Miranda K.; Rohloff P.; Moreno S. N. J. Acidocalcisomes - conserved from bacteria to man. Nat. Rev. Microbiol. 2005, 3, 251–261. 10.1038/nrmicro1097. [DOI] [PubMed] [Google Scholar]
- Hong-Hermesdorf A.; Miethke M.; Gallaher S. D.; Kropat J.; Dodani S. C.; Chan J.; Barupala D.; Domaille D. W.; Shirasaki D. I.; Loo J. A.; Weber P. K.; Pett-Ridge J.; Stemmler T. L.; Chang C. J.; Merchant S. S. Subcellular metal imaging identifies dynamic sites of Cu accumulation in Chlamydomonas. Nat. Chem. Biol. 2014, 10, 1034–1042. 10.1038/nchembio.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh H. C.; Collier S.; Guthrie J.; Robertson J. D.; Kornfeld K. Lysosome-related organelles in intestinal cells are a zinc storage site in C. elegans. Cell Metab. 2012, 15, 88–99. 10.1016/j.cmet.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Prieto A.; Alonso J.; Muñoz D.; Marcano L.; Abad Díaz de Cerio A.; Fernández de Luis R.; Orue I.; Mathon O.; Muela A.; Fdez-Gubieda M. L. On the mineral core of ferritin-like proteins: structural and magnetic characterization. Nanoscale 2016, 8, 1088–1099. 10.1039/C5NR04446D. [DOI] [PubMed] [Google Scholar]
- Tyler G. Rare earth elements in soil and plant systems - A review. Plant Soil 2004, 267, 191–206. 10.1007/s11104-005-4888-2. [DOI] [Google Scholar]
- Ramírez-Olvera S. M.; Trejo-Téllez L. I.; García-Morales S.; Pérez-Sato J. A.; Gómez-Merino F. C. Cerium enhances germination and shoot growth, and alters mineral nutrient concentration in rice. PLoS One 2018, 13, e0194691 10.1371/journal.pone.0194691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fall R.; Benson A. A. Leaf methanol - the simplest natural product from plants. Trends Plant Sci. 1996, 1, 296–301. 10.1016/S1360-1385(96)88175-0. [DOI] [Google Scholar]
- Fashui H.; Ling W.; Xiangxuan M.; Zheng W.; Guiwen Z. The effect of cerium(III) on the chlorophyll formation in spinach. Biol. Trace Elem. Res. 2002, 89, 263–276. 10.1385/BTER:89:3:263. [DOI] [PubMed] [Google Scholar]
- White P. J.; Broadley M. R. Calcium in Plants. Ann. Bot. 2003, 92, 487–511. 10.1093/aob/mcg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey J.; Guerinot M. L. Iron uptake and transport in plants: The good, the bad, and the ionome. Chem. Rev. 2009, 109, 4553–4567. 10.1021/cr900112r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abergel R. J.; Clifton M. C.; Pizarro J. C.; Warner J. A.; Shuh D. K.; Strong R. K.; Raymond K. N. The siderocalin/enterobactin interaction: a link between mammalian immunity and bacterial iron transport. J. Am. Chem. Soc. 2008, 130, 11524–11534. 10.1021/ja803524w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi O.; Kim J.; Kim J.-G.; Jeong Y.; Moon J. S.; Park C. S.; Hwang I. Pyrroloquinoline quinone is a plant growth promotion factor produced by Pseudomonas fluorescens B16. Plant Physiol. 2008, 146, 657–668. 10.1104/pp.107.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler P. K. Calcium: a central regulator of plant growth and development. Plant Cell 2005, 17, 2142–2155. 10.1105/tpc.105.032508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voiniciuc C.; Dean G. H.; Griffiths J. S.; Kirchsteiger K.; Hwang Y. T.; Gillett A.; Dow G.; Western T. L.; Estelle M.; Haughn G. W. FLYING SAUCER1 Is a transmembrane RING E3 ubiquitin ligase that regulates the degree of pectin methylesterification in Arabidopsis seed mucilage. Plant Cell 2013, 25, 944–959. 10.1105/tpc.112.107888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Li J.; Zhou Q.; Yang G.; Ding X. L.; Li X.; Cai C. X.; Zhang Z.; Wei H. Y.; Lu T. H.; Deng X. W.; Huang X. H. Rare earth elements activate endocytosis in plant cells. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12936–12941. 10.1073/pnas.1413376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.; Wang L.; He J.; Yang Z.; Huang X. Direct imaging of how lanthanides break the normal evolution of plants. J. Inorg. Biochem. 2018, 182, 158–169. 10.1016/j.jinorgbio.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Wang L.; He J.; Wei H.; Yang Z.; Huang X. Arabinogalactan proteins are the possible extracellular molecules for binding exogenous cerium(III) in the acidic environment outside plant cells. Front. Plant Sci. 2019, 10, 153. 10.3389/fpls.2019.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Gong X.; Ying W.; Chao L.; Hong M.; Wang L.; Fashui H. Cerium relieves the inhibition of chlorophyll biosynthesis of maize caused by magnesium deficiency. Biol. Trace Elem. Res. 2011, 143, 468–477. 10.1007/s12011-010-8830-y. [DOI] [PubMed] [Google Scholar]
- Sato K. Bacteriochlorophyll formation by facultative methylotrophs, Protaminobacter ruber and Pseudomonas AM 1. FEBS Lett. 1978, 85, 207–210. 10.1016/0014-5793(78)80456-6. [DOI] [PubMed] [Google Scholar]
- Yachandra V. K.; Yano J. Calcium in the oxygen-evolving complex: structural and mechanistic role determined by X-ray spectroscopy. J. Photochem. Photobiol., B 2011, 104, 51–59. 10.1016/j.jphotobiol.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui E. Y.; Tran R.; Yano J.; Agapie T. Redox-inactive metals modulate the reduction potential in heterometallic manganese–oxido clusters. Nat. Chem. 2013, 5, 293–299. 10.1038/nchem.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui E. Y.; Agapie T. Reduction potentials of heterometallic manganese–oxido cubane complexes modulated by redox-inactive metals. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10084–10088. 10.1073/pnas.1302677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakou A.; Ghanotakis D. F. Substitution of lanthanides at the calcium site(s) in Photosystem II affects electron transport from tyrosine Z to P680+. Biochim. Biophys. Acta, Bioenerg. 1993, 1141, 303–308. 10.1016/0005-2728(93)90057-M. [DOI] [Google Scholar]
- Redling K.Rare earth elements in agriculture with emphasis on animal husbandry. Ludwig-Maximilians-Universitat München, 2006. [Google Scholar]
- Edington S. C.; Gonzalez A.; Middendorf T. R.; Halling D. B.; Aldrich R. W.; Baiz C. R. Coordination to lanthanide ions distorts binding site conformation in calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3126–E3134. 10.1073/pnas.1722042115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edington S. C.; Halling D. B.; Bennett S. M.; Middendorf T. R.; Aldrich R. W.; Baiz C. R. Non-additive effects of binding site mutations in calmodulin. Biochemistry 2019, 58, 2730–2739. 10.1021/acs.biochem.9b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayshaw L. L.; Smith R. C. G.; Badaoui M.; Irving J. A.; Price S. R. Lanthanides compete with calcium for binding to cadherins and inhibit cadherin-mediated cell adhesion. Metallomics 2019, 11, 914–924. 10.1039/C8MT00317C. [DOI] [PubMed] [Google Scholar]