

Abstract

Direct synthetic routes to amidines are desired, as they are widely present in many biologically active compounds and organometallic complexes. N-Acyl amidines in particular can be used as a starting material for the synthesis of heterocycles and have several other applications. Here, we describe a fast and practical copper-catalyzed three-component reaction of aryl acetylenes, amines, and easily accessible 1,4,2-dioxazol-5-ones to N-acyl amidines, generating CO2 as the only byproduct. Transformation of the dioxazolones on the Cu catalyst generates acyl nitrenes that rapidly insert into the copper acetylide Cu–C bond rather than undergoing an undesired Curtius rearrangement. For nonaromatic dioxazolones, [Cu(OAc)(Xantphos)] is a superior catalyst for this transformation, leading to full substrate conversion within 10 min. For the direct synthesis of N-benzoyl amidine derivatives from aromatic dioxazolones, [Cu(OAc)(Xantphos)] proved to be inactive, but moderate to good yields were obtained when using simple copper(I) iodide (CuI) as the catalyst. Mechanistic studies revealed the aerobic instability of one of the intermediates at low catalyst loadings, but the reaction could still be performed in air for most substrates when using catalyst loadings of 5 mol %. The herein reported procedure not only provides a new, practical, and direct route to N-acyl amidines but also represents a new type of C–N bond formation.
Introduction
Amidines are useful substrates with a variety of applications. The amidine structure in 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is used frequently as a non-nucleophilic base in synthetic chemistry,1 and more complex amidines are even considered to be “superbases”.2 Amidines also have been used as ligands for organometallic complexes3 and as starting material for the synthesis of N-heterocycles.4−10 The structure itself is present in various natural products and biologically active compounds such as janoxepin, pentamidine, and sildenafil (see Figure 1).11,12
Figure 1.

Several amidine compounds and their applications.
Multiple methods exist for the synthesis of amidines in general, including the Pinner reaction,13 substitution of phenoxyimidates,14 Beckman-type rearrangements,15 and dehydrogenative coupling reactions,16,17 or by catalysis with palladium14,18−21 or nickel,22 as well as some multicomponent reactions.23−33 Chang developed strategies for the synthesis of sulfonated and phosphorylated amidines via traditional click chemistry with azides, followed by a thermal N2 exclusion, resulting in a net coupling of a nitrene to an alkyne.29,34−45 However, the general (kinetic) stability of most triazoles puts severe limitations on the applicability of other azides, and this method also proved to be unsuitable for the synthesis of N-benzoyl or N-acyl amidines.45,46 Nicasio and Pérez showed that a copper-catalyzed reaction of acetylenes with N-aroyl or N-acyl azides produces triazoles or oxazoles instead of N-acyl amidines.47,48 In fact, development of direct methods for the synthesis of N-acyl amidines remains an unsolved challenge. Current synthetic routes to N-acyl amidines usually rely on reactions of premade primary amides with amino acetals49 or acylation of amidines with stoichiometric coupling reagents.50−52 A direct, atom-efficient synthetic strategy is thus desired.
N-Acyl amidines are a particularly interesting class of compounds. They are important substructures in a variety of medicines53,54 and have been used as precursors for cyclization reactions,50,53 as ligands for catalysts and other molecular assemblies,54 or as substrates for reductive alkylation.55 We therefore focused on developing a new method for the direct synthesis of N-acyl amidines via a one-pot multicomponent reaction.
Since our group is focusing on C–H activation using metallocarbenes and metallonitrenes for the catalytic formation of new C–C and C–N bonds,56−62 we considered that a related acyl nitrene transfer strategy could be useful for the synthesis of N-acyl amidines. However, realizing new strategies to convert acyl nitrenes in a selective manner is rather troublesome. While amides are among the most stable nitrogen-containing moieties, acyl nitrenes are intrinsically unstable. Even when bound to a metal, alkyl migration via the Curtius rearrangement generally rapidly leads to an isocyanate (Scheme 1, left).63−65 Hence, in the context of seeking protocols to couple acyl nitrenes to C-nucleophiles, such Curtius rearrangements are undesired and thus represent a selectivity problem, in particular for aliphatic acyl nitrenes. The barrier for this rearrangement is lower for aliphatic acyl nitrenes than for aromatic derivatives, and the intrinsic instability of aliphatic acyl azides makes them challenging to isolate and unsuitable for prolonged storage.
Scheme 1. Acyl Nitrene Formation from 1,4,2-Dioxazol-5-ones, Followed by an Undesired Metal-Induced Curtius Rearrangement (Left) versus Desired Nitrene Insertion into an M–Cacetylide Bond (Right).

In addition to the intrinsic instability of acyl nitrenes, efficient formation of these reactive intermediates constitutes an additional challenge. Conventional precursors used for metallonitrene chemistry are iminoiodinanes, haloamines, organic azides, and hydroxylamines,66 most of which are difficult to prepare and display poor solubility, require elevated temperatures to activate, or are associated with selectivity and sustainability issues. Cyclic nitrene precursors were recently shown to be efficient nitrene precursors,67 which are more practical. In particular, 1,4,2-dioxazol-5-ones are noteworthy acyl nitrene precursors in this perspective. They are stable until at least 100 °C, have a low activation barrier upon coordination to a metal, and produce only CO2 as a side product.68 They can be synthesized by two practical steps from acids, esters, or acyl chlorides.69 Although these properties have been known for over 50 years, this class of substrates only have become popular for applications in catalysis since the work of Bolm in 2014.70
Still, the intrinsic instability of N-acyl metallonitrenes generated from these substrates is a major issue. Successful approaches to use N-acyl metallonitrenes in catalysis typically rely on directed C–H activation or generation of the acylnitrene on a metal center already containing a preactivated co-substrate.71−77
Inspired by the work of Wang on copper-catalyzed carbene insertion into C–H bonds,82 we anticipated that acyl nitrenes could be coupled to terminal alkynes as a new type of C–N bond formation. We envisioned that the activation of 1,4,2-dioxazol-5-ones on copper acetylides could readily form an acyl nitrene on copper that should rapidly insert into the Cu–C bond to furnish a C–N bond, avoiding decomposition by the Curtius rearrangement and thus providing a direct route to acylated amidines (Scheme 1, right).
Results and Discussion
In an initial attempt to couple acyl nitrenes to acetylenes, we added dioxazolone 1a to a mixture of copper bromide, chloroform, phenylacetylene (2a), and diisopropylamine (3a). The yellow color, typical for copper acetylide complexes, slowly turned dark green upon addition of the dioxazolone. Analysis of the crude reaction mixture by mass spectrometry suggested that a three-component reaction had taken place.
1H NMR analysis indeed confirmed the formation of the three-component reaction product 4aaa in 63% yield (Table 1). The reaction thus readily gives access to this N-acyl amidine. To optimize the reaction conditions, we explored the efficiency of the reaction using different Cu(I) catalysts and different solvents (ligands used in this study are shown in Figure 2). Most copper salts worked equally well (Table 1, entries 1–5). The [Cu(MeCN)4]+ salts appeared to work slightly better, which is commonly observed for copper-catalyzed reactions.83 We also employed some well-defined Cu(I)-ligand complexes, including complexes containing the Xantphos ligand (Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) (Table 1, entries 6–11).84−86 Surprisingly, while most complexes perform poorly, [Cu(OAc)(Xantphos)] gave a very high selectivity. The reaction works well in a variety of different solvents under aerobic conditions at room temperature (Table 1, entries 12–19). Results obtained in toluene and ethyl acetate show that the reaction is selective in both polar and apolar solvents. In methanol and acetonitrile, the desired product was not observed, which is suggestive of catalyst inhibition by coordination of these solvents to copper. In tetrahydrofuran (THF), however, full conversion toward the product is observed. Excellent yield is obtained with 5 mol % catalyst loading (or more), but the yield drops with lower catalyst loadings (Table 1, entries 20–24). Notably, when the reaction is diluted 10 times, formation of the product 4aaa was not observed (Table 1, entry 25). Instead, 1,1-diisopropyl-3-phenethylurea compound 5aa was formed, indicating a Curtius rearrangement of the acyl nitrene. Replacing the Xantphos ligand by 1,2-bis(diphenylphosphino)ethane (dppe) or 2 equiv of PPh3 results in a decreased yield of 4aaa (Table 1, entries 26 and 27).
Table 1. Optimization of Reaction Conditionsa.

| no. | catalyst (mol % Cu) | solvent | yield (%) |
|---|---|---|---|
| 1 | CuCl (10) | CHCl3 | 55 |
| 2 | CuBr (10) | CHCl3 | 63 |
| 3 | CuI (10) | CHCl3 | 62 |
| 4 | [Cu(NCMe)4]BF4 (10) | CHCl3 | 65 |
| 5 | [Cu(NCMe)4]PF6 (10) | CHCl3 | 75 |
| 6 | [Cu(TpBr3)(NCMe)] (10) | CHCl3 | 5 |
| 7 | [Cu(TpMs)(THF)] (10) | CHCl3 | 0 |
| 8 | [CuCl(IPr)] (10) | CHCl3 | 37 |
| 9 | [CuI(Xantphos)] (10) | CHCl3 | 0 |
| 10 | [Cu(OAc)(Xantphos)] (10) | CHCl3 | 98 |
| 11 | [Cu(NCMe)2(Xantphos)]BF4 (10) | CHCl3 | 72 |
| 12 | [Cu(OAc)(Xantphos)] (10) | DCM | 58 (97%b) |
| 13 | [Cu(OAc)(Xantphos)] (10) | THF | 95 |
| 15 | [Cu(OAc)(Xantphos)] (10) | EtOAC | 92 |
| 17 | [Cu(OAc)(Xantphos)] (10) | DCE | 74 |
| 19 | [Cu(OAc)(Xantphos)] (10) | toluene | 93 |
| 20 | [Cu(OAc)(Xantphos)] (5) | CHCl3 | 97 |
| 21 | [Cu(OAc)(Xantphos)] (2) | CHCl3 | 43 (94%b) |
| 22 | [Cu(OAc)(Xantphos)] (1) | CHCl3 | 10 (19%b) |
| 23 | [Cu(OAc)(Xantphos)] (0.5) | CHCl3 | 0 |
| 24 | [Cu(OAc)(Xantphos)] (0.1) | CHCl3 | 0 |
| 25c | [Cu(OAc)(Xantphos)] (5) | CHCl3 | 0 (98%b) |
| 26 | [Cu(OAc)] + dppe (5) | CHCl3 | 74%b |
| 27 | [Cu(OAc)] + 2 PPh3 (5) | CHCl3 | 82%b |
Reaction of 1a (0.5 mmol), 2a (2 equiv), 3a (2 equiv), and catalyst in solvent (1 mL), stirred overnight in a closed 4 mL vial at room temperature in air. Yields are determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as external standard.
Reactions performed in a N2-filled glovebox.
10 mL of chloroform was used.
Figure 2.

Ligands used in this work.
We hypothesized that aerobic oxidation of one or more reaction intermediates could inhibit catalysis. Indeed, performing the reactions under an N2 atmosphere led to high yields of the product (Table 1, entries 12, 21, and 25). Surprisingly, in the absence of a catalyst but in the presence of amine 3a full conversion of dioxazolone 1a to urea 5aa was observed at room temperature. Further screening (see the SI) demonstrated that secondary amines are able to activate and decompose 1,4,2-dioxazol-5-ones, resulting in the urea (Scheme 2). This side reaction and the Curtius rearrangement catalyzed by an oxidized copper complex likely explain the decreased yields when using lower catalyst concentrations under aerobic conditions (Table 1, entries 21 and 22).
Scheme 2. Desired Catalyzed Reaction vs the Uncatalyzed Side Reaction (Urea Formation).

The copper-catalyzed three-component reaction produces only CO2 as a byproduct. In view of the atom efficiency of the process, we tested the efficacy of this reaction with a lower ratio of phenylacetylene and diisopropylamine relative to the dioxazolone substrate (see the SI).
The use of fewer equivalents of acetylene has a larger effect on the yield than lowering the amount of diisopropylamine (56% and 84% yield, respectively). When one equivalent of all reactants was used, we still obtained 70% of the N-acyl amidine. It should be noted that a lower yield is obtained when using just one equivalent of phenylacetylene in combination with a higher concentration of diisopropylamine. This indicates that fast formation of a copper acetylide species is important and that this process must outcompete the diisopropylamine-assisted decomposition of the dioxazolone to obtain high yields of the desired N-acyl amidine.
Scope of the (Xantphos)Cu-Catalyzed Reaction
The catalytic reaction is not limited to the use of phenylethyl dioxazolone 1a (Table 2) and actually has a relatively large substrate scope. Longer dioxazolone substrates can be used without a decrease in yield (4baa–4daa), and double bonds are tolerated as well (4eaa). The yield dropped significantly for benzyl dioxazolone 1f and cyclohexyl dioxazolone 1g. Presumably, slower activation of the bulkier substrates on the copper acetylide species leads to faster aerobic decomposition of the catalyst. The fact that under an atmosphere of N2 products 4faa and 4gaa are obtained in good yields supports that hypothesis. An oxygen atom at the β-position of the dioxazolone is deleterious for the yield, as 62% of urea 5ia was obtained when 1i was used. We suspect that this is the result of chelation of the substrate or product to copper, inhibiting the formation of the acetylide on the metal. A first attempt to apply para-tolyl acetylene as a terminal alkyne in the copper-catalyzed three-component reaction yielded only 35% of N-acyl amidine 4aba. However, again under a protective atmosphere of nitrogen we observed high yields of the desired product (see Table 2). Para-, meta-, and ortho-tolyl acetylene all gave a similar high yield, suggesting a negligible influence of sterics at the acetylene (4aba–4ada). The electron-rich 4-ethynyl anisole gave slightly lower yields (4aea), which could be related to the lower acidity of the acetylene C–H bond, thus leading to less efficient formation of the copper acetylide complex. When using an alkyl acetylene (2j), which is even less acidic, product 4aja was not observed. Electron-withdrawing phenyl acetylenes, on the other hand, were converted with high efficiency (4afa–4aga). Even the use of the heteroaromatic acetylenes 3-ethynylpyridine and 3-ethynylthiophene resulted in efficient formation of the desired products 4aha and 4aja (79% and 98%, respectively).
Table 2. Scope of the [Cu(OAc)(Xantphos)]-Catalyzed Three-Component Reactiona.


Reaction of 1x (0.5 mmol), 2y (2 equiv), 3a (2 equiv), and [Cu(OAc)(Xantphos)] (5 mol %) in CHCl3 (1 mL), stirred for 10 min in a closed 4 mL vial at room temperature. Isolated yields are given. *Performed under an N2 atmosphere. #Quantitative formation of product according to 1H NMR with 1,3,5-trimethoxybenzene as internal standard. $The urea was formed as the main product.
Next, we explored the scope of using different amines as coupling partners in reactions catalyzed by [Cu(OAc)(Xantphos)] (Table 3). We initially were disappointed to see that benzylamine, isopropylamine, and diethylamine did not yield any N-acyl amidine (3b–3d). Dicyclohexylamine (3h), however, did show a very high yield for the formation of the desired product. We hypothesized that the less bulky amines might coordinate too strongly to the copper atom and compete with coordination of the dioxazolone and thereby inhibit the reaction. To verify our assumption, we further explored the effect of changing the steric bulk of the amine. Indeed, the less bulky amines N-ethylisopropylamine 3e and N-methyl-tert-butylamine 3f do not result in any desired product formation, while the bulkier amine N-ethyl-tert-butylamine 3g produced the desired product 4aag in high yield (96%). The change from 3f to 3g might seem abrupt, but note that in amine 3g the ethyl group is pushed away from the tert-butyl group when coordinated to Cu(I), thus making it much bulkier and hence a weaker donor. The combined results suggest that smaller amines poison the catalyst, while the bulkier amines fit poorly in the “Xantphos pocket”, resulting in negligible catalyst inhibition and thus a high product yield (see Figure 3). Upon increasing the steric bulk of the amine further, i.e., employing 2,2,6,6-tetramethylpiperidine (3i) as nucleophile, we observed formation of the desired product as the enamine tautomer (4aai, see the SI).
Table 3. Scope of Amines Used for the [Cu(OAc)(Xantphos)]-Catalyzed Three-Component Reactiona.


Reaction of 1a (0.5 mmol), 2a (2 equiv), 3z (2 equiv), and [Cu(OAc)(Xantphos)] (5 mol %) in CHCl3 (1 mL), stirred for 10 min in a closed 4 mL vial at rt. Isolated yields for 4aaz are given. *A tautomer (enamine) was isolated.
Figure 3.

Proposed catalyst poisoning by small amine substrates.
Mechanistic Investigations
To gain more information about the reaction mechanism, we studied the formation of copper species with 31P NMR spectroscopy, investigated the kinetics of the reaction, and performed supporting density functional theory (DFT) studies.
We used 31P NMR spectroscopy to study the formation of [Cu(CCPh)(Xantphos)] under optimized reaction conditions, both in air and under a nitrogen atmosphere. [Cu(OAc)(Xantphos)] in CDCl3 gave a 31P NMR signal at δ = −17 ppm (Figure 4A), which remained unchanged upon addition of phenylacetylene or diisopropylamine (Figure 4B/C). When both reactants were added to the solution of [Cu(OAc)(Xantphos)] in CDCl3 in air, a new signal was observed at δ = +29 ppm, which corresponds to Xantphos-P-dioxide, and a small broad peak at δ = −13 ppm emerged as well (Figure 4D). Under an N2 atmosphere, we did not observe ligand oxidation and detected a new singlet at δ = −15 ppm, along with a signal at δ = −19 ppm corresponding to free Xantphos (Figure 4E). Coordination of Xantphos to copper acetylide or salt metathesis of [CuCl(Xantphos)] with lithium phenylacetylide (Figure 4F) led to the same 31P NMR signal at δ = −15 ppm, supporting the formation of [Cu(CCPh)(Xantphos)]. Rapid formation of copper acetylide complex [Cu(CCPh)(Xantphos)] upon addition of a base to a reaction mixture containing both [Cu(OAc)(Xantphos)] and phenylacetylene is further associated with a characteristic color change from colorless to bright yellow.87 The influence of air on the yield of the catalytic reactions in the case of lower catalyst loadings or when sterically more demanding dioxazolones are used implies that [Cu(CCPh)(Xantphos)] reacts with water or dioxygen from air.
Figure 4.
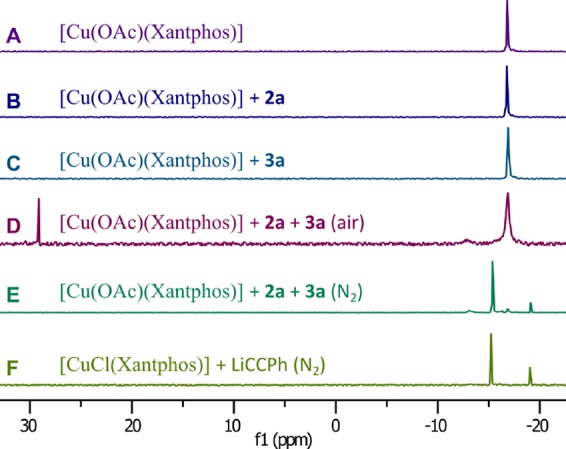
31P NMR studies on the formation of [Cu(CCPh)(Xant-phos)]. 31P NMR spectra measured under an N2 atmosphere in CDCl3. (A) [Cu(OAc)(Xantphos)]; (B) [Cu(OAc)(Xantphos)] with 40 equiv of phenylacetylene; (C) [Cu(OAc)(Xantphos)] with 40 equiv of iPr2NH; [Cu(OAc)(Xantphos)] with 40 equiv of both phenylacetylene and iPr2NH under (D) air or (E) N2; (F) [Cu(CCPh)(Xantphos)] formed from salt metathesis from [CuCl(Xantphos)] and lithium phenylacetylide.
Together with the formation of the acetylide complex, we could always observe a broad peak at δ = −13 ppm and free Xantphos, which might result from clustering of copper species. Dimers could be observed in mass spectrometry, and indication for the formation of [Cu3(CCPh)3(Xantphos)2] species was obtained by single-crystal X-ray diffraction (low-quality data set; see the SI for a connectivity plot).
To get more insight into the reaction mechanism, we investigated the kinetics of the reaction, following the pressure buildup caused by CO2 release during the reaction in a dedicated closed system as a measure of the substrate conversion over time.88,89 The observed rates are plotted as a function of the concentrations of the catalyst and each of the substrates 1a, 2a, and 3a (Figure 5A–D).
Figure 5.

Kinetic experiments and X-ray structure of the catalyst interacting with phenyl acetylene. We varied the concentration of the catalyst and all three substrates and calculated the slope of the steepest part in the graph as the reaction rate. These rates have been plotted over the related concentration to give graphs A, B, C, and D. Graph E shows the pressure during the reaction toward 4aaa for [Cu(OAc)(Xantphos)] (red) and [Cu(NCMe)2(Xantphos)]BF4 (blue). F shows the crystal structure obtained by crystallization in the presence of acetylene, showing a H-bonding interaction with the acetate.
The reaction is clearly first order in both [catalyst] and [dioxazolone] and (nearly) zero order in [iPr2NH]. The rate dependence of [PhCCH] (Figure 5C) is more complex and suggestive of saturation kinetics. This behavior is most likely caused by a (slightly endergonic) pre-equilibrium between [Cu(OAc)(Xantphos)] and [Cu(CCPh)(Xantphos)]. The existence of such an equilibrium could be confirmed by H/D exchange studies. Burk and co-workers showed with H/D scrambling that copper acetates and copper acetylides are in fast equilibrium.90 In a similar experiment, using [Cu(OAc)(Xantphos)] as the catalyst, we also observed H/D scrambling of phenylacetylene-D and 4-CF3-phenylacetylene (see the SI for details). Combined with the 31P NMR studies shown in Figure 4 and the small effect of [iPr2NH] on the rate, the data suggest that this pre-equilibrium shifts toward the acetylide complex in the presence of the amine.
From the catalyst screening studies presented in Table 1, it is clear that the “unsaturated” [Cu(NCMe)2(Xantphos)]BF4 complex is less selective than [Cu(OAc)(Xantphos)] for the formation of the N-acyl amidine, indeed confirming that the acetate plays a prominent role in the catalytic cycle. When comparing the conversion over time between the reaction catalyzed by [Cu(OAc)(Xantphos)] and the [Cu(NCMe)2(Xantphos)]BF4 (Figure 5E), we observed a very large difference in rate, confirming that the acetate is important for rapid formation of the acetylide complex.91 In fact, the X-ray structure of the [Cu(OAc)(Xantphos)]-HCCPh adduct, obtained by crystallization of [Cu(OAc)(Xantphos)] in the presence of an excess of phenylacetylene, reveals a direct hydrogen-bonding (H-bonding) interaction between the copper-bound κ2-acetate fragment and the acidic proton of the acetylene (Figure 5F).
Further mechanistic information was obtained using DFT calculations (Figure 6, see the SI for details). While rapid formation of acetylide complex B was observed experimentally during the NMR studies, DFT calculations show that the acetylene activation pre-equilibrium step is endergonic by about +3 kcal/mol. This small discrepancy is likely a medium effect, a result of the reaction being performed under nonstandard concentration conditions or a combination thereof.92N-Coordination and subsequent activation of the dioxazolone substrate on the Cu center, leading to CO2 dissociation, over TS1 produces acyl nitrene acetylide intermediate D. Interestingly, two possible transition states need to be considered for the C–N bond formation step, involving acyl nitrene insertion into the Cu–C bond of the acetylide fragment. In TS2-1, the acyl nitrene is κ1-N coordinated and C–N coupling leads to a η1-N coordination of the amide moiety. In TS2-2, the acyl nitrene coordinates in a κ2-N,O fashion and C–N bond formation leads to a chelating coordination of the carbonyl and the triple bond.
Figure 6.

DFT (B3LYP-D3-TZ2P)-computed energies of the acyl nitrene activation and insertion.
The transition state barriers of these processes (from D) are very similar (∼7.5 kcal mol–1). The resulting intermediates, E for TS2-1 and F for TS2-2, are different by only 3.5 kcal/mol and connected by a low energy barrier transition state TS3 and thus can both be present during the reaction. The undesired Curtius rearrangement from intermediate D was calculated to be ∼3 kcal higher in energy than TS2-1 and TS2-2, which is in line with the experimental observations (see the SI for further details). For the protodemetalation of the vinylideneamide by an incoming equivalent of acetylene, acetic acid, or even diisopropylamine, many pathways can be considered from either E or F. The protodemetalation was therefore not included in the DFT studies.
The combined information gathered from the catalytic studies and the mechanistic investigations led us to the proposed catalytic cycle shown in Scheme 3. The reaction sequence involves activation of acetylene 2a by complex A to form acetylide complex B, most likely assisted by the amine. Subsequently, B activates the dioxazolone to form acyl nitrene intermediate D, which leads to either E or F, followed by protodemetalation to regenerate A and to form vinylideneamide G. Finally, reaction of the amine with the electrophilic intermediate G produces the N-acylamidine product 4.
Scheme 3. Proposed Catalytic Cycle.

Expanding the Scope to Benzoyl Amidines Using CuI
As shown in Table 2, phenyl dioxazolone 1h is not converted by [Cu(OAc)(Xantphos)], thus preventing the one-pot three-component synthesis of benzoyl amidines when using this catalyst. However, we argued that it might well be possible to expand the substrate scope of the newly discovered one-pot Cu-catalyzed three-component reaction from alkyl dioxazoles to aryl dioxazolones if we would use a less bulky Cu catalyst. Such reactions are desirable, as (just like for N-acyl amidines) there are currently no practical or high-yielding reactions available for the efficient one-pot (multicomponent) synthesis of benzoyl amidines. Sharpless and Chang published a method to synthesize the corresponding amidine from a benzoyl azide, but the desired compound was obtained in only very low yield (9% based on 1H NMR spectroscopy).44 Hence, we considered that it remained important to develop new catalytic methods to convert aryl dioxazolones to benzoyl amidines. Therefore, we decided to investigate the reactivity of less bulky, phosphine-free copper salts in reactions using these substrates. Gratifyingly, CuI proved to be a suitable catalyst to extend the protocol to the synthesis of N-benzoyl amidine derivatives. Using 10 mol % of CuI as the catalyst and phenyl dioxazolone as the substrate yielded the desired product in 68% isolated yield (see Table 4). This method works for electron-rich as well as electron-poor aryl dioxazolones (4jaa, 4kaa). The reaction still seems to be affected by sterics, but o-phenyl dioxazolone still produced 4laa in 34% isolated yield.
Table 4. Synthesis of N-Benzoyl Amidines from Aryl-1,4,2-dioxazol-5-ones Catalyzed by CuIa.


Reaction of 1a (0.5 mmol), 2a (2 equiv), 3z (2 equiv), and CuI (10 mol %) in CHCl3 (1 mL), stirred for 30 min in a closed 4 mL vial at room temperature. Isolated yields are shown.
Conclusion
In this work we have presented a new, fast, efficient, and practical three-component reaction of an acyl nitrene precursor catalyzed by copper to produce N-acylamidines and benzoyl amidines. To the best of our knowledge, this is the first reported catalytic procedure to synthesize these types of products. This atom-efficient reaction uses dioxazolone substrates that are easily accessible and only generates CO2 as byproduct under mild conditions, making this an attractive methodology for both small- and large-scale organic synthesis.
Acknowledgments
This research was supported by The Netherlands Organization for Scientific Research (NWO), the Holland Research School of Molecular Chemistry (HRSMC), and the UvA Research Priority Area “Sustainable Chemistry”. We would like to thank Lukas Wolzak, Eduard O. Bobylev, and Ed Zuidinga for the mass spectrometry measurements and Xavier Caumes for leading us to the [Cu(OAc)(Xantphos)] complex.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07140.
The authors declare no competing financial interest.
Supplementary Material
References
- Nand B.; Khanna G.; Chaudhary A.; Lumb A.; M. Khurana J. 1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU): A Versatile Reagent in Organic Synthesis. Curr. Org. Chem. 2015, 19, 790–812. 10.2174/1385272819666150402221133. [DOI] [Google Scholar]
- Ishikawa T.; Kumamoto T.. Amidines in Organic Synthesis. In Superbases for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, UK, pp 49–91. [Google Scholar]
- Collins S. Polymerization Catalysis with Transition Metal Amidinate and Related Complexes. Coord. Chem. Rev. 2011, 255, 118–138. 10.1016/j.ccr.2010.07.005. [DOI] [Google Scholar]
- McGowan M. A.; McAvoy C. Z.; Buchwald S. L. Palladium-Catalyzed N-Monoarylation of Amidines and a One-Pot Synthesis of Quinazoline Derivatives. Org. Lett. 2012, 14, 3800–3803. 10.1021/ol301700y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. F.; Zhu X.; Chiba S. Copper-Catalyzed Aerobic [3 + 2]-Annulation of N-Alkenyl Amidines. J. Am. Chem. Soc. 2012, 134, 3679–3682. 10.1021/ja2120629. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang J.; Lu P.; Wang Y. Copper-Catalyzed Cascade Preparation of Dihydropyrimidin-4-Ones from N-(Prop-2-Yn-1-Yl)Amides and Azides. J. Org. Chem. 2013, 78, 8816–8820. 10.1021/jo401094j. [DOI] [PubMed] [Google Scholar]
- Li S.; Li Z.; Yuan Y.; Li Y.; Zhang L.; Wu Y. Gold(I)-Catalyzed Aminohalogenation of Fluorinated N′-Aryl-N-Propargyl Amidines for the Synthesis of Imidazole Derivatives under Mild Conditions. Chem. - Eur. J. 2013, 19, 1496–1501. 10.1002/chem.201202402. [DOI] [PubMed] [Google Scholar]
- Rajagopal B.; Chen Y.-Y.; Chen C.-C.; Liu X.-Y.; Wang H.-R.; Lin P.-C. Cu(I)-Catalyzed Synthesis of Dihydropyrimidin-4-Ones toward the Preparation of β- and β3-Amino Acid Analogues. J. Org. Chem. 2014, 79, 1254–1264. 10.1021/jo402670d. [DOI] [PubMed] [Google Scholar]
- Deibl N.; Ament K.; Kempe R. A Sustainable Multicomponent Pyrimidine Synthesis. J. Am. Chem. Soc. 2015, 137, 12804–12807. 10.1021/jacs.5b09510. [DOI] [PubMed] [Google Scholar]
- Li J.; Tang M.; Zang L.; Zhang X.; Zhang Z.; Ackermann L. Amidines for Versatile Cobalt(III)-Catalyzed Synthesis of Isoquinolines through C-H Functionalization with Diazo Compounds. Org. Lett. 2016, 18, 2742–2745. 10.1021/acs.orglett.6b01199. [DOI] [PubMed] [Google Scholar]
- Greenhill J. V.; Lue P. Amidines and Guanidines in Medicinal Chemistry. Prog. Med. Chem. 1993, 30, 203–326. 10.1016/S0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- Sprogøe K.; Manniche S.; Larsen T. O.; Christophersen C. Janoxepin and Brevicompanine B: Antiplasmodial Metabolites from the Fungus Aspergillus Janus. Tetrahedron 2005, 61, 8718–8721. 10.1016/j.tet.2005.06.086. [DOI] [Google Scholar]
- Wang Z.Pinner Reaction. In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp 2237–2240. [Google Scholar]
- Saluste C. G.; Whitby R. J.; Furber M. Palladium-Catalysed Synthesis of Imidates, Thioimidates and Amidines from Aryl Halides. Tetrahedron Lett. 2001, 42, 6191–6194. 10.1016/S0040-4039(01)01201-1. [DOI] [Google Scholar]
- Fleury L. M.; Wilson E. E.; Vogt M.; Fan T. J.; Oliver A. G.; Ashfeld B. L. Amine-Free Approach toward N-Toluenesulfonyl Amidine Construction: A Phosphite-Mediated Beckmann-Like Coupling of Oximes and p-Toluenesulfonyl Azide. Angew. Chem., Int. Ed. 2013, 52, 11589–11593. 10.1002/anie.201305141. [DOI] [PubMed] [Google Scholar]
- Rouzi A.; Hudabaierdi R.; Wusiman A. Synthesis of N -Sulfonylformamidines by Tert-Butyl Hydroperoxide-Promoted, Metal-Free, Direct Oxidative Dehydrogenation of Aliphatic Amines. Tetrahedron 2018, 74, 2475–2481. 10.1016/j.tet.2018.03.074. [DOI] [Google Scholar]
- Zheng Y.; Mao J.; Chen J.; Rong G.; Liu D.; Yan H.; Chi Y.; Xu X. Unexpected C = N Bond Formation via NaI-Catalyzed Oxidative de-Tetra-Hydrogenative Cross-Couplings between N,N-Dimethyl Aniline and Sulfamides. RSC Adv. 2015, 5, 50113–50117. 10.1039/C5RA06773A. [DOI] [Google Scholar]
- DeKorver K. A.; Johnson W. L.; Zhang Y.; Hsung R. P.; Dai H.; Deng J.; Lohse A. G.; Zhang Y.-S. N-Allyl-N-Sulfonyl Ynamides as Synthetic Precursors to Amidines and Vinylogous Amidines. An Unexpected N-to-C 1,3-Sulfonyl Shift in Nitrile Synthesis. J. Org. Chem. 2011, 76, 5092–5103. 10.1021/jo200780x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluste C. G.; Crumpler S.; Furber M.; Whitby R. J. Palladium Catalysed Synthesis of Cyclic Amidines and Imidates. Tetrahedron Lett. 2004, 45, 6995–6996. 10.1016/j.tetlet.2004.07.151. [DOI] [Google Scholar]
- Tetala K. K. R.; Whitby R. J.; Light M. E.; Hurtshouse M. B. Palladium-Catalysed Three Component Synthesis of α,β-Unsaturated Amidines and Imidates. Tetrahedron Lett. 2004, 45, 6991–6994. 10.1016/j.tetlet.2004.07.150. [DOI] [Google Scholar]
- Saluste C. G.; Whitby R. J.; Furber M. A Palladium-Catalyzed Synthesis of Amidines from Aryl Halides. Angew. Chem., Int. Ed. 2000, 39, 4156–4158. 10.1002/1521-3773(20001117)39:22<4156::AID-ANIE4156>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Garduño J. A.; Garcia J. J. Synthesis of Amidines and Benzoxazoles from Activated Nitriles with Ni(0) Catalysts. ACS Catal. 2015, 5, 3470–3477. 10.1021/acscatal.5b00348. [DOI] [Google Scholar]
- Zhou M.; Li J.; Tian C.; Sun X.; Zhu X.; Cheng Y.; An G.; Li G. A Metal-Free Three-Component Reaction of Trans-β-Nitrostyrene Derivatives, Dibromo Amides, and Amines Leading to Functionalized Amidines. J. Org. Chem. 2019, 84, 1015–1024. 10.1021/acs.joc.8b02998. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Huang B.; Qiao G.; Zhu L.; Xiao F.; Chen F.; Fu B.; Zhang Z. Tandem Coupling of Azide with Isonitrile and Boronic Acid: Facile Access to Functionalized Amidines. Angew. Chem., Int. Ed. 2017, 56, 4320–4323. 10.1002/anie.201700539. [DOI] [PubMed] [Google Scholar]
- Shojaei S.; Ghasemi Z.; Shahrisa A. Three-Component Synthesis of N -Sulfonylformamidines in the Presence of Magnetic Cellulose Supported N-Heterocyclic Carbene-Copper Complex, as an Efficient Heterogeneous Nanocatalyst. Tetrahedron Lett. 2017, 58, 3957–3965. 10.1016/j.tetlet.2017.08.075. [DOI] [Google Scholar]
- Kim J.; Stahl S. S. Cu-Catalyzed Aerobic Oxidative Three-Component Coupling Route to N-Sulfonyl Amidines via an Ynamine Intermediate. J. Org. Chem. 2015, 80, 2448–2454. 10.1021/jo5029198. [DOI] [PubMed] [Google Scholar]
- Dai Q.; Jiang Y.; Yu J. T.; Cheng J. Palladium-Catalyzed Three-Component Reaction of N-Tosyl Hydrazones, Isonitriles and Amines Leading to Amidines. Chem. Commun. 2015, 51, 16645–16647. 10.1039/C5CC06771E. [DOI] [PubMed] [Google Scholar]
- Xu X.; Gao J.; Cheng D.; Li J.; Qiang G.; Guo H. Copper-Catalyzed Highly Efficient Multicomponent Reactions of Terminal Alkynes, Acid Chlorides, and Carbodiimides: Synthesis of Functionalized Propiolamidine Derivatives. Adv. Synth. Catal. 2008, 350, 61–64. 10.1002/adsc.200700333. [DOI] [Google Scholar]
- Seok H. K.; Doo Y. J.; Chang S. Phosphoryl Azides as Versatile New Reaction Partners in the Cu-Catalyzed Three-Component Couplings. J. Org. Chem. 2007, 72, 9769–9771. 10.1021/jo7016247. [DOI] [PubMed] [Google Scholar]
- Bae I.; Han H.; Chang S. Highly Efficient One-Pot Synthesis of N-Sulfonylamidines by Cu-Catalyzed Three-Component Coupling of Sulfonyl Azide, Alkyne, and Amine. J. Am. Chem. Soc. 2005, 127, 2038–2039. 10.1021/ja0432968. [DOI] [PubMed] [Google Scholar]
- Keung W.; Bakir F.; Patron A. P.; Rogers D.; Priest C. D.; Darmohusodo V. Novel α-Amino Amidine Synthesis via Scandium(III) Triflate Mediated 3CC Ugi Condensation Reaction. Tetrahedron Lett. 2004, 45, 733–737. 10.1016/j.tetlet.2003.11.051. [DOI] [Google Scholar]
- Zhu S.; Xu Y.; Jin G. A Novel Synthesis of N -Fluoroalkanesulfonylamidines Using a Three-Component Reaction. Can. J. Chem. 2003, 81, 265–268. 10.1139/v03-032. [DOI] [Google Scholar]
- McFarland J. W. Reactions of Cyclohexylisonitrile and Isobutyraldehyde with Various Nucleophiles and Catalysts. J. Org. Chem. 1963, 28, 2179–2181. 10.1021/jo01044a006. [DOI] [Google Scholar]
- Husmann R.; Na Y. S.; Bolm C.; Chang S. Copper-Catalyzed One-Pot Synthesis of α-Functionalized Imidates. Chem. Commun. 2010, 46, 5494–5496. 10.1039/c0cc00941e. [DOI] [PubMed] [Google Scholar]
- Yoo E.; Chang S. Copper-Catalyzed Multicomponent Reactions: Securing a Catalytic Route to Ketenimine Intermediates and Their Reactivities. Curr. Org. Chem. 2009, 13, 1766–1776. 10.2174/138527209789630497. [DOI] [Google Scholar]
- Kim J. Y.; Kim S. H.; Chang S. Highly Efficient Synthesis of α-Amino Amidines from Ynamides by the Cu-Catalyzed Three-Component Coupling Reactions. Tetrahedron Lett. 2008, 49, 1745–1749. 10.1016/j.tetlet.2008.01.073. [DOI] [Google Scholar]
- Hwang S. J.; Cho S. H.; Chang S. Evaluation of Catalytic Activity of Copper Salts and Their Removal Processes in the Three-Component Coupling Reactions. Pure Appl. Chem. 2008, 80, 873–879. 10.1351/pac200880050873. [DOI] [Google Scholar]
- Cho S. H.; Hwang S. J.; Chang S. Copper-Catalyzed Three-Component Reaction of 1-Alkynes, Sulfonyl Azides, and Water: N-(4-Acetamidophenylsulfonyl)-2-Phenylacetamide. Org. Synth. 2008, 85, 131–137. 10.1002/0471264229.os085.14. [DOI] [Google Scholar]
- Yoo E. J.; Chang S. A New Route to Indolines by the Cu-Catalyzed Cyclization Reaction of 2-Ethynylanilines with Sulfonyl Azides. Org. Lett. 2008, 10, 1163–1166. 10.1021/ol800049b. [DOI] [PubMed] [Google Scholar]
- Kim J.; Lee S. Y.; Lee J.; Do Y.; Chang S. Synthetic Utility of Ammonium Salts in a Cu-Catalyzed Three-Component Reaction as a Facile Coupling Partner. J. Org. Chem. 2008, 73, 9454–9457. 10.1021/jo802014g. [DOI] [PubMed] [Google Scholar]
- Cho S. H.; Chang S. Room Temperature Copper-Catalyzed 2-Functionalization of Pyrrole Rings by a Three-Component Coupling Reaction. Angew. Chem., Int. Ed. 2008, 47, 2836–2839. 10.1002/anie.200705940. [DOI] [PubMed] [Google Scholar]
- Yoo E. J.; Bae I.; Cho S. H.; Han H.; Chang S. A Facile Access to N-Sulfonylimidates and Their Synthetic Utility for the Transformation to Amidines and Amides. Org. Lett. 2006, 8, 1347–1350. 10.1021/ol060056j. [DOI] [PubMed] [Google Scholar]
- Cho S. H.; Yoo E. J.; Bae I.; Chang S. Copper-Catalyzed Hydrative Amide Synthesis with Terminal Alkyne, Sulfonyl Azide, and Water. J. Am. Chem. Soc. 2005, 127, 16046–16047. 10.1021/ja056399e. [DOI] [PubMed] [Google Scholar]
- Yoo E. J.; Ahlquist M.; Bae I.; Sharpless K. B.; Fokin V. V.; Chang S. Mechanistic Studies on the Cu-Catalyzed Three-Component Reactions of Sulfonyl Azides, 1-Alkynes and Amines, Alcohols, or Water: Dichotomy via a Common Pathway. J. Org. Chem. 2008, 73, 5520–5528. 10.1021/jo800733p. [DOI] [PubMed] [Google Scholar]
- Yoo E. J.; Ahlquist M.; Kim S. H.; Bae I.; Fokin V. V.; Sharpless K. B.; Chang S. Copper-Catalyzed Synthesis of N-Sulfonyl-1,2,3-Triazoles: Controlling Selectivity. Angew. Chem., Int. Ed. 2007, 46, 1730–1733. 10.1002/anie.200604241. [DOI] [PubMed] [Google Scholar]
- Yoo E. J.; Ahlquist M.; Bae I.; Sharpless K. B.; Fokin V. V.; Chang S. Mechanistic Studies on the Cu-Catalyzed Three-Component Reactions of Sulfonyl Azides, 1-Alkynes and Amines, Alcohols, or Water: Dichotomy via a Common Pathway. J. Org. Chem. 2008, 73, 5520–5528. 10.1021/jo800733p. [DOI] [PubMed] [Google Scholar]
- Cano I.; Álvarez E.; Nicasio M. C.; Pérez P. J. Regioselective Formation of 2,5-Disubstituted Oxazoles via Copper(I)-Catalyzed Cycloaddition of Acyl Azides and 1-Alkynes. J. Am. Chem. Soc. 2011, 133, 191–193. 10.1021/ja109732s. [DOI] [PubMed] [Google Scholar]
- Haldón E.; Álvarez E.; Carmen Nicasio M.; Pérez P. J. 1,2,3-Triazoles from Carbonyl Azides and Alkynes: Filling the Gap. Chem. Commun. 2014, 50, 8978–8981. 10.1039/C4CC03614J. [DOI] [PubMed] [Google Scholar]
- Barber C. G.; Blakemore D. C.; Chiva J. Y.; Eastwood R. L.; Middleton D. S.; Paradowski K. A. 1-Amido-1-Phenyl-3-Piperidinylbutanes - CCR5 Antagonists for the Treatment of HIV: Part 2. Bioorg. Med. Chem. Lett. 2009, 19, 1499–1503. 10.1016/j.bmcl.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Li E.; Wang M.; Wang Z.; Yu W.; Chang J. NBS-Mediated Practical Cyclization of N-Acyl Amidines to 1,2,4-Oxadiazoles via Oxidative N-O Bond Formation. Tetrahedron 2018, 74, 4613–4618. 10.1016/j.tet.2018.07.036. [DOI] [Google Scholar]
- Gupta P. K.; Hussain M. K.; Asad M.; Kant R.; Mahar R.; Shukla S. K.; Hajela K. A Metal-Free Tandem Approach to Prepare Structurally Diverse N-Heterocycles: Synthesis of 1,2,4-Oxadiazoles and Pyrimidinones. New J. Chem. 2014, 38, 3062–3070. 10.1039/C4NJ00361F. [DOI] [Google Scholar]
- Castanedo G. M.; Seng P. S.; Blaquiere N.; Trapp S.; Staben S. T. Rapid Synthesis of 1,3,5-Substituted 1,2,4-Triazoles from Carboxylic Acids, Amidines, and Hydrazines. J. Org. Chem. 2011, 76, 1177–1179. 10.1021/jo1023393. [DOI] [PubMed] [Google Scholar]
- Asad M.; Gupta P. K.; Jaiswal S. K.; Hajela K. Iodine-induced Oxidative Cyclisation of N-Acyl Amidines: A Rapid Synthesis of 3,5-Disubstituted-1,2,4-Oxadiazoles. Chemistry Select 2016, 1, 4753–4757. 10.1002/slct.201601194. [DOI] [Google Scholar]
- Clodt J. I.; Fröhlich R.; Eul M.; Würthwein E. U. Metallomacrocyclic Complexes by Self-Assembly of Ni II and Cu II Ions and Chelating Bis(N-Acylamidines). Eur. J. Inorg. Chem. 2012, 2012, 1210–1217. 10.1002/ejic.201101350. [DOI] [Google Scholar]
- Ronson T. O.; Renders E.; Van Steijvoort B. F.; Wang X.; Wybon C. C. D.; Prokopcová H.; Meerpoel L.; Maes B. U. W. Ruthenium-Catalyzed Reductive Arylation of N-(2-Pyridinyl)Amides with Isopropanol and Arylboronate Esters. Angew. Chem., Int. Ed. 2019, 58, 482–487. 10.1002/anie.201810947. [DOI] [PubMed] [Google Scholar]
- te Grotenhuis C.; de Bruin B. Radical-Type Reactions Controlled by Cobalt: From Carbene Radical Reactivity to the Catalytic Intermediacy of Reactive o-Quinodimethanes. Synlett 2018, 29, 2238–2250. 10.1055/s-0037-1610204. [DOI] [Google Scholar]
- Chirila A.; van Vliet K. M.; Paul N. D.; de Bruin B. [Co(MeTAA)] Metalloradical Catalytic Route to Ketenes via Carbonylation of Carbene Radicals. Eur. J. Inorg. Chem. 2018, 2018, 2251–2258. 10.1002/ejic.201800101. [DOI] [Google Scholar]
- Karns A. S.; Goswami M.; de Bruin B. Catalytic Synthesis of Indolines by Hydrogen Atom Transfer to Cobalt(III)-Carbene Radicals. Chem. - Eur. J. 2018, 24, 5253–5258. 10.1002/chem.201704626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Grotenhuis C.; van den Heuvel N.; van der Vlugt J. I.; de Bruin B. Catalytic Dibenzocyclooctene Synthesis via Cobalt(III)-Carbene Radical and Ortho-Quinodimethane Intermediates. Angew. Chem., Int. Ed. 2018, 57, 140–145. 10.1002/anie.201711028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers P. F.; van der Vlugt J. I.; Schneider S.; de Bruin B. Nitrene Radical Intermediates in Catalytic Synthesis. Chem. - Eur. J. 2017, 23, 13819–13829. 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami M.; de Bruin B. Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes. Molecules 2018, 23, 1052. 10.3390/molecules23051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami M.; Geuijen P.; Reek J. N. H.; de Bruin B. Application of [Co(Corrole)] - Complexes in Ring-Closing C-H Amination of Aliphatic Azides via Nitrene Radical Intermediates. Eur. J. Inorg. Chem. 2018, 2018, 617–626. 10.1002/ejic.201701343. [DOI] [Google Scholar]
- Ghosh A. K.; Brindisi M.; Sarkar A. The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. ChemMedChem 2018, 13, 2351–2373. 10.1002/cmdc.201800518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Wu T.; Liang K.; Xia C. Curtius-like Rearrangement of an Iron-Nitrenoid Complex and Application in Biomimetic Synthesis of Bisindolylmethanes. Org. Lett. 2016, 18, 2228–2231. 10.1021/acs.orglett.6b00864. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Leogane O. Boc-Protected Amines via a Mild and Efficient One-Pot Curtius Rearrangement. Org. Lett. 2005, 7, 4107–4110. 10.1021/ol051428b. [DOI] [PubMed] [Google Scholar]
- Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- Shimbayashi T.; Sasakura K.; Eguchi A.; Okamoto K.; Ohe K. Recent Progress on Cyclic Nitrenoid Precursors in Transition Metal Catalyzed Nitrene Transfer Reactions. Chem. - Eur. J. 2018, 25, 3156–3180. 10.1002/chem.201803716. [DOI] [PubMed] [Google Scholar]
- Sauer J.; Mayer K. K. Thermolyse Und Photolyse von 3-Subtituierten Δ2-1.4.2-Dioxazolinonen-(5), Δ2-1.4.2-Dioxazolin-Thionen-(5) Und 4-Substituierten Δ3-1.2.5.3-Thiadioxazolin-s-Oxiden. Tetrahedron Lett. 1968, 9, 319–324. 10.1016/S0040-4039(01)98753-2. [DOI] [Google Scholar]
- Hong S. Y.; Park Y.; Hwang Y.; Kim Y. B.; Baik M.-H.; Chang S. Selective Formation of γ-Lactams via C-H Amidation Enabled by Tailored Iridium Catalysts. Science 2018, 359, 1016–1021. 10.1126/science.aap7503. [DOI] [PubMed] [Google Scholar]
- Bizet V.; Buglioni L.; Bolm C. Light-Induced Ruthenium-Catalyzed Nitrene Transfer Reactions: A Photochemical Approach towards N-Acyl Sulfimides and Sulfoximines. Angew. Chem., Int. Ed. 2014, 53, 5639–5642. 10.1002/anie.201310790. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Luo G.; Yang J.; Luo Y. Cobalt-Catalysed Unactivated C(Sp3)-H Amination: Two-State Reactivity and Multi-Reference Electronic Character. Catal. Sci. Technol. 2019, 9, 1879–1890. 10.1039/C9CY00239A. [DOI] [Google Scholar]
- Shi H.; Dixon D. J. Dithiane-Directed Rh(III)-Catalyzed Amidation of Unactivated C(Sp3)-H Bonds. Chem. Sci. 2019, 10, 3733–3737. 10.1039/C8SC05225E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H.; Rovis T. Ir-Catalyzed Intermolecular Branch-Selective Allylic C-H Amidation of Unactivated Terminal Olefins. J. Am. Chem. Soc. 2019, 141, 2268–2273. 10.1021/jacs.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa S.; Kato Y.; Tanaka R.; Kojima M.; Yoshino T.; Matsunaga S. Enantioselective C(Sp3)-H Amidation of Thioamides Catalyzed by a Cobalt III /Chiral Carboxylic Acid Hybrid System. Angew. Chem., Int. Ed. 2019, 58, 1153–1157. 10.1002/anie.201812215. [DOI] [PubMed] [Google Scholar]
- Huang D. Y.; Yao Q. J.; Zhang S.; Xu X. T.; Zhang K.; Shi B. F. Amide-Directed Cobalt(III)-Catalyzed C-H Amidation of Ferrocenes. Org. Lett. 2019, 21, 951–954. 10.1021/acs.orglett.8b03938. [DOI] [PubMed] [Google Scholar]
- Xu L.; Li T.; Wang L.; Cui X. Rh(III)-Catalyzed One-Pot Synthesis of Benzimidazoquinazolines via C-H Amidation-Cyclization of N-LG-2-Phenylbenzoimidazoles. J. Org. Chem. 2019, 84, 560–567. 10.1021/acs.joc.8b02396. [DOI] [PubMed] [Google Scholar]
- In a few recent examples alkyl-1,4,2-dioxazol-5-one was converted without co-substrate preactivation, enabled by catalyst optimization to inhibit the Curtius rearrangement78−80 or stabilization of the acyl nitrene by a genetically engineered enzyme.81
- Park Y.; Chang S.. Asymmetric Formation of γ-Lactams via C-H Amidation Enabled by Chiral Hydrogen-Bond-Donor Catalysts. Nat. Catal. 2019, 2, 219. 10.1038/s41929-019-0230-x. [DOI] [Google Scholar]
- Hwang Y.; Park Y.; Kim Y. B.; Kim D.; Chang S. Revisiting Arene C(sp2)-H Amidation by Intramolecular Transfer of Iridium Nitrenoids: Evidence for a Spirocyclization Pathway. Angew. Chem., Int. Ed. 2018, 57, 13565–13569. 10.1002/anie.201808892. [DOI] [PubMed] [Google Scholar]
- Hong S. Y.; Son J.; Kim D.; Chang S. Ir(III)-Catalyzed Stereoselective Haloamidation of Alkynes Enabled by Ligand Participation. J. Am. Chem. Soc. 2018, 140, 12359–12363. 10.1021/jacs.8b08134. [DOI] [PubMed] [Google Scholar]
- Cho I.; Jia Z. J.; Arnold F. H. Site-Selective Enzymatic C-H Amidation for Synthesis of Diverse Lactams. Science 2019, 364, 575–578. 10.1126/science.aaw9068. [DOI] [PubMed] [Google Scholar]
- Xu S.; Chen R.; Fu Z.; Gao Y.; Wang J.. Cu(I)-Catalyzed Coupling of Bis(Trimethylsilyl)Diazomethane with Terminal Alkynes: A Synthesis of 1,1-Disilyl Allenes. J. Org. Chem. 2018, 83, 6186–6192 10.1021/acs.joc.8b00651. [DOI] [PubMed] [Google Scholar]
- Panera M.; Díez J.; Merino I.; Rubio E.; Gamasa M. P. Synthesis of Copper(I) Complexes Containing Enantiopure Pybox Ligands. First Assays on Enantioselective Synthesis of Propargylamines Catalyzed by Isolated Copper(I) Complexes. Inorg. Chem. 2009, 48, 11147–11160. 10.1021/ic901527x. [DOI] [PubMed] [Google Scholar]
- Gonell S.; Caumes X.; Orth N.; Ivanović-Burmazović I.; Reek J. N. H. Self-Assembled M12L24 Nanospheres as a Reaction Vessel to Facilitate a Dinuclear Cu(I) Catalyzed Cyclization Reaction. Chem. Sci. 2019, 10, 1316–1321. 10.1039/C8SC03767A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer P. C. J.; Van Leeuwen P. W. N. M.; Reek J. N. H. Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis. Acc. Chem. Res. 2001, 34, 895–904. 10.1021/ar000060+. [DOI] [PubMed] [Google Scholar]
- Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium-Catalyzed Hydroformylation: Effect of the Bite Angle. Organometallics 1995, 14, 3081–3089. 10.1021/om00006a057. [DOI] [Google Scholar]
- De Boer S. Y.; Gloaguen Y.; Lutz M.; Van Der Vlugt J. I. Cu I Click Catalysis with Cooperative Noninnocent Pyridylphosphine Ligands. Inorg. Chim. Acta 2012, 380, 336–342. 10.1016/j.ica.2011.10.037. [DOI] [Google Scholar]
- Chirila A.; Brands M. B.; de Bruin B. Mechanistic Investigations into the Cyclopropanation of Electron-Deficient Alkenes with Ethyl Diazoacetate Using [Co(MeTAA)]. J. Catal. 2018, 361, 347–360. 10.1016/j.jcat.2018.02.013. [DOI] [Google Scholar]
- Luconi L.; Osipova E. S.; Giambastiani G.; Peruzzini M.; Rossin A.; Belkova N. V.; Filippov O. A.; Titova E. M.; Pavlov A. A.; Shubina E. S. Amine Boranes Dehydrogenation Mediated by an Unsymmetrical Iridium Pincer Hydride: (PCN) vs (PCP) Improved Catalytic Performance. Organometallics 2018, 37, 3142–3153. 10.1021/acs.organomet.8b00488. [DOI] [Google Scholar]
- Kalvet I.; Tammiku-Taul J.; Mäeorg U.; Tämm K.; Burk P.; Sikk L. NMR and DFT Study of the Copper(I)-Catalyzed Cycloaddition Reaction: H/D Scrambling of Alkynes and Variable Reaction Order of the Catalyst. ChemCatChem 2016, 8, 1804–1808. 10.1002/cctc.201600176. [DOI] [Google Scholar]
- We believe that the induction period visible in the graph is due to a delayed pressure buildup because of solubility of CO2 in the solvent.
- When one equivalent of both iPr2NH and PhCCH were used compared to [(Xantphos)Cu(OAc)], under identical reaction conditions, we did not observe the conversion of this complex (see the SI).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.