

Abstract
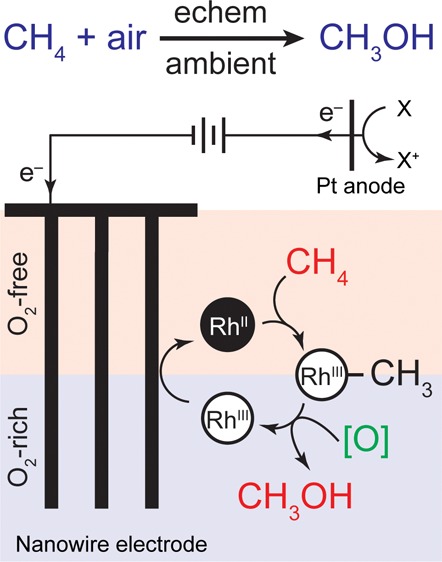
Direct chemical synthesis from methane and air under ambient conditions is attractive yet challenging. Low-valent organometallic compounds are known to activate methane, but their electron-rich nature seems incompatible with O2 and prevents catalytic air oxidation. We report selective oxidation of methane to methanol with an O2-sensitive metalloradical as the catalyst and air as the oxidant at room temperature and ambient pressure. The incompatibility between C–H activation and O2 oxidation is reconciled by electrochemistry and nanomaterials, with which a concentration gradient of O2 within the nanowire array spatially segregated incompatible steps in the catalytic cycle. An unexpected 220 000-fold increase of the apparent reaction rate constants within the nanowire array leads to a turnover number up to 52 000 within 24 h. The synergy between nanomaterials and organometallic chemistry warrants a new catalytic route for CH4 functionalization.
Short abstract
Nanowire array promotes the separation of incompatible reaction steps to allow air-sensitive molecules to catalytically oxidize natural gas to alcohols under ambient conditions.
Introduction
It is attractive to directly use air and natural gas, mostly methane (CH4), as raw materials for the synthesis of methanol (CH3OH),1−4 an important commodity chemical. High-valent, electron-deficient organometallic compounds have been attempted as the centers for C–H activation and the immediate oxidants, presuming that the metal complexes can be reoxidized by air to fulfill a catalytic cycle.5−8 Because of the low reactivity of its C–H bond, CH4 functionalization proceeds at elevated temperatures which incurs possible overoxidation into other products.9−11 Alternatively, electron-rich organometallic compounds are capable of selectively activating CH4 at low temperature.2,4,12 This intrigues us to establish a hypothetical catalytical cycle at ambient conditions, in which a reductive or homolytic step of CH4 activation is followed by air oxidation to yield CH3OH with minimal overoxidation. However, as the step of CH4 activation may not be favored thermodynamically and O2 can oxidatively quench the catalytic species (Figure 1A), external energy input is needed for catalyst regeneration, and a spatial control of these incompatible reactions is required.
Figure 1.
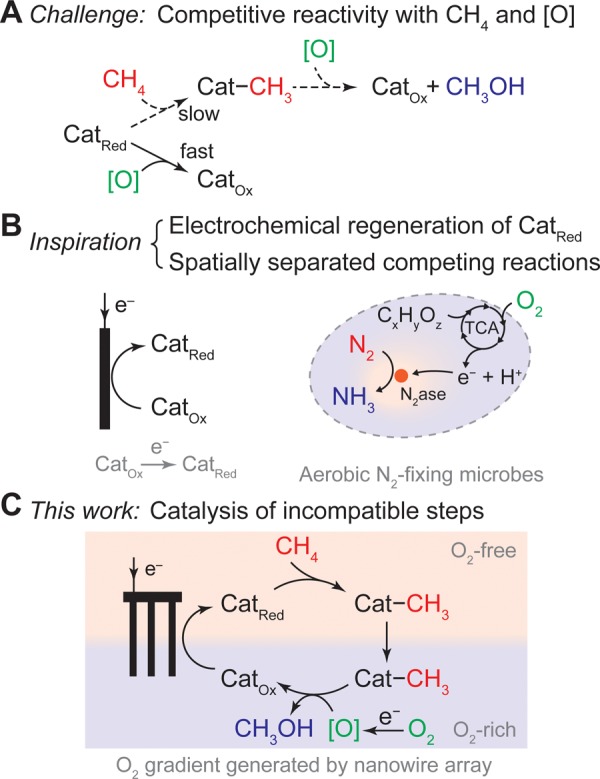
Motivation toward a catalytic cycle for ambient air oxidation of CH4 to CH3OH. (A) The incompatibility of low-valent, electron-rich organometallic compounds for CH4 functionalization with O2-derived oxidants ([O]). (B) The inspirations to address such an incompatibility from examples in biology and (C) the proposed approach reported in this work.
In biology, incompatible biochemical reactions coexist within one organelle by localizing conflicting reactions. One example is the fixation of dinitrogen (N2) in aerobic bacteria (Figure 1B). O2-sensitive nitrogenase for N2 fixation is powered by the reducing equivalents generated from the tricarboxylic acid (TCA) cycle with O2 as the terminal electron acceptor.13 The tandem reactions of aerobic respiration and N2 fixation are only possible with the buildup of an O2 gradient, where the O2-sensitive nitrogenase is positioned in a local anaerobic part of cytoplasm and the TCA cycle in an aerobic one.14 Inspired by the strategies employed in biology, we propose that in order to fulfill a catalytic cycle, the steps of C–H activation and air oxidation should be connected for the catalysis yet spatially separated with mitigated oxidative quenching (Figure 1C). While these requirements are challenging in a homogeneous solution, we posit that they can be satisfied with the use of a nanowire array electrode and electrochemistry. When an electrode is biased at a potential more negative than the redox potentials of O2 and the catalyst, redox-active catalysts can be regenerated by electrochemistry.15 Moreover, the electrochemical reduction of O2 will establish a local O2 gradient in the solution near the electrode surface. This effect is much more pronounced for nanomaterials and porous electrodes in general,16,17 effectively creating an O2-free domain within nanomaterials suitable for chemical steps incompatible with O2. In support of this argument, our previous work demonstrated that a nanowire array electrode can create an O2-free domain that allows anaerobic microbial reduction of CO2.16 Establishing a similar O2 gradient and regenerating the CH4-activating catalyst with electrochemically active nanowires (Figure 1C), here we report a catalytic cycle for ambient air oxidation of CH4 to CH3OH with O2-sensitive, electron-rich RhII tetramesityl porphyrin metalloradical, (TMP)RhII (1a, Figure 2A), as the catalyst.18−20
Figure 2.
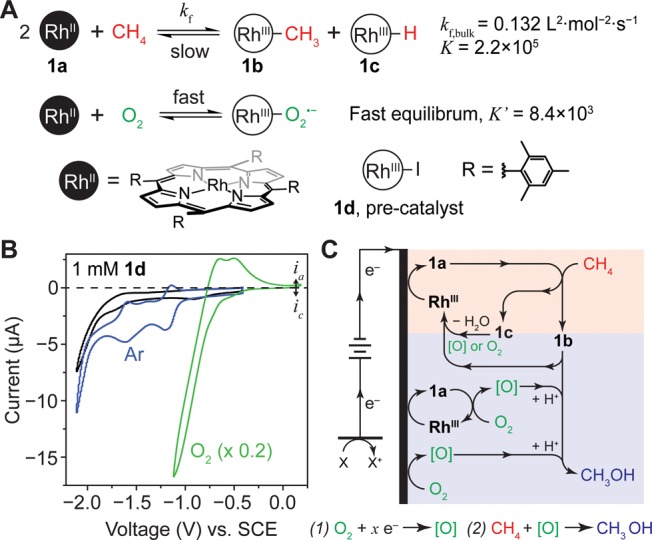
Electrochemical characterization and proposed catalytic cycle utilizing (TMP)RhII, 1a, as the catalyst. (A) The reactivities of RhII metalloradical with CH4 and O2. (B) Cyclic voltammograms of 1 mM 1d with 0.1 M TBAClO4 in 1,2-DFB under Ar (blue) and air (green) environment. Black, blank solution without 1d. 100 mV/s; Pt working electrode for blank and in Ar, glassy carbon electrode in O2. The current in O2 is multiplied by a factor of 0.2. (C) Schematic of the proposed catalytic cycle with 1d as the precatalyst. Upon CH3OH formation, RhIII is generated whose charge is balanced by the perchlorate anion (ClO4–) in solution, which has been omitted for clarity. Oxidant [O] signifies reactive oxygen species such as hydrogen peroxide and superoxide. The proposed reaction is displayed below the catalytic cycle. x = 2.1 on average based on experimental data (entries 2–6 in Table S1).
Results and Discussion
At ambient conditions, 2 equiv of 1a reversibly activate 1 equiv of CH4 with a large equilibrium constant (K = 2.2 × 105 at 298 K), which yields the methylated and hydride species ((TMP)Rh–CH3, 1b; (TMP)Rh–H, 1c, respectively) (Figure 2A).19 The sterically bulky TMP ligand and the requirement of a four-centered transition state warrant a selectivity toward CH4 by two orders of magnitude versus other larger substrates including CH3OH.21 However, in a homogeneous solution, such a reactivity with CH4 is not translatable to catalysis when paired with oxidants such as O2. 1a and O2 react to form a RhIII superoxo species under a fast equilibrium (K′ = 8.4 × 103 at 298 K, Figure 2A),22 and this reaction outcompetes the kinetically slow process of CH4 activation (kf,bulk = 0.132 M–2·s–1 at 296 K).19 Despite this, we argue that electrochemistry can regenerate 1a in situ from its oxidized counterparts thereby potentially allowing the activation of CH4 with 1a in air. RhIII tetramesityl porphyrin iodide ((TMP)Rh–I, 1d) was synthesized based on literature (Figure S1).19 In an argon (Ar) environment and noncoordinating solvent, 1,2-difluorobenzene (1,2-DFB),23 with 0.1 M tetrabutylammonium perchlorate (TBAClO4), a cyclic voltammogram (CV) of 1d on a platinum (Pt) working electrode displays quasi-reversible behavior at an electrode potential Eappl= −1.26 V vs standard calomel electrode (SCE) (Figure 2B), consistent with a previous report that the RhII species can be regenerated by electrochemistry.24,25 In the presence of O2, a catalytic cathodic wave was observed on a glassy carbon electrode preceding the RhIII/RhII redox couple (Eappl < – 0.7 V vs SCE) (green trace in Figure 2B), while the CV trace in the absence of 1d yielded no such activity (Figure S2). Previous literature report the generation of superoxide and peroxide as the immediate products both in solution or electrochemically when O2 and RhII porphyrin are in a stoichiometric ratio.22,26,27 Here our experimental data in air suggest that additional catalytic irreversible reduction of O2 is feasible when the amount of O2 is in surplus.
The capability of generating reactive oxygen species [O] electrochemically with Rh porphyrin leads us to explore whether those [O] can activate 1b and yield CH3OH. Stoichiometric reactions between different hydroperoxide species and 1b, a stable compound prepared in air (Figure S3), were performed at a 1:1 ratio under ambient conditions (see Supporting Information). The reaction between t-butyl hydroperoxide and 1b was tracked via 1H NMR and indicated the formation of CH3OH at the expense of the methyl group in 1b (Figure S4). This suggests that the methylated species 1b is capable of releasing CH3OH by hydroperoxide. Moreover, we found that a 3-h electrolysis of 1b at Eappl= −1.4 V vs SCE yielded a stoichiometric amount of CH3OH (Table S1, entry 1). A gas chromatograph equipped with a mass spectrometer (GC-MS), allowing for a clear separation of electrolyte and catalyst from product determination, was used to detect the product after establishing a calibration curve (Figure S5). This indicates that the electrochemically generated [O] from O2 reduction can be a serendipitous oxidant which yields CH3OH after the step of CH4 activation. During the aforementioned electrolysis, on average 2.3 equiv of electrons are consumed per CH3OH molecule synthesized, indicating that [O] is possibly of a hydroperoxide nature. However, not all of the generated [O] will lead to CH3OH formation, conveying that the value of 2.3 electrons acts as an upper boundary for the reaction during electrolysis.
Given the literature and our experimental data, we propose to establish a solution catalytic cycle of incompatible reactions at ambient conditions in air (Figure 2C), which is impossible in homogeneous solution but potentially feasible when combining electrochemistry and nanomaterials. A silicon (Si) nanowire array was proposed to offer a similar and even enhanced effect as that of a porous electrode with respect to induced concentration gradients.16,17 By utilizing nanowire array morphology as the working electrode in the presence of 1d and O2, the oxidant [O] will be electrochemically generated in situ from O2 with the creation of an O2 gradient. The created O2 gradient enables a nanoscopic separation of incompatible reaction steps. In a localized anaerobic environment near the base of the wire array (pink area in Figure 2C), electrochemically regenerated 1a activates CH4 and yields 1b, which diffuses out and oxidatively hydroxylates to yield CH3OH in the aerobic region (blue area in Figure 2C).
Numerical simulations based on electrochemistry models support the feasibility of the proposed catalytic cycle in the wire array. Finite-element simulations using the COMSOL Multiphysics program were conducted for different electrode geometries based on the experimentally available information (see Supporting Information),16,17 including the fast electrochemical equilibrium of RhIII/RhII redox couple, the reported chemical reactivities,19,22 and the molecular diffusion coefficients determined by diffusion ordered spectroscopy (DOSY) with 1H nuclear magnetic resonance (NMR) (Figure S6). Figure 3A displays the calculated concentrations of 1a, 1d, and O2, denoted as [1a], [1d], and [O2], respectively, versus the distance away from electrode surface (z) on a planar electrode at Eappl = −1.4 V vs SCE. An anaerobic domain of predominantly CH4-reactive 1a, pink colored in Figure 3A, is minimal as compared to the extensive aerobic domain (light blue) where CH4-unreactive 1d is predominant. In contrast, for an exemplary wire array of 50 μm length, 4 μm diameter, and 15 μm periodicity (i.e., distance between adjacent wires) under the same condition, an extended anaerobic region is visible toward the base of the array and potentially favors CH4 activation (Figure 3B). These results support our hypothesis that a nanowire array electrode can spatially define an anaerobic region for CH4 activation, which is microscopically adjacent to an aerobic one ready for CH3OH formation. Variation of the physical parameters such as the reactivities between O2 and 1a as well as the charge-transfer rate of O2 reduction (Figure S7) does not alter the effectiveness of the wire array for establishing concentration gradients, indicating the robustness of this design.
Figure 3.

Numerical simulations and experimental validation of a microscopic concentration gradient for CH4 activation. (A, B) Simulated concentration gradients of O2, 1a, and 1d ([O2], [1a], and [1d], respectively) near a planar (A) and wire array (B) electrode. z, distance away from electrode surface; Eappl= −1.5 V vs SCE. (C) Jablonski diagram illustrating potential phosphorescence emission of 1a and 1d. The triplet state lifetime (τT) of 1a is much shorter than the one of 1d. I/I0, normalized emission intensity of phosphorescence. (D) Experimentally determined I/I0 versus z for planar (black) and wire array (red). 0.1 mM 1d in the bulk solution, 0.1 M TBAClO4 in 1,2-DFB, Eappl= −1.5 V vs SCE. (E, F) The corresponding cross-sectional heatmaps of unnormalized phosphorescence intensity without (E) and with (F) Eappl. The surface of the Si wire array is delineated in yellow. Scale bar, 15 μm.
Spatially resolved optical measurements confirmed the predicted concentration gradients of 1a and 1d within the wire array electrode. The Si wire array, used as a model system, was prepared by reactive ion etching after photolithography (Figure S8, Supporting Information).28 The geometry was based on the same one used in the numerical simulation (Figure 3B) to help validate the conclusions drawn from the simulations. Electrochemical characterizations suggest that the prepared Si wire arrays are electrochemically active toward O2 reduction with the presence of 1d (Figure S9). As the lifetime of the excited triplet (τT) for 1d (>2 μs) is much longer than the one of 1a (∼200 ns),24 under optical excitation 1d exhibits much stronger phosphorescence emission from 630 to 750 nm as compared to 1a (Figure S10). Thus, in a mixed solution containing both 1a and 1d, the local concentration percentage of 1d, and subsequently the percentage of 1a, can be tracked by monitoring the phosphorescence intensity after normalizing to the intensity when only 1d is in the solution (I/I0) (Figure 3C). An electrochemical setup was constructed under a confocal microscope with 526 nm excitation to in situ map the phosphorescence intensity near the electrode surface in air (Figure S11, Supporting Information). Figure 3D displays the values of I/I0 at different z values for both planar (black) and wire array (red) Si electrodes when Eappl= −1.5 V vs SCE. Near a planar electrode, the values of I/I0 remain constant, and it suggests that the local concentration of 1d was not significantly perturbed (Figures 3D and S12). For a Si wire array (Figure S8) that possesses the exact same geometry simulated in Figure 3B,28 the values of I/I0 decrease toward the base of wire array, indicating a local depletion of 1d and subsequently an accumulation of 1a. The accumulation of CH4-reactive 1a is also suggested in the steady-state cross-sectional heatmaps of phosphorescence. A distinguishably lower emission intensity profile was observed when Eappl= −1.5 V vs SCE in the wire array (Figure 3E), as compared to the case at the open-circuit condition (Figure 3F). The fidelity between simulation (Figure 3A,B) and experimental results (Figure 3E,F) confirms that the wire array spatially generates an O2-free domain in air with a localized accumulation of 1a, which is reactive toward ambient CH4 activation.
Selective ambient air oxidation of CH4 to CH3OH was observed with 1d as the precatalyst when Eappl= −1.4 V vs SCE on a Si wire array electrode. A Si nanowire array, prepared by electroless wet etching, with a wire length of ∼15 μm and diameter of ∼100 nm (Figure 4A),29 was applied as the working electrode for a three-electrode configuration in a customized electrochemical reactor (Figure S13). A gas mixture with a defined ratio between CH4 and air (PCH4/Pair) was delivered at a constant rate under ambient pressure. In a 1,2-DFB solution of 1 mM 1d and PCH4/Pair = 35, a 3-h bulk electrolysis on a Si nanowire electrode (Eappl= −1.4 V vs SCE) yielded 0.37 ± 0.20 mM CH3OH (n = 3, Figure 2B, Table S1, entry 2). The observed CH3OH can be directly attributed to the reactivity of 1a and not the platinum (Pt) counter electrode as a similar performance is observed when a graphitic carbon cloth is substituted as the counter electrode (Table S1, entry 3). Longer electrolysis up to 24 h led to a higher concentration of CH3OH up to 6.45 ± 0.92 mM (Table S1, entries 4–6). Since experimentally there was some fluctuation of electrochemical current as the electrolysis was conducted at a constant potential, a fairer comparison between experiments of different durations is based on the moles of CH3OH normalized to the average electrochemical current. The CH3OH yield normalized by the current (nCH3OH/I) is a near linear function of the electrolysis duration (Figure 4C), which suggests a continuous catalytic reaction without much catalyst degradation. On average, 2.1 ± 0.3 equiv of electrons, a value averaged based on entries 2–6 in Table S1, corresponds to the formation of 1 equiv of CH3OH. The calculated value in the bulk electrolysis of 1d in a CH4/air mixture is lower than the theoretical value of 4 should hydroperoxide be the only reactive oxygen species. This suggests that reactive oxygen species other than hydroperoxide, such as superoxide, likely contribute to the oxidation of 1b and the formation of CH3OH. As hydroperoxide is known to react reversibly with 1a in a similar fashion as O2,30 a spatial distribution of reactive oxygen species generated by the O2 also contributes to the observed reactivity. Interestingly, no other C1 or C2 liquid products were observed, and the generation of CO or CO2 was not detectable in the outgas by GC-MS (Figure S14). While overoxidation may pose an issue since 1a is known to activate CH3OH,17 the absence of other products but CH3OH formation in the electrolysis suggests a strong selectivity for CH4, possibly due to the 100-times faster rate of CH4 activation as compared to CH3OH by 1a,21,31 as well as the relatively high solubility of CH4 in the solvent (9.54 mM at 1 bar CH4 based on 1H NMR).
Figure 4.

Ambient air oxidation of alkanes to primary alcohols enabled by nanomaterials and electrochemistry. (A) Si nanowire array imaged by a scanning electron microscope. Scale bar, 2 μm. (B) General conditions used for catalytic ambient air CH4 oxidation to CH3OH. (C) The amount of generated CH3OH normalized to the average electric current (nCH3OH/I), as a function of the electrolysis duration. (D) Mass spectra for the electrolyte solution after 3-h bulk electrolysis. Red, 13CH4 as the substrate; blue, CH4 of natural isotope abundance. (E) Catalytic reactivities for different alkane substrates. BDE, bond dissociation energy; TON, turnover number based on catalyst in solution; TON′, turnover number based on catalyst in reaction layer; kf,nano and kf,bulk, kinetic rate constants of C–H activation by 1a calculated in nanowire array and reported in the literature,19 respectively. (F) The relationship between nCH3OH/I in a 3-h electrolysis and the lengths of nanowire array. A planar electrode was considered as an array of 0 μm wire length.
Electrolysis in the absence of either 1d, air, or CH4 led to the disappearance of CH3OH formation (Table S1, entries 7–9, respectively). Introducing 13C-labeled CH4 as the substrate in lieu of the one with natural 13C abundance resulted in the surge of m/z = 33 peak in the mass spectrum (Figure 4D, Table S1, entry 10). This suggests the formation of a 13CH3OH•+ fragment in the spectrum from the yielded 13CH3OH (Figure S15). Our observations are consistent with a selective catalysis of CH3OH formation with CH4 as the substrate and O2 as the oxidant. The turnover number (TON), defined as the ratio between product concentration and the concentration of precatalyst 1d in solution, was calculated to be 0.37 for the 3-h electrolysis and 6.45 for the 24-h electrolysis (Figure 4E and Table S1). Such a definition of TON values obviates the fact that only the catalyst molecules within the nanowire array are responsible for CH4 activation in our proposed mechanism. Therefore, we also calculated an alternative turnover number (TON′), which is defined as the ratio between the moles of generated product and the moles of 1d precatalyst within the nanowire array. This TON′ relevant to electrochemical catalysis15 was found to be 2972 for the 3-h electrolysis and up to 51 807 for a 24-h experiment (Figure 4E and Table S1). The calculated values of turnover numbers are comparable to those reported values of other catalysts for CH4 functionalization (Tables S2 and S3), while our process is operating at room temperature and ambient pressure with air as the oxidant and CH3OH as the product.
We further applied this ambient catalytic system to other substrates including ethane (C2H6), propane (C3H8), and toluene (PhCH3). In all cases, selective oxidation to primary alcohols was observed (Table S1, entries 11–13), and their corresponding TON and TON′ are shown in Figure 4E. When t-butylbenzene was introduced as the substrate, no oxidation products were observed, which is in line with a previous report about the reactivity of RhII porphyrin species32 (Table S1, entry 14). The reaction kinetics for different substrates was also compared in the developed catalytic system. As catalytic reactions of different substrates were conducted under different substrate concentrations in solution (see Supporting Information), the observed kinetic rate constants other than the turnover numbers were employed for evaluation. Given that the step of C–H activation is shown to be turnover-limiting (vide infra), we calculated the rate constants of C–H activation in a nanowire array, kf,nano, based on the observed rate of alcohol accumulation (Figure 4E). Despite the large differences of bond dissociation energies (BDE) of the cleaved C–H bonds (Figure 4E), kf,nano, which is independent of substrate concentration, appears to decrease even as BDE is simultaneously decreasing. Such a dependence of kf,nano over different substrates conveys the significant effect of steric constraint from 1a as reported before.18,19
Electrochemically generated 1a is the active species for CH4 activation, and the nanowire array is responsible for 1a’s sustained presence and activity in air. We found that halving the concentration of 1d in bulk electrolysis led to a decrease of reaction rate by 4.3 times (Table S1, entry 15). This is consistent with the second-order kinetics on 1a for CH4 activation (Figure 2A) and implies that C–H activation is turnover-limiting in the proposed catalytic cycle (Figure 2A). When we substituted the precatalyst 1d in the bulk electrolysis with a RhIII octaethyl porphyrin iodide ((OEP)Rh-I, 2) synthesized based on the literature (Figure S16),33 no CH3OH was produced (Table S1, entry 16). While 2 exhibits a similar electrochemical response as 1d with a slight shift of redox potential (Figure S17), the less bulky OEP supporting ligand is reported to favor the formation of the [(OEP)RhII]2 dimer, which is unreactive toward CH4.18 The observed difference of reactivities between 1d and 2 as precatalysts suggests that it is the electrochemically generated 1a that activates CH4. Moreover, the catalytic ambient air oxidation of CH4 to CH3OH stopped, and no CH3OH was observed when the nanowire array electrode was replaced with a planar wireless electrode, a wire array with larger spacing among wires, or an increased O2 partial pressure at PCH4/Pair = 1 (Table S1, entry 17, 18, and 19, respectively). Such observations are indeed consistent with our simulation results that a higher concentration of O2, planar wireless electrode, or a less dense nanowire array all mitigate the anaerobic domain, the population of 1a, and thereby the reactivity toward CH4 (Figure S18). Moreover, on the other hand, a 3-h electrolysis with PCH4 /Pair > 1000 yielded 0.25 mM CH3OH (Table S1, entry 20), illustrating the existence of a fine window of O2 partial pressure, which will result in optimal CH3OH generation. These control experiments also indirectly support previous reports regarding the incompatibility of 1a with the O2 in air.22
Along the same lines, the effect of nanowire length was also investigated to ascertain its role in catalysis and CH3OH formation. Additional nanowire arrays of 10 and 27 μm in length were prepared (Figure S19). The yields of CH3OH for a 3-h electrolysis were 0, 0.19 (Table S1, entry 21), 0.37, and 0.45 (Table S1, entry 22) for a planar electrode and nanowires of 10, 15, and 27 μm, respectively. The corresponding nCH3OH/I values are plotted as a function of nanowire length in Figure 4F. As the length of the nanowire increases, the anaerobic domain in which C–H activation occurs expands, resulting in accelerated catalysis and subsequently more CH3OH formation. Such an increase of reaction rate plateaued between nanowires of 15 and 27 μm in length, suggesting that the system reached the intrinsic limit based on its mechanism, and an additional length of nanowire is not beneficial for reaction productivity anymore. Lastly, a spent nanowire electrode, defined as a nanowire array that was previously utilized for a CH3OH-yielding electrolysis, exhibited no activity toward CH4 (Table S1, entry 23), and measurement of X-ray photoelectron spectroscopy (Figure S20) found no residual Rh species on the nanowire’s surface after electrolysis. It shows that the catalytic system is robust with minimal catalyst degradation, and any possible Rh nanoparticle formation on the surface of the nanowires is not responsible for the observed reactivity.
Interestingly, the rate of CH4 activation by 1a was significantly increased in the nanowire array as compared to the one in bulk solution. kf,nano = 2.9 × 104 L2·mol–2·s–1 for CH4 activation, about 220 000 times the value in bulk solution (kf,bulk = 0.132 L2·mol–2·s–1).19 A similar enhancement, by a factor of about 870 000, was observed when toluene was the substrate. As the C–H activation step of 1a undergoes an entropically disfavored four-centered transition state,18,19 high concentration and favorable orientation between two Rh centers will increase the reaction kinetics of CH4 activation.21 We speculated that the negative charges from the native oxide on the Si nanowire’s surface as well as the relatively low dielectric constant of 1,2-DFB23 promote the adsorption of precatalyst 1d near the nanowire’s surface. While this effect will not alter the reactivity between 1a and O2 as suggested in our experiments (comparing entry 2, 19, and 20 in Table S1), it will lead to a high local concentration of 1a, potentially create favorable intermolecular orientation between neighboring Rh centers, and subsequently increase its rate of CH4 activation. Such a putative argument is supported by the observation that the rate of CH3OH formation was minimal when the negative charges on the Si surface were passivated with terminal trimethylsilyl groups34 (Table S1, entry 24). It implies that confining homogeneous organometallic reactions within the space of a nanowire array can accelerate the reaction rate significantly, an effect possibly similar to the one observed when an organometallic catalyst is encapsulated in a supramolecular cavity.35 Overall, the introduction of electrochemistry and nanomaterials enables a catalytic ambient air oxidation of CH4 to CH3OH with the use of a low-valent electron-rich organometallic compound that is otherwise unsuitable as a catalyst in a homogeneous solution. The concept of spatially separating incompatible reaction steps at the nanoscale for a complete catalytic cycle provides new options for designing catalysis for a broad range of chemical transformations.
Acknowledgments
We would like to acknowledge Greg Khitrov for the use of GC-MS, Xun Guan for the SEM images, Manisha Swain from the Kwon Group for synthesizing 2-(1-hydroperoxy-1-methoxyethyl)-5-methylcyclohexan-1-ol, and Paula Diaconescu for constructive discussions. We thank the Molecular Instrumentation Center lab at the University of California, Los Angeles for sample characerizations. E.D.C. acknowledges the UCLA undergraduate summer research program; C.L. acknowledges the startup fund from the University of California, Los Angeles and the financial support of the Jeffery and Helo Zink Endowed Professional Development Term Chair.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00625.
Full experimental details, detailed synthetic procedures, electrochemical characterizations, numerical simulation, product quantification, and additional tables and figures (PDF)
Author Contributions
C.L. supervised the project. C.L. and B.S.N. designed experiments and wrote the paper. B.S.N. synthesized the compounds with the assistance from E.D.C. and J.C.Q. B.S.N. conducted electrochemical characterizations and product quantification. L.S. conducted numerical simulations and experiments of confocal microscopy. All the authors discussed the results and assisted during the manuscript preparation.
The authors declare no competing financial interest.
Supplementary Material
References
- Gunsalus N. J.; et al. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–857. 10.1021/acs.chemrev.6b00739. [DOI] [PubMed] [Google Scholar]
- Labinger J. A.; Bercaw J. E. Understanding and exploiting C-H bond activation. Nature 2002, 417, 507–514. 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- Caballero A.; Perez P. J. Methane as raw material in synthetic chemistry: the final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. 10.1039/c3cs60120j. [DOI] [PubMed] [Google Scholar]
- Shilov A. E.; Shul’pin G.. Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes. In Activation of C-H Bonds by Low-Valent Metal Complexes (“The Organometallic Chemistry”); Kluwer Academic Publishers, 2002; pp 127–129. [Google Scholar]
- Periana R. A.; et al. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. 10.1126/science.259.5093.340. [DOI] [PubMed] [Google Scholar]
- Periana R. A.; et al. Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. 10.1126/science.280.5363.560. [DOI] [PubMed] [Google Scholar]
- Jones C.; et al. Selective Oxidation of Methane to Methanol Catalyzed, with C–H Activation, by Homogeneous, Cationic Gold. Angew. Chem. 2004, 116, 4726–4729. 10.1002/ange.200461055. [DOI] [PubMed] [Google Scholar]
- O’Reilly M. E.; Kim R. S.; Oh S.; Surendranath Y. Catalytic Methane Monofunctionalization by an Electrogenerated High-Valent Pd Intermediate. ACS Cent. Sci. 2017, 3, 1174–1179. 10.1021/acscentsci.7b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latimer A. A.; Kakekhani A.; Kulkarni A. R.; Nørskov J. K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. 10.1021/acscatal.8b00220. [DOI] [Google Scholar]
- Schwarz H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem., Int. Ed. 2011, 50, 10096–10115. 10.1002/anie.201006424. [DOI] [PubMed] [Google Scholar]
- Labinger J. A. Selective alkane oxidation: hot and cold approaches to a hot problem. J. Mol. Catal. A: Chem. 2004, 220, 27–35. 10.1016/j.molcata.2004.03.051. [DOI] [Google Scholar]
- Janowicz A. H.; et al. Oxidative addition of soluble iridium and rhodium complexes to carbon-hydrogen bonds in methane and higher alkanes. Pure Appl. Chem. 1984, 56, 13–23. 10.1351/pac198456010013. [DOI] [Google Scholar]
- Dixon R.; Kahn D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2004, 2, 621–631. 10.1038/nrmicro954. [DOI] [PubMed] [Google Scholar]
- Tsoy O. V.; Ravcheev D. A.; Čuklina J.; Gelfand M. S. Nitrogen Fixation and Molecular Oxygen: Comparative Genomic Reconstruction of Transcription Regulation in Alphaproteobacteria. Front. Microbiol. 2016, 7, 1343. 10.3389/fmicb.2016.01343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savéant J. M.Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry; John Wiley & Sons, Inc., 2006. [Google Scholar]
- Liu C.; et al. Nanowire–Bacteria Hybrids for Unassisted Solar Carbon Dioxide Fixation to Value-Added Chemicals. Nano Lett. 2015, 15, 3634–3639. 10.1021/acs.nanolett.5b01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J. S.; Tobias C. W. Theoretical Analysis of Current Distribution in Porous Electrodes. J. Electrochem. Soc. 1962, 109, 1183–1191. 10.1149/1.2425269. [DOI] [Google Scholar]
- Sherry A. E.; Wayland B. B. Metalloradical activation of methane. J. Am. Chem. Soc. 1990, 112, 1259–1261. 10.1021/ja00159a064. [DOI] [Google Scholar]
- Wayland B. B.; Ba S.; Sherry A. E. Activation of methane and toluene by rhodium(II) porphyrin complexes. J. Am. Chem. Soc. 1991, 113, 5305–5311. 10.1021/ja00014a025. [DOI] [Google Scholar]
- Thompson S. J.; Brennan M. R.; Lee S. Y.; Dong G. Synthesis and applications of rhodium porphyrin complexes. Chem. Soc. Rev. 2018, 47, 929–981. 10.1039/C7CS00582B. [DOI] [PubMed] [Google Scholar]
- Zhang X.-X.; Wayland B. B. Rhodium(II) Porphyrin Bimetalloradical Complexes: Preparation and Enhanced Reactivity with CH4 and H2. J. Am. Chem. Soc. 1994, 116, 7897–7898. 10.1021/ja00096a057. [DOI] [Google Scholar]
- Cui W.; Wayland B. B. Superoxo, Peroxo, and Hydroperoxo Complexes Formed from Reactions of Rhodium Porphyrins with Dioxygen: Thermodynamics and Kinetics. J. Am. Chem. Soc. 2006, 128, 10350–10351. 10.1021/ja0628755. [DOI] [PubMed] [Google Scholar]
- O’Toole T. R.; Younathan J. N.; Sullivan B. P.; Meyer J. T. 1,2-Difluorobenzene: a relatively inert and noncoordinating solvent for electrochemical studies on transition-metal complexes. Inorg. Chem. 1989, 28, 3923–3926. 10.1021/ic00319a032. [DOI] [Google Scholar]
- Vitols S. E.; Friesen D. A.; Williams D. S.; Melamed D.; Spiro T. G. Excited State Dynamics of Rh(II) Tetramesityl Porphyrin Monomer from Nanosecond Transient Absorption and Emission Spectroscopy. J. Phys. Chem. 1996, 100, 207–213. 10.1021/jp952003h. [DOI] [Google Scholar]
- Grass V.; Lexa D.; Momenteau M.; Savéant J.-M. Reductive Electrochemistry of Rhodium Porphyrins. Disproportionation of Intermediary Oxidation States. J. Am. Chem. Soc. 1997, 119, 3536–3542. 10.1021/ja964023i. [DOI] [Google Scholar]
- Wayland B. B.; Newman A. R. Dioxygen and nitric oxide complexes of rhodium porphyrins. Inorg. Chem. 1981, 20, 3093–3097. 10.1021/ic50223a067. [DOI] [Google Scholar]
- Anderson J. E.; Yao C. L.; Kadish K. M. Electroreduction of the dioxygen adduct of rhodium tetraphenylporphyrin: (TTP)Rh(O2). Inorg. Chem. 1986, 25, 3224–3228. 10.1021/ic00238a027. [DOI] [Google Scholar]
- Liu C.; Tang J.; Chen H. M.; Liu B.; Yang P. A Fully Integrated Nanosystem of Semiconductor Nanowires for Direct Solar Water Splitting. Nano Lett. 2013, 13, 2989–2992. 10.1021/nl401615t. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Geyer N.; Werner P.; de Boor J.; Gösele U. Metal-Assisted Chemical Etching of Silicon: A Review. Adv. Mater. 2011, 23, 285–308. 10.1002/adma.201001784. [DOI] [PubMed] [Google Scholar]
- Choi K. S.; Lai T. H.; Lee S. Y.; Chan K. S. Reduction of Rhodium (III) Porphyrin Hydroxide to Rhodium(II) Porphyrin. Organometallics 2011, 30, 2633–2635. 10.1021/om200075f. [DOI] [Google Scholar]
- Cui W.; Wayland B. B. Activation of C–H/H–H Bonds by Rhodium(II) Porphyrin Bimetalloradicals. J. Am. Chem. Soc. 2004, 126, 8266–8274. 10.1021/ja049291s. [DOI] [PubMed] [Google Scholar]
- Del Rossi K. J.; Wayland B. B. Formation and thermal reactions of rhodium-carbon bonds derived from the reactions of octaethylporphyrin-rhodium(III) dimer with alkyl carbon-hydrogen bonds in alkyl aromatics. J. Am. Chem. Soc. 1985, 107, 7941–7944. 10.1021/ja00312a023. [DOI] [Google Scholar]
- Collman J. P.; Boulatov R. Synthesis and Reactivity of Porphyrinatorhodium(II)–Triethylphosphine Adducts: The Role of PEt3 in Stabilizing a Formal Rh(II) State. J. Am. Chem. Soc. 2000, 122, 11812–11821. 10.1021/ja001364u. [DOI] [Google Scholar]
- Plummer J. D.; Deal M. D.; Griffin P. B.. Silicon VLSI Technology: Fundamentals, Practice and Modeling; Prentice Hall, Inc., 2000. [Google Scholar]
- Fiedler D.; Leung D. H.; Bergman R. G.; Raymond K. N. Selective Molecular Recognition, C–H Bond Activation, and Catalysis in Nanoscale Reaction Vessels. Acc. Chem. Res. 2005, 38, 349–358. 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.