

Abstract
Having a compact yet robust structurally based identifier or representation system is a key enabling factor for efficient sharing and dissemination of research results within the chemistry community, and such systems lay down the essential foundations for future informatics and data-driven research. While substantial advances have been made for small molecules, the polymer community has struggled in coming up with an efficient representation system. This is because, unlike other disciplines in chemistry, the basic premise that each distinct chemical species corresponds to a well-defined chemical structure does not hold for polymers. Polymers are intrinsically stochastic molecules that are often ensembles with a distribution of chemical structures. This difficulty limits the applicability of all deterministic representations developed for small molecules. In this work, a new representation system that is capable of handling the stochastic nature of polymers is proposed. The new system is based on the popular “simplified molecular-input line-entry system” (SMILES), and it aims to provide representations that can be used as indexing identifiers for entries in polymer databases. As a pilot test, the entries of the standard data set of the glass transition temperature of linear polymers (Bicerano, 2002) were converted into the new BigSMILES language. Furthermore, it is hoped that the proposed system will provide a more effective language for communication within the polymer community and increase cohesion between the researchers within the community.
Short abstract
BigSMILES, a line notation that supports intrinsically stochastic molecules on top of the simplified molecular-input line-entry system (SMILES), is presented to pave the way for polymer informatics.
1. Introduction
Line notations that encode the connectivity of a molecule into a line of text are a very popular choice for storing chemical structures owing to their memory compactness, their simultaneous human readability and machine-friendliness, and their compatibility with most software and input systems.1 In synergy with the advances in machine learning and data mining algorithms, a good line notation can enable data-driven research and materials discovery.2−4 For small molecules, many line notations have been developed, including the simplified molecular-input line-entry system (SMILES),5,6 the SYBYL line notation (SLN),7 the Wiswesser line notation (WLN),8 ROSDAL,9 the modular chemical descriptor language (MCDL),10 or more recently the international chemical identifier (InChI).11 Among them, SMILES is the most popular linear notation, and it is generally considered the most human-readable variant, with by far the widest software support.1 In practice, SMILES provides a simple set of representations that are suitable as labels for chemical data and as a memory compact identifier for data exchange between researchers. Moreover, SMILES and its extensions serve as descriptive codes that allow rapid generation of graphical objects that could be searched for chemical structures with tools such as Open Babel.12 Furthermore, as a text-based system, SMILES is also a natural fit to many text-based machine learning algorithms. When combined with string kernels, SMILES strings can be used with kernelized learning methods such as the support vector machine.13 These superior characteristics have made SMILES a perfect tool for translating chemistry knowledge into a machine-friendly form, and it has been successfully applied for small molecule property prediction14−16 and computer-aided synthesis planning.2,17,18
However, polymers have resisted description by these structural languages. This is because most structural languages such as SMILES have been designed to describe molecules or chemical fragments that are well-defined atomistic graphs. Since polymers are stochastic molecules, they do not have unique SMILES representations. As discussed by Audus and de Pablo in a recent viewpoint,19 this lack of a unified naming or identifier convention for polymer materials is one of the major hurdles slowing down the development of the polymer informatics field. While pioneering efforts on polymer informatics such as the Polymer Genome project20 have demonstrated the usefulness of SMILES extensions in polymer informatics, the fast development of new chemistry and the rapid development of materials informatics and data-driven research make the need for a universally applicable naming convention for polymers ever more urgent.20−28
Recently, several notable schemes have been proposed as a potential solution to this issue: the hierarchical editing language for macromolecules (HELM) developed by the Pistoia Alliance,29 the International Union of Pure and Applied (IUPAC) international chemical identifier (InChI),11 and the CurlySMILES language,30 an extension of SMILES that aims to provide support for polymers, composite materials, and crystals. However, while HELM is useful in describing macromolecules and biopolymers with well-defined structures, it is not designed to capture the stochastic nature of polymers. On the other hand, InChI is not specifically designed for polymers and does not support branched polymers, and CurlySMILES primarily focuses on polymers where structural features, such as the head–tail configuration, are already well-defined. Moreover, CurlySMILES requires the introduction of many new parameters to accompany its annotation syntax, which significantly increase the complexity of the language and reduce its readability. Finally, CurlySMILES does not support the encoding of randomly branched polymers. As such, this means that the need for a flexible structurally based identifier system that supports a wide variety of different polymeric structures remains extremely pressing.
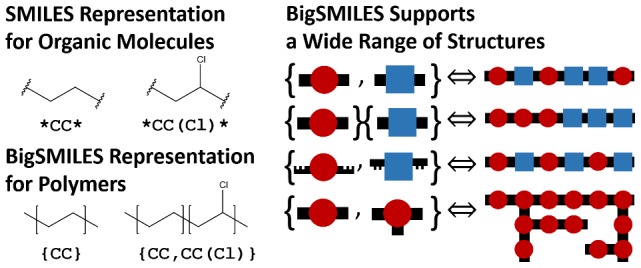
Here, a new structurally based construct is proposed as an addition to the highly successful SMILES representation that can treat the stochastic nature of polymer materials. Since polymers are high-molar-mass molecules, this construct is named BigSMILES. In BigSMILES, polymeric fragments are represented by a list of repeating units enclosed by curly brackets. The chemical structures of the repeating units are encoded using normal SMILES syntax, but with additional bonding descriptors that specify how different repeating units are connected to form polymers. As depicted in Figure 1, this simple design of syntax would enable the encoding of macromolecules over a wide range of different chemistries, including homopolymer, random copolymers and block copolymers, and a variety of molecular connectivities, ranging from linear polymers to ring polymers to even branched polymers. Except for a handful of new additional rules and operators, all syntax of BigSMILES follows the same syntax as the original SMILES. This means that, as in SMILES, BigSMILES representations are compact, self-contained text strings. Furthermore, a multitude of polymer structures that are more complicated than the examples schematically illustrated in Figure 1 can be constructed through the composition of three new basic operators and original SMILES symbols. This is demonstrated in detail along with the discussion of BigSMILES syntax in the next section.
Figure 1.
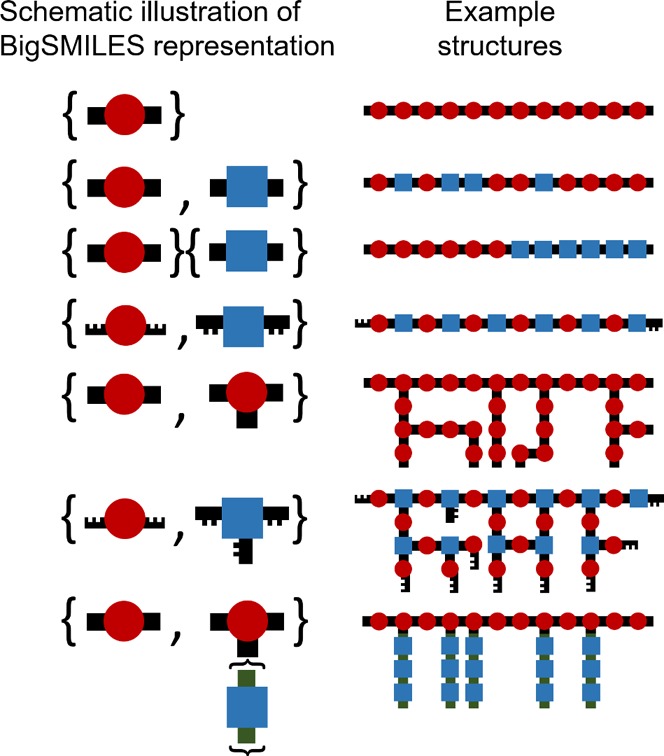
Schematic illustration of the syntax of BigSMILES and some of the structures that can be encoded using BigSMILES. Polymeric fragments are represented by a list of repeating units enclosed within curly brackets. Repeating units are composed of SMILES strings (represented by the red circles and blue squares in the left panel) with additional descriptors (black structures on the left panel) that give the details of the connectivity pattern between repeating units.
2. Syntax
The major extension of BigSMILES to SMILES is the introduction of an additional stochastic object that represents a fragment of a molecule that is intrinsically stochastic in its structure. Unlike small molecules, for which each string corresponds to a single chemical structure, references to the BigSMILES string refer to a group of molecules that have a distribution of structural features and properties. In analogy with statistical physics, this ensemble of polymer molecules consists of many molecular states, where each molecular state is an arrangement of atoms into a molecule that could possibly be realized. Each molecular state has some probability of occurrence, which is determined by the specific rules of chemistry governing the system as the molecules were formed. The rules determining the probability of observing a given molecule in a polymer may be extremely complex, involving changes in the probability of forming a given molecular configuration as a function of both time and space within a reactive system.
While the exact quantification of structural features and properties can be difficult, the monomer and mechanism of polymerization used restrict the set of possible chemical structures present in the ensemble based on the generally known rules of connectivity. Exploiting this, a stochastic object is defined as a machine-friendly representation of the molecular ensemble, without specifying the probability of occurrence of any individual molecular state. The stochastic objects resemble the widely used structural formula representation that is commonly used to describe macromolecules. The object is identified by a pair of curly brackets around a comma-delimited list of repeating units of the polymer:
![]()
The curly bracket is used to avoid conflict with other notation in the existing SMILES syntax. In this representation, each repeating unit within the object essentially resembles the repeating units that are bracketed within the parentheses in a structural formula. The comparison between a BigSMILES stochastic object and a corresponding structural formula is illustrated in Figure 2. In BigSMILES, the entire object, which is bracketed by the curly brackets, symbolizes a piece of a molecular fragment that has a random structure. Since BigSMILES is an extension of the SMILES language, the SMILES syntax for specifying chiral centers (namely, the use of “@” and “@@” symbols), aromatic atoms (the use of lowercase letters), electric charges, ring closures, and many other features5 is retained to provide means for encoding detailed molecular structures. Section SVI (p S17) and Section SVII (p S18) in the SI contain more details on the treatment of tacticity and polyelectrolytes.
Figure 2.
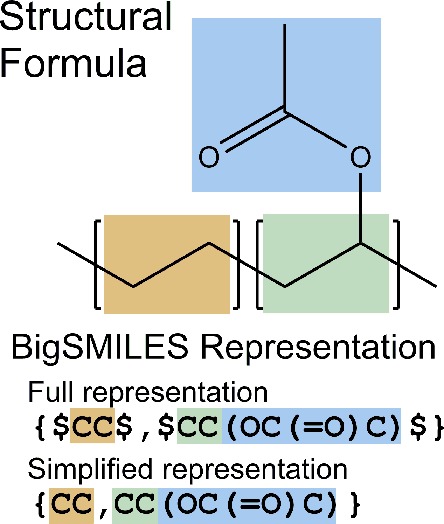
Comparison between the structural formula (top) of poly(ethylene-vinyl acetate) (EVA) and the BigSMILES representation (bottom) of EVA. The representations of ethylene monomer shaded in orange and vinyl acetate monomer shaded in green in the structural formula are very similar to the machine-friendly BigSMILES stochastic object representation. Note that the BigSMILES representation exists as both a simplified representation, in which “$” are omitted, and a full representation, where bonding sites to other repeating units are explicitly indicated. While it is conventional to draw the structural formula with the repeating units in their canonical orientation, this does not imply that all the repeating units are oriented in this specific configuration within the polymer chain; the orientation may be head-to-tail or head-to-head in many cases depending on the nature of the polymerization. Here, the nature of vinyl polymerization is captured by the BigSMILES representation by allowing the units to take both orientations.
2.1. Bonding Descriptor Syntax
In simple linear polymer segments, the repeat units may be written in a way such that the strings for each repeat unit may be directly concatenated together in any order or orientation to form a representation of a polymer molecule. Figure 2 includes a representation of one such hypothetical polymer segment; however, in many cases (such as polymers synthesized via ring-opening polymerization with repeating units always in a specific orientation, or if there exist multiple sets of orthogonal reactions that prohibit the formation of a certain connection between some repeating units), more complex bonding patterns can arise. To differentiate and clearly specify different connectivity patterns between repeating units, two types of bonding descriptors are introduced.
The first type of connection is AA type bonding, where connections can occur between any two bonding moieties within a group of possible moieties. This is commonly found in the bonds formed from chain polymerization of vinyl monomers, where each polymerized vinyl carbon can in principle connect to any other polymerized vinyl carbon found in other repeat units (allowing for head-to-head, tail-to-tail, and head-to-tail addition). Section SI (p S2) in the Supporting Information gives a more detailed discussion of this feature. For this type of connectivity, the “$” notation is used. For example, for a linear polymer segment formed from vinyl monomers ethylene and 1-butene, the stochastic object reads
![]()
As illustrated by the example, in general, there are multiple equally valid representations for each repeating unit, and the bonding site to other repeating units (the position of symbol “$”) can be placed at any position in the repeating unit. Furthermore, there can be more than two such sites per repeat unit, which becomes useful when the notation is generalized to represent branched polymers. For example, Figure S2a (p S11, Section SIV in the SI) gives a representation for branched polyethylene using branching units with three or more connection sites to the other repeating units. It should be emphasized that the list in the BigSMILES stochastic object is defined based on repeating units rather than monomers. Therefore, for monomers such as isoprene, which may have up to four isomerization states upon polymerization,31 each isomerization state is treated as a distinct repeating unit in the stochastic object, as illustrated in Figure 3b.
Figure 3.
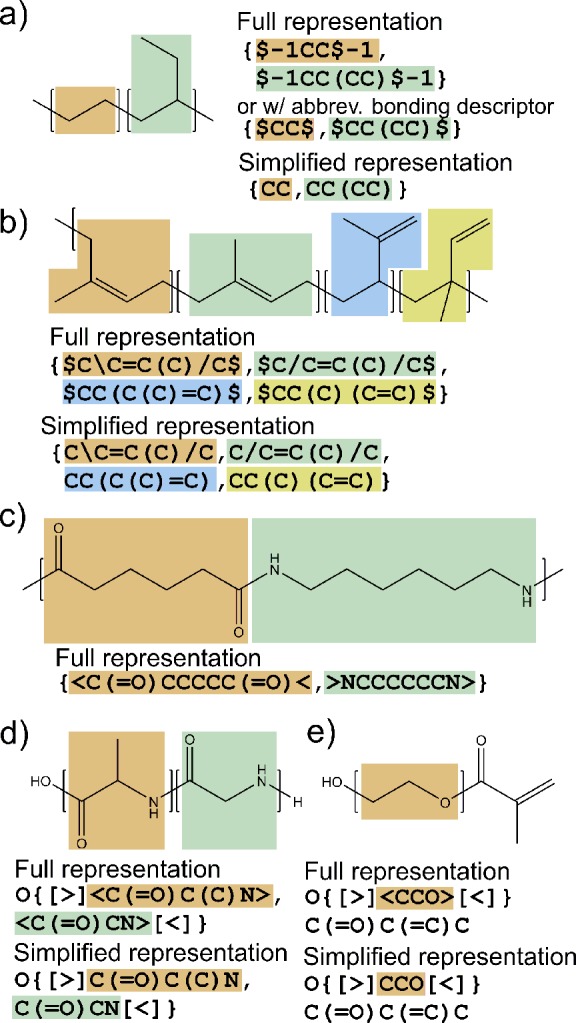
Examples to illustrate the syntax of BigSMILES for polymers synthesized via different chemistries. (a) Vinyl copolymer poly(ethylene-co-1-butene) formed from chain polymerization, (b) four distinct isomerization states for polyisoprene (1,2-addition isomer is retained for completeness despite the fact that its amount can be negligibly small in natural rubber), (c) step polymerized nylon-6,6, (d) step polymerized poly(alanine-co-glycine), and (e) poly(ethylene glycol) methacrylate formed from polymerization of epoxides.
If multiple orthogonal sets of AA type connections exist within the same molecule, the symbol “$” can be appended with a positive integer n into “$n” to distinguish between different sets of connections. By default, “$” represents a single bond connection; however, if the repeating units are connected by other bonds, the bond type or bond order can be specified by using the SMILES bond order representation, with “$=n” for double bonds, “$#n” for triple bonds, and “$\n” and “$/n” for explicitly specifying the cis–trans isomerization states of single bonds directly adjacent to double bonds, respectively. Note that integer IDs n should serve as unique identifiers for the different sets of bonds and therefore not be reused within the same stochastic object. Since the scope of this identifier is only within the stochastic object (between the curly brackets), identifiers within different stochastic objects are distinct even if the IDs appear to be identical. Furthermore, while explicitly stating the additional bond order in every bonding descriptor enhances clarity, the first occurrence of the bond of a particular ID is treated as the definition for the connectivity pattern associated with the ID, and the details of the bond order can be omitted for simplicity in later occurrences. If there is only one group of bonds within the stochastic object, the integer ID can be dropped for simplicity if no additional descriptor (such as the bond order) is needed for the bond. In the special case where there are just two connective sites per repeat unit, and only one type of AA bond of bond order one exists, if the repeat unit is written such that these two sites are at the termini of the repeat unit, the symbol “$” may be omitted altogether, as in the case illustrated in Figure 3a,b. This provides a substantial simplification for a very wide range of common polymers and is referred to as the “simplified representation.”
In the $ representation of AA bonding, any bond indicated by “$n” can be joined to any other bond “$n”, and the repeating unit in the polymeric structure need not connect in the orientation specified in the repeat unit list. Therefore, structures with repeat units in the flipped orientation are implicitly included. For instance, this bonding descriptor can be used for representing vinyl polymers, for which both the head-to-head and the head-to-tail configurations need to be included so the overall BigSMILES representations capture the full ensemble of the possible configuration of the polymer (Figure S1, p S2). Including both configurations is especially important in describing polymers such as poly(vinyl alcohol) or fluorinated vinyl polymers, for which there are known to be a significant number of head-to-head oriented pairs along the chain.32 However, it is emphasized that while the bonding descriptor specifies the ensemble of possible configurations, it does not provide information on the relative weights for each of the configurations.
For the second type of bonding, AB type bonding, a bonding moiety cannot connect directly to other moieties within the same group but can only connect to moieties in another conjugate group. This is commonly seen in monomers polymerized with condensation reactions. For example, in a polyamide, the amide bonds between monomers are always between an acid moiety and an amine moiety but never between two acid or two amine moieties. In this case, angle brackets “<” and “>” are used to indicate the bonds, where bonds must form between conjugate pairs of brackets. For example, the polymeric segment of nylon-6,6, as shown in Figure 3c, may be represented in BigSMILES as

As the asymmetric bonding descriptor represents bonds and connectivity resulting from the reaction of a pair of conjugate end groups, such as polymers synthesized from the polycondensation reaction of a pair of end groups, conjugate symbols are selected for each of the two bonds. For instance, in the nylon-6,6 example, all the amine ends are denoted by the symbol “>”, whereas the carboxyl ends are denoted by the opposite symbol “<”; similarly, the amine ends on the polypeptide in Figure 3d share the symbol “>”, and the carboxyl ends are denoted by the opposite symbol “<”. Similar to the “$” symbol, if multiple groups of AB type bonds exist, or higher bond order is needed, the notation can be extended to “<bn” or and “>bn”, where b is either “–”, “=”, “#”, “\”, or “/” depending on the bond order or bond type, and n is a positive integer. Again, for single bonds, where b is “–”, b can be omitted for simplicity. Practical examples on the usage of the bonding descriptors and common errors in encoding BigSMILES strings are, respectively, provided in Section SIX (pp S45–S53) and Section SII (pp S6–S9) of the Supporting Information.
2.2. Fragment Name Definition Notation
In BigSMILES syntax, repeating units are represented by an extended version of SMILES strings. While this design ensures that BigSMILES strings are standalone and self-descriptive, in some cases it might be more beneficial to have some portions of the BigSMILES representations be replaced by more abstract but compact proxies, for example, the names of repeating units. This is especially helpful when the structure is complex, and the resulting BigSMILES representation becomes long. To facilitate understanding, a definition of molecular fragments that associate user-defined names with partial BigSMILES strings is allowed in BigSMILES using the following syntax:

The definition of repeat unit names is placed at the end of the entire BigSMILES string, with each definition of fragment enclosed by curly brackets and delimited by periods. When fragments are used within the original BigSMILES object, a square bracket should be enclosed to avoid potential confusion of # with triple bonds. Note that the fragments should conform to the BigSMILES syntax and produce a syntactically valid BigSMILES string when embedded within the original BigSMILES object through a substring replacement. In addition, while having complete (fully bracketed on both sides) BigSMILES stochastic objects within the fragment definition is allowed, no bonding descriptors (except for those within fully bracketed stochastic objects) should be included within the fragment definition, so that all occurrences of the bonding to other repeating units appear explicitly in the BigSMILES stochastic objects. In many cases, the fragment definition notation can significantly increase the readability of the BigSMILES strings; two such examples are illustrated in Figure 4. Fragment notation also provides a way of introducing monomer libraries to improve the readability of BigSMILES. An initial library illustrating a wide variety of examples is provided in the Supporting Information (Section SI, p S3, Table S1).
Figure 4.
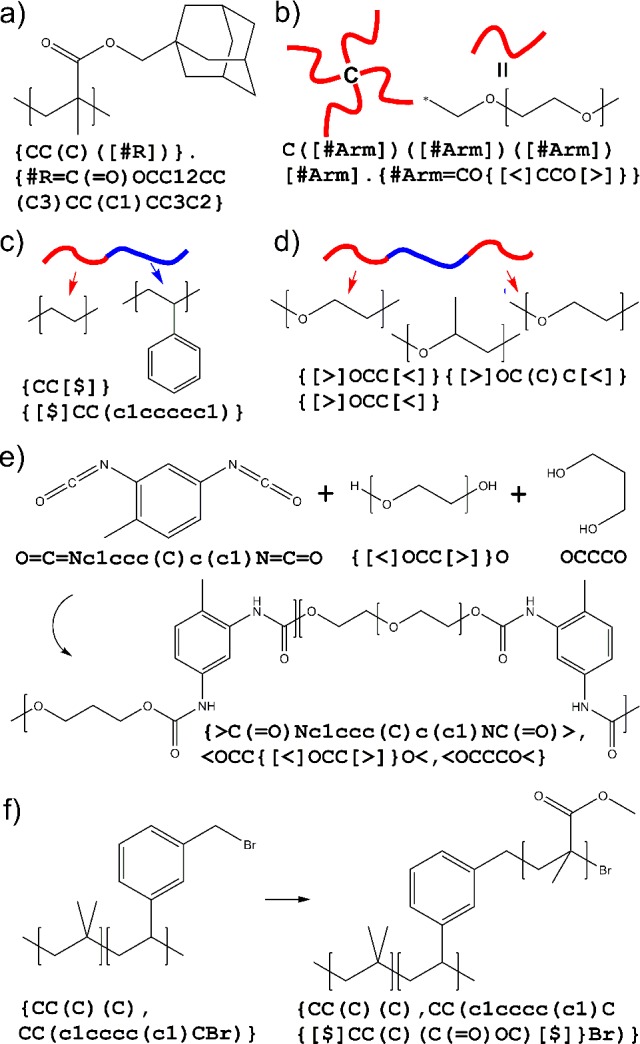
Examples to illustrate useful features of BigSMILES syntax. First, pendant groups (a) or arms (b) can be replaced by user-defined names to improve readability. (c, d) Second, direct concatenation of BigSMILES stochastic objects provides simple representation for block copolymers. Finally, nesting of stochastic object becomes useful in representing copolymers with oligomer chain extenders (e) or polymer grafts (f).
2.3. Concatenation and Nesting of Stochastic Objects
The BigSMILES stochastic object defined earlier represents polymeric fragments. In principle, as in the SMILES language, the adjacent strings outside the stochastic object concatenated to the string within the stochastic object form a continuous chemical structure. However, to ensure chemical validity, how the termini connect to exterior strings should be specified using leading and trailing bonding descriptors within the curly brackets:

The additional bonding descriptors indicate how the exterior atoms are connected to the fragment. Therefore, they should be conjugates to the specific desired terminal; i.e., the additional descriptor should be “>nb” if the desired terminal bond type is “<nb”. This syntax is especially useful for explicitly specifying how polymers with AB type bonds connect to the exterior. For example, if the end groups bracketing the terminals of the nylon-6,6 in Figure 3c are explicitly specified, with both ends terminated by carboxylate group, additional “>” would need to be added to both ends of the stochastic object to indicate that the terminal bonds are of connected to the carbon on the carboxylate group rather than the nitrogen on the amine group:
![]()
It should
be noted that this concatenation syntax only allows up
to two connections to the exterior. In some cases, because of the
nature of the repeating unit, by specifying the ending bonding orientation,
the bond type at the beginning of the object is also determined. This
is common in polycondensation of AB type monomers or ring opening
polymerization, where the connectivity on one end completely determines
the connectivity pattern on the other end. For example, if the end
groups OH were to be positioned on the left of the stochastic object
representing a glycine alanine copolymer, only the C-to-N orientation
of the polyamide makes sense given the placement of the end group.
In these cases where at least one end of the polymer is capped by
external groups, if all repeating units within the stochastic objects
have only a pair of conjugate connective sites belonging to the same
AB bond group, and all repeating units are written so that the sites
are placed at the termini with the same orientation, then “<”
and “>” at the termini of the repeating units may
be
omitted to simplify the representation. With this simplification,
the PEG example in Figure 3e may be simplified as ![]() whereas the previous glycine alanine copolymer
can be simplified
as
whereas the previous glycine alanine copolymer
can be simplified
as ![]() Although the N-terminus of the polymer seems uncapped, there
is an implicit hydrogen that terminates the polymer. Collectively,
these simplifications for AB bonding are also referred to as the “simplified
representation.”
Although the N-terminus of the polymer seems uncapped, there
is an implicit hydrogen that terminates the polymer. Collectively,
these simplifications for AB bonding are also referred to as the “simplified
representation.”
The SMILES feature that allows string
concatenation to represent
a continuous chemical structure enables blocks of polymeric structure
in a copolymer to be written as the direct concatenation of several
stochastic objects. For example, a polyethylene-block-polystyrene structure shown in Figure 4c can be easily encoded by concatenating
the two polymers segments  Similarly, this representation can be
generalized to represent
multiblock copolymers, such as the triblock poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene
glycol) (PEG-b-PPG-b-PEG) illustrated
in Figure 4d. Note
that, in this triblock copolymer example, the syntax is greatly simplified
with the omission of the terminal “<” and “>”
in the repeating units.
Similarly, this representation can be
generalized to represent
multiblock copolymers, such as the triblock poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene
glycol) (PEG-b-PPG-b-PEG) illustrated
in Figure 4d. Note
that, in this triblock copolymer example, the syntax is greatly simplified
with the omission of the terminal “<” and “>”
in the repeating units.
In the BigSMILES syntax, it is possible
to nest multiple levels
of stochastic objects within a stochastic object to create more complex
structures. To illustrate the syntax of nesting, consider synthesis
of a polyurethane through polycondensation of 1,3-propanediol, ethylene
glycol oligomers, and toluene diisocyanate (TDI), as illustrated in Figure 4e. The ethylene glycol
oligomers are encoded as one stochastic object, and this can be nested
in a second stochastic object representing the overall polyurethane
polymer:  This example can be easily generalized to describe polymers
resulting from the polycondensation of more than two types of oligomers
or repeating units.
This example can be easily generalized to describe polymers
resulting from the polycondensation of more than two types of oligomers
or repeating units.
Another scenario that demonstrates the convenience
of nesting is
the representation of graft polymers. Consider polyisobutene-graft-poly(methyl methacrylate) (PIB-g-PMMA)
synthesized by grafting from the linear copolymer of poly[isobutene-co-(m-bromomethylstyrene)], illustrated
in Figure 4f, as an
example. With the polymer graft nested within the backbone, the graft
polymer can be represented by  or, separately defining the polymer graft
with the syntax
provided in the previous section, the polymer can also be represented
as
or, separately defining the polymer graft
with the syntax
provided in the previous section, the polymer can also be represented
as  When possible, readability and ease of comprehension will
usually benefit from encoding a polymer in a non-nested way.
When possible, readability and ease of comprehension will
usually benefit from encoding a polymer in a non-nested way.
2.4. Branched Polymers and Polymer Networks
Up to now, all examples have been focused on linear polymer segments, where each repeat unit has two attachment points corresponding to the start and end of its SMILES string. However, the stochastic object can also be generalized to represent randomly branching polymers. For example, consider a low-density polyethylene (LDPE) molecule with long chain branching (Figure S2a, p S11, Section SIV). Its BigSMILES representation is

Unlike other repeating units discussed up to this point, the second and third repeating units each have functionality larger than two (and therefore the “$” symbols cannot be omitted). Therefore, they serve as branching points, and the entire stochastic object represents a randomly branching structure, which resembles the structure of LDPE. Note that while linear segments of the LDPE molecule can have an odd number of carbons because of branching, the overall linear backbone of LDPE must have an even number of carbons. Hence, the repeating units in this case consist of molecular fragments with two carbons. In practical cases, the fraction of the last repeat unit should be very small compared to the other two repeating units, and this unit is retained in the list here for completeness. Other branched polymers or polymer networks can also be encoded similarly. In Section SIII (pp S10 and S11) of the Supporting Information, more examples, including hyperbranched polymers, end-linked polymer networks, and vulcanized networks, are given; additional discussion on noncovalent or dynamic networks can be found in Section SIV, pp S12 and S13 of the SI.
2.5. End Groups
In BigSMILES, there are two valid ways of specifying end groups. The first way is to explicitly append the end groups around the polymeric fragment represented by the stochastic object; this method allows specification of a deterministic end group. This was used in the previous section to specify the structure for a methacrylate terminated PEG, as illustrated in Figure 3e. The other way is to append the list of possible end groups as a comma-delimited list to the end of the list of repeating units, separated by a semicolon:

The end groups
are represented as if they are also repeating units,
with the same bonding descriptors “$nb”,
“<nb”, or “>nb” as repeating units that indicate the allowed connectivity
patterns between repeating units and the end groups. However, the
nature of end groups dictates that they should have only one possible
bond to another repeating unit, to terminate the structure. For example,
in the nylon-6,6 case, two different end groups are possible![]() where the carboxylic
and amine end groups are included within
the list of repeating units. Note that, in this example, hydrogen
atoms are explicitly written for clarity. When end groups are specified
using this representation, it means that all the unconnected bonds
on the molecular fragment generated using the list of repeating units
with two or more connections to other repeating units are capped with
the specified end groups. This representation can be especially useful
when there are multiple possible end groups. For instance, the variability
of the end groups on the two ends of nylon-6,6 synthesized from polycondensation
of adipic acid and hexamethylenediamine is implicitly considered by
using this representation. The effectiveness of the latter representation
is especially demonstrated by the following example. Consider linear
polystyrene synthesized from AIBN initiated radical polymerization.
It could have three different end groups depending on the route of
termination:
where the carboxylic
and amine end groups are included within
the list of repeating units. Note that, in this example, hydrogen
atoms are explicitly written for clarity. When end groups are specified
using this representation, it means that all the unconnected bonds
on the molecular fragment generated using the list of repeating units
with two or more connections to other repeating units are capped with
the specified end groups. This representation can be especially useful
when there are multiple possible end groups. For instance, the variability
of the end groups on the two ends of nylon-6,6 synthesized from polycondensation
of adipic acid and hexamethylenediamine is implicitly considered by
using this representation. The effectiveness of the latter representation
is especially demonstrated by the following example. Consider linear
polystyrene synthesized from AIBN initiated radical polymerization.
It could have three different end groups depending on the route of
termination:

The possible terminal structures are illustrated in Figure 5a. In this example, the SMILES string leading to the random fragment is synonymous with specifying that the fragment already has one of its two ends capped by an initiator, indicated by the leading [$]. Therefore, it leaves only one unconnected bond on the fragment. The other end group can be one of the three end groups trailing the ethylene monomer in the list. The first possible case is that the other end group is also the initiator, which corresponds to the second entry on the list (first one in the end group list). This happens when termination by coupling takes place. The styrene repeating unit within the end group is written in a reversed orientation to emphasize the preferred configuration in polymerization. On the other hand, when termination from disproportionation occurs, the end of the polymer can be capped by either of the two groups at the end of the list.
Figure 5.
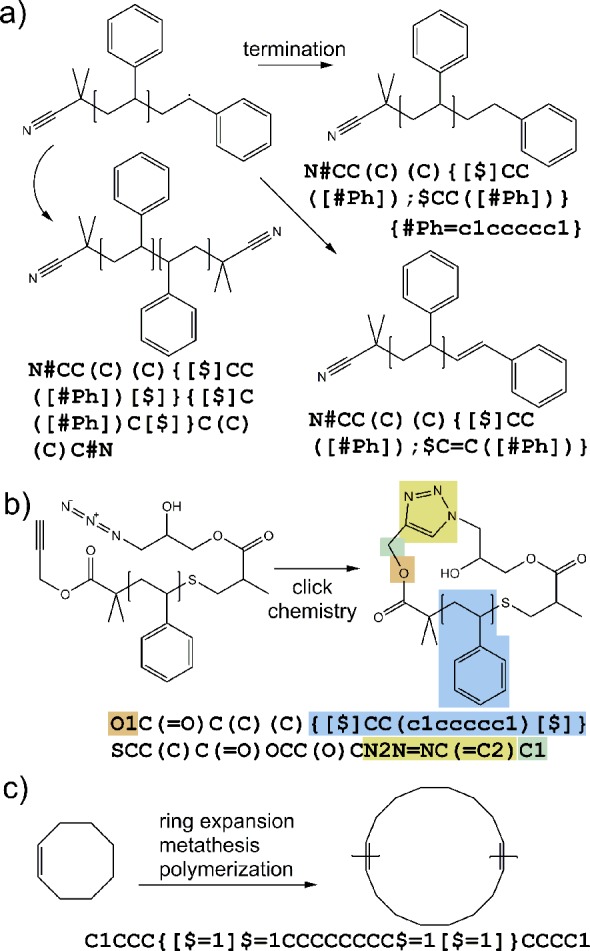
(a) Illustration of possible termination products for free radical polymerization of polystyrene. (b) Polystyrene ring polymer synthesized from azide–alkyne click chemistry. Since the rings and cycles within the repeating units are independent of the macrocycle (that lead to the formation of the ring polymer), the ring closure integer identifier within the stochastic object is independent of the identifiers outside of the object even if the numbers were the same. (c) Ring polymer synthesized from ring expansion metathesis polymerization (REMP).
When randomly branched polymers
are considered, the representation
that includes end groups into the list of repeating units has large
advantages. Consider the hyperbranched polymer example in Figure S2b (p S11, Section SIV); if the end group
for the #B moiety is #E, then the
end groups of the hyperbranched polymer can be easily specified using
the following representation: ![]() Note that, in this case, it is impossible
to explicitly
append end group #E to the polymer fragment, because
different members of the ensemble of molecules represented by the
stochastic fragment have different numbers of unclosed bonds.
Note that, in this case, it is impossible
to explicitly
append end group #E to the polymer fragment, because
different members of the ensemble of molecules represented by the
stochastic fragment have different numbers of unclosed bonds.
2.6. Macrocycles
For macrocycles that
are well-defined, such as the cycle structures in ring polymers, the
macrocycles are encoded using the usual syntax for describing cycles
in SMILES. To illustrate this, consider a polystyrene ring synthesized
through alkyne azide click chemistry, as illustrated in Figure 5b. In this case, since rings
and cycles within repeating units do not extend beyond a single repeating
unit, the macrocycle associated with the ring polymer can be treated
with the usual SMILES ring closure syntax. The integer identifier
that was used within the repeating units for ring closure is considered
to be independent of any ring closure ID that was used in other parts
of the BigSMILES string. Therefore, the BigSMILES representation for
this polymer reads where the ring closure for the macrocycle is selected to
be between the sulfur atom and its neighboring atom. Meanwhile, the
ring closure denoted by 2 and 3 describes the ring closure in the
phenyl group and the ring with nitrogen atoms. It should be emphasized
that, similar to the ID used in bonding descriptors, the scope for
ring indices within a stochastic object is local
to the object, and independent of other ring indices not within the
stochastic object. In principle, other well-defined, nonstochastic
cycles can be encoded in a similar manner. For example, a ring polymer
synthesized with ring expansion metathesis polymerization (REMP) developed
by Grubbs and co-workers33 can also be
encoded with similar syntax, as illustrated in Figure 5c.
where the ring closure for the macrocycle is selected to
be between the sulfur atom and its neighboring atom. Meanwhile, the
ring closure denoted by 2 and 3 describes the ring closure in the
phenyl group and the ring with nitrogen atoms. It should be emphasized
that, similar to the ID used in bonding descriptors, the scope for
ring indices within a stochastic object is local
to the object, and independent of other ring indices not within the
stochastic object. In principle, other well-defined, nonstochastic
cycles can be encoded in a similar manner. For example, a ring polymer
synthesized with ring expansion metathesis polymerization (REMP) developed
by Grubbs and co-workers33 can also be
encoded with similar syntax, as illustrated in Figure 5c.
On the other hand, randomly formed cycles, such as the random loops in polymer networks, cannot be explicitly enumerated because each cycle requires indexing in the SMILES language. While the examples shown in Figure S2b–d (p S11, Section SIV in the SI) do not explicitly present the possibility of macrocycle formation, the rules of connectivity implicitly allow it, and enumeration of molecular states represented by the BigSMILES structure according to algorithms for generating gel connectivity, such as the algorithms adopted by Stepto and co-workers,34 Eichinger and co-workers,35 or Olsen and co-workers,36,37 will include the formation of these cyclic structures. Examples of BigSMILES strings for such structures are included in Section SIV (pp S10–S11) of the Supporting Information.
2.7. Ladder Polymers and Repeating Units with Multiatom Connections
The syntax up to this point assumes that neighboring repeating units are always connected through a single pair of atoms. However, for some materials, such as ladder polymers, this condition does not hold. To represent ladder polymers or other polymers with multiple connections between a single monomer pair, the bonding descriptors are nested by the following syntax:
![]()
The outer layer (everything except the part bracketed by “[...]”) encodes the connectivity between the repeating units with the same syntax as detailed in previous sections. Atoms on a repeating unit connecting to the same neighboring repeating units are indicated by an identical outer layer bond type, bond order b and bond ID n. For detailed examples of the use of nested bonding descriptors, please refer to p S14, Section SV in the Supporting Information.
3. Discussion
BigSMILES provides a well-defined, compact, and machine-friendly extension to the SMILES language that allows stochastic polymer structures to be represented. In this stochastic sense, a polymeric material is actually a set of molecules which may be conceptualized as an ensemble of different chemical states (defined by the bonding pattern of atoms), each with a probability of occurrence within the set of molecules that represents the material. BigSMILES enables, in a compact form, the ensemble of different chemical states to be represented; however, it does not provide information on the probability of observing any given chemical state. This is conceptually similar to the chemical structure of a polymer, which does not specify, for example, the molar mass distribution.
In principle, information about the probability of observing each molecular configuration within the ensemble can be quantified by measurement of physiochemical properties, such as the molar mass distribution, tacticity, or monomer reactivity ratios and feed ratios. However, developing an identifier notation by using a fixed set of property descriptors is challenging in practice. In most practical settings, only a few of the chemical structural features and properties of the macromolecules are characterized experimentally. Furthermore, the literature lacks consensus on how to treat this problem: researchers typically do not measure the same data using constant methods for each polymer, and data required to fully define molecular probabilities is usually missing. In some cases, measurements may not even be possible. This means that any form of encoding that relies on describing the macromolecules using a predefined set of properties will not meet the needs of the macromolecular community, nor will it be universally accepted. There are also substantial issues with data uncertainty and disagreements about evidence that have the potential to cause controversy. Therefore, to make the representation general and universally applicable, a syntax is developed that clearly separates the definition of the ensemble of molecular states accessible in a polymerization, a relatively noncontroversial topic, from the probability of achieving a given molecular state, a topic around which there is much greater debate and uncertainty of measurement. This is analogous to defining an ensemble of states in statistical mechanics without assigning the Boltzmann weights. While both are important for property calculation, by separating the two tasks it is possible to provide concrete molecular identifiers. Alternately, the demarcation of stochastic objects with curly brackets could enable additional specifications to be included in the list of elements beyond repeat units and end groups, providing an additional forum for the specification of certain additional chemical properties.
In the current form, a single polymer can be represented by multiple distinct yet equally valid BigSMILES representations. For practical purposes, canonicalization of BigSMILES to provide a unique representation for each distinct polymer would be essential for the application of BigSMILES to polymer informatics. Software packages to accompany BigSMILES are also of prime importance for practical purposes because they would serve both as a standard representation generator and a tool that could help eliminate human errors. The developments of both the canonicalization scheme and the supporting software are currently in progress and will be reported in the near future. In its current form, BigSMILES can still be used as structural identifiers in applications such as a data entry identifier in polymer databases. To demonstrate its general applicability, the entries of a well-known data set38 of glass transition data are converted into BigSMILES representations (cf. Section SVIII, pp S19–S44, in the Supporting Information). In addition to being used as identifiers, BigSMILES representations are designed in a way in which it can also be used as the basis of a chemical fingerprint generator. By considering pairs or triplets of repeating units and higher-order structures, chemical motifs with different levels of complexity and detail can be easily generated with the representation. These motifs can be used in cheminformatics applications to construct feature vectors that are fed into supervised learning models for property predictions. Furthermore, these generated motifs can also be used in chemical fragment search or chemical structure search. Finally, the structures of BigSMILES representations also allow generic chemical pattern searches. Queries such as “find polymers that are linear”, “find linear copolymers that have two components”, or “find branched polymers with trifunctional junctions” can be straightforwardly processed with regular expression or other pattern matching languages. These aforementioned features, including the generator for chemical fingerprints, chemical fragment search, and generic structural feature queries, will be implemented and demonstrated in future work. This capability enables access and searching of polymer materials from multiple levels of abstraction, which we believe will be highly convenient for the community.
4. Summary
In this work, a new text-based structural representation system designed to accommodate the stochastic nature of polymers is proposed. By adding a novel stochastic object to the widely used simplified molecular-input line-entry system, the features of SMILES can now be applied to polymers through BigSMILES. As the new representation system adds only a few elementary rules to the original syntax of SMILES while maintaining full compatibility with SMILES, most of the advantages of SMILES, including memory compactness, machine friendliness, and wide applicability, are retained in BigSMILES. Therefore, BigSMILES representations are excellent candidates for indexing identifiers in a polymer database system, as well as structural descriptors that could be used to search for polymer materials. Furthermore, as the chemical spaces represented by the BigSMILES strings can be straightforwardly probed with iterative generation of molecular fragments of varying sizes, BigSMILES representations can be readily used to automatically extract chemical subgraphs and generate molecular fingerprints. This feature can provide a convenient foundation for the generation of data sets that could be used along with machine learning models to fuel data-driven research. Ultimately, BigSMILES benefits the polymer community and increases cohesion between studies by providing a common language that is more effective and suitable for polymers.
Acknowledgments
This work was funded by the Center for the Chemistry of Molecularly Optimized Networks, a National Science Foundation (NSF) Center for Chemical Innovation (CHE-1832256). H.M. was supported by Furukawa Electric Co. Ltd. The authors would like to thank Ísis Biembengut (Braskem) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00476.
Discussion on orientation of repeating units, list of common repeating units and their equivalent string replacement, representation for charged polymers and tacticity; common mistakes in encoding BigSMILES strings; examples of BigSMILES encodings, including branched polymers, polymer networks, ladder polymers, and other more complex polymers; and discussion on treating dynamic, topological, and physical bonds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- O’Boyle N. M. Towards a Universal SMILES Representation-A Standard Method to Generate Canonical SMILES Based on the InChI. J. Cheminf. 2012, 4 (1), 22. 10.1186/1758-2946-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. Machine Learning in Computer-Aided Synthesis Planning. Acc. Chem. Res. 2018, 51 (5), 1281–1289. 10.1021/acs.accounts.8b00087. [DOI] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4 (2), 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer S.; DeRaedt L.; Helma C. In Molecular Feature Mining in HIV Data, Proceedings of the seventh ACM SIGKDD international conference on Knowledge discovery and data mining; 2001; pp 136–143.
- Weininger D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28 (1), 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Weininger D.; Weininger A.; Weininger J. L.SMILES. 2. Algorithm for Generation of Unique SMILES Notation. J. Chem. Inf. Model. 1989, 29 (2), 97–101. 10.1021/ci00062a008. [DOI] [Google Scholar]
- Ash S.; Cline M. A.; Homer R. W.; Hurst T.; Smith G. B. SYBYL Line Notation (SLN): A Versatile Language for Chemical Structure Representation. J. Chem. Inf. Comput. Sci. 1997, 37 (1), 71–79. 10.1021/ci960109j. [DOI] [Google Scholar]
- Vollmer J. J. Wiswesser Line Notation: An Introduction. J. Chem. Educ. 1983, 60 (3), 192. 10.1021/ed060p192. [DOI] [Google Scholar]
- Rohbeck H.-G.Representation of Structure Description Arranged Linearly. In Software Development in Chemistry 5; Springer: Berlin, Heidelberg, 1991; pp 49–58. [Google Scholar]
- Gakh A. A.; Burnett M. N. Modular Chemical Descriptor Language (MCDL): Composition, Connectivity, and Supplementary Modules. J. Chem. Inf. Comput. Sci. 2001, 41 (6), 1494–1499. 10.1021/ci000108y. [DOI] [PubMed] [Google Scholar]
- Heller S. R.; McNaught A.; Pletnev I.; Stein S.; Tchekhovskoi D. InChI, the IUPAC International Chemical Identifier. J. Cheminf. 2015, 7 (1), 23. 10.1186/s13321-015-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3 (1), 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D.-S.; Zhao J.-C.; Yang Y.-N.; Zhao C.-X.; Yan J.; Liu S.; Hu Q.-N.; Xu Q.-S.; Liang Y.-Z. In Silico Toxicity Prediction by Support Vector Machine and SMILES Representation-Based String Kernel. SAR QSAR Environ. Res. 2012, 23 (1–2), 141–153. 10.1080/1062936X.2011.645874. [DOI] [PubMed] [Google Scholar]
- Napolitano F.; Zhao Y.; Moreira V. M.; Tagliaferri R.; Kere J.; D’Amato M.; Greco D. Drug Repositioning: A Machine-Learning Approach through Data Integration. J. Cheminf. 2013, 5 (1), 30. 10.1186/1758-2946-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montavon G.; Rupp M.; Gobre V.; Vazquez-Mayagoitia A.; Hansen K.; Tkatchenko A.; Müller K.-R.; VonLilienfeld O. A. Machine Learning of Molecular Electronic Properties in Chemical Compound Space. New J. Phys. 2013, 15 (9), 95003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9 (2), 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Jin W.; Rogers L.; Jamison T. F.; Jaakkola T. S.; Green W. H.; Barzilay R.; Jensen K. F. A Graph-Convolutional Neural Network Model for the Prediction of Chemical Reactivity. Chem. Sci. 2019, 10 (2), 370–377. 10.1039/C8SC04228D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Struble T. J.; Coley C. W.; Wang Y.; Green W. H.; Jensen K. F. Using Machine Learning To Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4 (11), 1465–1476. 10.1021/acscentsci.8b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audus D. J.; dePablo J. J. Polymer Informatics: Opportunities and Challenges. ACS Macro Lett. 2017, 6 (10), 1078–1082. 10.1021/acsmacrolett.7b00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Chandrasekaran A.; Huan T. D.; Das D.; Ramprasad R. Polymer Genome: A Data-Powered Polymer Informatics Platform for Property Predictions. J. Phys. Chem. C 2018, 122 (31), 17575–17585. 10.1021/acs.jpcc.8b02913. [DOI] [Google Scholar]
- Huan T. D.; Mannodi-Kanakkithodi A.; Ramprasad R. Accelerated Materials Property Predictions and Design Using Motif-Based Fingerprints. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92 (1), 14106. 10.1103/PhysRevB.92.014106. [DOI] [Google Scholar]
- Mannodi-Kanakkithodi A.; Pilania G.; Huan T. D.; Lookman T.; Ramprasad R. Machine Learning Strategy for Accelerated Design of Polymer Dielectrics. Sci. Rep. 2016, 6, 20952. 10.1038/srep20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa S.; Al-Naamani E.; Saeki A. Computer-Aided Screening of Conjugated Polymers for Organic Solar Cell: Classification by Random Forest. J. Phys. Chem. Lett. 2018, 9 (10), 2639–2646. 10.1021/acs.jpclett.8b00635. [DOI] [PubMed] [Google Scholar]
- Cravero F.; Schustik S.; Martínez M. J.; Barranco C. D.; Díaz M. F.; Ponzoni I. In Feature Selection and Polydispersity Characterization for QSPR Modelling: Predicting a Tensile Property, International Conference on Practical Applications of Computational Biology & Bioinformatics; 2018; pp 43–51.
- Tchoua R. B.; Chard K.; Audus D. J.; Ward L. T.; Lequieu J.; DePablo J. J.; Foster I. T. In Towards a Hybrid Human-Computer Scientific Information Extraction Pipeline, , 2017 IEEE 13th International Conference on e-Science; 2017; pp 109–118.
- Pilania G.; Wang C.; Jiang X.; Rajasekaran S.; Ramprasad R. Accelerating Materials Property Predictions Using Machine Learning. Sci. Rep. 2013, 3, 2810. 10.1038/srep02810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Wang C.; Lorenzini R. G.; Ma R.; Zhu Q.; Sinkovits D. W.; Pilania G.; Oganov A. R.; Kumar S.; Sotzing G. A.; et al. Rational Design of All Organic Polymer Dielectrics. Nat. Commun. 2014, 5, 4845. 10.1038/ncomms5845. [DOI] [PubMed] [Google Scholar]
- Peerless J. S.; Milliken N. J. B.; Oweida T. J.; Manning M. D.; Yingling Y. G. Soft Matter Informatics: Current Progress and Challenges. Adv. Theory Simulations 2019, 2, 1800129. 10.1002/adts.201800129. [DOI] [Google Scholar]
- Zhang T.; Li H.; Xi H.; Stanton R. V.; Rotstein S. H.HELM: A Hierarchical Notation Language for Complex Biomolecule Structure Representation. J. Chem. Inf. Model. 2012, 52 (10), 2796–2806. 10.1021/ci3001925. [DOI] [PubMed] [Google Scholar]
- Drefahl A. CurlySMILES: A Chemical Language to Customize and Annotate Encodings of Molecular and Nanodevice Structures. J. Cheminf. 2011, 3 (1), 1. 10.1186/1758-2946-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H.; Tanaka H.; Yamasaki K.; Hashimoto T. Bicontinuous Microdomain Morphology of Block Copolymers. 1. Tetrapod-Network Structure of Polystyrene-Polyisoprene Diblock Polymers. Macromolecules 1987, 20 (7), 1651–1662. 10.1021/ma00173a036. [DOI] [Google Scholar]
- Odian G.Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, 2004. [Google Scholar]
- Bielawski C. W.; Benitez D.; Grubbs R. H. An “Endless” Route to Cyclic Polymers. Science 2002, 297 (5589), 2041–2044. 10.1126/science.1075401. [DOI] [PubMed] [Google Scholar]
- Rolfes H.; Stepto R. F. T. A Development of Ahmad-Stepto Gelation Theory. Makromol. Chem., Macromol. Symp. 1993, 76, 1–12. 10.1002/masy.19930760103. [DOI] [Google Scholar]
- Leung Y.-K.; Eichinger B. E. Computer Simulation of End-Linked Elastomers. I. Trifunctional Networks Cured in the Bulk. J. Chem. Phys. 1984, 80 (8), 3877–3884. 10.1063/1.447169. [DOI] [Google Scholar]
- Wang R.; Alexander-Katz A.; Johnson J. A.; Olsen B. D. Universal Cyclic Topology in Polymer Networks. Phys. Rev. Lett. 2016, 116 (18), 188302. 10.1103/PhysRevLett.116.188302. [DOI] [PubMed] [Google Scholar]
- Lin T.-S.; Wang R.; Johnson J. A.; Olsen B. D. Topological Structure of Networks Formed from Symmetric Four-Arm Precursors. Macromolecules 2018, 51 (3), 1224–1231. 10.1021/acs.macromol.7b01829. [DOI] [Google Scholar]
- Bicerano J.Prediction of Polymer Properties; cRc Press: Boca Raton, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.