

Abstract
Background:
It has been reported that smoking is one of the strongest positive risk factors for abdominal aortic aneurysms (AAAs). Although many studies have been directed to decipher the effect of smoking on AAA, its effect on macrophage activation has not yet been explored.
Objectives:
We have reported the importance of osteoclastogenesis (OCG) in aneurysm formation. Therefore, we examined the effect of cigarette smoking on OCG and arterial aneurysmal formation by using cigarette smoke extract (CSE) in this study.
Methods:
Macrophage cell lines were stimulated with CSE, and their activation and differentiation were examined in vitro. Since macrophages activated through the OCG pathway are identified by tartrate-resistant acid phosphatase (TRAP) expression, these cells are referred to as TRAP-positive macrophages (TPMs) in this study. We also applied CSE-contained PBS in the calcium chloride-induced mouse carotid aneurysm model in vivo.
Results:
Macrophages stimulated with CSE expressed significantly higher levels of nuclear factor of activated T-cells cytoplasmic 1 (NFATcl), TRAP, cathepsin K, matrix metalloproteinase-9 and membrane-type metalloproteinase (MT1-MMP). CSE-treated mouse aneurysms showed increased aneurysm size with increased TPM infiltration and protease expression compared to non-CSE-treated mouse aneurysms.
Conclusions:
These results suggest that CSE intensifies OCG in macrophages and promotes arterial aneurysmal progression.
Keywords: Aneurysm, Cigarette smoke extract, Tartrate-resistant acid phosphatase, Tartrate-resistant acid phosphatase-positive macrophage, Osteoclastogenesis
Introduction
Abdominal aortic aneurysm (AAA) is one of the most common aneurysmal diseases, and a Swedish study showed that the prevalence of AAAs was 2.2% [1]. This ratio can be lowered by the modification of several risk factors for AAA [2]. There is a great need for noninvasive therapeutic strategies for AAA since invasive surgical repair, by open or endovascular surgery, is still the gold standard for AAA therapy [3, 4]. Therefore, it is important to understand the pathophysiology of AAA to elucidate potential therapeutic targets. Aneurysmal degeneration of the aortic wall is caused by the degradation of elastin [3]. One of the known pathogenic features underlying this process is the upregulation of proteases, mainly from macrophages [4].
Cigarette smoking is identified as a strong positive risk factor for AAA [5]. Many studies have been directed at understanding the effect of cigarette smoke on the progression of arterial plaque and calcification typically found in coronary artery disease and peripheral artery disease patients [6–8]. Previous studies have shown that cigarette smoke extract (CSE) promotes the activation of macrophages through several kinds of pathways [9, 10]. Previously, studies demonstrated the capacity of whole cigarette smoke, CSE, or components of cigarette smoke, such as nicotine and 3,4-benzopryene, to stimulate aneurysm formation [11–13]. Therefore, several components of cigarette smoke alone, or in combination could affect the initiation of AAA. Our group has focused on the dysregulation of proteases in AAA through osteoclastogenesis (OCG) in activation of macrophages [14–17]. During OCG, receptor activator of NF-κB (RANK) ligand (RANKL) can activate macrophages through binding to its receptor, RANK, on the surface of macrophages. This RANKL-RANK complex subsequently accelerates down-stream signaling pathways, which lead to the expression of OCG-related genes, such as tartrate-resistant acid phosphatase (TRAP), matrix metalloproteinase-9 (MMP-9), and cathepsin K [18, 19]. TRAP expression is the main characteristic of OCG, therefore, we refer to these activated macrophages as TRAP-positive macrophages (TPMs) in this article.
To evaluate the effect of CSE on OCG and arterial aneurysmal progression, our current study tests the effect of CSE on macrophages and their protease expression in vitro. In addition, we applied CSE to the mouse calcium chloride (CaCl2)-induced carotid aneurysm model and examined its effect on the progression of arterial aneurysms and OCG in vivo.
Materials and Methods
Cell Culture and Treatments
A mouse macrophage cell line (RAW 264.7) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM containing 10% FBS, 100 IU/ mL penicillin, and 100 μg/mL streptomycin. For macrophage activation, macrophage cells were maintained in MEMα supplemented with 10% charcoal-stripped FBS, penicillin and streptomycin, and stimulated with or without CSE. To induce the OCG, we treated macrophages with 50 ng/mL recombinant murine soluble RANKL (sRANKL) (Peprotech, Rocky Hill, NJ, USA). Furthermore, 100 ng/mL of anti-mouse RANKL neutralizing monoclonal antibody (Oriental Yeast Co., Tokyo, Japan) was added to neutralize the effect of sRANKL.
Methylthiazol Tetrazolium Assay
RAW 264.7 cells were seeded in 96-well plates (10,000 cells per well) 1 day before treatment and were cultured. Cells were then treated with 1–10% CSE or untreated for 24 h. After 24 h, cell viability was analyzed with the methylthiazol tetrazolium (MTT) cell proliferation assay kit (Cayman chemical, Ann Arbor, MI, USA). Briefly, 10 μg of MTT reagent was added to each well, mixed gently, and allowed to incubate for 4 h at 37 ° C in a CO2 incubator. Next, 100 μL of dimethylsulfoxide (Sigma-Aldrich, St. Louis, MO, USA) was added to each well, and the absorbance was read at 570 nm using a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Preparation of CSE and Treatment
CSE protocol was prepared using a modification of a previous report [20]. One full-strength Marlboro cigarette (Philip Morris USA, Richmond, VA, USA) with a filter was directly connected to one end of a tube, and the other end of the tube was emerged in 10 mL of PBS in a vacuum glass vessel. The cigarette was ignited, and the smoke allowed to permeate the PBS by application of a vacuum to the vessel. The CSE-PBS solution was filtered through a 0.22-μm filter (Millipore, Bedford, MA, USA). The CSE was prepared immediately before each experiment. An optical density was read at 320 nm, and 1.0 of optical density was defined as 100% CSE. Moreover, a plot of the optical density versus CSE was examined and shown to be linear. We added the CSE solution to make the various concentrations of CSE in culture medium. We also added PBS without CSE to the culture medium of cells as control.
Western Blotting
Protein extraction was performed at 0–4 ° C. Protein from cultured cells was extracted with a radioimmunoprecipitation assay buffer with protease inhibitor cocktail (Cell Signaling Technology, Danvers, MA, USA). Twenty-microgram samples were separated on 8 or 12% polyacrylamide gels in Laemmli buffer and transferred electrophoretically onto polyvinylidene difluoride membranes. Primary antibodies used for Western blotting included mouse NFATc1 antibody (MA3–024; Thermo Fisher Scientific, Rockford, IL, USA), rabbit TRAP antibody (ab191406; Abcam, Cambridge, UK), mouse cathepsin K antibody (sc-48353; Santa Cruz Biotechnology), rabbit MMP-9 antibody (sc-10737; Santa Cruz Biotechnology), rabbit MT1-MMP antibody (#13130; Cell Signaling Technology), and mouse a-tubulin antibody (sc-23948; Santa Cruz Biotechnology). These primary antibodies were detected by horseradish peroxidase-conjugated secondary antibodies and visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Relative protein expression levels for Western blotting were quantified and compared between the groups.
Quantitative Reverse Transcription Polymerase Chain Reaction
Total RNA was extracted from the RAW 264.7 cells using PureLink RNA Mini Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Concentrations of RNA were determined by reading the absorbance at 260 nm. Reverse transcription for RNA to make cDNA was performed using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). Quantitative realtime PCR was performed with SYBR Green dye using the 7500 Fast Real-Time PCR instrument (ABI, Foster City, CA, USA). Sequences of primers used were as follows: TRAP, F: 5’-TCC TGG CTC AAA AAG CAG TT-3’, and R: 5’-ACA TAG CCC ACA CCG TTC TC-3’; MMP-9, F: 5’-CAT TCG CGT GGA TAA GGA GT-3’, and R: 5’-GTT CAC CTC ATG GTC CAC CT-3’; MT1-MMP, F: 5’-AGA GAG TCT GGT GGC TTG GA-3’, and R: 5’-CTT CAG GTC CTG CTG TCT CC-3’; NFATcl, F: 5’-TGG CTA CCG ACA TGT GTT GT-3’, and R: 5’-GAC CAG GGG AGC TAT GAA CA-3’; GAPDH, F: 5’-AAC TTT GGC ATT GTG GAA GG-3’, and R: 5’-ACA CAT TGG GGG TAG GAA CA-3’. The expression level for each gene was normalized to the GAPDH expression level in the same sample and analyzed by the 2−ΔΔCt method.
Flow Cytometry
For in vitro experiments, RAW 264.7 cells were cultured as previously described, and 2.5 × 105 cells were stained for flow cytometric analysis. For in vivo experiments, mouse carotid arteries were harvested and incubated at 37 ° C with shaking for 1 h in digestion buffer containing 450 U/mL collagenase I (Sigma-Aldrich), 125 U/mL collagenase XI (Sigma-Aldrich), 60 U/ mL hyaluronidase (Worthington Biochemical, Lakewood, NJ, USA), and 60 U/mL deoxyribonuclease I (Worthington), as previously described [21]. The digested tissue was then dissociated to obtain a single-cell suspension by passage through a 70 μm cell strainer, and 2.5 × 105 cells were stained for flow cytometric analysis as follows. Cells were washed with FACS wash buffer (PBS, 3% FBS) and simultaneously stained for surface expression of CD11b and live/dead stained with anti-CD11b-BV711 antibody (563168; BD Biosciences, Franklin Lakes, NJ, USA) and Ghost 510 (Tonobo, San Diego, CA, USA), respectively, for 30 min at 4° C. Cells were then washed and resuspended in fixation buffer (Invitrogen, San Diego, CA, USA), and allowed to fix for 15 min at room temperature. Cells were washed in permeabilization buffer (Invitrogen), and resuspended in permeabilization buffer containing the following primary antibodies: anti-TRAP-Alexa Fluor 488 (ab216934; Abcam), anti-cathepsinK-Alexa Fluor 647 (sc-48353; Santa Cruz Biotechnology), and anti-MMP-9-PE (sc-13520; Santa Cruz Biotechnology). Primary antibodies and cells were incubated for 30 min at room temperature. Following incubation, the cells were washed with permeabilization buffer and resuspended in FACS wash buffer. Cells were analyzed on an Attune NxT Flow Cytometer (Invitrogen). Fluorescence minus one controls were prepared and analyzed for each fluorochrome used.
Osteoclast Formation Assay
To evaluate osteoclast formation, we performed TRAP staining. Briefly, 3,000 RAW 264.7 macrophages per well were seeded in 96-well plates, and these macrophages were cultured in MEMα with 10% cFBS. CSE or 50 ng/mL of sRANKL were added to induce OCG for 5 days. Then, cells were stained with a commercialized TRAP staining kit (Sigma-Aldrich) according to the manufacturer’s instruction. We defined red and multinucleated (>3 nuclei) cells as TRAP-positive cells and counted the number of osteoclasts in each well.
The Mouse Arterial Aneurysmal Model and Treatments
Ten-week-old male C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The procedures for creating our modified CaCl2-induced mouse model of arterial aneurysm were previously described [22]. Briefly, 0.5 M CaCl2-soaked gauze was applied perivascularly for 20 min on the carotid artery as indicated. The gauze was replaced with another PBS-soaked or CSE-contained PBS-soaked gauze for 10 min, and the incised area was sutured. The mice were divided into two groups; CaCl2 and PBS-soaked model (n = 10), and CaCl2 and CSE-contained PBS-soaked model (n = 10). The mice were sacrificed 7 days after surgery, and the arteries were measured with an electronic digital caliper (VWR International, West Chester, PA, USA) and collected for histological examinations after fixing by perfusion with 4% PFA. For the flow cytometric analysis, the mice were also divided into two groups; CaCl2 and PBS-soaked model (n = 5), and CaCl2 and CSE-contained PBS-soaked model (n = 5). To obtain live cells for flow cytometry, the arteries were not fixed, but perfused with DMEM. All animal procedures were conducted in accordance with experimental protocols that were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin, Madison (Protocol M005383).
Statistical Analysis
Data are reported as the means ± standard deviation. Statistical analysis was performed with the GraphPad Prism program, version 7.0 (GraphPad Software Inc., San Diego, CA, USA). Differences between the groups were compared by the Student’s t test or one-way analysis of variance (one-way ANOVA) with repeated measures followed by Turkey’s comparison test for multiple comparisons. p values less than 0.05 were accepted as statistically significant.
Results
CSE Affects Cell Viability
Examination of macrophage cell viability following exposure to various concentrations of CSE showed that CSE was toxic to cells in a dose-dependent manner as evaluated by the MTT assay (Fig. 1). While most of the macrophages (84.7 ± 13.4%) survived in 2% CSE medium, only half of cells (55.1 ± 4.18%) survived in 3% of CSE medium. Therefore, we decided to stimulate RAW 264.7 cells using 2% CSE, unless otherwise noted.
Fig. 1.
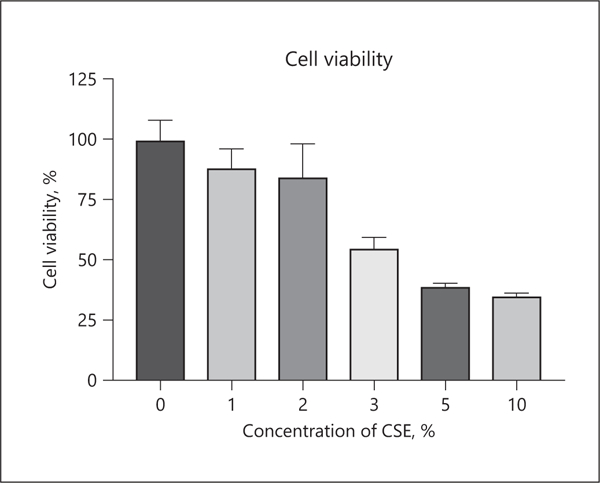
Effects of CSE on the cell viability of RAW 264.7 cells. RAW 264.7 cells were cultured in various concentrations of CSE for 24 h. To examine the cytotoxicity of CSE, an MTT assay was performed. Values are presented as mean ± SD for 4 replicates. CSE, cigarette smoke extract; SD, standard deviation.
CSE Treatment Induces an OCG-Associated Transcription Factor, with Increases in OCG-Associated Protein and mRNA Expression
One of the master transcription factors of macrophage activation is nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) [23]. There are other proteins involved in OCG, including TRAP, cathepsin K and MMP-9. Furthermore, it has been reported that membrane-type 1 MMP (MT1-MMP) plays one of the key roles to induce OCG [24], and infiltrating macrophages show higher expression of MT1-MMP [25]. Therefore, we examined the expression levels of these proteins after stimulating macrophages with 2% CSE. The protein expression level of NFATcl was significantly higher in the CSE-treated group than the control group (10.6 ± 1.75 vs. 1.00 ± 0.42, p < 0.01) (Fig. 2a). Furthermore, there was significantly higher TRAP expression in the CSE-treated group compared to control (1.97 ± 0.48 vs. 1.00 ± 0.11, p < 0.05) (Fig. 2b). The expressions of cathepsin K, MMP-9, and MT1-MMP were also significantly higher in the CSE-treated group compared to the control group (1.70 ± 0.28 vs. 1.00 ± 0.04, p < 0.05, 1.97 ± 0.48 vs. 1.00 ± 0.11, p < 0.01, and 5.95 ± 1.37 vs. 1.00 ± 0.34, p < 0.01, respectively) (Fig. 2c–e). The expression level of mRNA was significantly higher in the CSE-treated group (2.68 ± 0.35 vs. 1.00 ± 0.14, p < 0.01) (Fig. 2f). There were also statistically significant differences in the mRNA expression levels of MMP-9 and MT1-MMP between the control and CSE-treated groups (1.00 ± 0.10 vs. 11.9 ± 1.64, p < 0.01, and 1.00 ± 0.17 vs. 18.3 ± 2.93, p < 0.01, respectively) (Fig. 2g, h). These results showed that CSE treatment promotes OCG, which was demonstrated by increased expression of NFATc1, TRAP, cathepsin K, MMP-9, and MT1-MMP.
Fig. 2.
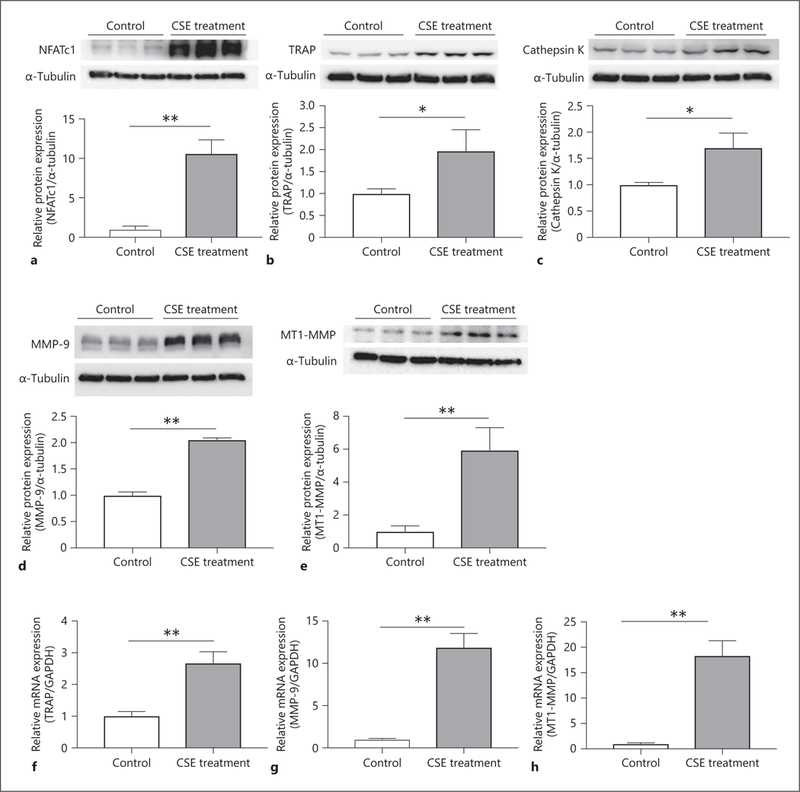
CSE stimulates the protein and mRNA expression correlated with macrophage activation. (a) RAW 264.7 cells were incubated in medium with or without 2% CSE for 2 days for Western blotting analysis. Relative protein expression level of NFATc1 in the CSE-treated group showed a statistically significant difference compared with the untreated group. Macrophage cells were cultured with or without 2% CSE to examine TRAP protein (b), cathepsin K protein (c), and MMP-9 protein (d) levels for 3 days, and MT1-MMP for 2 days (e) by Western blotting analysis. The mRNA expression of TRAP (f) and MMP-9 (g) were examined after being cultured in 2% CSE-contained medium for 3 days, and MT1-MMP for 2 days (h). Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. CSE, cigarette smoke extract; NFATc1, nuclear factor of activated T-cells cytoplasmic 1; TRAP, tartrate-resistant acid phosphatase; MMP-9, matrix metalloproteinase-9; MT1-MMP, membrane-type 1 matrix metalloproteinase; SD, standard deviation.
CSE Treatment Stimulates TPM Protease Expression
To delineate the protease expression of TPMs compared to non-TPMs, we performed flow cytometric analysis. CSE treatment induced a statistically significant increase in the percentage of TPMs compared with the control group in vitro (4.94 ± 2.26 vs. 0.27 ± 0.27%, p < 0.05) (Fig. 3a). Furthermore, the percentage of cathepsin K- and MMP-9-positive cells were higher in the CSE-treated group than controls in vitro (3.65 ± 1.70 vs. 0.24 ± 0.19%, p < 0.05 and 5.42 ± 1.78 vs. 0.54 ± 0.20%, p < 0.05, respectively) (Fig. 3b, c). Next, we compared the fluorescence intensity of cathepsin K and MMP-9 between TPMs (CD11b+, TRAP+) and non-TPMs (CD11b+, TRAP) in the CSE-treated group. Even though there was no significant difference in the median fluorescence intensity (MFI) of cathepsin K between non-TPMs and TPMs (4,817 ± 1,092 vs. 8,534 ± 2,412, p = 0.07), we found the trend that TPMs showed a higher level of expression of cathepsin K than non-TPMs (Fig. 3d). Moreover, TPMs exhibited a higher MFI of MMP-9 compared to non-TPMs (3,450 ± 553.7 vs. 2,094 ± 449.4, p < 0.05) (Fig. 3e).
Fig. 3.
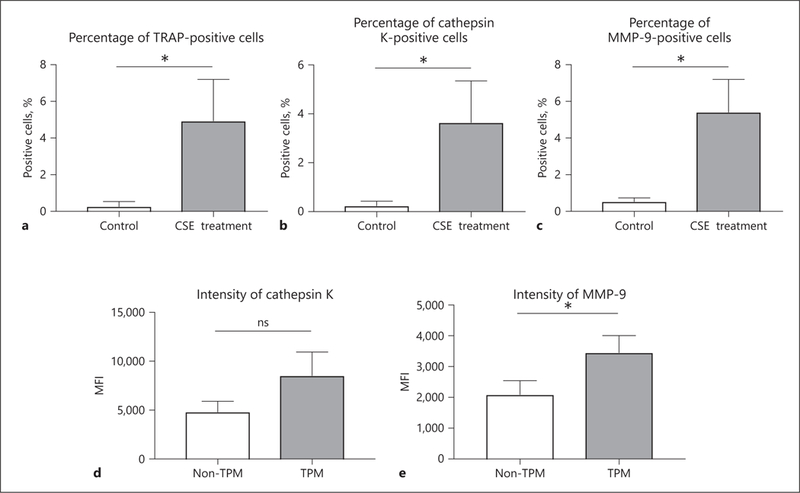
CSE treatment promotes the activation of macrophage cells. Flow cytometric analysis showed that the percentages of live cells positive for TRAP (a), cathepsin K (b), and MMP-9 (c) were higher in the CSE treatment group (cultured for 5 days) than in the nontreated group. In the CSE treatment group, TPM showed higher levels of expression of cathepsin K (d) and MMP-9 (e) compared to non-TPM. Values are presented as means ± SD for at least 3 replicates. * p < 0.05. ns, not significant; CSE, cigarette smoke extract; TRAP, tartrate-resistant acid phosphatase; MMP-9, matrix metalloproteinase-9; TPM, TRAP-positive macrophage; MFI, median fluorescence intensity; SD, standard deviation.
The Effect of Anti-RANKL Neutralizing Antibody on CSE-Treated TPM Activation
As mentioned above, classical OCG is induced through RANKL-RANK pathway. Therefore, macrophages were stimulated with CSE or sRANKL under the anti-RANKL neutralizing antibody. Even though the protein expression of NFATc1 in the sRANKL-treated group was significantly inhibited by neutralizing antibody (1.58 ± 0.27 vs. 19.5 ± 3.79, p < 0.01) (Fig. 4a), CSE-induced TPM activation was not inhibited by anti-RANKL antibody (CSE treatment group vs. anti-RANKL antibody treatment group, 14.1 ± 4.33 vs. 11.3 ± 2.41, p = 0.49) (Fig. 4b). The mRNA expression of NFATc1 in the anti-RANKL antibody treatment group was statistically lower compared to the sRANKL treatment group (5.95 ± 0.05 vs. 7.71 ± 1.07, p < 0.05) (Fig. 4c); however, the CSE-induced mRNA expression of NFATc1 was not attenuated by anti-RANKL antibody (CSE treatment group vs. anti-RANKL antibody treatment group, 2.51 ± 0.19 vs. 2.36 ± 1.07, p = 0.93) (Fig. 4d). Furthermore, the number of osteoclasts in the sRANKL treated group was statistically lowered by anti-RANKL neutralizing antibody (0.50 ± 0.55 vs. 140.0 ± 18.2, p < 0.01); however, the number of TPMs in the CSE treatment group did not change with neutralizing antibody (CSE treatment group vs. anti-RANKL antibody treatment group, 22.0 ± 7.38 vs. 21.0 ± 10.9, p = 0.99) (Fig. 4e). These results indicate that CSE-stimulated OCG is not dependent on the RANKL-RANK pathway. We have already reported that there could be a mechanism to induce OCG independently of the RANKL-RANK pathway [15]. Therefore, CSE might involve an alternative mechanism to induce OCG for macrophages.
Fig. 4.
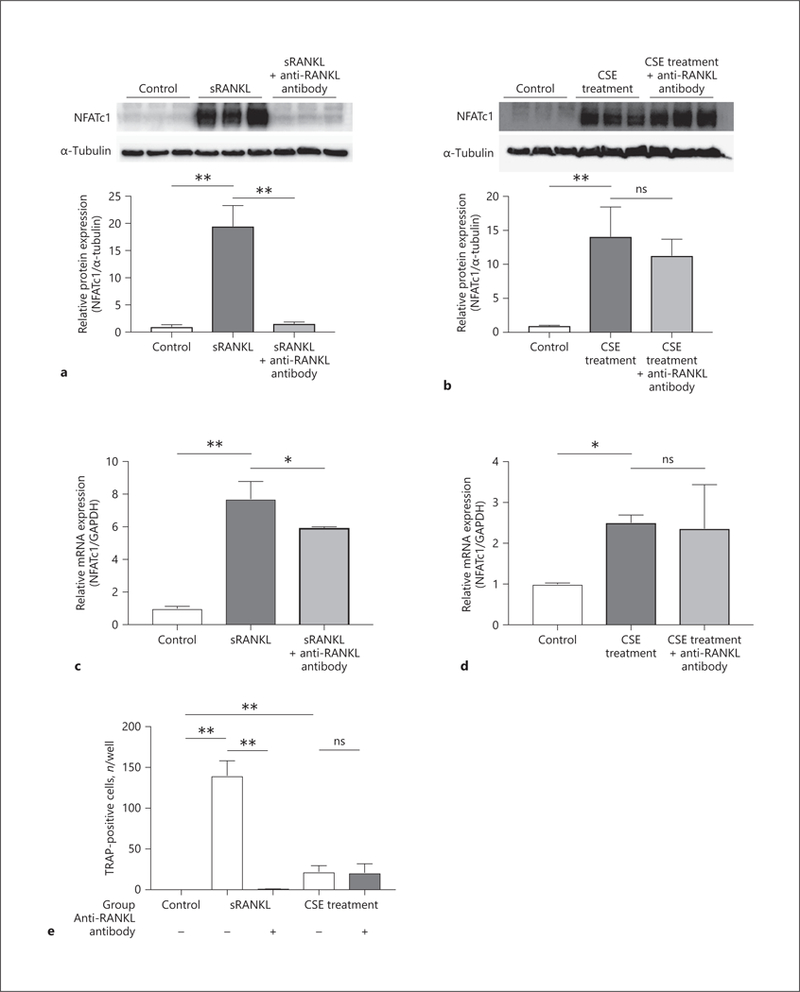
Effect of anti-RANKL neutralizing antibody on osteoclastogenic macrophage activation. To induce osteoclastogenesis, RAW 264.7 cells were incubated in medium with 50 ng/mL of sRANKL or 2% CSE for 2 days. Furthermore, 100 ng/mL of anti-mouse RANKL neutralizing monoclonal antibody was added to inhibit the RANKL-RANK pathway. By Western blotting analysis, the protein expression levels of NFATc1 were compared in the sRANKL treatment group (a) and CSE treatment group (b). NFATcl gene expression levels were evaluated in the sRANKL treatment group (c) and CSE treatment group (d). Macrophages were also cultured and stimulated by sRANKL or CSE with or without anti-RANKL antibody for 5 days, and the number of TRAP-positive macrophages was counted (e). Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; RANK, receptor activator of NF-κB; RANKL, receptor activator of NF-κB ligand; sRANKL, soluble RANKL; CSE, cigarette smoke extract; NFATc1, nuclear factor of activated T-cells cytoplasmic 1; TRAP, tartrate-resistant acid phosphatase; SD, standard deviation.
CSE Promotes Aneurysm Formation in the CaCl2-Induced Model
We examined the effect of CSE on carotid arterial aneurysmal formation using the mouse CaCl2-induced model [15]. In brief, we applied CaCl2 perivascularly for 20 min, followed by CSE-contained PBS, or PBS only as a control, perivascularly for 10 min. As shown in Figure 5a–c, CSE induced the progression of arterial aneurysm compared to control. The fold increase of aneurysm diameter was significantly higher in the CSE-treated group compared to the control group (2.23 ± 0.30 vs. 1.56 ± 0.13, p < 0.01).
Fig. 5.
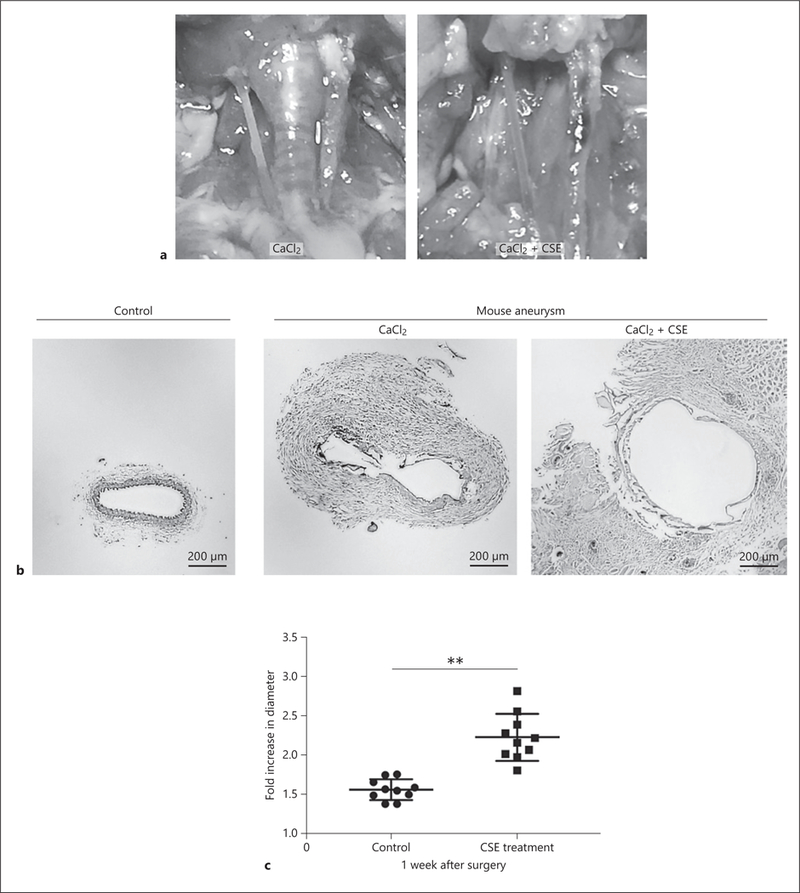
Mice were subjected to carotid aneurysm induction via the CaCl2 or CaCl2+CSE models. a Aneurysmal dilatation of left carotid arteries via CaCl2 or CaCl2+CSE induction. b Representative images of hematoxylin and eosin-stained control and aneurysmal mouse carotid sections. c The fold increase in diameter 1 week after surgery was higher in the CaCl2+CSE mouse aneurysmal model than the CaCl2 aneurysmal model. Values are presented as means ± SD for at least 3 replicates. ** p < 0.01. Scale bar, 200 μm (b). CSE, cigarette smoke extract; SD, standard deviation.
Addition of CSE to the CaCl2 Aneurysmal Model Induces Enhanced TPM Activation
To further investigate the promotive effect of CSE in aneurysm formation, we harvested aneurysmal tissues from mice, and used flow cytometric analysis. First, we investigated myeloid cell infiltration via the presence of CD11b-positive cells. There was no significant difference in the number of CD11b-positive cells between the control and CSE-treated groups (3.41 ± 1.37% vs. 5.61 ± 2.58%, p = 0.13) (Fig. 6a). This may indicate that CSE treatment does not increase the recruitment of myeloid cells, but functions to augment their activation, as there were more TRAP-positive cells in the CSE-treated group than the conventional CaCl2-induced model (3.30 ± 1.50% vs. 1.61 ± 0.53%, p < 0.05) (Fig. 6b). Furthermore, we analyzed the direct effect of CSE by treating carotids only with CSE (no CaCl2). Treatment with CSE alone was unable to increase the number of TRAP-positive cells compared to the CaCl2− and CaCl2+CSE-treated groups (Fig. 6b). Even though CSE alone was not enough to induce aneurysmal change and TPM activation, exposure to CSE after degenerative remodeling of arteries by CaCl2 treatment might promote the enhancement of aneurysmal change. In further examinations, we evaluated the expression of cathepsin K and MMP-9 in our treatment groups. The percentage of cathepsin K positive cells was also higher in the CSE-treated group compared to the control group (7.69 ± 1.64% vs. 3.87 ± 1.08%, p < 0.01) (Fig. 6c). Interestingly, the expression of MMP-9 was not different between the control and CSE-treated groups (11.1 ± 5.90% vs. 11.5 ± 4.59%, p = 0.92) (Fig. 6d). Furthermore, we compared the fluorescence intensity of cathepsin K and MMP-9 in TPMs between the control and CSE-treated groups. The CSE-treated group showed a higher MFI of cathepsin K and MMP-9 compared to the control group (7,513 ± 1,648 vs. 4,603 ± 902.3, p < 0.01, and 10,227 ± 2,484 vs. 5,863 ± 1,808, p < 0.05, respectively) (Fig. 6e, f).
Fig. 6.
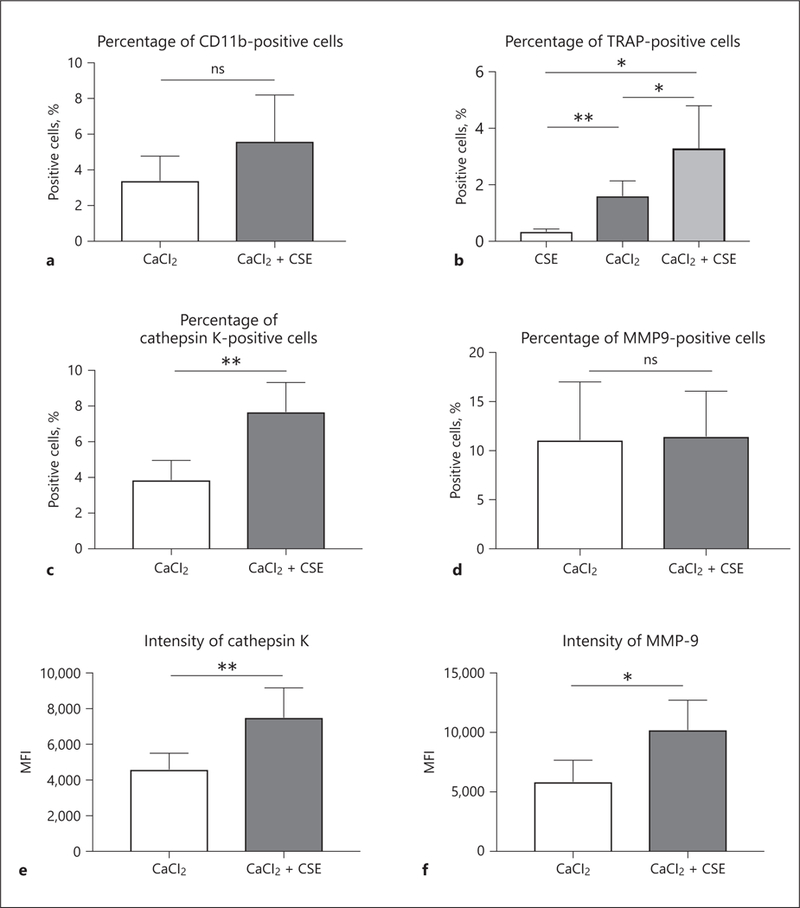
Dissociated cells from mouse aneurysmal samples collected 1 week after induction surgery were examined by flow cytometry. a To investigate myeloid cell infiltration, we used CD11b as surface marker. b To evaluate the effect of CSE, we applied CSE without CaCl2 to carotid arteries to compare TRAP-positive cells with CaCl2− and CaCl2+CSE-treated groups. In further analysis, the percentages of live cells positive for cathepsin K (c) were higher in the CaCl2+CSE group than the CaCl2 group. d On the other hand, there was no significant difference in the percentage of MMP-9-positive cells between the groups. Among the TPMs, the expression levels of cathepsin K (e) and MMP-9 (f) were higher in the CaCl2+CSE group than in the CaCl2 group. Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; CSE, cigarette smoke extract; TRAP, tartrate-resistant acid phosphatase; MMP-9, matrix metalloproteinase-9; MFI, median fluorescence intensity; SD, standard deviation.
Discussion
It has been shown that cigarette smoking is one of the strongest positive risk factors for arterial disease, including AAA [5]. Our results showed that CSE stimulated the activation of macrophages both in vitro and in vivo. We have shown that CSE stimulation induced OCG with up-regulation of NFATc1 and promoted expression of the OCG-related proteases TRAP, cathepsin K, and MMP-9.
Several reports have showed that CSE induces proinflammatory cytokines and proteases which lead to macrophage activation. Yang et al. [26] reported that CSE augments an inflammatory response via the NF-κB pathway. Chen et al. [27] also reported that macrophages cultured in CSE-supplemented medium increased the expression of monocyte chemotactic protenin-1, MMP-9, IL-8, and TNF-α at both protein and mRNA levels. These proinflammatory mediators and proteases are closely related to OCG [28]. Therefore, we hypothesized that CSE promotes OCG and the induction of arterial aneurysms, including AAA, through the activation of OCG-related proteases and proinflammatory mediators. Interestingly, Ghosh et al. [29] reported that the cell culture medium from CSE-treated aortic endothelial cells or aortic smooth muscle cells induced the production of MMP-9 in macrophages. However, this result did not demonstrate the direct effect of CSE on macrophages. Furthermore, TRAP and cathepsin K are more osteoclast-specific proteases than MMP-9. Therefore, we added CSE solution directly to macrophages and examined its effect on OCG. Our in vitro study proved that CSE treatment by itself was sufficient to induce OCG as indicated by the expression of the osteoclast-specific proteases TRAP and cathepsin K [27]. Furthermore, we showed that topical application of CSE enhanced aneurysmal progression in the mouse CaCl2 arterial aneurysm model. These results proved our hypothesis that CSE induces OCG with increased TPMs that produce higher levels of proteases, which are correlated with the induction of arterial aneurysms. This is the first study to prove the causal nexus of pathogenesis behind the effect of cigarette smoking on macrophage activation in arterial aneurysms.
Among the many proteases, MMP-9 has proven to be one of the most important in aneurysm formation and development [30]. Interestingly, our in vivo study did not show significant difference in the number of MMP-9 positive cells by CSE treatment. However, the MFI of MMP-9 in TPMs induced by CSE was higher compared to the control in vivo, and we observed the same result in vitro. The level of MFI also provided the expression levels of proteases, including MMP-9 [31–33]. Therefore, these results showed that CSE significantly increased the expression of MMP-9 in TPMs, which might enhance aneurysm formation compared to the non-CSE condition. Furthermore, CSE treatment induced a higher number of cathepsin K positive cells and higher MFI of cathepsin K in TPMs in vitro and in vivo. Cathepsin K is known to be one of the strongest proteases typically produced by osteoclasts. Of the many inhibitors of cathepsin K available, odanacatib was an attractive option for use in humans to treat osteoporosis. Odanacatib reduces the resorptive activity of osteoclasts without loss of function for bone remodeling [34]. Therefore, future studies to explore the possibility of targeting cathepsin K as a novel pharmacological therapy for arterial aneurysmal formation could be interesting.
There are some limitations of this study. First, we treated the macrophage cells with 2% CSE. There are some papers in which several concentrations of CSE were added as treatment [20, 35], however, it is unclear how CSE concentration relates to human exposure through cigarette smoking. Therefore, 2% CSE might not be the most appropriate concentration to approximate exposure through smoking. In the animal experiments, we used a modified CaCl2-induced carotid mouse model of aneurysm instead of the CaCl2-induced AAA mouse model in this study. We have already shown that the modified CaCl2-induced carotid mouse model could induce aneurysmal change in the carotid artery, and OCG [14, 15]. This modified CaCl2-induced carotid aneurysmal model is much easier and safer to induce than the CaCl2-induced AAA mouse model. The abdominal aorta is located on the inferior vena cava, which can make the common CaCl2-induced AAA mouse model more difficult to induce without injuries or experimental bias. Several authors have also previously attempted to apply this type of CaCl2-induced carotid arterial aneurysmal model to arterial aneurysmal models including AAA [36, 37]; however, there might be possible pathophysiological differences between abdominal and carotid aneurysmal models and it is a limitation of this study. CD11b is a surface marker that is not exclusive to macrophages, therefore, other cell types such as natural killer cells, neutrophils, granulocytes, and B cells might be included in the representation of CD11b+ cells in our in vivo flow cytometry experiments [38, 39]. However, CD11b is a reliable murine and human marker for cells of the myeloid lineage, which are the precursors of the osteoclast-like cells central to this study [40]. Furthermore, CD11b expression is found on both resident and inflammatory monocyte populations in mice and humans, therefore, CD11b represents a reliable marker of osteoclast precursors that may be derived from either tissue-resident or migratory monocyte populations [41]. Additionally, commonly used macrophage markers such as CD68 and F4/80 may not be ideal markers for our purposes due to demonstrations that their expression may inhibit OCG [42, 43]. We also rely on coexpression of CD11b and TRAP to identify TPMs in this study, as cells of the monocyte/macrophage lineage are the primary cell type known to express high levels of TRAP [44].
Conclusion
In this study, we showed that: (1) CSE treatment promotes TPM activation with higher levels of proteases; (2) CSE treatment induced enhanced aneurysm progression and increased TPM activation; (3) TPMs stimulated by CSE showed higher expression of cathepsin K and MMP-9. Further studies are necessary to elucidate the signaling pathways by which CSE induces OCG.
Acknowledgments
Funding Sources
This work was supported by Uehara Memorial Foundation Research Fellowship Award (Kimihiro Igari), and the Vascular Surgery Research Training Grant (T32HL110853) (Matthew J. Kelly).
Footnotes
Statement of Ethics
All animal experiments complied with the Animal Research: Reporting of in vivo Experiments (ARRIVE) guidelines, developed by the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs). All animal procedures were conducted in accordance with experimental protocols that were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin, Madison (Protocol M005383).
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Svensjö S, Björck M, Gürtelschmid M, Djavani Gidlund K, Hellberg A, Wanhainen A. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation. 2011. September;124(10):1118–23. [DOI] [PubMed] [Google Scholar]
- 2.Kent KC. Clinical practice. Abdominal aortic aneurysms. N Engl J Med. 2014. November;371(22): 2101–8. [DOI] [PubMed] [Google Scholar]
- 3.Takayama T, Yamanouchi D. Aneurysmal disease: the abdominal aorta [viii.]. Surg Clin North Am. 2013. August;93(4):877–91. [DOI] [PubMed] [Google Scholar]
- 4.Kurosawa K, Matsumura JS, Yamanouchi D. Current status of medical treatment for abdominal aortic aneurysm. Circ J. 2013;77(12): 2860–6. [DOI] [PubMed] [Google Scholar]
- 5.Kent KC, Zwolak RM, Egorova NN, Riles TS, Manganaro A, Moskowitz AJ, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg. 2010. September;52(3):539–48. [DOI] [PubMed] [Google Scholar]
- 6.Cheezum MK, Kim A, Bittencourt MS, Kassop D, Nissen A, Thomas DM, et al. Association of tobacco use and cessation with coronary atherosclerosis. Atherosclerosis. 2017. February;257:201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yankelevitz DF, Henschke CI, Yip R, Boffetta P, Shemesh J, Cham MD, et al. ; FAMRI-IEL-CAP Investigators. Second-hand tobacco smoke in never smokers is a significant risk factor for coronary artery calcification. JACC Cardiovasc Imaging. 2013. June;6(6):651–7. [DOI] [PubMed] [Google Scholar]
- 8.Szarejko-Paradowska A, Gluba-Brzózka A, Pietruszyński R, Rysz J. Assessment of the relationship between selected cardiovascular risk factors and the indices of intima-media thickness and coronary artery calcium score in various stages of chronic kidney disease. Int Urol Nephrol. 2015. December;47(12):2003–12. [DOI] [PubMed] [Google Scholar]
- 9.Yuan F, Fu X, Shi H, Chen G, Dong P, Zhang W. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PLoS One. 2014. September; 9(9):e107063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birrell MA, Wong S, Catley MC, Belvisi MG. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J Cell Physiol. 2008. January; 214(1):27–37. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, et al. Activation of AMP-activated protein kinase a2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012. June;18(6):902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji K, Zhang Y, Jiang F, Qian L, Guo H, Hu J, et al. Exploration of the mechanisms by which 3,4-benzopyrene promotes angiotensin II-induced abdominal aortic aneurysm formation in mice. J Vasc Surg. 2014. February;59(2):492–9. [DOI] [PubMed] [Google Scholar]
- 13.Norman PE, Curci JA. Understanding the effects of tobacco smoke on the pathogenesis of aortic aneurysm. Arterioscler Thromb Vasc Biol. 2013. July;33(7):1473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T, Takei Y, Yamanouchi D. Hyperglycemia suppresses calcium phosphate-induced aneurysm formation through inhibition of macrophage activation. J Am Heart Assoc. 2016. March;5(3):e003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takei Y, Tanaka T, Kent KC, Yamanouchi D. Osteoclastogenic differentiation of macrophages in the development of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2016. September;36(9): 1962–71. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T, Kelly M, Takei Y, Yamanouchi D. RANKL-mediated osteoclastogenic differentiation of macrophages in the abdominal aorta of angiotensin II-infused apolipoprotein E knockout mice. J Vasc Surg. 2018. December;68(6S): 48S–59S.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamanouchi D, Takei Y, Komori K. Balanced mineralization in the arterial system: possible role of osteoclastogenesis/osteoblastogenesis in abdominal aortic aneurysm and stenotic disease. Circ J. 2012;76(12):2732–7. [DOI] [PubMed] [Google Scholar]
- 18.Kwak HB, Jin HM, Ha H, Kang MJ, Lee SB, Kim HH, et al. Tumor necrosis factor-alpha induces differentiation of human peripheral blood mononuclear cells into osteoclasts through the induction of p21(WAF1/Cip1). Biochem Biophys Res Commun. 2005. May; 330(4):1080–6. [DOI] [PubMed] [Google Scholar]
- 19.Kanazawa K, Kudo A. TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J Bone Miner Res. 2005. May;20(5):840–7. [DOI] [PubMed] [Google Scholar]
- 20.Di YP, Zhao J, Harper R. Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J Biol Chem. 2012. August; 287(33):27948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006. May; 203(5):1273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanouchi D, Morgan S, Stair C, Seedial S, Lengfeld J, Kent KC, et al. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg. 2012. August;56(2):455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurihara C, Tanaka T, Yamanouchi D. Hyperglycemia attenuates receptor activator of NF-κB ligand-induced macrophage activation by suppressing insulin signaling. J Surg Res. 2017. June;214:168–75. [DOI] [PubMed] [Google Scholar]
- 24.Uemura T, Liu YK, Kuboki Y. Preliminary communication. MRNA expression of MT1-MMP, MMP-9, cathepsin K, and TRAP in highly enriched osteoclasts cultured on several matrix proteins and ivory surfaces. Biosci Biotechnol Biochem. 2000. August;64(8):1771–3. [DOI] [PubMed] [Google Scholar]
- 25.Xiong W, Knispel R, MacTaggart J, Greiner TC, Weiss SJ, Baxter BT. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J Biol Chem. 2009. January;284(3):1765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and post-translational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006. July;291(1):L46–57. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Guan XJ, Peng XH, Cui ZL, Luan CY, Guo XJ. Acetylation of lysine 9 on histone H3 is associated with increased pro-inflammatory cytokine release in a cigarette smoke-induced rat model through HDAC1 depression. Inflamm Res. 2015. July;64(7):513–26. [DOI] [PubMed] [Google Scholar]
- 28.Yavropoulou MP, Yovos JG. Osteoclastogenesis—current knowledge and future perspectives. J Musculoskelet Neuronal Interact. 2008. Jul-Sep;8(3):204–16. [PubMed] [Google Scholar]
- 29.Ghosh A, Pechota LV, Upchurch GR Jr, Eliason JL. Cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke: the effects on MMP2 and 9. Mol Cell Biochem. 2015. December; 410(1–2):75–84. [DOI] [PubMed] [Google Scholar]
- 30.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002. September; 110(5): 625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medeiros NI, Fares RC, Franco EP, Sousa GR, Mattos RT, Chaves AT, et al. Differential expression of matrix metalloproteinases 2, 9 and cytokines by neutrophils and monocytes in the clinical forms of Chagas disease. PLoS Negl Trop Dis. 2017. January;11(1):e0005284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cowden Dahl KD, Symowicz J, Ning Y, Gutierrez E, Fishman DA, Adley BP, et al. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008. June;68(12):4606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Yang W, Pei F, Ma W, Wang Y. Downregulation of matrix metalloproteinases contributes to the inhibition of cell migration and invasion in HepG2 cells by sodium valproate. Oncol Lett. 2015. July;10(1):531–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drake MT, Clarke BL, Oursler MJ, Khosla S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr Rev. 2017. August;38(4):325–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One. 2013; 8(3):e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallo A, Saad A, Ali R, Dardik A, Tellides G, Geirsson A. Circulating interferon-γ-inducible Cys-X-Cys chemokine receptor 3 ligands are elevated in humans with aortic aneurysms and Cys-X-Cys chemokine receptor 3 is necessary for aneurysm formation in mice. J Thorac Cardiovasc Surg. 2012. March; 143(3):704–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pimiento JM, Maloney SP, Tang PC, Muto A, Westvik TS, Fitzgerald TN, et al. Endothelial nitric oxide synthase stimulates aneurysm growth in aged mice. J Vasc Res. 2008;45(3): 251–8. [DOI] [PubMed] [Google Scholar]
- 38.Pinhas N, Sternberg-Simon M, Chiossone L, Shahaf G, Walzer T, Vivier E, et al. Murine peripheral NK-cell populations originate from site-specific immature NK cells more than from BM-derived NK cells. Eur J Immunol. 2016. May;46(5):1258–70. [DOI] [PubMed] [Google Scholar]
- 39.Zemans RL, Briones N, Young SK, Malcolm KC, Refaeli Y, Downey GP, et al. A novel method for long term bone marrow culture and genetic modification of murine neutrophils via retroviral transduction. J Immunol Methods. 2009. January;340(2):102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003. May;423(6937):337–42. [DOI] [PubMed] [Google Scholar]
- 41.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005. December;5(12):953–64. [DOI] [PubMed] [Google Scholar]
- 42.Jackson MF, Scatena M, Giachelli CM. Osteoclast precursors do not express CD68: results from CD68 promoter-driven RANK transgenic mice. FEBS Lett. 2017. March;591(5):728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang JH, Sim JS, Zheng T, Yim M. F4/80 inhibits osteoclast differentiation via downregulation of nuclear factor of activated T cells, cytoplasmic 1. Arch Pharm Res. 2017. April; 40(4):492–9. [DOI] [PubMed] [Google Scholar]
- 44.Oddie GW, Schenk G, Angel NZ, Walsh N, Guddat LW, de Jersey J, et al. Structure, function, and regulation of tartrate-resistant acid phosphatase. Bone. 2000. November;27(5):575–84. [DOI] [PubMed] [Google Scholar]