

Abstract
Background:
Even though hypoxia-inducible factor-1α (HIF-1α) is among the transcriptional factors demonstrated to contribute to the formation of abdominal aortic aneurysms (AAAs), the precise mechanism has been unclear. Digoxin is known as an inhibitor of HIF-1α, and shows a protective effect against the progression of AAAs.
Objectives:
We tested the effect of digoxin on osteoclastogenesis (OCG) and examined the pathway through which digoxin exerts inhibition of HIF-1α.
Materials and Methods:
RAW 264.7 macrophage cells were cultured and stimulated by soluble receptor activator of NF-κB ligand (sRANKL) with or without digoxin. First, we tested the effect of digoxin to attenuate macrophage activation, which led to OCG, characterized by tartrate-resistant acid phosphatase (TRAP)-positive macrophages (TPMs).
Results:
The activation of TPMs stimulated by sRANKL was attenuated by digoxin treatment. Further-more, the receptor activator of NF-κB (RANK)/receptor activator of NF-κB ligand (RANKL) complex signaling pathway, which is stimulated by HIF-1α, was downregulated by digoxin treatment.
Conclusions:
These results show that digoxin attenuates OCG. By inhibition of HIF-1α, digoxin decreases OCG through the downregulation of the RANK/RANKL signaling pathway. Therefore, digoxin is a potential candidate for medical treatment of AAAs.
Keywords: Aneurysm, Digoxin, Hypoxia-inducible factor-1α, Osteoclastogenesis, Macrophage
Introduction
Abdominal aortic aneurysm (AAA) is one of the most common diseases affecting the elderly population, with an incidence of 3.9–7.2% in males, and 1.0–1.3% in females over 50 years old in the USA [1]. Some patho-physiologic features have been shown to be the underlying process of AAA, including upregulation of proteases, especially from macrophages [2]. Inflammation and matrix degradation in the vascular structure are crucial for the initiation and formation of AAAs. Even though there have been significant advances in understanding the pathophysiologic features of AAAs, surgical repair is the only approved treatment for large AAAs (aortic diameter >5.5 cm) [3]. Patients with small AAAs (<5.5 cm) are not recommended for surgical treatment, rather watchful waiting, and there is no pharmacologic treatment for these patients [4]. Therefore, the need exists for a non-surgical therapeutic intervention for patients with AAA.
Hypoxia-inducible factor-1α (HIF-1α) is a transcriptional factor that plays an important role in the regulation of cellular responses under hypoxic conditions to enable cell survival [5]. The overexpression of HIF-1α was detected from human AAA samples [6], and HIF-1α has been implicated in several vascular pathologies, including AAA and atherosclerosis [7]. Even though HIF-1α critically modulates vascular diseases, the mechanism by which HIF-1α contributes to the pathophysiology of AAA has remained poorly understood. It has been reported that HIF-1α can also contribute to the production of pro-inflammatory cytokines, which promotes macro-phage activation as well as phagocytic function [8]. Our group has reported that several kinds of proteases are produced by osteoclastogenesis (OCG), which is a specialized activation of macrophages, and promote AAA formation [9–12]. During OCG, receptor activator of NF-κB ligand (RANKL) can activate macrophages through binding to receptor activator of NF-κB (RANK), which is found on the surface of macrophages. The RANK/RANKL complex promotes transcription of the OCG-related gene, nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) [13]. NFATc1 is one of the master transcriptional factors related to macrophage activation, and promotes the expression of OCG-related genes such as tartrate-resistant acid phosphatase (TRAP), matrix metal-loproteinase-9 (MMP-9), and cathepsin K [14, 15]. Expression of these proteases is characteristic of osteoclasts; therefore, we will evaluate the expression levels of NFATc1 and proteases to identify osteoclastogenic macrophage activation. Furthermore, the RANK/RANKL complex is induced by the activation of HIF-1α [5]. Therefore, we hypothesize that cross-talk between HIF-1α and the RANK/RANKL complex might contribute to the formation of AAA.
It has been reported that digoxin is a powerful inhibitor of HIF-1α [16]. Digoxin improves cardiac stroke by suppressing the function of sodium/potassium pumps in the plasma membrane and increasing intracellular calcium concentration [17]. In addition, digoxin can attenuate the in vivo progression of AAA in a mouse model [18]. Therefore, we hypothesize that digoxin has a potential role as non-surgical treatment for AAA.
In this study, we examine the effect of digoxin on OCG in macrophages. Furthermore, we investigate the pathway by which digoxin inhibits OCG, and evaluate the potential of digoxin as an OCG-specific pharmacological treatment for AAA.
Materials and Methods
Cell Culture and Treatments
A murine monocytic cell line (RAW 264.7) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium containing 100 IU/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS). For osteoclast differentiation, RAW 264.7 cells were maintained in minimal essential medium-alpha (MEMα) supplemented with 10% charcoal-stripped FBS (cFBS), penicillin, and streptomycin. Those cells were stimulated by 30 ng/mL murine soluble RANKL (sRANKL; Peprotech, Rocky Hill, NJ, USA) with or without 0.5 μM digoxin (Selleck Chemicals, Houston, TX, USA).
TRAP Staining
RAW 264.7 cells were seeded in 96-well plates (3,000 cells per well) 1 day before treatment and were cultured. Macrophage cells were cultured in MEMα with 10% cFBS and stimulated by 30 ng/mL murine sRANKL with or without 0.5 μM digoxin for 5 days. After incubation, cells were fixed with 4% paraformaldehyde for 15 min, and stained for TRAP activity using a commercialized TRAP staining kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. TRAP-positive cells were defined as red, multinucleated (>3 nuclei) cells and counted in each well (at least 3 wells).
Quantitative Reverse Transcription PCR
The total RNA from the RAW 264.7 cells was isolated by Pure-Link RNA Mini Kit (Thermo Scientific) according to the manufacturer’s instructions. The concentrations of RNA were determined by reading the absorbance at 260 nm. One microgram of total RNA was reverse transcribed to cDNA with an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Quantitative real-time PCR was performed with SYBR Green dye using the 7500 Fast Real-Time PCR instrument (ABI, Foster City, CA, USA). The PCR primer sequences used were as follows: TRAP, F: 5′-TCC TGG CTC AAA AAG CAG TT-3′, and R: 5′-ACA TAG CCC ACA CCG TTC TC-3′; NFATc1, F: 5′-TGG CTA CCG ACA TGT GTT GT-3′, and R: 5′-GAC CAG GGG AGC TAT GAA CA-3′; cathepsin K, F: 5′-CGA AAA GAG CCT AGC GAA CA-3′, and R: 5′-TGG GTA GCA GCA GAA ACT TG-3′; MMP-9, F: 5′-CAT TCG CGT GGA TAA GGA GT-3′, and R: 5′-GTT CAC CTC ATG GTC CAC CT-3′; RANK, F: 5′-TTG TGG CAG GGG ACT TTA AC-3′, and R: 5′-ATT GTC ATC CTG CCC TCA AC-3–; RANKL, F: 5′-TCG TGG AAC ATT AGC ATG GA-3′, and R: 5′-CCT CTC CCA ATC TGG TTC AA-3′; GAPDH, F: 5′-AAC TTT GGC ATT GTG GAA GG-3′, and R: 5′-ACA CAT TGG GGG TAG GAA CA-3′. The relative mRNA expression levels for each of the target genes were calculated using the 2−ΔΔCt method and normalized against the GAPDH house-keeping gene.
Western Blotting
Proteins were extracted from RAW 264.7 cells, using the radioimmunoprecipitation assay buffer with protease inhibitor cocktail (Cell Signaling Technology, Danvers, MA, USA) at 0–4 ° C. The concentrations of proteins were determined and equal amounts (20 μg) of protein were loaded on 8 or 12% polyacrylamide gels in Laemmli buffer. Subsequently, the proteins were transferred electrophoretically onto polyvinylidene difluoride membranes. These membranes were blocked with Tris-buffered saline containing 0.05% Tween-20 (TBS-T), supplemented with 5% skimmed milk for 1 h on a rocking shaker at room temperature. The primary antibodies used for Western blotting were as follows: rabbit HIF-1α antibody (1:1,000, NB100–449, Novus Biologicals, Littleton, CO, USA), mouse NFATc1 antibody (1: 2,000, sc-7294, Santa Cruz Biotechnology, CA, USA), mouse cathepsin K antibody (1:1,000, sc-48353, Santa Cruz Biotechnology), mouse RANK antibody (1:1,000, sc-374360, Santa Cruz Biotechnology), mouse RANKL antibody (1:1,000, ab45039, Abcam, Cambridge, UK), and mouse α-tubulin antibody (1:10,000, sc-23948, Santa Cruz Biotechnology). These primary antibodies were diluted in 1% bovine serum albumin, and incubated with the membranes at 4 ° C overnight. After incubation with primary antibodies, the membranes were washed with TBS-T, and incubated with secondary antibodies for 1 h at room temperature. The immunoactive proteins were visualized with SuperSignal West Fem-to Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL, USA). The relative expression levels of proteins for Western blotting were quantified.
Flow Cytometry
For flow cytometric analysis, macrophage cell lines were cultured, and stimulated by 30 ng/mL sRANKL with or without 0.5 μM digoxin for 3 days. After the incubation, 2.5 × 105 cells in each group were stained for analysis. First, cells were blocked with 100 μg/mL of purified mouse IgG (Jackson Immuno-Research, West Grove, PA, USA) for 20 min at 4 ° C. Then cells were washed with FACS wash buffer (PBS, 3% FBS) and stained for viability with Ghost 510 (Tonobo, San Diego, CA, USA) for 30 min at 4 °C. The cells were then washed with FACS wash buffer, resuspended in fixation buffer (Invitrogen, San Diego, CA, USA), and allowed to fix for 15 min at room temperature. Cells were washed in permeabilization buffer (Invitrogen), and resuspended in permeabilization buffer containing the following primary antibodies: anti-TRAP-Alexa Fluor 488 (ab216934, Abcam), anti-cathepsin K-Al-exa Fluor 647 (sc-48353, Santa Cruz Biotechnology), and anti-MMP-9-PE (sc-13520, Santa Cruz Biotechnology). Primary antibodies and cells were incubated for 30 min at room temperature. Following incubation, the cells were washed with permeabilization buffer and resuspended in FACS wash buffer. Cells were analyzed on an Attune NxT Flow Cytometer (Invitrogen). Fluorescence minus one controls were prepared and analyzed for each fluorochrome used.
Statistical Analysis
Data are presented as the means ± standard deviation. Differences between the groups were compared by the GraphPad Prism program, version 7.0 (GraphPad Software Inc., San Diego, CA, USA). Comparisons between two groups were performed using the Student t test. Multiple comparisons among the groups were analyzed using one-way analysis of variance with repeated measures followed by Turkey multiple comparison test. Differences between the groups were considered significant at a value of p < 0.05.
Results
Digoxin Inhibits OCG Stimulated by sRANKL
First, we examined the ability of digoxin to attenuate the OCG induced by sRANKL. TRAP expression is a marker for osteoclasts, and we refer to these stimulated, activated macrophages as TRAP-positive macrophages (TPMs) in this study. We stained TPMs for TRAP to visualize the morphological changes affected by digoxin. Even though there were some multinucleated cells that were positive for TRAP staining in the digoxin-treated group (Fig. 1a), there was a statistically significant difference of the number of TPMs between the sRANKL-stimulated group and the digoxin treatment group (101.2 ± 12.7 vs. 66.0 ± 10.6, p < 0.05; Fig. 1b). Furthermore, we examined the expression level of mRNA of TRAP to evaluate the inhibitive effect of digoxin. Digoxin significantly suppressed the expression of TRAP mRNA induced by sRANKL stimulation compared to the untreated group (25.2 ± 10.1 vs. 220.5 ± 30.8).
Fig. 1.
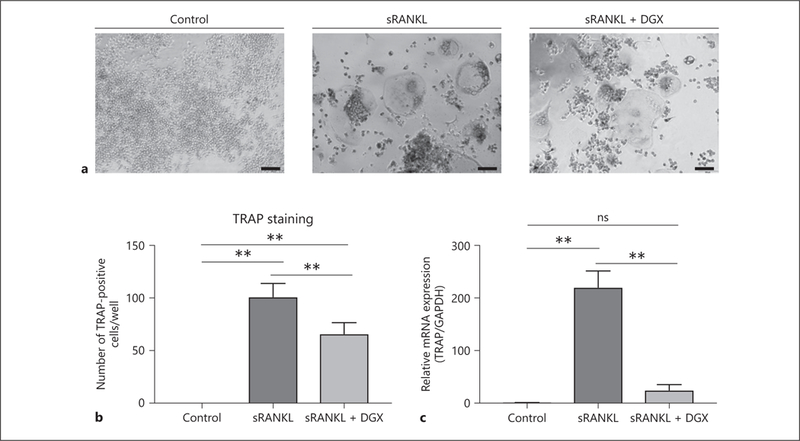
Effects of digoxin on the activity of TPMs stimulated by sRANKL. a RAW 264.7 cells were cultured in MEMα (control group), MEMα and 30 ng/mL murine sRANKL (sRANKL group), and MEMα and 30 ng/mL murine sRANKL and 0.5 μM digoxin (sRANKL + DGX group) for 5 days. Representative images of TRAP-staining in each group are shown. b The number of TRAP-positive cells was greater in the sRANKL-stimulated group than the digoxin treatment group. Scale bars, 50 μm. c RAW 264.7 cells were cultured for 2 days and examined. The relative mRNA expression level of TRAP in the digoxin treatment group showed a statistically significant lower level than the sRANKL-stimulated group. Values are presented as means ± SD for at least 3 replicates. ** p < 0.01. sRANKL, soluble receptor activator of NF-κB ligand; DGX, digoxin; TRAP, tartrate-resistant acid phosphatase.
Digoxin Treatment Reduces OCG-Associated mRNA and Protein Expression
There are several transcriptional factors and proteases that characterize OCG macrophage activation, such as NFATc1, cathepsin K, and MMP-9. Therefore, we examined the mRNA expression of these OCG-related genes to evaluate the effect of digoxin. The mRNAs of NFATc1 (Fig. 2a), cathepsin K (Fig. 2b), and MMP-9 (Fig. 2c) were significantly induced by sRANKL treatment (3.51 ± 0.91, 100.1 ± 11.9, and 508.6 ± 69.9, respectively). With digoxin treatment, the mRNA expression levels of NFATc1 (Fig. 2a), cathepsin K (Fig. 2b), and MMP-9 (Fig. 2c) were significantly lower compared to the sRANKL treatment group (1.07 ± 0.09 vs. 3.51 ± 0.91, p < 0.05, 12.9 ± 4.43 vs. 100.1 ± 11.9, p < 0.05, and 363.3 ± 32.1 vs. 508.6 ± 69.9, p < 0.05, respectively).
Fig. 2.
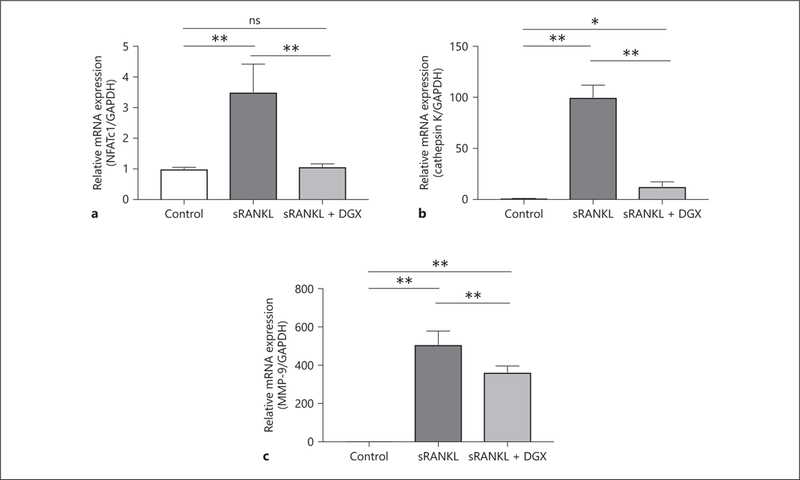
Digoxin reduces the mRNA expression related to macro-phage activation. Macrophages were separated into 3 groups (control, sRANKL, and sRANKL + DGX) and cultured for 2 days to examine the mRNA expression levels of NFATc1 (a), cathepsin K (b), and MMP-9 (c). NFATc1, cathepsin K, and MMP-9 all showed statistically lower mRNA expression levels in the sRANKL + DGX groups than sRANKL groups. Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; sRANKL, soluble receptor activator of NF-κB ligand; DGX, digoxin; NFATc1, nuclear factor of activated T-cells cytoplasmic 1; MMP-9, matrix metalloproteinase-9.
In addition to the examination of mRNA expression levels, we evaluated the expression level of proteins. It is known that digoxin inhibits HIF-1α protein translation without affecting the transcription of HIF-1α mRNA [19]. Therefore, we examined the protein expression level of HIF-1α in groups stimulated by sRANKL or additionally treated with digoxin. Cell stimulation with sRANKL resulted in increased HIF-1α protein expression compared to the untreated control group (1.68 ± 0.67 vs. 1.00 ± 0.29; Fig. 3a). With additional treatment by digoxin, the protein expression level of HIF-1α was lower than the sRANKL treatment group (0.46 ± 0.06 vs. 1.68 ± 0.67; Fig. 3a). Therefore, it was confirmed that sRANKL stimulation induces HIF-1α protein expression, and digoxin reduces HIF-1α protein expression stimulated by sRANKL. Not only HIF-1α, but also the protein expression levels of NFATc1 (Fig. 3b) and cathepsin K (Fig. 3c) were significantly higher in sRANKL-stimulated groups than control groups (10.1 ± 3.46 vs. 1.00 ± 0.16, p < 0.05, and 7.38 ± 0.68 vs. 1.00 ± 0.03, p < 0.05, respectively). Protein expression levels of NFATc1 (Fig. 3b) and cathepsin K (Fig. 3c) were suppressed by digoxin treatment compared to sRANKL-stimulated groups (3.49 ± 0.55 vs. 10.1 ± 3.46, p < 0.05, and 1.96 ± 0.25 vs. 7.38 ± 0.68, p < 0.05, respectively). These results showed that digoxin attenuated the expression levels of OCG-related mRNAs and proteins.
Fig. 3.
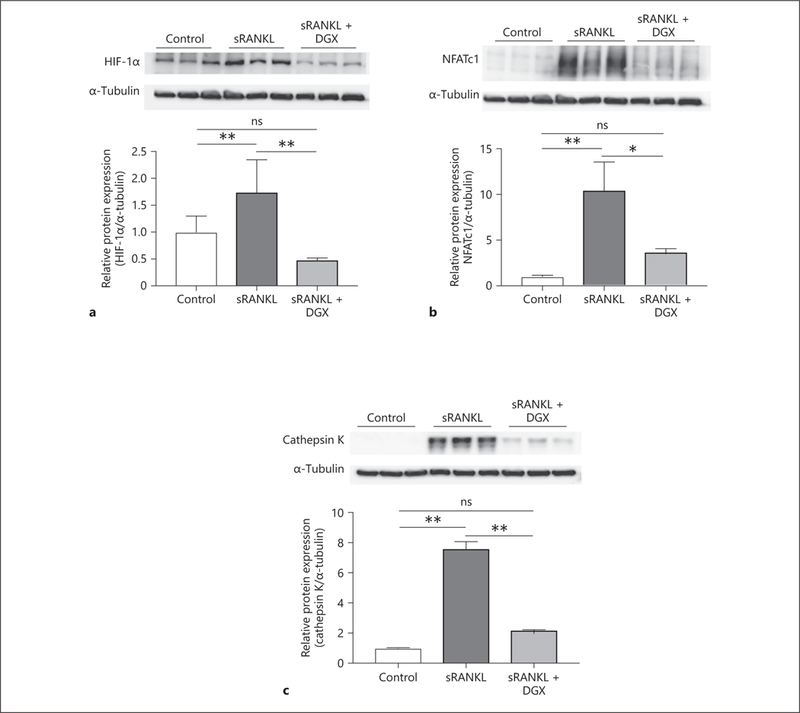
Digoxin attenuates the protein expression induced by sRANKL stimulation. Macrophages were separated into 3 groups (control, sRANKL, and sRANKL + DGX) and cultured to examine the protein expression of HIF-1α (a) and NFATc1 (b) for 2 days, and cathepsin K (c) for 3 days. All protein levels were significantly lower in the sRANKL + DGX groups than sRANKL groups. Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; sRANKL, soluble receptor activator of NF-κB ligand; DGX, digoxin; HIF-1α, hypoxia-inducible factor-1α; NFATc1, nuclear factor of activated T-cells cytoplasmic 1.
Digoxin Treatment Attenuates TPM Protease Expression
To compare the protease expression of sRANKL-treated macrophages to digoxin-treated macrophages, flow cytometric analysis was conducted. TRAP, cathepsin K, and MMP-9 are known as important proteases in OCG related to AAA formation [20]. Therefore, we examined the percentage of TRAP-, cathepsin K-, and MMP-9-positive cells via flow cytometry. There were more TRAP-, cathepsin K-, and MMP-9-positive cells in the sRANKL treatment group compared to the control group (3.29 ± 0.76 vs. 0.83 ± 0.16%, p < 0.05, 0.90 ± 0.17 vs. 0.55 ± 0.18%, p < 0.05, and 9.24 ± 2.67 vs. 5.21 ± 01.82%, p < 0.05, respectively; Fig. 4a–c). When we treated sRANKL-stimulated macrophages with digoxin, the percentage of TRAP-, cathepsin K-, and MMP-9-positive cells decreased in the digoxin-treated group compared to the sRANKL-treated group (1.31 ± 0.15 vs. 3.29 ± 0.76%, p < 0.05, 0.51 ± 0.13 vs. 0.90 ± 0.17%, p < 0.05, and 4.99 ± 0.86 vs. 9.24 ± 2.67%, p < 0.05, respectively; Fig. 4a–c). These results demonstrated that digoxin treatment decreased the expression level of TPM-related proteases.
Fig. 4.
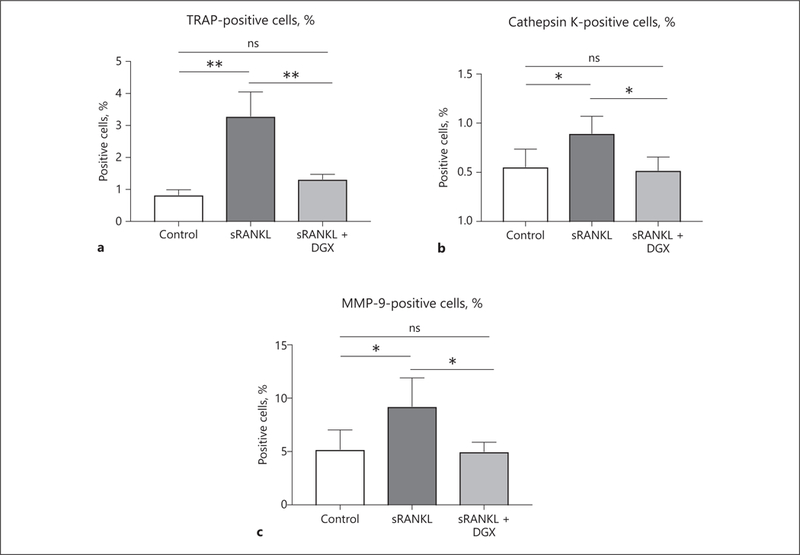
Digoxin treatment decreases the activation of macrophages. Flow cytometric analysis showed that the percentage of live cells positive for TRAP (a), cathepsin K (b), and MMP-9 (c) were higher in the sRANKL treatment group (cultured for 3 days) than the digoxin treatment group. Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; sRANKL, soluble receptor activator of NF-κB ligand; DGX, digoxin; TRAP, tartrate-resistant acid phosphatase; MMP-9, matrix metalloproteinase-9.
Digoxin Attenuates OCG through the Pathway of RANK/RANKL Complex
It is known that the RANK/RANKL complex plays a key role in initiating OCG, and is induced through the activation of HIF-lα [5]. We hypothesize that digoxin attenuates the initiation of AAA by the downregulation of the RANK/RANKL complex through the inhibition of HIF-lα. The protein expression levels of RANK (Fig. 5a) and RANKL (Fig. 5b) were significantly lower in the digoxin treatment groups than sRANKL-stimulated groups (0.91 ± 0.07 vs. 1.16 ± 0.11, p < 0.05, and 1.12 ± 0.02 vs. 1.62 ± 0.19, p < 0.05, respectively). Moreover, digoxin significantly decreased the mRNA expression levels of RANK (Fig. 5c) and RANKL (Fig. 5d) compared to sRANKL-stimulated groups (1.42 ± 0.06 vs. 2.47 ± 0.09, p < 0.05, and 1.27 ± 0.17 vs. 2.53 ± 0.18, p < 0.05, respectively). Therefore, digoxin appears to attenuate OCG through the inhibition of the RANK/RANKL complex pathway.
Fig. 5.
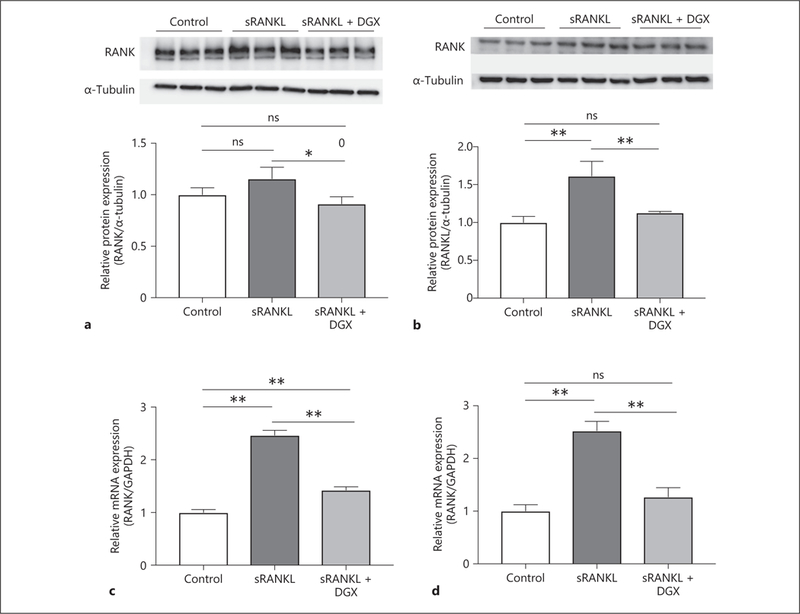
Digoxin inhibits the HIF-1α-induced RANK/RANKL complex pathway stimulated by sRANKL. RAW 264.7 cells were divided into 3 groups (control, sRANKL, and sRANKL + DGX) and cultured for 3 days to examine the protein and mRNA expression levels. The protein expression levels of RANK (a) and RANKL (b) were statistically lower in the sRANKL + DGX groups than sRANKL groups. Furthermore, the mRNA expression levels of RANK (c) and RANKL (d) were significantly lower in the sRANKL + DGX groups than sRANKL groups. Values are presented as means ± SD for at least 3 replicates. * p < 0.05, ** p < 0.01. ns, not significant; sRANKL, soluble receptor activator of NF-κB ligand; DGX, digoxin; RANK, receptor activator of NF-κB; RANKL, receptor activator of NF-κB ligand.
Discussion
In this study, we demonstrated that digoxin, an inhibitor of HIF-1α, attenuated OCG through the downregulation of the RANK/RANKL complex pathways. We stimulated RAW 264.7 macrophages with sRANKL, with or without digoxin. Several transcriptional factors and proteases, including HIF-1α, were upregulated by sRANKL stimulation, and these upregulations were inhibited by digoxin. Furthermore, digoxin treatment decreased the mRNA and protein expression of RANK and RANKL, which led to the inhibition of OCG stimulated by sRANKL. Therefore, we showed that the use of digoxin for treatment of AAA warrants further investigation.
It is known that HIF-1α is the master transcriptional factor driving cellular adaptation to hypoxic conditions, and plays a key role in osteogenesis [21]. Furthermore, it has been reported that HIF-1α is involved in osteoclast differentiation and activity, which affects bone metabolism [22]. We have already reported the significant correlation between bone metabolism, especially OCG, and the initiation of AAA [9–12]. Therefore, we hypothesized that HIF-1α activation induces OCG, which are involved in AAA formation. To test this hypothesis, we treated macrophages with sRANKL, which led to OCG, and simultaneously treated with digoxin to inhibit the activity of HIF-1α. In this experiment, we showed that the expression levels of protein and mRNA of RANK and RANKL were lowered by digoxin treatment, which proved that digoxin attenuated OCG stimulated by sRANKL through the downregulation of the RANK/RANKL complex signaling pathway. There have been some reports showing HIF-1α activation related to the NF-κB pathway and mitogen-activated protein kinase (MAPK) pathway [23, 24]. These NF-κB and MAPK pathways were controlled by RANK/RANKL complex interaction [25]. Therefore, the RANK/RANKL complex and downstream signaling pathways, including NF-κB and MAPK pathway, might play one of the key roles in inhibiting OCG by digoxin in our experimental model.
Digoxin is known as a selective inhibitor of HIF-1α translation, and has a suppressive effect of initiation and progression of AAAs in in vivo models [17, 26]. Recent studies have shown that digoxin inhibits the differentiation of T helper 17 (Th17) cells, and production of interleukin (IL)-17A [27, 28]. The AAA wall contains several kinds of inflammatory cells, such as macrophages and T lymphocytes, especially Th17 cells [29]. It has been reported that digoxin attenuates the Th17/IL-17A-related inflammatory response, which leads to protection from AAA formation [30]. Even though there have been some reports that digoxin exerts an anti-inflammatory effect, to our knowledge, there has been no report that digoxin attenuates OCG. Therefore, this is the first report to show the inhibition of OCG by digoxin. Furthermore, it has been reported that digoxin increases blood glucose levels [31, 32]. We have already reported that hyperglycemia attenuates the activation of macrophages through the activation of the glucose-sensing nuclear receptor [9], and suppression of insulin signaling [13]. Even though hyperglycemic conditions induced by digoxin might be one of the protective effects for AAA, we have not tested this hypothesis yet. Therefore, further experiments are necessary to prove this hypothesis.
There are some limitations of this study. First, we applied only our in vitro OCG model to this study. We have not examined our hypothesis that digoxin attenuates OCG, one of the initiators of AAA formation, through the downregulation of the RANK/RANKL signaling pathway in in vivo models, yet. Furthermore, we have only examined in vitro osteoclastogenic changes by culturing and stimulating the RAW 264.7 cell line. It has been reported that most of the macrophages accumulating in the aneurysmal aortic wall derive from circulating monocytes, which are produced in the bone marrow [33]. Therefore, we plan to examine OCG not only in RAW 264.7 cells, but also in bone marrow-derived macrophages. We showed that digoxin inhibited OCG stimulated by sRANKL through the inhibition of the RANK/RANKL pathway. Additionally, we should evaluate the ability of digoxin to inhibit HIF-1α upon hypoxic stimulation, which typically induces HIF-1α.
Conclusions
In this report, we demonstrated that: (1) digoxin treatment attenuates OCG stimulated by sRANKL; (2) digoxin decreases the activation of HIF-1α, and (3) digoxin inhibits OCG through the downregulation of the RANK/RANKL complex signaling pathway. Further studies, including in vivo experiments, are needed to evaluate the effect of digoxin on AAA formation as it relates to OCG. Hypoxic stimulation also induces HIF-1α activation, and the effect of digoxin should be examined under hypoxic conditions. Through these future experiments, we intend to show the ability of digoxin to attenuate the formation and progression of AAAs, and its potential as a medical treatment.
Acknowledgments
Funding Sources
This work was supported by a Uehara Memorial Foundation Research Fellowship Award (to K.I.), and the Vascular Surgery Research Training Grant (T32HL110853; to M.J.K.).
Footnotes
Disclosure Statement
The authors have no conflicts of interest to declare.
Statement of Ethics
No human or animal subjects were used in the experiments.
References
- 1.LeFevre ML; U.S. Preventive Services Task Force. Screening for abdominal aortic aneurysm: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2014. August;161(4):281–90. [DOI] [PubMed] [Google Scholar]
- 2.Kurosawa K, Matsumura JS, Yamanouchi D. Current status of medical treatment for abdominal aortic aneurysm. Circ J. 2013;77(12): 2860–6. [DOI] [PubMed] [Google Scholar]
- 3.Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2005. February;142(3):203–11. [DOI] [PubMed] [Google Scholar]
- 4.Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, et al. Genetic and pharmacologic disruption of interleukin-1β signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2013. February;33(2):294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang ZN, Zhang F, Tang P, Qi XW, Jiang J. Hypoxia induces RANK and RANKL expression by activating HIF-1α in breast cancer cells. Biochem Biophys Res Commun. 2011. May;408(3):411–6. [DOI] [PubMed] [Google Scholar]
- 6.Choke E, Cockerill GW, Dawson J, Chung YL, Griffiths J, Wilson RW, et al. Hypoxia at the site of abdominal aortic aneurysm rupture is not associated with increased lactate. Ann N Y Acad Sci. 2006. November;1085(1):306–10. [DOI] [PubMed] [Google Scholar]
- 7.Kasivisvanathan V, Shalhoub J, Lim CS, Shepherd AC, Thapar A, Davies AH. Hypoxia-inducible factor-1 in arterial disease: a putative therapeutic target. Curr Vasc Pharmacol. 2011. May;9(3):333–49. [DOI] [PubMed] [Google Scholar]
- 8.Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008. April; 180(7):4697–705. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka T, Takei Y, Yamanouchi D. Hyperglycemia suppresses calcium phosphate-induced aneurysm formation through inhibition of macrophage activation. J Am Heart Assoc. 2016. March;5(3):e003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takei Y, Tanaka T, Kent KC, Yamanouchi D. Osteoclastogenic differentiation of macrophages in the development of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2016. September;36(9): 1962–71. [DOI] [PubMed] [Google Scholar]
- 11.Yamanouchi D, Takei Y, Komori K. Balanced mineralization in the arterial system: possible role of osteoclastogenesis/osteoblastogenesis in abdominal aortic aneurysm and stenotic disease. Circ J. 2012;76(12):2732–7. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka T, Kelly M, Takei Y, Yamanouchi D. RANKL-mediated osteoclastogenic differentiation of macrophages in the abdominal aorta of angiotensin II-infused apolipoprotein E knockout mice. J Vasc Surg. 2018. December; 68(6S)48–59.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurihara C, Tanaka T, Yamanouchi D. Hyperglycemia attenuates receptor activator of NF-κB ligand-induced macrophage activation by suppressing insulin signaling. J Surg Res. 2017. June;214:168–75. [DOI] [PubMed] [Google Scholar]
- 14.Kwak HB, Jin HM, Ha H, Kang MJ, Lee SB, Kim HH, et al. Tumor necrosis factor-alpha induces differentiation of human peripheral blood mononuclear cells into osteoclasts through the induction of p21(WAF1/Cip1). Biochem Biophys Res Commun. 2005. May; 330(4):1080–6. [DOI] [PubMed] [Google Scholar]
- 15.Kanazawa K, Kudo A. TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J Bone Miner Res. 2005. May;20(5):840–7. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008. December;105(50):19579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt TA, Allen PD, Colucci WS, Marsh JD, Kjeldsen K. No adaptation to digitalization as evaluated by digitalis receptor (Na,K-ATPase) quantification in explanted hearts from donors without heart disease and from digitalized recipients with end-stage heart failure. Am J Cardiol. 1993. January; 71(1):110–4. [DOI] [PubMed] [Google Scholar]
- 18.Tsai SH, Huang PH, Hsu YJ, Peng YJ, Lee CH, Wang JC, et al. Inhibition of hypoxia inducible factor-1α attenuates abdominal aortic aneurysm progression through the down-regulation of matrix metalloproteinases. Sci Rep. 2016. July;6(1):28612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, et al. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA. 2012. January;109(4):1239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002. September; 110(5): 625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araldi E, Schipani E. Hypoxia, HIFs and bone development. Bone. 2010. August;47(2): 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knowles HJ, Athanasou NA. Acute hypoxia and osteoclast activity: a balance between enhanced resorption and increased apoptosis. J Pathol. 2009. June;218(2):256–64. [DOI] [PubMed] [Google Scholar]
- 23.Brito C, Stavroullakis A, Oliveira T, Prakki A. Cytotoxicity and potential anti-inflammatory activity of velutin on RAW 264.7 cell line differentiation: implications in periodontal bone loss. Arch Oral Biol. 2017. November;83:348–56. [DOI] [PubMed] [Google Scholar]
- 24.Sala MA, Chen C, Zhang Q, Do-Umehara HC, Wu W, Misharin AV, et al. JNK2 up-regulates hypoxia-inducible factors and contributes to hypoxia-induced erythropoiesis and pulmonary hypertension. J Biol Chem. 2018. January;293(1):271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen XW, Liu WT, Wang YX, Chen WJ, Li HY, Chen YH, et al. Cyclopropanyldehydro-costunolide LJ attenuates high glucose-induced podocyte injury by suppressing RANKL/RANK-mediated NF-κB and MAPK signaling pathways. J Diabetes Complications. 2016. July;30(5):760–9. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Xu B, Xuan H, Ge Y, Wang Y, Wang L, et al. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J Vasc Surg. 2018. November;68(5):1538–1550.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. 2011. April;472(7344):486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita-Sato S, Ito S, Isobe T, Ohyama T, Wak-abayashi K, Morishita K, et al. Structural basis of digoxin that antagonizes RORgamma t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J Biol Chem. 2011. September;286(36):31409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006. May;26(5):987–94. [DOI] [PubMed] [Google Scholar]
- 30.Wei Z, Wang Y, Zhang K, Liao Y, Ye P, Wu J, et al. Inhibiting the Th17/IL-17A-related inflammatory responses with digoxin confers protection against experimental abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2014. November;34(11):2429–38. [DOI] [PubMed] [Google Scholar]
- 31.Spigset O, Mjorndal T. Increased glucose intolerance related to digoxin treatment in patients with type 2 diabetes mellitus. J Intern Med. 1999. October;246(4):419–22. [DOI] [PubMed] [Google Scholar]
- 32.Krusteva E. Effect of digoxin on experimental adrenaline-induced hyperglycemia and insulin-induced hypoglycemia. Folia Med (Plovdiv). 1992;34(3–4):14–6. [PubMed] [Google Scholar]
- 33.Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017. August;14(8):457–71. [DOI] [PubMed] [Google Scholar]