

Abstract
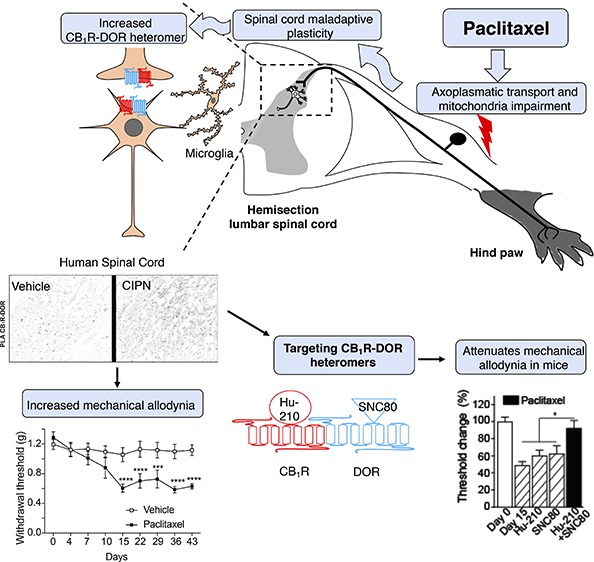
Cannabinoid 1 (CB1R) and delta opioid receptors (DOR) associate to form heteromers that exhibit distinct pharmacological properties. Not much is known about CB1R-DOR heteromer location or signaling along the pain circuit in either animal models or patients with chemotherapy-induced peripheral neuropathy (CIPN). Here, we use paclitaxel to induce CIPN in mice and confirm the development of mechanical allodynia. Under these conditions, we find significant increases in CB1R-DOR heteromers in the dorsal spinal cord of mice with CIPN as well as in postmortem spinal cords from human subjects with CIPN compared to controls. Next, we investigated receptor signaling in spinal cords of mice with CIPN and found that treatment with a combination of low signaling doses of CB1R and DOR ligands leads to significant enhancement in G-protein activity that could be selectively blocked by the CB1R-DOR antibody. Consistent with this, administration of subthreshold doses of a combination of ligands (CB1R agonist, Hu-210, and DOR agonist, SNC80) leads to significant attenuation of allodynia in mice with CIPN that is not seen with the administration of individual ligands, and this could be blocked by the CB1R-DOR antibody. Together, these results imply that CB1R-DOR heteromers upregulated during CIPN-associated mechanical allodynia could serve as a potential target for treatment of neuropathic pain including CIPN.
Keywords: CB1R-DOR, heteromer, chemotherapy-induced peripheral neuropathy, allodynia, paclitaxel, pain
Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is a debilitating and dose-dependent side effect caused by anticancer agents that interferes with cancer therapy regimens and affects long-term quality of life.1,2 Even though the epidemiology of CIPN is unclear, the overall incidence is estimated to be approximately 38% in patients treated with multiple chemotherapy agents,3 although this percentage varies depending on the dose, chemotherapy cocktails used, and duration of exposure.2 Taxanes, vinca alkaloids, platinum derivatives, bortezomib, and thalidomide are the most frequent agents causing CIPN.4 These drugs predominantly impair afferent sensory fibers with a symmetric, distal, length-dependent “glove and stocking” distribution.5 Patients report either positive (e.g., paresthesia, dysesthesia, tingling, burning, stabbing, or aching pain), negative (e.g., sensory loss) symptoms or evoked pain such as allodynia, that is, pain due to a stimulus that does not normally provoke pain.6 Although the mechanisms underlying the changes leading to allodynia are not fully understood, several studies have suggested a three-hit hypothesis involving structural plasticity of Aβ afferents, loss of spinal inhibitory neurons, and changes in afferent drive or neuron-glia signaling (reviewed in ref (7)). Paclitaxel is a taxane-based drug commonly used alone or in combination with platinum compounds for the treatment of solid tumors such as breast, lung, or ovarian cancer.8 It is believed that the antineoplastic properties of paclitaxel are due to its ability to increase the stability of tubulin polymers, thus inhibiting cellular replication.9 Although the mechanism by which paclitaxel damages peripheral sensory fibers is not fully understood, it has been suggested that paclitaxel impairs axoplasmatic transport,10 causes a dysfunction in neuronal mitochondria,11 and promotes epithelial damage leading to axonal degeneration.12 In addition, human studies have revealed that mechanical transmission related pathways (i.e., Aβ and Aδ fibers) are preferentially impaired in CIPN patients treated with paclitaxel.13 This leads to patients experiencing CIPN, and to date there are no effective treatments for this condition.
Cannabinoids and opioids are among the drugs known to induce analgesia. Furthermore, cannabinoid and opioid receptors and their endogenous ligands share similar anatomical and cellular distribution through sensory nuclei and related pathways (e.g., dorsal spinal cord) and receptor activation leads to pain attenuation.14 However, the clinical utility of ligands targeting these receptors is limited by a compromise between efficacy and time-related side-effects such as tolerance (i.e., need to increase a dose to maintain the analgesic effects) and undesired effects in nonrelated pain regions (i.e., gastrointestinal disturbances or depression), especially for μ-opioid receptor (MOR) agonists.15,16 In the case of δ-opioid receptor (DOR) agonists, studies show that they have markedly different properties from MOR agonists17 in that they are less effective as analgesics in acute pain models but appear to be efficacious in the treatment of neuropathic pain, specifically mechanical allodynia.18−21 Moreover, DOR agonists do not induce the same types of adverse events as MOR agonists (e.g., depression and constipation)22,23 and their chronic administration does not cause hyperalgesia.24 However, to date drugs targeting either opioid or cannabinoid receptors have had limited clinical success in the treatment of neuropathic pain.
Growing evidence demonstrates that cannabinoid and opioid receptors, such as other G-protein coupled receptors (GPCRs) can physically interact with each other to form receptor heteromers. This interaction leads to a switch in signaling, trafficking, and pharmacological properties (i.e., allosteric modulation) of each functional receptor unit (i.e., monomer or protomer).25 This challenges the classic notion that GPCRs function solely as independent monomers and demonstrates their functional complexity. Allosteric modulation induced by the binding of a ligand to the orthosteric site of one receptor (e.g., CB1R) can modify the affinity and/or intrinsic efficacy of the ligand for the orthosteric site of the partner receptor (e.g., DOR), ultimately leading to a functional response, independently of its affinity for the receptor.26 Not surprisingly, a combination of low doses of drugs targeting cannabinoid and opioid receptors or the use of novel therapeutic approaches (e.g., use of bivalent drugs) that take advantage of this phenomenon have shown promising results in animal models and in humans.27,28 In an attempt to explore the CB1R-DOR interaction we previously showed in heterologous cells that CB1R and DOR associate to form heteromers that exhibit distinct pharmacological properties.29 However, the main challenge in the field of GPCR heteromers is demonstrating their presence and functional significance not only in native tissues, and even more challenging in vivo but also the clinical consequences in human patients. By using an animal model of sciatic nerve injury (SCI), we demonstrated a disease-specific regulation in the prefrontal cortex of animals with neuropathic pain.30 However, little is known about the specific location, regulation, and behavioral effects of CB1R-DOR heteromers in animal models of CIPN and in human disease.
In this study we used anatomical, proximity-based, pharmacological and behavioral techniques to demonstrate the presence of CB1R-DOR heteromers in vitro (i.e., heterologous cells), ex vivo by using an animal model of paclitaxel-induced peripheral neuropathy and tissue samples from human patients affected with CIPN, and in vivo by targeting the CB1R-DOR heteromer. Our findings suggest that CB1R-DOR-mediated signaling and allodynia can be modulated by a combination of ligands targeting both receptors. Thus, targeting CB1R-DOR heteromers in CIPN is a novel therapeutic approach to treat this condition.
Material and Methods
Ethics Statement
Animal studies were carried out according to protocols approved by the Institutional Animal Care and Use Committees at the Icahn School of Medicine at Mount Sinai (IACUC-2014-0291) and Utah State University (IACUC 2775).
All described human samples (controls and patients) were obtained from the biorepository and pathology core brain bank at Mount Sinai Hospital. This study was performed in compliance with Mount Sinai Hospital ethics committee guidelines. For this type of study formal consent was not required.
Animals
Mice. Adult male C57BL/6J mice (12 weeks) were purchased from Jackson laboratories (Ban Harbor, ME) and used for the experimental groups (vehicle and paclitaxel). Animals were maintained on a 12-h light/dark cycle and were acclimatized to their environment for 1 week prior to experimentation. For antibody testing, DOR–/– mice were generated at the Mount Sinai Mouse Genetics Research Facility as described previously.29
Postmortem Human Samples
The Mount Sinai Data Warehouse provided the information regarding the human autopsies that met the following criteria: cancer patients who suffered pain in the limbs in the context of treatment with paclitaxel and/or platinum. We included both paclitaxel and platinum treatments for the following reasons: (i) both have poor penetration through the blood-brain barrier (BBB) so their central effects are thought to be preceded by a peripheral sensitization,31,32 (ii) both cause a similar length and dose-dependent sensory axonal neuropathy,4 and (iii) both exhibit similar CB1R and DOR protein level profiles in the spinal cord (see Results section). Patients with advanced or poorly controlled diabetes mellitus, alcohol abuse, evidence of metastatic disease that involves the disruption of the BBB, systemic diseases causing pain (e.g., rheumatoid arthritis), compressive or hereditary neuropathy, consumption or treatment with cannabinoid or opiates were excluded. Spinal cords from 10 subjects with no history of pain or any neurological disease were used as controls. Patients and controls were matched for age. The study was based on 19 human patients. Formalin-fixed paraffin-embedded (FFPE) lumbar spinal cord sections were collected from the Brain and Tissue Repository of Mount Sinai. Nine cases (six males, three females) treated with paclitaxel and/or platinum chemotherapy with a clinically confirmed diagnosis of CIPN according to the medical records and 10 controls (seven males, three females) were included in this study. Neuropathological examination of the spinal cord was initially performed at the Department of Pathology to confirm the absence of macroscopic changes.
Assessment of Mechanical Allodynia (Von Frey Filament Test) In Mice
All animals (vehicle- and paclitaxel-treated) were placed onto a testing platform containing a metal, perforated floor (Stoelting Co., Wood Dale, IL, USA). Animals were acclimatized to the testing chamber for 3 days (40 min/session). After verifying that the mice were calm, mechanical allodynia was assessed by applying von Frey filaments to the midplantar region of both hindpaws for approximately 2 s per stimulus using calibrated filaments (Stoelting Co., Wood Dale, IL, USA). All trials began with the 1 g filament, and proceeded using an up–down trial design.33,34 Response (i.e., sudden paw withdrawal, sudden flinching, or sudden paw licking) in two out of three trials was regarded as a positive response. A negative response was followed by the use of a larger filament. The average score was used as the recorded value. Paw mechanical withdrawal threshold was expressed in g, Figure 3B and in % where 100% represent the baseline values (before vehicle or paclitaxel was administered), and subsequent measurements were expressed as relative to baseline values, Figure 3C.
Figure 3.

Co-administration of low doses of CB1R and DOR ligands potentiates the antiallodynic effect of each drug alone. (A) Schematic representation of vehicle and paclitaxel treatment as well as of administration of CB1R and DOR agonists. C57BL/6 male mice were intraperitoneally injected with paclitaxel or vehicle on day 0 after behavioral testing (Von Frey filament test). Paclitaxel or vehicle injections were repeated on days 2, 4, and 6. Behavioral assessments (Von Frey filament test) were conducted on days 4, 7, 10, and 15. Vehicle- and paclitaxel-mediated neuropathic pain animals were randomly distributed into different groups and subjected to different drug treatments (either Hu-210, SNC80, a combination of Hu-210, and SNC80 also referred as combo, anti-CB1R-DOR antibody and combo or anti-Flag antibody and combo) on day 16. After a washout period of 7 days animals received a different treatment. The same strategy was carried out for the third, fourth, and fifth treatments (day 23, 30, 37, and 44) so every group received every treatment. Animals were subjected to behavioral testing (Von Frey filament test) on the day before and the day after the different treatments. (B) Mechanical allodynia in paclitaxel-treated mice develops by Day 7 and persists up to Day 43 of testing. The threshold is normalized to each animal’s response on Day 0, prior to commencement of paclitaxel administration. Paclitaxel produced a significant decrease in mechanical threshold compared to vehicle controls F(1, 25) = 25.48, p < 0.0001. (C) Changes in mechanical threshold following drug treatment. Combined administration of Hu-210 and SNC80 can produce greater alleviation of neuropathic pain compared to Hu-210 or SNC80 alone. This effect was not seen in the absence of neuropathic pain (F (1, 194) = 39.14, p < 0.0001). In addition, pretreatment with the anti-CB1R-DOR antibody significantly decreased the analgesic response of the combined administration of Hu-210 and SNC80, whereas pretreatment with anti-Flag antibody did not (F(6, 109) = 7.828, p < 0.0001). Data on left panel was analyzed using a two-way ANOVA and Sidak’s multiple comparison test while data on right panel was analyzed by one-way ANOVA with Tukey’s multiple comparison test. Data represents mean ± SEM (n = 18, paclitaxel; n = 20, vehicle). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s.= not significant.
Paclitaxel, Analgesic Drugs, and Monoclonal Antibodies
Paclitaxel (Sigma-Aldrich, MO, USA) stocks were dissolved in DMSO (50 mg/mL). The CB1R agonist Hu-210 (Tocris bioscience, MN, USA) and the DOR agonist SNC80 (Tocris Bioscience) were reconstituted in DMSO (10 mM stock solutions). Paclitaxel was diluted in a vehicle which comprised Kolliphor EL (Sigma-Aldrich, MO, USA)/ethanol/0.9% saline in a 1:1:18 ratio, and administered intraperitoneally (i.p.; 4 mg/kg) to 7 mice in a total volume of 100 μL on four alternate days (cumulative dose 16 mg/kg) to induce neuropathy.35 The control group received the same vehicle containing DMSO instead of the drug. Development of paclitaxel-induced mechanical allodynia was assessed on days 0, 4, 7, and 15 from the first injection. A second set of animals was treated with vehicle (n = 20) or paclitaxel (n = 18) in a similar way. Animals were then split into different groups and randomized to receive Hu-210 (0.5 μg/kg), SNC80 (0.1 mg/kg), a combination of both (also referred as combo) dissolved in 0.9% saline (100 μL; i.p.), combo + 5 μL of intrathecal anti-Flag monoclonal antibody (Flag M2 mAb, Millipore-Sigma) or combo + 5 μL of intrathecal anti-CB1R-DOR monoclonal antibody (see CB1R-DOR generation in a subsequent section) at day 16. Following a washout period of 7 days, animals were tested again for mechanical allodynia. The next day (day 22) each group received a different drug treatment. The same strategy was carried out for the third, fourth, and fifth treatments (day 23, 30, 37, and 44) so that every group received every treatment (Figure 3A).
Intrathecal Administration of Monoclonal Antibodies
A dose of 5 μL of 1 mg/mL anti-CB1R-DOR monoclonal antibody or 1 mg/mL anti-Flag antibody was injected intrathecally. This volume avoids redistribution of the antibody through the cerebrospinal fluid to the basal cisterns of the brain.36 Animals were lightly restrained on a flat bench surface and percutaneus intrathecal injections were made into the L4–L5 or L5–L6 intervertebral space of unanaesthetized mice using a 50 μL Hamilton syringe connected to a 30-gauge needle as previously described.37 Intrathecal placement was suggested by a lateral tail flick as the needle penetrated into the subarachnoid space. After 5 min, drug treatment was administered as discussed in the previous section.
Membrane Preparation
Membranes were prepared from spinal cords of vehicle- and paclitaxel-treated mice. Animals were euthanized with CO2 gas and then rapidly decapitated. Spinal cords were removed, and the lumbar section dissected out. To prepare membranes, dissected tissue was manually homogenized in cold buffer containing 20 mM Tris, 2 mM EGTA, 250 mM MgCl2, and 250 mM sucrose (pH 7.4). The homogenate was centrifuged at 27 000g for 15 min at 4 °C, and the supernatant was discarded. The resulting pellet was resuspended in the same cold buffer and incubated for 10 min at 4 °C, followed by centrifugation at 27 000g for 15 min at 4 °C. The supernatant was discarded, and the resultant pellet was resuspended in buffer containing 2 mM Tris, 2 mM EGTA, and 10% glycerol (pH 7.4). Membranes were stored in aliquots at −80 °C.
CB1R-DOR Antibody Generation
The generation and characterization of the CB1R-DOR antibody was described previously.30 Briefly, mice were first made tolerant to immunogenic epitopes in N2A cell membranes endogenously expressing CB1R. This was followed by i.p. injections of membranes from N2A cells expressing DOR, along with three boosters (one booster injection every 15 days). Spleen cells from animals giving a high titer by enzyme-linked immunosorbent assay (ELISA) with membranes from N2A cells expressing DOR were used to generate monoclonal antibodies. Clones secreting monoclonal antibodies were screened by ELISA as described38 against the following cells lines: untransfected N2A cell membranes that endogenously expressed CB1R, N2A cell membranes coexpressing CB1R with either DOR, MOR, KOR, CB2R, or AT1R and HEK293 membranes expressing either DOR, or coexpressing KOR and DOR or MOR and DOR. Hybridoma supernatants from clones that gave a signal only with CB1R-DOR were further purified as described in ref (38) and screened for specificity against cortical membranes from wild-type, CB1R–/–, and DOR–/– animals.
ELISA
Quantification of levels for receptor homomers (commercial antibodies) or heteromers (our antibody) in vehicle- and paclitaxel-administered animals was carried out by ELISA.38 Membranes (from n = 7 animals/group) were diluted in PBS (5 μg/100 μL) and coated onto 96-well plates (5 μg/well). All experiments were carried out in triplicate. Membranes were fixed with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) for 20 min and then washed five times in PBS, followed by incubation in blocking buffer (1% bovine serum albumin (BSA) in PBS) for 90 min. Membranes were incubated overnight with primary antibody. Primary antibodies used included rabbit polyclonal antibody against N-terminus CB1R (Thermo Scientific, Rockford, IL, USA) (1:1000), a goat polyclonal antibody directed against the C-terminus of DOR (ProSci, Poway, CA, USA) (1:1000) and a mouse monoclonal antibody directed against CB1R-DOR (1:100). After the membranes were washed in PBS, horseradish peroxidase conjugated secondary antibody was added for 90 min. After final washes in PBS, colorimetric substrate was added, and the plate was scanned at 490 nm.
[35S]GTPγS Binding
Assays were carried out as described previously39−41 in membranes from CHO cells stably expressing DOR or CHO–DOR cells transiently expressing CB1R (DORCB1R), and lumbar spinal cord membranes from vehicle and paclitaxel animals (n = 7/group) 16 days after the first paclitaxel injection. The membranes were incubated with two concentrations (0.1 and 1 nM) of the DOR agonist SNC80 in the presence of 2 mM GDP and 0.5 mM [35S] GTPγS for 1 h at 30 °C. Basal binding was determined in the presence of GDP and absence of the agonist. Nonspecific binding was determined by adding 10 μM GTPγS to a parallel set of tubes. Membrane bound radioactivity was collected by filtration and detected using a scintillation counter. In experiments examining the effect of the CB1R antagonist, SR141716 or the monoclonal antibodies to CB1R-DOR (1 μg) and CB1R-AT1R (1 μg), membranes were preincubated with these agents for 15 min prior to carrying out the assay. In experiments examining the effect of the combination of SNC80 and the CB1R agonist, Hu-210, both drugs were added together.
Immunohistofluorescence Assay
To characterize cell subtypes, the following primary antibodies (1:200) raised in mouse were used: Neu-N Alexa 488 for neuronal nuclei (Millipore, MA, USA), Bassoon for presynaptic location (Enzo, CA, USA), GFAP for astrocytes (Cell Signaling, MA, USA) or Iba-1 for microglia (Millipore, MA, USA). An antimouse Alexa 488 secondary antibody (1:200) was used to detect the above-mentioned primary antibodies except for Neu-N.
In Situ Proximity Ligation Assay (PLA)
PLA was carried out on CHO cells transiently expressing DOR or coexpressing CB1R and DOR, on postfixed mice and FFPE human lumbar spinal cord sections using the Duolink in situ kit (Sigma-Aldrich, MO, USA) as described.42 Primary antibody dilution was 1:200 for rabbit anti-CB1R (Thermo Scientific, Rockford, IL, USA) and for goat anti-DOR (ProSci, Poway, CA, USA). For antibody validation (CB1R and DOR) WT, CB1R and DOR knockout mice were used (Suppl. Figure 1).
Confocal Visualization Settings and Automated Dot Counting
Stained samples (immunohistofluorescence and PLA signal) were inspected under a Leica SP5 DMI laser-scanning confocal microscope using a 63 × /1.4 oil objective and sequential scanning with narrow band-pass filters (420–480 nm for DAPI, 505–530 nm for Alexa 488, and 560–615 nm for PLA signal). Dot counting was performed as described elsewhere.42
Statistical Analyses
Animals were randomly assigned to experimental conditions. Paw withdrawal thresholds (mechanical allodynia) were calculated for each paw and averaged. Data were analyzed using ANOVA for repeated measures or one-way ANOVA as appropriate. Two-way ANOVA was used to identify the source of significant analgesic effects at each time point and to compare postinjection responses with the values of the previous day, followed by Sidak’s multiple comparisons test. The impact of paclitaxel on CB1R, DOR, or CB1R-DOR levels was analyzed using two-tailed t tests. For the analysis of [35S]GTPγS binding results, values from each experimental condition (e.g., SNC80 dose response curve ±10 nM SR141716) were compared using a one-way ANOVA. Statistical analyses were performed using Prism 7 (GraphPad Software, San Diego, California). p < 0.05 was considered significant.
Results
CB1R and DOR Form Heteromers in Heterologous Cells
We carried out a proximity ligation assay (PLA) to investigate if CB1R and DOR are in close proximity for interaction, and can form heteromers. We find a PLA signal only in cells coexpressing CB1R and DOR, and not in cells expressing only DOR (Figure 1A), confirming that CB1R and DOR are in close proximity (criteria 1 for receptor heteromerization).43,44 To demonstrate that the CB1R-DOR heteromer exhibits a unique biochemical fingerprint (criteria 2 for receptor heteromerization)43,44 we used CHO cells individually expressing or coexpressing CB1R and DOR. We examined signaling by treating cells with either 1 nM SNC80 or Hu-210 or with a combination of both. We find that a combination of SNC80 and Hu-210 leads to a significantly greater [35S]GTPγS binding as compared to each agonist alone in cells coexpressing both receptors (Figure 1B, right panel). In addition, we find that this increase in signaling is significantly blocked by the CB1R-DOR heteromer selective antibody, but not by the CB1R-AT1R antibodies (Figure 1B, right panel). These results suggest that the ligand-occupied CB1R allosterically modulates the associated DOR activity. To test this further, we examined if the CB1R antagonist, SR141716, can allosterically modulate the associated DOR signaling. As seen with the agonist, SR141716 increased SNC80-mediated signaling in cells coexpressing CB1R and DOR. This increase in signaling could be blocked by the CB1R-DOR, but not by the CB1R-AT1R heteromer selective antibodies (Figure 1 middle and left panels); the antibodies have no effect on signaling in cells expressing only DOR (Figure 1 left panel). These results indicate that the CB1R-DOR heteromers also fulfill criteria 3 for receptor heteromerization in that a heteromer specific reagent (CB1R-DOR heteromer selective antibody) blocks heteromer-specific properties (i.e., potentiation of DOR signaling observed in the presence of a CB1R ligand). Taken together, these studies argue for the notion that CB1R and DOR form heteromers in heterologous cells.
Figure 1.

CB1R-DOR heteromer selective monoclonal antibodies block heteromer-mediated signaling in cells coexpressing both receptors. (A) Schematic representation of proximity ligation assay (PLA) used to demonstrate the presence of CB1R-DOR heteromers (left side). Positive PLA signal (red dots, left panel figure) is seen only with cells coexpressing CB1R and DOR and not in cells where anti-receptor primary antibodies were not used (middle panel figure) or that lack CB1R (right panel figure). (B) [35S]GTPγS binding assay using membranes from CHO cells expressing DOR (left panel) or coexpressing CB1R and DOR (middle and right panels) following treatment with SNC80 (0.1 nM or 1 nM) in the absence or presence of either 10 nM SR141716 (left and middle panels) or 1 nM Hu-210 (right panel). The effect of heteromer-selective antibodies on [35S]GTPγS binding was examined by preincubating membranes with either CB1R-DOR (black bars) or CB1R-AT1R (lined bars) heteromer selective monoclonal antibodies. Basal [35S]GTPγS binding observed in the absence of ligand treatment was taken as 100%. Data represent mean ± SEM (n = 3). * p < 0.05, ***p < 0.001, **** p < 0.0001.
Paclitaxel-Induced Mechanical Allodynia Increases the Abundance of CB1R-DOR Receptor Heteromers in the Superficial Posterior Region of the Lumbar Spinal Cord of Mice
Next, we investigated the involvement of CB1R-DOR complexes in paclitaxel-induced neuropathy. For this, we first treated C57BL/6 male mice with paclitaxel and assessed them for development of mechanical allodynia. We find that treatment with paclitaxel induces a significant decrease in mechanical threshold (mechanical allodynia) that could be detected even 15 days after the first paclitaxel injection (Figure 2A). In contrast, vehicle-treated mice exhibit a slightly higher mechanical threshold throughout the same period in agreement with observations made previously by other investigators.35 Next, we examined the abundance of CB1R and DOR in the lumbar spinal cord of paclitaxel-treated mice and compared it to vehicle-treated mice. There is a significant increase of CB1R (99%) and DOR (40%) protein levels in paclitaxel-treated mouse spinal cords as measured by ELISA. Interestingly, using a CB1R-DOR heteromer selective antibody we also find a significant increase in the levels of this complex (125%) in paclitaxel-treated animals compared to vehicle-treated controls (Figure 2B). These results support the notion that paclitaxel treatment leads to an increase in CB1R-DOR heteromers in the lumbar spinal cord.
Figure 2.

Paclitaxel treatment induces mechanical allodynia and increases CB1R-DOR heteromers levels and signaling in the lumbar spinal cord of mice. (A) Treatment with paclitaxel induces mechanical allodynia in C57BL/6 mice. At day 15 the difference in the mechanical threshold is significantly lower in animals treated with paclitaxel. Vehicle-injected animals show an increase in the mechanical threshold during the study period. ****p < 0.0001 versus vehicle-injected, Student’s test (n = 7 animals/group).(B) ELISA using rabbit polyclonal anti-CB1R antibody (left panel), goat polyclonal anti-DOR antibody (middle panel), and mouse monoclonal anti-CB1R-DOR heteromer selective antibody (right panel) in lumbar sections of the spinal cord of mice treated with paclitaxel to induce neuropathic pain. Vehicle-treated animals were used as controls. Data are mean ± SE (n = 7/group). Statistically significant differences between vehicle and paclitaxel groups are indicated ** p < 0.01, ****p < 0.0001, Student’s t test. (C) CB1R-DOR heteromers (red dots) are detected in the posterior region of the lumbar spinal cord of vehicle- and paclitaxel-treated animals. No PLA signal is detected in the absence of primary anti-receptor antibodies. Quantification of the PLA signal detects a significant increase in CB1R-DOR heteromers in paclitaxel-treated mice; *p < 0.05, Student’s t test (n = 7 animals/group). (D) Membranes from lumbar spinal cords of vehicle- and paclitaxel-treated mice were treated with 1 nM SNC80, 1 nM Hu-210 or both, in the presence or absence of either CB1R-DOR or CB1R-AT1R heteromer selective monoclonal antibodies. Basal [35S]GTPγS binding in the absence of ligand treatment were taken as 100%. Data represent mean ± SEM (n = 7). *p < 0.05, ***p < 0.001, ****p < 0.0001 Student’s t test for comparison between vehicle and paclitaxel, otherwise one-way ANOVA.
To examine if CB1R and DOR are in close proximity for direct interactions in the spinal cord (criteria 1 for heteromerization),43,44 we carried out the proximity ligation assay (PLA) using antibodies to CB1R and DOR (Figure 2C). We find that animals treated with paclitaxel exhibit an increased abundance of the PLA signal compared to vehicle-treated controls (132.4 ± 25.9 vs 55.86 ± 7.7, p < 0.01) (Figure 2C, left and middle panels); this increased PLA signal occurs particularly in somatodendritic and presynaptic regions of neurons and astrocytes (Suppl. Figure 2). Negative controls carried out in the absence of primary antibodies did not detect the presence of a PLA signal (Figure 2C, right panel). These results are consistent with the notion that in the spinal cord of paclitaxel-treated mice there is an increase in the levels of CB1R and DOR, and that the receptors are in close proximity for direct receptor–receptor interactions.
We examined whether the interaction between CB1R and DOR affected receptor signaling. For this we compared agonist-mediated changes in G protein activity in membranes from vehicle-treated and paclitaxel-treated animals, and found comparable levels of [35S]GTPγS binding mediated by 1 nM Hu-210 or 1 nM SNC80 in both groups (Figure 2D). Interestingly, a combination of 1 nM Hu-210 and 1 nM SNC80 led to a significant increase in [35S]GTPγS binding to membranes from paclitaxel-treated animals as compared to vehicle-treated controls (Figure 2D). These results suggest that coactivation of CB1R and DOR in paclitaxel-treated animals leads to potentiation in receptor signaling responses (criteria 2 for receptor heteromerization). Next, we tested the contribution of the heteromer-mediated signaling using CB1R-DOR heteromer selective antibodies. We find that incubation of spinal cord membranes with the CB1R-DOR antibody, but not CB1R-AT1R heteromer selective antibody, selectively blocked the enhanced signaling seen with the combination of Hu-210 and SNC80 (criteria 3 for receptor heteromerization) (Figure 2D). A residual activity of 8 ± 6% was detected in the presence of the CB1R-DOR antibody which was not statistically different from that observed with each drug alone (Figure 2D). Together, these results suggest that paclitaxel treatment increases not only CB1R-DOR heteromer levels but also signaling via the heteromer, and that the agonist-occupied CB1R allosterically modulates the associated DOR activity.
Treatment with a Combination of Low Doses of CB1R and DOR Agonists Attenuates Paclitaxel-Induced Mechanical Allodynia and This Effect Is Blocked by the CB1R-DOR Selective Antibody
Next, we tested if a combination of low doses of Hu-210 and SNC80 could alleviate mechanical allodynia induced by paclitaxel. For this, animals were treated with vehicle or paclitaxel as described previously. Using a within-group treatment design, on day 15 animals were divided into different groups to receive either Hu-210 (i.p. 0.5 μg/kg; treatment 1), SNC80 (i.p. 0.1 mg/kg; treatment 2), a combination of both (i.p.; treatment 3), a combination of both preceded by 5 μL of i.t. anti-CB1R-DOR or anti-Flag antibody (Figure 3A). The treatment order was randomly assigned with a washout period of 7 days after each treatment. Mechanical allodynia was assessed 24 h before (Figure 3B) and 40 min after each individual treatment. Using this paradigm of treatment we find that treatment with Hu-210 or SNC80 alone had a mild effect in the % change of mechanical threshold in vehicle- and paclitaxel-treated animals (10.7 vs 11.2% in the case of Hu-210, and 12.4 vs 13.5% in the case of SNC80). However, a combination of Hu-210 and SNC80 resulted in a statistically significant mechanical threshold difference in the paclitaxel group (43.3%) vs the vehicle group (12.3%), p < 0.018 (Figure 3C). In order to assess if the difference in mechanical threshold by the combination of drugs is mediated by the CB1R-DOR heteromer, we used the CB1R-DOR selective antibody. Pretreatment with the anti-CB1R-DOR antibody significantly decreased the analgesic effect of the combination of the two drugs in the paclitaxel group (40.2%) versus the vehicle group (12.1%), p < 0.05. On the other hand, pretreatment with the anti-Flag antibody (used as control) did not produce a significant decrease in the analgesic effect of both drugs in the paclitaxel group compared to the vehicle group. Taken together, these results support the idea that the CB1R-DOR heteromer could serve as a potential therapeutic target for the treatment of paclitaxel-induced mechanical allodynia, and that a combination of CB1R and DOR agonists could be used to treat paclitaxel-induced neuropathy.
Paclitaxel and Platinum Derivatives Induce an Increase of CB1R and DOR Levels in the Superficial Region of the Human Lumbar Spinal Cord
For CB1R-DOR heteromers to be considered a therapeutic target for the treatment of paclitaxel-induced neuropathy in humans, it is important to first assess the levels of this heteromer in the spinal cord. To do this, we obtained post-mortem human spinal cord sections from human control subjects and from patients diagnosed with CIPN (paclitaxel and/or platinum compounds) (Suppl. Table 1). We carried out PLA to investigate the presence of CB1R-DOR in the superficial region of the lumbar spinal cord. We observed a significant increase in the PLA signal for CB1R-DOR (p < 0.0003) between control (218 ± 31.3 PLA dots) and CIPN (503 ± 55.3 PLA dots) groups (Figure 4 and Suppl. Table 1). In addition, CB1R-DOR PLA levels in the CIPN group did not depend on the type of tumor present or on the drug regimen received (Suppl. Table 1). Taken together, these results demonstrate that CB1R-DOR levels are increased in the spinal cord of cancer patients treated with paclitaxel and/or platinum compounds. This finding supports this heteromer as a novel target for the treatment of neuropathies associated with the use of these compounds.
Figure 4.

CB1R-DOR heteromer levels are significantly increased in patients with chemotherapy-induced peripheral neuropathy. (A) Axial section of the lumbar spinal cord of one deceased subject stained with hematoxylin and eosin (H&E). Upper black square highlights the dorsal region that was used for PLA; lower black square highlights the ventral horn region. (B) Quantification of PLA dots for CB1R-DOR in control (left panel of C) and CIPN patients (right panel of C). ***p < 0.001 control vs paclitaxel Student’s t test (n = 10 controls or 9 CIPN patients). (C) CB1R-DOR heteromers (red dots in color panels, black dots in inverted color panels) are detected in the dorsal region of the lumbar spinal cord of control and chemotherapy-induced neuropathy patients. No PLA signal is detected in the ventral horn of the spinal cord of either control or CIPN patients.
Discussion
Neuropathic pain is a complex disorder that includes a wide variety of causes (e.g., systemic vs isolated diseases), all of them accompanied by injury to the somatosensory system. Contrary to other neuropathies, CIPN has a definite onset time, clear identification and exposure to the toxic drug, and different pathogenic mechanisms leading to neurotoxicity.45 Previous studies have shown that drugs targeting either CB1R or DOR in the spinal cord could attenuate neuropathic pain.46,47 Thus, CB1R and DOR are increasingly becoming an important target for investigation of new drugs to treat pain (reviewed in refs (48 and 49)). However, although the location of CB1R in pain-related nuclei makes it a promising target for pain treatments, its ubiquitous presence throughout the CNS50 makes it difficult for selective targeting to specific regions. In addition, some clinical trials have shown that cannabinoid treatment side-effects could outweigh the benefits provided.51 In the case of DOR, it exhibits a similar distribution and/or higher analgesic profile as MOR; however, the use of moderate to high doses of the DOR agonist, SNC80, has been reported to trigger seizures.52 Given that heteromers would have a restricted tissue distribution compared to receptor homomers, it is possible that targeting the CB1R-DOR heteromer for the development of therapeutics could avoid many of the side-effects observed with drugs targeting individual receptors. In this study we demonstrate that (1) CB1R-DOR heteromers can be identified in the lumbar spinal cord of CIPN mice and human patients, (2) the CB1R-DOR heteromer is mostly located in neurons, (3) CB1R acts as an allosteric modulator of DOR within the CB1R-DOR heteromer leading to a potentiation of signaling when ligands for both receptors are added, (4) that targeting the CB1R-DOR heteromer leads to increased attenuation of neuropathic pain compared to individual receptor–ligand pairs, and (5) that the CB1R-DOR heteromer in the spinal cord plays a crucial role in paclitaxel-induced mechanical allodynia. These results suggest that the CB1R-DOR heteromer constitutes a promising target for the treatment of CIPN.
Previous studies examining levels of CB1R and DOR in the spinal cord during neuropathic pain did not detect an increase in the level of DOR.53−55 In contrast, in the current study we found an increase in the levels of DOR and CB1R-DOR in the dorsal spinal cord of both animals and human patients. This could be due to the different models used to induce neuropathic pain [i.e., nerve ligation model (previous studies) versus CIPN model (current study)]. These findings would be in agreement with observations that CIPN might have a disease-specific signature compared to other causes of neuropathic pain45 and point out the importance of CB1R and DOR in this disorder. Although CB1R and DOR have been reported to be present at the membrane or on fibers impinging upon dorsal horn neurons,56,57 astrocytes, and microglia,58−61 there is limited information about the location of CB1R-DOR heteromers. In this study, we made use of PLA and found that CB1R and DOR are in close enough proximity for direct interactions in neurons, presynaptic fibers, and astroglial cells within the superficial posterior horn of the lumbar spinal cord of mice. Moreover following CIPN, we detect significant increases in CB1R-DOR heteromer levels not only in the dorsal region of the spinal cord (part of the sensory pathway) but also in the basolateral amygdala (BLA) (participates in the processing of polymodal nociceptive information and development of chronic pain state,62Suppl. Figure 3). Interestingly, our results with CIPN human spinal cord samples confirm the significant increase of CB1R-DOR levels seen with tissue from mice treated with paclitaxel. Taken together our observations point to a significant relevance of the CB1R-DOR heteromer in the regulation of neuropathic pain processing.63
Both CB1R and DOR couple to Gαi/o proteins, and the activation of either receptor leads to the inhibition of adenylyl cyclase activity, membrane depolarization, and neurotransmitter release leading to physiological responses such as pain attenuation.14 We previously showed in heterologous cells coexpressing both receptors that the addition of Hu-210 leads to a decrease in Gαi/o signaling engaging both PLC- and arrestin2-mediated pathways to phosphorylate ERK.29 In this study we show that the addition of SNC80 causes a decrease in DOR-mediated Gαi/o signaling when CB1R and DOR are coexpressed, suggesting that CB1R acts as a negative allosteric modulator for DOR signaling. Interestingly, we find that the coadministration of SNC80 with a CB1R agonist (Hu-210) or an antagonist (SR141716) disrupts the negative allosteric modulation of DOR signaling. This is blocked by the CB1R-DOR heteromer-selective antibody. These results would suggest that the CB1R-DOR heteromer is in a distinct conformation when both binding sites are simultaneously occupied; this could explain the differential ability of each receptor to allosterically modulate the partner receptor’s binding and intrinsic efficacy similar to what has been reported for the A2AR-D2R heteromer.64 Moreover, we find that coadministration of low doses of SNC80 in combination with Hu-210 can significantly attenuate neuropathic pain in paclitaxel-treated animals. Although this study focused on the changes within the spinal cord, we cannot exclude an effect of the activation of CB1R-DOR heteromers in the BLA (that are increased following paclitaxel treatment) on the increased attenuation of mechanical allodynia observed with a combination of ligands targeting both receptors. Taken together our results indicate that the CB1R-DOR heteromer could be a novel therapeutic target for the treatment of neuropathic pain associated with CIPN.
Acknowledgments
We thank the Department of Pathology at Mount Sinai Hospital, those who collected the human samples used in this study, as well as patients and their families, whose help and participation made this work possible. We thank Dr. José Luis Lanciego and Elvira Roda for kindly providing images for the CB1R antibody testing. Microscopy analysis was performed at the Microscopy CORE at the Icahn School of Medicine at Mount Sinai. Figures 1, 3, and abstract graphic were composed using Motifolio.com PPT Drawing Toolkits Biology Bundle. Postimaging analysis (i.e., deconvolution) was performed as described42 with ImageJ64 [version 1.48].
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00008.
Validation of antibodies; detection of CB1R-DOR heteromers in the dorsal region of the lumbar spinal cord of paclitaxel treated mice using PLA; CB1R-DOR heteromers increased in the basolateral amygdala of paclitaxel treated mice; summary information on control and patients with chemotherapy-induced peripheral neuropathy (PDF)
Author Present Address
# Department of Physiology & Biophysics, Molecular Medicine Research Building, Virginia Commonwealth University, 1220 East Broad Street Richmond, VA, 23298. Email: Salvador.Sierra@vcuhealth.org.
This work was supported by National Institute of Health Awards DA008863 and NS026880 to L.A.D. S.S. was supported by a grant from Alfonso Martin Escudero Foundation.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Cavaletti G.; Marmiroli P. (2015) Chemotherapy-induced peripheral neurotoxicity. Curr. Opin. Neurol. 28, 500–507. 10.1097/WCO.0000000000000234. [DOI] [PubMed] [Google Scholar]
- Hershman D. L.; Weimer L. H.; Wang A.; Kranwinkel G.; Brafman L.; Fuentes D.; Awad D.; Crew K. D. (2011) Association between patient reported outcomes and quantitative sensory tests for measuring long-term neurotoxicity in breast cancer survivors treated with adjuvant paclitaxel chemotherapy. Breast Cancer Res. Treat. 125, 767–774. 10.1007/s10549-010-1278-0. [DOI] [PubMed] [Google Scholar]
- Cavaletti G.; Zanna C. (2002) Current status and future prospects for the treatment of chemotherapy-induced peripheral neurotoxicity. Eur. J. Cancer 38, 1832–1837. 10.1016/S0959-8049(02)00229-0. [DOI] [PubMed] [Google Scholar]
- Grisold W.; Cavaletti G.; Windebank A. J. (2012) Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro-oncology 14 (Suppl 4), iv45–54. 10.1093/neuonc/nos203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershman D. L.; Lacchetti C.; Dworkin R. H.; Lavoie Smith E. M.; Bleeker J.; Cavaletti G.; Chauhan C.; Gavin P.; Lavino A.; Lustberg M. B.; Paice J.; Schneider B.; Smith M. L.; Smith T.; Terstriep S.; Wagner-Johnston N.; Bak K.; Loprinzi C. L. (2014) American Society of Clinical Oncology. (2014, June 20) Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 32, 1941. 10.1200/JCO.2013.54.0914. [DOI] [PubMed] [Google Scholar]
- Baranowski A., Abrams P., Berger R., Buffington T., Collett B., Emmanuel A., Fall M., Hanno P., Howard F., Hughes J., Nickel C., Nordling J., Tripp D., Vincent K., Wesselmann U., and Williams A. C. d. C. (2018) Classification of Chronic Pain, 2nd ed. revised, IASP Task Force on Taxonomy, Seattle, WA. [Google Scholar]
- Kuner R.; Flor H. (2017) Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 18, 20–30. 10.1038/nrn.2016.162. [DOI] [PubMed] [Google Scholar]
- E R. (2003) Microtubule-Targeting Natural Products, in Cancer Medicine 6 (DW K., JF H., and E F., Eds.) 6 ed., p 2699. B.C. Decker. [Google Scholar]
- Schiff P. B.; Horwitz S. B. (1980) Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. U. S. A. 77, 1561–1565. 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatters S. J. L.; Bennett G. J. (2006) Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 122, 245–257. 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe N. E.; Morfini G.; Brady S. T.; Feinstein S. C.; Wilson L.; Jordan M. A. (2013) Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. NeuroToxicology 37, 231–239. 10.1016/j.neuro.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisse T. S.; Middleton L. J.; Pellegrini A. D.; Martin P. B.; Spaulding E. L.; Lopes O.; Brochu E. A.; Carter E. V.; Waldron A.; Rieger S. (2016) Paclitaxel-induced epithelial damage and ectopic MMP-13 expression promotes neurotoxicity in zebrafish. Proc. Natl. Acad. Sci. U. S. A. 113, E2189–98. 10.1073/pnas.1525096113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty P. M.; Cata J. P.; Cordella J. V.; Burton A.; Weng H.-R. (2004) Taxol-induced sensory disturbance is characterized by preferential impairment of myelinated fiber function in cancer patients. Pain 109, 132–142. 10.1016/j.pain.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Howlett A. C.; Barth F.; Bonner T. I.; Cabral G.; Casellas P.; Devane W. A.; Felder C. C.; Herkenham M.; Mackie K.; Martin B. R.; Mechoulam R.; Pertwee R. G. (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 54, 161–202. 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Ashton C. H. (1999) Adverse effects of cannabis and cannabinoids. Br. J. Anaesth. 83, 637–649. 10.1093/bja/83.4.637. [DOI] [PubMed] [Google Scholar]
- Konturek S. J. (1980) Opiates and the gastrointestinal tract. Am. J. Gastroenterol. 74, 285–291. [PubMed] [Google Scholar]
- Pradhan A. A.; Smith M. L.; Zyuzin J.; Charles A. (2014) δ-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br. J. Pharmacol. 171, 2375–2384. 10.1111/bph.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabli N.; Cahill C. M. (2007) Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 127, 84–93. 10.1016/j.pain.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Petrillo P.; Angelici O.; Bingham S.; Ficalora G.; Garnier M.; Zaratin P. F.; Petrone G.; Pozzi O.; Sbacchi M.; Stean T. O.; Upton N.; Dondio G. M.; Scheideler M. A. (2003) Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8aalpha,8abeta, 12bbeta)]7,10-Dimethyl-1-methoxy-11-(2-methylpropyl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[2,3-g]isoquinoline hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J. Pharmacol. Exp. Ther. 307, 1079–1089. 10.1124/jpet.103.055590. [DOI] [PubMed] [Google Scholar]
- Scherrer G.; Imamachi N.; Cao Y.-Q.; Contet C.; Mennicken F.; O’Donnell D.; Kieffer B. L.; Basbaum A. I. (2009) Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137, 1148–1159. 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R.; Tawfik V. L.; Wang D.; François A.; Solorzano C.; Shuster S. A.; Choudhury P.; Betelli C.; Cassidy C.; Smith K.; de Nooij J. C.; Mennicken F.; O’Donnell D.; Kieffer B. L.; Woodbury C. J.; Basbaum A. I.; MacDermott A. B.; Scherrer G. (2014) Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 81, 1312–1327. 10.1016/j.neuron.2014.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsy-Roy J. A.; Marson L.; Van Loon G. R. (1989) Sympathoadrenal, cardiovascular and blood gas responses to highly selective mu and delta opioid peptides. J. Pharmacol. Exp. Ther. 251, 1096–1103. [PubMed] [Google Scholar]
- Porreca F.; Mosberg H. I.; Hurst R.; Hruby V. J.; Burks T. F. (1984) Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharmacol. Exp. Ther. 230, 341–348. [PubMed] [Google Scholar]
- Pradhan A. A. A.; Walwyn W.; Nozaki C.; Filliol D.; Erbs E.; Matifas A.; Evans C.; Kieffer B. L. (2010) Ligand-directed trafficking of the δ-opioid receptor in vivo: two paths toward analgesic tolerance. J. Neurosci. 30, 16459–16468. 10.1523/JNEUROSCI.3748-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C.; Gomes I.; Devi L. A. (2006) mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br. J. Pharmacol. 148, 387–395. 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S.; Casadó V.; Devi L. A.; Filizola M.; Jockers R.; Lohse M. J.; Milligan G.; Pin J.-P.; Guitart X.; Mattson M. P. (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol. Rev. 66, 413–434. 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narang S.; Gibson D.; Wasan A. D.; Ross E. L.; Michna E.; Nedeljkovic S. S.; Jamison R. N. (2008) Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J. Pain 9, 254–264. 10.1016/j.jpain.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Le Naour M.; Akgün E.; Yekkirala A.; Lunzer M. M.; Powers M. D.; Kalyuzhny A. E.; Portoghese P. S. (2013) Bivalent ligands that target μ opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J. Med. Chem. 56, 5505–5513. 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R. R.; Bushlin I. I.; Gomes I. I.; Tzavaras N. N.; Gupta A. A.; Neves S. S.; Battini L. L.; Gusella G. L. G.; Lachmann A. A.; Ma’ayan A. A.; Blitzer R. D. R.; Devi L. A. L. (2012) Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS One 7, e29239-e29239 10.1371/journal.pone.0029239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushlin I.; Rozenfeld R.; Devi L. A. (2010) Cannabinoid-opioid interactions during neuropathic pain and analgesia. Curr. Opin. Pharmacol. 10, 80–86. 10.1016/j.coph.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimans J. J.; Vermorken J. B.; Wolbers J. G.; Eeltink C. M.; Meijer O. W.; Taphoorn M. J.; Beijnen J. H. (1994) Paclitaxel (Taxol) concentrations in brain tumor tissue. Ann. Oncol. 5, 951–953. 10.1093/oxfordjournals.annonc.a058736. [DOI] [PubMed] [Google Scholar]
- McKeage M. J.; Hsu T.; Screnci D.; Haddad G.; Baguley B. C. (2001) Nucleolar damage correlates with neurotoxicity induced by different platinum drugs. Br. J. Cancer 85, 1219–1225. 10.1054/bjoc.2001.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan S. R.; Bach F. W.; Pogrel J. W.; Chung J. M.; Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Richner M.; Bjerrum O. J.; Nykjaer A.; Vaegter C. B. (2011) The spared nerve injury (SNI) model of induced mechanical allodynia in mice. J. Visualized Exp. e3092-e3092 10.3791/3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Guindon J.; Cornett B. L.; Makriyannis A.; Mackie K.; Hohmann A. G. (2015) Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol. Psychiatry 77, 475–487. 10.1016/j.biopsych.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieselbach R. E.; Di Chiro G.; Freireich E. J.; Rall D. P. (1962) Subarachnoid distribution of drugs after lumbar injection. N. Engl. J. Med. 267, 1273–1278. 10.1056/NEJM196212202672502. [DOI] [PubMed] [Google Scholar]
- Hylden J. L.; Wilcox G. L. (1980) Intrathecal morphine in mice: a new technique. Eur. J. Pharmacol. 67, 313–316. 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Gomes I. I.; Gupta A. A.; Devi L. A. L. (2013) G-protein-coupled heteromers: regulation in disease. Methods Enzymol. 521, 219–238. 10.1016/B978-0-12-391862-8.00012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I.; Filipovska J.; Devi L. A. (2003) Opioid receptor oligomerization. Detection and functional characterization of interacting receptors. Methods Mol. Med. 84, 157–183. 10.1385/1-59259-379-8:157. [DOI] [PubMed] [Google Scholar]
- Gomes I.; Jordan B. A.; Gupta A.; Trapaidze N.; Nagy V.; Devi L. A. (2000) Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 20, RC110. 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Mulder J.; Gomes I.; Rozenfeld R.; Bushlin I.; Ong E.; Lim M.; Maillet E.; Junek M.; Cahill C. M.; Harkany T.; Devi L. A. (2010) Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci. Signaling 3, ra54–ra54. 10.1126/scisignal.2000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I.; Sierra S.; Devi L. A. (2016) Detection of Receptor Heteromerization Using In Situ Proximity Ligation Assay. Curr. Protoc. Pharmacol. 75, 2.16.1–2.16.31. 10.1002/cpph.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre S.; Baler R.; Bouvier M.; Caron M. G.; Devi L. A.; Durroux T.; KK F.; George S. R.; Javitch J. A.; Lohse M. J.; Mackie K.; Milligan G.; Pfleger K. D.; Pin J. P.; Volkow N. D.; Waldhoer M.; Woods A. S.; Franco R. (2009) Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 5, 131–134. 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I.; Ayoub M. A.; Fujita W.; Jaeger W. C.; Pfleger K. D. G.; Devi L. A. (2016) G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 56, 403–425. 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmiroli P.; Cavaletti G. (2016) Drugs for the treatment of peripheral neuropathies. Expert Opin. Pharmacother. 17, 381–394. 10.1517/14656566.2016.1120719. [DOI] [PubMed] [Google Scholar]
- Aceves M.; Mathai B. B.; Hook M. A. (2016) Evaluation of the effects of specific opioid receptor agonists in a rodent model of spinal cord injury. Spinal Cord 54, 767–777. 10.1038/sc.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Tai L.; Qiu Q.; Mitchell R.; Fleetwood-Walker S.; Joosten E. A.; Cheung C. W. (2017) Endocannabinoid activation of CB1 receptors contributes to long-lasting reversal of neuropathic pain by repetitive spinal cord stimulation. Eur. J. Pain 21, 804–814. 10.1002/ejp.983. [DOI] [PubMed] [Google Scholar]
- Spahn V.; Stein C. (2017) Targeting delta opioid receptors for pain treatment: drugs in phase I and II clinical development. Expert Opin. Invest. Drugs 26, 155–160. 10.1080/13543784.2017.1275562. [DOI] [PubMed] [Google Scholar]
- Whiting P. F.; Wolff R. F.; Deshpande S.; Di Nisio M.; Duffy S.; Hernandez A. V.; Keurentjes J. C.; Lang S.; Misso K.; Ryder S.; Schmidlkofer S.; Westwood M.; Kleijnen J. (2015) Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA 313, 2456–2473. 10.1001/jama.2015.6358. [DOI] [PubMed] [Google Scholar]
- Herkenham M.; Lynn A. B.; Little M. D.; Johnson M. R.; Melvin L. S.; de Costa B. R.; Rice K. C. (1990) Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U. S. A. 87, 1932–1936. 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R.; Kristensen P. K.; Bartels E. M.; Bliddal H.; Astrup A. (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370, 1706–1713. 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- Chung P. C. S.; Boehrer A.; Stephan A.; Matifas A.; Scherrer G.; Darcq E.; Befort K.; Kieffer B. L. (2015) Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav. Brain Res. 278, 429–434. 10.1016/j.bbr.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosier B.; Doyen P. J.; Brolet A.; Muccioli G. G.; Ahmed E.; Desmet N.; Hermans E.; Deumens R. (2015) Inhibition of the regulator of G protein signalling RGS4 in the spinal cord decreases neuropathic hyperalgesia and restores cannabinoid CB1 receptor signalling. Br. J. Pharmacol. 172, 5333–5346. 10.1111/bph.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I.; Parkitna J. R.; Korostynski M.; Makuch W.; Kaminska D.; Przewlocka B.; Przewlocki R. (2009) Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 141, 283–291. 10.1016/j.pain.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Stevens C. W.; Kajander K. C.; Bennett G. J.; Seybold V. S. (1991) Bilateral and differential changes in spinal mu, delta and kappa opioid binding in rats with a painful, unilateral neuropathy. Pain 46, 315–326. 10.1016/0304-3959(91)90114-D. [DOI] [PubMed] [Google Scholar]
- Mansour A.; Fox C. A.; Akil H.; Watson S. J. (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 18, 22–29. 10.1016/0166-2236(95)93946-U. [DOI] [PubMed] [Google Scholar]
- Veress G.; Meszar Z.; Muszil D.; Avelino A.; Matesz K.; Mackie K.; Nagy I. (2013) Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct. Funct. 218, 733–750. 10.1007/s00429-012-0425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral G. A.; Marciano-Cabral F. (2005) Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J. Leukocyte Biol. 78, 1192–1197. 10.1189/jlb.0405216. [DOI] [PubMed] [Google Scholar]
- Caruso P.; Naccarato M.; Faoro V.; Pracella D.; Borando M.; Dotti I.; Koscica N.; Stanta G.; Pizzolato G.; Manganotti P. (2016) Expression of the Endocannabinoid Receptor 1 in Human Stroke: An Autoptic Study. J. Stroke Cerebrovasc Dis 25, 2196–2202. 10.1016/j.jstrokecerebrovasdis.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Duan Y.-L.; Wang S.-Y.; Zeng Q.-W.; Su D.-S.; Li W.; Wang X.-R.; Zhao Z. (2011) Astroglial reaction to delta opioid peptide [D-Ala2, D-Leu5] enkephalin confers neuroprotection against global ischemia in the adult rat hippocampus. Neuroscience 192, 81–90. 10.1016/j.neuroscience.2011.06.067. [DOI] [PubMed] [Google Scholar]
- Shrivastava P.; Cabrera M. A.; Chastain L. G.; Boyadjieva N. I.; Jabbar S.; Franklin T.; Sarkar D. K. (2017) Mu-opioid receptor and delta-opioid receptor differentially regulate microglial inflammatory response to control proopiomelanocortin neuronal apoptosis in the hypothalamus: effects of neonatal alcohol. J. Neuroinflammation 14, 83. 10.1186/s12974-017-0844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitler A.; Kamoun N.; Goyon S.; Wahis J.; Charlet A.; Poisbeau P.; Darbon P. (2016) Favouring inhibitory synaptic drive mediated by GABA(A) receptors in the basolateral nucleus of the amygdala efficiently reduces pain symptoms in neuropathic mice. Eur. J. Neurosci 43, 1082–1088. 10.1111/ejn.13217. [DOI] [PubMed] [Google Scholar]
- Tsuda M. (2018) Modulation of Pain and Itch by Spinal Glia. Neurosci. Bull. 82, 522. 10.1007/s12264-017-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventura J.; Navarro G.; Casadó-Anguera V.; Azdad K.; Rea W.; Moreno E.; Brugarolas M.; Mallol J.; Canela E. I.; Lluis C.; Cortés A.; Volkow N. D.; Schiffmann S. N.; Ferré S.; Casadó V. (2015) Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc. Natl. Acad. Sci. U. S. A. 112, E3609–18. 10.1073/pnas.1507704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.