

Abstract
Objectives
Blood biomarkers for cerebral tissue ischemia are lacking. The goal was to identify a blood transcriptomic signature jointly identified in the ischemic brain.
Methods
A nonhuman primate model with middle cerebral artery (MCA) territory infarction was used to study gene expression by microarray during acute ischemic cerebral stroke in the brain and the blood. Brain samples were collected in the infarcted and contralateral non‐infarcted cortex as well as blood samples before and after occlusion. Gene expression was compared between the two brain locations to find differentially expressed genes. The expressions of these genes were then compared in the blood pre‐ and post‐occlusion.
Results
Hierarchical clustering of brain expression data revealed strong independent clustering of ischemic and nonischemic brain samples. The top five enriched, up‐regulated gene sets in the brain were TNF α signaling, apoptosis, P53 pathway, hypoxia, and UV response up. A comparison of differentially expressed genes in the brain and blood revealed a significant overlap of gene expression patterns. Stringent analysis of blood expression data from pre‐ and post‐occlusion samples in each monkey identified nine genes highly differentially expressed in both the brain and the blood. Many of these up‐regulated genes belong to pathways involved in cell death and DNA damage repair.
Interpretation
Common gene expression profile can be identified in the brain and blood and clearly differentiates ischemic from nonischemic conditions. Therefore, specific blood transcriptomic signature may represent a surrogate for brain ischemic gene expression.
Introduction
Blood‐based biomarkers of ischemic stroke present a considerable challenge as blood lacks direct contact with the brain. With the exception of circumventricular organs characterized by their high permeability capillaries and including midline structures around the third and fourth ventricle, the brain has the blood brain barrier (BBB) which consists of two major elements: A BBB for solutes is formed by specialized capillary endothelial cells that are joined by complex tight junctions and the BBB that regulates the entry of immune cells into the central nervous system (CNS) in postcapillary venules.1
Biomarkers refer to imaging, chemical, or other biological tests that can be used to measure the presence or progress of disease or the effects of treatment.
Increasing evidence suggests that peripheral proteins, nucleic acids, or lipids can be used to confirm diagnosis of ischemic stroke and to monitor disease progression. To date, however, none have been implemented in clinical practice. Protein biomarkers have been more widely investigated, but no diagnostic test has proven to be perfectly accurate, because of low sensitivity or specificity.2
The use of RNA in blood as a diagnostic marker is an emerging field that is supported by its clinical application in the diagnosis of breast cancer, coronary artery disease, and infectious disease.3
Brain transcriptomic analysis has been used in many experimental studies with cerebral ischemia in the brain to measure gene expression changes. Most experimental transcriptomic studies were performed in rat and mouse on focal or global brain ischemia models.4 However, to our knowledge few experiments were performed in nonhuman primates.5, 6
Blood transcriptomic studies in patients with stroke has already been performed from peripheral blood mononuclear cells and whole blood with different goals7 (1) to differentiate ischemic from controls8, 9, 10, 11; (2) to identify different mechanisms of stroke, that is, large vessel versus embolic stroke, lacunar versus non‐lacunar stroke, to identify cryptogenic strokes12, 13, 14; (3) to differentiate transient ischemic attacks (TIA) from controls.15
Combined brain and blood transcriptomics in humans with focal ischemic stroke in the same individual is not feasible for obvious reasons but would theoretically provide valuable information on the potential to identify a common transcriptomic signature, therefore opening the possibility to identify potential blood biomarkers as a surrogate for brain ischemia mechanisms.
To test this hypothesis, we studied the gene expression pattern in tissue undergoing cerebral ischemia as well as the gene expression in the blood of nonhuman primates.
Primates are unique models to study brain ischemia since they have highly similar genomes to human as well as anatomical homology; for example, they both have non‐lissencephalic brain as opposed to rodents. Furthermore, the transcriptomes of the cerebral cortex, in both human and chimpanzee, are very similar to each other and differ more between individuals than among cerebral regions within an individual.16
Methods
Animal experiment
Experiments were performed in two male rhesus macaques (Macaca mulatta) aged 12–13 years. An experimental protocol was submitted to the Regional Ethics Committee for Animal Experimentation (Normandy) and approval was granted to conduct the study (referral No. N/02‐03‐08/03/02‐11). Experiments were performed by licensed investigators (C.O.) and in accordance with French ethical laws (act No. 87‐848; Ministère de l’Agriculture et de la Forêt) and European Communities Council Directives (2010/63/EU) guidelines for the care and use of laboratory animals. During the course of the present studies, the monkeys were housed at the Cyceron Research Centre (Establishment for Animal Experimentation, agreement No. B14118001) in individual cages maintained at 24°C with 50% relative humidity on a 12‐h/12‐h light/dark cycle and were fed with commercial chow supplemented with fresh fruits and water ad libitum. Throughout the duration of these studies, a veterinary surgeon was available to oversee the well‐being of the animals.
Thrombotic Rhesus Macaque model
Experiments were performed as previously described.17
In vivo MRI acquisition
Monkeys were studied in a 3T clinical MRI (Philips Sense Flex M). Imaging was performed in the axial and coronal plane and included the following sequences: 3D‐time‐of‐flight angiography, T2‐weighted, fluid attenuation inversion recovery (FLAIR), diffusion‐weighted imaging (DWI), and pre‐ and post‐contrast T1‐weighted and perfusion‐weighted imaging (PWI).
Blood samples
Blood samples were obtained at T0 before surgery but after general anesthesia, and at T1:2H00, T2:3H30, and T3:4H30 after MCA occlusion. For each blood sample, 2.5 mL was drawn for RNA.
Brain samples
Twelve brain samples were taken from each monkey. In each monkey, six samples were taken near the infarction and six samples from the corresponding location in the other hemisphere. A total of 12 ischemic and 12 nonischemic brain samples were analyzed. Two monkeys were operated and sacrificed around 5:30 h after the onset of occlusion (S3: 5:12; S2: 5:35). An MRI was performed around 3 h after occlusion onset (S3: 03:10; S2: 3:05). Animal was infused by intracardiac injection after thoracotomy by a total volume of 8 L of 4°C saline serum. The brain was then removed after craniotomy and placed into a mold specifically designed for coronal brain slicing. Each cut of the brain was further placed onto a grid to be able to identify X and Y coordinates on the ischemic and contralateral hemisphere. Arbitrarily, on the grid, the Roman numerals were for the right hemispheric ischemic side and Arabic numerals for left nonischemic hemisphere side. Ischemia was visible to the naked eye (Fig. 1D) and confirmed by hypometabolic ischemic tissue treated by tetrazolium (2,3,5‐triphenyltetrazolium Chloride 1%) which appeared white compared to the pinkish staining obtained with the nonischemic contralateral hemisphere. Three ischemic cortical samples per animal were prepared: one potentially corresponding to the core, and two from the edge, corresponding potentially to a penumbral region. Three homologous samples from the contralateral hemisphere per animal were also prepared.
Figure 1.
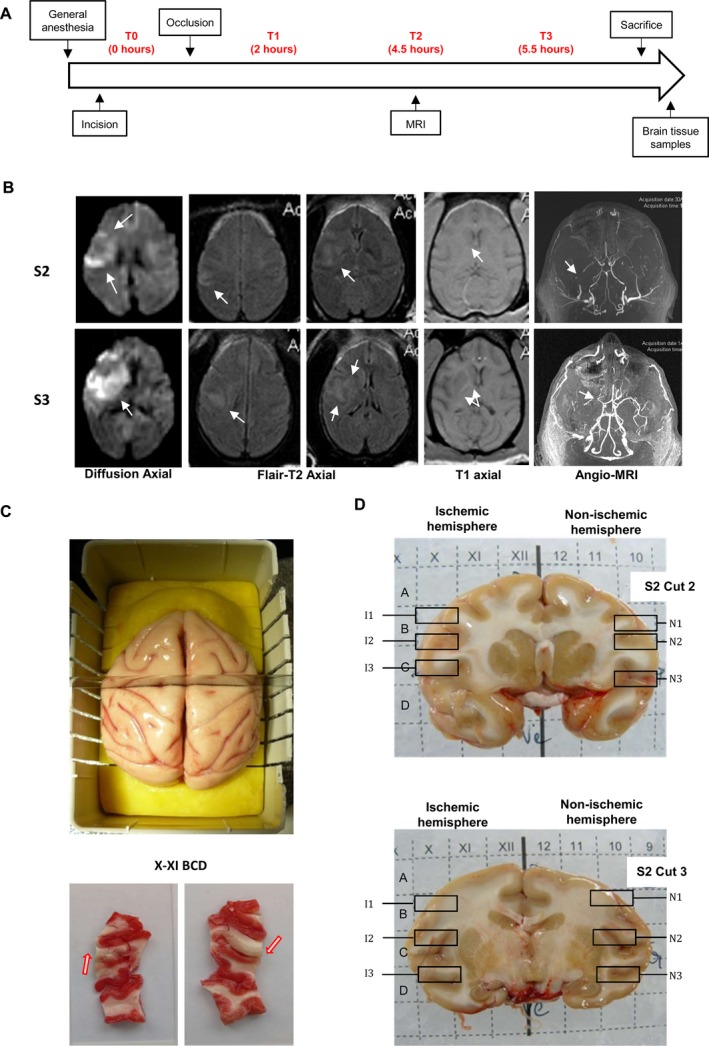
Experimental design, cerebral MRI, and tissue processing. (A) Experimental design. T0 blood sample before MCA (Middle cerebral artery) occlusion and represents the time right before incision of the animal but after general anesthesia. T1, T2, and T3 were blood sample after MCA occlusion. (B) Magnetic Resonance Imaging was performed 3 h after occlusion and includes Diffusion‐weighted imaging (DWI), Flair‐T2, T1, and angio‐MRI. Note on S3 animal the hypersignal (arrow) in the cortex and caudate nucleus on DWI and Flair‐T2 imaging. On S2 animal, the volume appears to be smaller compared to S3 either on DWI or Flair‐T2. Angio‐MRI of Willis circle showed the absence of arterial flux on right MCA artery (arrow) compared the left one. (C) Tissue processing. Upper part: brain placed into a mold for coronal brain slicing; Lower part: Adjacent cut of cortex (between column X and XI extending on three lines: BCD) studied for metabolic activity by triphenyltetrazolium chloride (TTC). Healthy tissue appeared in pink and ischemic cortex in white (arrow). Absence of TTC reflecting ischemia is differentiated from juxtacortical white matter (myelin). (D) Brain samples for Microarrays. Two coronal cuts of S2 brain (C2 and C3), taken as example, were placed onto a grid to identify X and Y coordinates. Arbitrarily, the Roman numeral was for the right ischemic brain and Arabic numerals for the left nonischemic hemisphere. (A–D) represent the different line. I1 (XB), I2 (XC) and I3 (XD) represent brain sample from the ischemic cortex and N1, N2, N3 represent the homologous nonischemic cortex samples.
Total RNA extraction
RNA was isolated from cerebral cortex using the RNeasy Microarray Tissue kit following the manufacturer's instructions (Qiagen). RNA was isolated from whole blood samples using PAXgene Blood RNA Kit (PreAnalytix). Globin mRNA was removed from total RNA using the GlobinClear kit (Ambion).
Macaca expression microarray and choice of samples
RNA from cerebral cortex (30 ng) and from blood sample (30 ng) were labelled using Low Input Quick Amp WT Labeling kit (Agilent Technologies). RNA spike‐in controls were used to adjust possible dye effects. RNA was converted to cDNA. T7 RNA polymerase was used for the synthesis and labelling of cRNA with Cy3. An equal amount (3.75 μg) of Cy3 cRNA probes was hybridized on 4 × 44 k Agilent DNA chip (catalogue# G2519F, Macacca mulatta). Hybridization was performed for 17 h. Hybridization images were obtained using Agilent DNA microarray scanner and intensity data were extracted using Feature Extraction software (Agilent Technologies). This array contains 43,803 rhesus macaque monkey probes. These probes are sourced from RefSeq (Release 37, Oct 2009), Unigene (Release 13, Oct 2009), UCSC MRNA (Oct 2009), Ensembl (Release 56, Sep 2009), UCSC RheMac2 (Jan 2006). Many probes are predicted based on orthologous human genes. Additionally, some probes are annotated as only Macaca mulatta cDNA, and others can be inferred from homology with human genes . Analysis was performed in collaboration with Genosplice, a company specializing in transcriptomic analysis.
Microarray analysis
Differential expression analysis of the Agilent microarray expression data was performed using limma18 from the Bioconductor project. Raw data were normalized by first performing background correction and then normalizing between arrays for all brain and blood samples also using limma. Probes were annotated using Agilent’s array information provided on Array Express (https://www.ebi.ac.uk/arrayexpress/arrays/A-GEOD-9861/?ref=E-GEOD-20043) and gene information provided by Genosplice. The limma package performs differential expression analysis by first fitting the expression data of each gene to a linear model. It then utilizes Empirical Bayes to borrow information across genes, which allows us to perform analysis across a small number of arrays.19
Accession Code
The raw microarray expression data are available on GEO. Accession code: GSE107452.
Statistics
-
(A) Step 1: Comparison of highly expressed genes in the brain and the blood.
(a) The brains of the two monkeys were analyzed separately. Genes were labeled as “highly differentially expressed” if they had a fold change of more than 2 (log2 fold change greater than 1) in both monkeys when comparing ischemic and nonischemic brain samples, and had a fold change of less than 2 when comparing ischemic samples between monkeys or when comparing nonischemic samples between monkeys (Fig. 2B). The fold change is ischemic expression divided by nonischemic expression. Thus a fold change of 2 means the ischemic expression was twice as high as nonischemic.
(b) Blood samples were analyzed by comparing the pre‐occlusion sample to each post‐occlusion sample. The two monkeys were analyzed separately. The time point with the largest fold change was selected for further analysis and was called “S3 max blood” and “S2 max blood” (Table 1). Genes were labeled as “highly differentially expressed” if they had a fold change more than 1.5 in both monkeys when comparing the pre‐occlusion sample to “S3 max blood” and “S2 max blood”. Only highly differentially expressed genes in both ischemic brain and post‐occlusion blood samples as defined above were compared and used to identify common highly differentially expressed genes (Fig. 6).
(B) Step 2: Comparison of highly differentially expressed genes in the brain and blood considering that all brain samples are independent, ignoring monkey effect.
Figure 2.
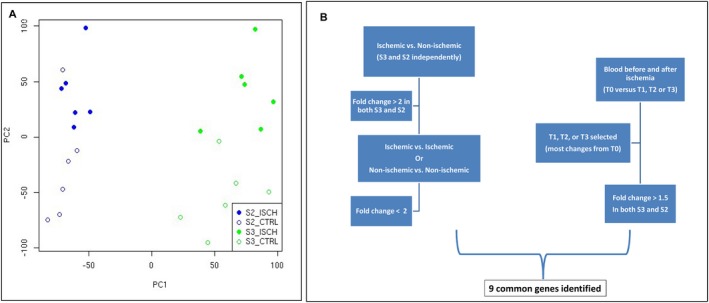
Strategy of analysis and Principal component analysis. Comparison of transcriptome in the brain and the blood. Genes were labeled as “highly differentially expressed” if they had a fold change of more than 2 (log2 fold change greater than 1) in both monkeys when comparing ischemic and nonischemic brain samples, and had a fold change of less than 2 when comparing ischemic samples between monkeys or when comparing nonischemic samples between monkeys. Of the genes identified as highly differentially expressed in the brain, nine were also differentially expressed in the blood (fold change ≥ 1.5). Principal component analysis of the expression data on the two monkeys. The first two axes of variation (PC1 and PC2) are shown. The plots corresponding to the different monkeys are in different colors with the data from the ischemic side of the brain shown in filled circles.
Table 1.
Highly differentiated genes in ischemic brain and blood samples S3 and S2.
| Gene symbol | S3 brain | S2 brain | S3 max blood | S3 timepoint | S2 max blood | S2 timepoint |
|---|---|---|---|---|---|---|
| HSPA1B | 4.67 | 5.40 | 1.86 | s3.T0vsT3 | 0.99 | s2.T0vsT1 |
| LOC720054 (heat shock 70 kDa protein 1) | 4.51 | 5.25 | 1.70 | s3s3.T0vsT3 | 0.53 | s2.T0vsT1 |
| NPAS4 | 3.57 | 2.65 | 0.05 | s3.T0vsT2 | 0.06 | s2.T0vsT2 |
| LOC718890 (DNAJB1) | 3.37 | 4.16 | 1.12 | s3.T0vsT1 | 0.22 | s2.T0vsT1 |
| ATF3 | 2.99 | 3.38 | 0.27 | s3.T0vsT1 | −0.04 | s2.T0vsT1 |
| HSPB1 | 2.85 | 3.30 | −0.63 | s3.T0vsT2 | −0.15 | s2.T0vsT3 |
| RRAD | 2.74 | 4.26 | 0.81 | s3.T0vsT3 | −0.03 | s2.T0vsT1 |
| NR4A1 | 2.72 | 2.88 | 0.03 | s3.T0vsT1 | −0.27 | s2.T0vsT3 |
| CYR61 | 2.61 | 2.34 | 0.11 | s3.T0vsT2 | 0.17 | s2.T0vsT3 |
| C‐FOS | 2.53 | 3.77 | 0.17 | s3.T0vsT2 | 0.45 | s2.T0vsT2 |
| GADD45G | 2.25 | 2.04 | 0.44 | s3.T0vsT3 | 0.87 | s2.T0vsT3 |
| RGS3 | 2.01 | 2.04 | 0.23 | s3.T0vsT1 | 0.57 | s2.T0vsT2 |
| LOC714407 | 1.84 | 1.76 | −0.19 | s3.T0vsT2 | 0.28 | s2.T0vsT3 |
| ARC | 1.84 | 1.90 | 0.27 | s3.T0vsT3 | 0.08 | s2.T0vsT3 |
| PTGS2 | 1.84 | 1.41 | 3.09 | s3.T0vsT2 | 3.78 | s2.T0vsT3 |
| RGS2 | 1.81 | 1.79 | 0.10 | s3.T0vsT2 | 0.81 | s2.T0vsT2 |
| CCL3 | 1.78 | 2.18 | −0.49 | s3.T0vsT1 | 0.00 | s2.T0vsT3 |
| BAG3 | 1.70 | 2.97 | 0.99 | s3.T0vsT1 | 0.71 | s2.T0vsT1 |
| HSPA4L | 1.68 | 1.64 | −0.13 | s3.T0vsT1 | 0.30 | s2.T0vsT3 |
| EGR2 | 1.68 | 2.09 | −0.12 | s3.T0vsT2 | −0.16 | s2.T0vsT3 |
| ADM | 1.66 | 2.59 | 2.60 | s3.T0vsT3 | 0.86 | s2.T0vsT3 |
| TM4SF1 | 1.61 | 2.42 | 0.65 | s3.T0vsT3 | 1.01 | s2.T0vsT3 |
| EGR1 | 1.56 | 2.10 | 0.22 | s3.T0vsT1 | 0.07 | s2.T0vsT3 |
| DUSP1 | 1.43 | 1.34 | 2.08 | s3.T0vsT3 | 1.87 | s2.T0vsT2 |
| BTG2 | 1.42 | 1.38 | 0.39 | s3.T0vsT1 | 0.14 | s2.T0vsT2 |
| LOC715456 | 1.36 | 1.14 | 1.33 | s3.T0vsT1 | 0.15 | s2.T0vsT3 |
| HMOX1 | 1.30 | 1.13 | 2.81 | s3.T0vsT3 | 0.83 | s2.T0vsT3 |
| highly similar to human LDLR [CN641580] | 1.29 | 1.12 | 2.60 | s3.T0vsT1 | 1.56 | s2.T0vsT1 |
| DNAJA4 | 1.28 | 1.65 | 0.56 | s3.T0vsT3 | −0.03 | s2.T0vsT1 |
| MCL1 | 1.24 | 1.52 | 0.56 | s3.T0vsT3 | 0.34 | s2.T0vsT1 |
| LOC720001 | 1.20 | 1.07 | 0.31 | s3.T0vsT1 | 0.08 | s2.T0vsT1 |
| GADD45B | 1.08 | 2.17 | 0.46 | s3.T0vsT2 | 0.94 | s2.T0vsT2 |
| IL6 | 1.08 | 1.53 | −0.47 | s3.T0vsT3 | −0.10 | s2.T0vsT2 |
| ADFP | 1.08 | 1.25 | 0.61 | s3.T0vsT1 | 0.34 | s2.T0vsT1 |
| HES4 | 1.08 | 1.18 | −0.04 | s3.T0vsT1 | 0.13 | s2.T0vsT1 |
| DUSP5 | 1.05 | 1.67 | −0.30 | s3.T0vsT1 | −0.78 | s2.T0vsT1 |
| GEM | 1.04 | 1.55 | 0.39 | s3.T0vsT2 | 0.19 | s2.T0vsT1 |
| LOC717581 (similar to G0S2) | 1.00 | 1.96 | 1.90 | s3.T0vsT2 | 1.47 | s2.T0vsT3 |
Of the genes identified as highly differentially expressed in the brain, nine were also differentially expressed in the blood (fold change ≥ 1.5; highlighted in blue). All nine of these genes occur within the most significant hypergeometric overlap (bolded). T0 refers to pre‐occlusion, T1, T2 or T3 refer to time after occlusion (see Fig. 1). HSPA1B and LOC720054 represent duplicate probes for the same gene.
Figure 6.
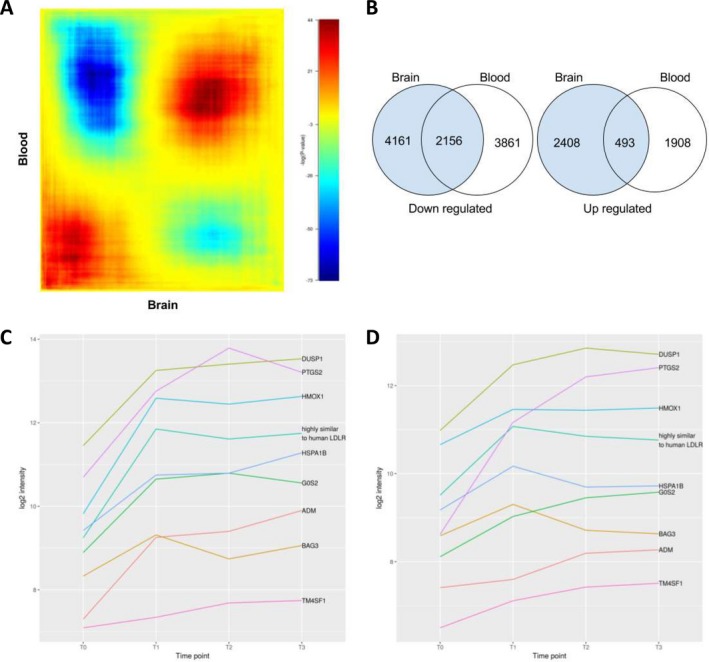
Overlaps of expressed genes in the brain and blood. (A) Heatmap of all possible overlaps of brain differentially expressed genes and blood differentially expressed genes (step size of 10,000).The log10‐transformed hypergeometric P‐values are indicated in the color scale bar with negative values indicating under enrichment. (B) Venn diagram of the most significant overlap between the two gene sets. Fisher's exact test based on the numbers displayed in the Venn diagram of Figure 6B found the overlap between the down‐regulated genes in the two tissues (2156 genes) highly significant (P = 5.43e‐20) as well as the overlap between the up‐regulated genes (493 genes, P = 7.79e‐19). For both tests, we used a background value of 20,217 (i.e., number of distinct genes on the array). Blood gene intensity kinetic in the blood for the 9 top overlapping expressed genes in the brain and blood in S3 (C) and S2 (D) between different time points: before (TO) and after (T1, T2 and T3) ischemia.
We also analyzed the data by considering that the different samples taken from a same monkey were independent. The same limma differential expression analysis was applied to identify differentially expressed genes between ischemic (12 samples) and nonischemic brain samples (12 samples). For blood, analysis was performed by comparing the six samples taken after occlusion against the two samples taken at T0. The results were corrected for multiple testing using the Benjamini–Hochberg method (Tables S1 and S2).
Quality control of data
Principal component analysis (PCA)
PCA was performed on the expression data to see how they cluster using the R prcomp function (R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria).
Internal validation in the brain
A probe‐specific two‐step TaqMan R Gene Expression Assay was used to validate microarray results (Applied Biosystems). Genes for validation were chosen based on ranking of differential expression and biological annotation relevant to ischemia. Since SMC2 showed a constant expression in ischemic and nonischemic samples we chose it for normalization of all target genes. Gene Expression Assay probe IDs were as follows: HSPA1B (A 01 P010726), GADD45G (A 01 P018040), CDKN1A (A 01 P002585), SMC2 (A 01 P019124). One hundred nanograms of total RNA of each sample was used to generate cDNA using the SuperScript® III First‐Strand Synthesis Kit (ThermoFisher scientific) following the manufacturer’s protocol. Real‐time PCR reactions were carried out on the Roche LightCycler R 480 System. Gene expressions were compared between ischemic and nonischemic tissues using the comparative CT method (∆∆CT Method) with the Mann–Whitney U test (Wilcoxon), utilizing Prism software v6.0c (GraphPad, La Jolla, CA).
External validation
We attempted to verify our results using data from a study by Cook et al.5 This study also examined stroke transcriptomics in gyrencephalic primate, however they used cynomolgus macaques, a close relative of Macaca mulatta.20 In order to validate the results found in Macaca mulatta, we analyzed placebo and nonischemic transcriptomic data from the Cook et al. study. The placebo ischemic and nonischemic data were accessed from GEO (accession: GSE35589) and analyzed using the methods described above.
Brain–blood gene overlap
Significant overlap between brain and blood differential expression results was determined using the rank–rank hypergeometric overlap test.21 This method performs a hypergeometric test on all possible overlaps of the sorted lists of genes in order to identify the cutoff at which the overlap between the two sets is most significant. The full gene lists were sorted by the fold change values of all the ischemic samples versus all the nonischemic samples for the two monkeys. In order to examine specific genes whose expression changed in the brain and the blood, we produced a filtered list of highly differentially expressed genes in the brain (see methods above) and calculated the max fold change value for each of these genes in blood.
A Venn diagram showing the overlap of differentially expressed genes from the different groups is provided as well as the P‐value of the Fisher exact test indicating significance of the overlap between both tissues.
Gene set enrichment analysis and interaction
Gene set enrichment analysis of was performed using the GSEA command line tool.22 This tool provides a predefined set of genes and determines whether each set is enriched near the top or bottom of the sorted experimental list, which is indicative of a phenotypic role. GSEA calculates an enrichment score for each gene set evaluated, this value indicates the degree to which the set is overrepresented at the top or bottom of the sorted experimental gene list. The normalized enrichment score (NES) is normalized by the size of the gene set.
Results from brain and blood differential expression analysis were sorted by log fold change. These lists were passed to GSEA pre‐ranked function. The analysis was run using 10,000 permutations, and gene sets with fewer than 10 genes were excluded. The resulting gene sets were sorted by the normalized enrichment score (NES). Empirical P‐values obtained based on the 10,000 permutations are provided.
The interactions between genes in these gene sets were visualized using the STRING database.23 Genes were entered into String if they had a fold change of at least 2 in the brain, and 1.5 in the blood. The thickness of the lines indicates the confidence that a relationship exists and the large nodes indicate proteins for which there is information about the tertiary structure.
Results
Animals were sacrificed about 5.5 h after ischemia onset (Table S3). Axial MRI diffusion‐weighted imaging showed a MCA focal ischemia in both animals (Fig. 1B). Analysis of the volume of ischemia in the two different animals showed that the volume of infarction was highly variable. S3 had a visibly larger infarction volume than S2. Superficial and deep infarctions were observed for animal S3 and S2. The infarction was also visible on T2 Flair image in both animals. Willis Angio‐MRI showed that MCA was proximally occluded for animal S3 while it was less clear for animal S2. Infarction was also visible on T1 sequence at the level of basal ganglia in animals S3 and S2.
Gene expression changes: ischemic versus nonischemic brain tissues
In this study, changes in expression between ischemic and nonischemic brain tissues were measured for individual genes, and the effect of these changes was examined at the level of gene sets. A differential expression analysis was performed comparing expression data from ischemic brain tissue to data from the corresponding region of the contralateral hemisphere, which did not undergo ischemia.
This analysis revealed that the expression patterns of ischemic and nonischemic brain samples are visibly different (Fig. 3A). When hierarchical clustering was applied to the top differentially expressed genes and the most variable genes regardless of the experimental condition, the ischemic samples clearly cluster independently from the nonischemic samples. Furthermore, monkey S3 had a more profound up‐regulation than monkey S2 in line with S3 having a larger infarction volume than S2.
Figure 3.
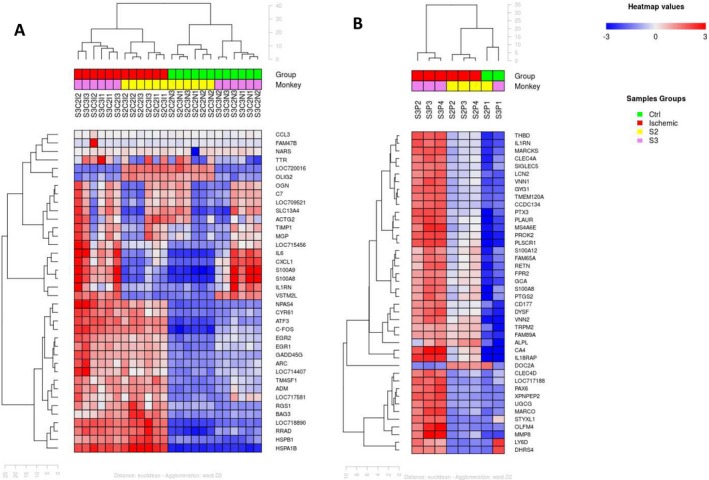
Hierarchical clustering in the brain and the blood. Hierarchical clustering of the top variant genes (regardless of experimental condition) was performed for both brain (A) and blood (B). Ischemic and nonischemic brain samples cluster independently (A). Pre‐occlusion (T0) and post‐occlusion (T1, T2, T3) samples clearly cluster independently –B) for both animals (S2 and S3). For each monkey (S2 & S3), “I” represents ischemic sample, “N” represents nonischemic sample, “C” represents the level of the cut (2 or 3) and 1, 2 or 3 designate different cortical areas. For example: S2C3I2 represents monkey S2, cut C3, ischemia and region 2 of the cortex (XC) (see Figure 1).
All of the highly differentially expressed genes were up‐regulated (Table 1). Pooled analysis of the brain data from both monkeys (Table S1) also found 35 of these 37 genes to be significantly differentially expressed even after adjustment for multiple testing.
Principal component analysis
Principal component analysis of the whole gene expression in the brain showed that the two animals could be differentiated according to the first axis of variation while the second axis was able to differentiate ischemic from nonischemic conditions except for one outlier (blue empty plot) (Fig. 2A).
Internal validation
To confirm the robustness of the microarray results, mRNA levels of three ischemic‐sensitive genes (HSPA1B, GADD45G, CDKN1A) were quantified by RT‐qPCR using samples from the animals. SMC2, which was found to exhibit invariant expression level across all test samples, was used as internal reference (housekeeping). Three reverse transcriptions were performed for each RNA samples followed by three independent qPCR runs, with replicate assay measurements for both target and reference genes. For all genes, the direction and magnitude of the change agreed well with the microarray data (Fig. 4).
Figure 4.
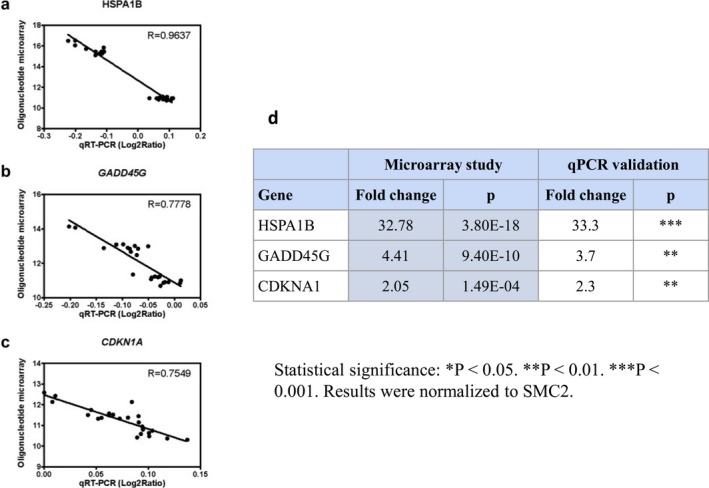
Internal validation. Correlation between mRNA levels and microarray intensity values for HSPA1B (A), GADD45G (B) and CDKN1A (C) by qRT‐PCR. (A–C) Semi‐logarithmic scale; X axis: Log2 ratio of qRT‐PCR mRNA normalized for SMC2; Y axis: Oligonucleotide microarray signal. P value from Mann–Whitney U test. (D) Comparison of fold change between microarray study and qRT‐PCR mRNA of HSPA1B, GADD45G, and CDKN1A.
External validation
Analysis of transcriptome data from the Cook et al. study of stroke5 in Macaca fascicularis produced very different results than what has been described here. Whereas our analysis of Macaca mulatta revealed many up‐regulated genes in the tissues affected by stroke, the data from Macaca fascicularis showed mainly down‐regulated genes. This is mostly likely due to the very different method for inducing stroke in the primates. The study by Cook et al.5 used surgical middle cerebral artery occlusion (MCAO), which they acknowledge produces a more severe stroke than is usually observed in human. It is likely that this more severe stroke model caused massive cell death resulting in mainly down‐regulated genes.
Gene sets enrichment
Gene sets that are significantly enriched in ischemic brain tissue were identified using the Broad Institute’s Gene Set Enrichment Analysis (GSEA).22 The top five enriched gene sets from GSEA’s Hallmark gene sets for the brain were: TNF‐α signaling via NF‐κB, P53 pathway, apoptosis, hypoxia, and UV response up, all of which were up‐regulated (Table 2).
Table 2.
The top five gene sets resulting from GSEA analysis differential expression results in the brain and blood.
| Gene set name | Set size | Genes present (%) | NES | FWER P‐value |
|---|---|---|---|---|
| In the brain | ||||
| TNF‐α signaling via NF‐κB | 108 | 60 (55.56%) | 2.9711735 | <2.10‐4 |
| P53 pathway | 112 | 28 (25%) | 2.5203888 | <2.10‐4 |
| Apoptosis | 98 | 26 (26.53%) | 2.4833248 | <2.10‐4 |
| Hypoxia | 118 | 33 (27.97%) | 2.4749684 | <2.10–4 |
| UV response up | 96 | 31 (32.29%) | 2.425803 | <2.10‐4 |
| In the blood | ||||
| Hypoxia | 118 | 42 (25.59%) | 1.9014156 | 7.0 .10‐4 |
| TNF‐α signaling via NF‐κB | 108 | 37 (34.36%) | 1.8912005 | 8.0E‐4 |
| IL6 JAK STAT3 signaling | 43 | 20 (46.5%) | 1.8102732 | 0.0035 |
| Inflammatory response | 110 | 35 (31.82%) | 1.7960621 | 0.0046 |
| Hedgehog signaling | 24 | 10 (41.67%) | 1.7309599 | 0.0162 |
NES, normalized enrichment score; GSEA, Gene Set Enrichment Analysis 10; FWER, Family‐wise error rate.
Interaction of genes products
The STRING database23 was used in order to visualize the interaction of the gene products of genes that are highly differentially expressed in the brain and blood within the top gene sets identified in the brain (fold change ≥ 2 in the brain, fold change ≥ 1.5 in the blood) (Fig. 5). It was noted that IL6 is a highly connected member in the networks representing TNF‐α signaling via NF‐κB, apoptosis, and hypoxia. The gene ATF3 also appears in all four networks and it interacts with IL6 in the three networks mentioned above. The fifth ranking gene set in the brain (Table 2), UV response up, is not shown because none of the top differentially expressed genes in this set have any known interactions.
Figure 5.
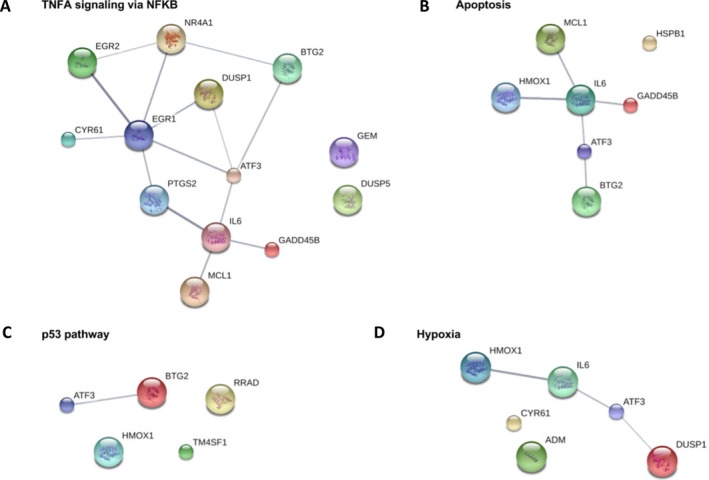
Relationships of the top differentially expressed genes within the top GSEA gene sets. The gene set UV response up is not shown because none of the top differentially expressed genes from this set interact. Line thickness indicates STRING DB’s confidence of the interaction. Filled nodes indicate that information is known about the protein’s tertiary structure. (A) TNF‐α signaling via NF‐κB (B) Apoptosis (C) p53 pathway (D) Hypoxia.
Gene expression changes: pre‐occlusion versus post‐occlusion blood samples
Differentially expressed genes in the blood during cerebral ischemia were identified by comparing all pre‐occlusion blood samples to all post‐occlusion blood samples. When hierarchical clustering was applied to the top differentially expressed genes, pre‐occlusion and post‐occlusion samples cluster separately (Fig. 3B). There also appears to be differing expression patterns in the blood of the two macaques based on this gene clustering. Monkey S3 has a more profound up‐regulation of these genes than monkey S2 possibly due to the fact that S3 had a larger infarction volume than S2 as it is the case for the brain (Fig. 3A).
As with the brain samples, gene set enrichment of the blood differential expression results was analyzed. The top five significantly enriched gene sets are hypoxia, TNF‐α signaling via NF‐κB, IL6 JAK‐SAT signaling, inflammatory response and Hedgehog signaling for the blood (Table 2).
Differentially expressed genes common between the brain and blood
Differential expression results from the brain and blood were examined for significant overlap when genes were sorted by fold change. Figure 6A shows that the two sorted gene sets are highly similar, particularly at the two extremes: the most up‐regulated (the red area in the lower left) and most down‐regulated (the red area in the upper right) genes during ischemia. Figure 6B shows the overlapping genes from the heatmap that are most significant. About 2156 down‐regulated genes significantly overlap between brain and blood samples, and 493 up‐regulated genes significantly overlap. The majority of these genes has a relatively low expression fold change, however the overlap of differentially expressed genes is very high between brain and blood samples.
Although many genes in these overlapping sets were not highly differentially expressed but some were expresses, and they have a very high change in expression in the brain that we were most interested in. Of the genes identified as highly differentially expressed in the brain, nine were also differentially expressed in the blood (fold change ≥ 1.5; Table 1, highlighted in blue). All nine of these genes occur within the most significant hypergeometric overlap (bolded). These genes are PTGS2, G0S2‐like, DUSP1, LDLR‐like, HMOX1, HSPA1B, BAG3, ADM, and TM4SF1. The majority of these genes has a sharp increase in expression in the blood, which levels off over time (Fig. 6C and D).
Discussion
A nonhuman primate model17 with human thrombin injection in the MCA artery was used to study patterns of gene expression changes during ischemic cerebral stroke. This is an embolic model of focal ischemia with a partial MCA ischemia. A time frame of 6 h was used because this is the window of time where therapeutic interventions (thrombolysis (<4:30) or thrombectomy (<6:00)) are possible in humans, although recent studies widen, in certain situations, the therapeutic window to 24 h24, 25 for thrombectomy. An early biomarker would be useful for an early diagnosis to differentiate cerebral ischemia from intracerebral hemorrage in order to use thrombolysis treatment, or to assist prehospital decision to orientate stroke patients to comprehensive stroke centers for those suitable for thrombectomy in case of proximal vessel occlusion (M1 segment of middle cerebral artery, or terminal internal carotid artery).
Our study of macaque microarray expression data from brain and blood revealed that ischemic and nonischemic samples can be distinguished based on their expression profiles, and the majority of highly differentially expressed genes is up‐regulated after ischemia. Many of these up‐regulated genes belong to pathways involved in cell death and DNA damage repair. A comparison of genes differentially expressed in the brain and blood revealed a significant overlap of gene expression patterns. These results indicate the potential to identify ischemic stroke through transcriptomics in the brain and the blood.
Brain and blood samples from only two different animals were available for this study making it difficult to rely on P‐values to select differentially expressed gene. We therefore adopted a robust strategy to analyze the data and select the gene probes based on fold changes between the ischemic and non‐ischemic brains that were concordant between the two monkeys. We also compared the different replicates obtained from different parts of the monkey brains on the ischemic and non‐ischemic sides. Although these samples are not independent as they were taken from the same monkey, the fact that they provide concordant results in different brain locations gives us confidence that they are not artifacts. Larger studies will be needed to support these results.
Gene expression in ischemic brain
Quality of the data was confirmed by PCA analysis showing that it was possible to differentiate both animal and ischemia from nonischemia by gene expression. Furthermore internal validation further supported generated results.
Hierarchical clustering of Macaca mulatta brain expression data revealed strong independent clustering of ischemic and nonischemic brain samples (Fig. 3). These results indicate a potential for identifying ischemic brain tissue based on expression profiles. Furthermore, different levels of RNA expression between monkeys of different ischemic severity in the brain and blood suggest that blood biomarkers of severity could be also identified in the blood.
The most significantly differentially expressed genes were up‐regulated, which is consistent with many previous studies of ischemia in mouse.26, 27 This study defined highly differentially expressed genes in the brain as genes that have a minimum fold change of 2 in both monkeys, and are not differentially expressed when comparing nonischemic samples between monkeys or when comparing ischemic samples between monkeys (fold change not greater than 2). The results of our analysis found that 37 genes were up‐regulated and none were down‐regulated in the brain. These results are similar to those of Buttner et al. which found 115 up‐regulated and 19 down‐regulated genes after 6 h.26 The larger number of genes identified in the Buttner et al. can be explained by the larger expression profile array used and expression differences between species.
All highly differentially expressed genes in ischemic brain tissue were up‐regulated. Many of these genes have been previously identified as up‐regulated genes in ischemic stroke for a variety of functions, notably: stress response, apoptosis, and signal transduction.
Gene expression changes in the blood
Hierarchical clustering of blood expression levels revealed that pre‐ and post‐occlusion samples cluster independently (Fig. 3B).
The expression patterns observed in the blood were similar to that of the brain, however there were some limitations in this study regarding the blood transcriptomics. Due to the relatively small number of samples (one sample per time point for each monkey) time course analysis was not feasible. In lieu of a time course analysis, samples collected before occlusion were compared to each post‐occlusion sample in a pairwise manner. In order to perform gene set analysis, the differential expression results from the comparison that yielded the largest change in expression were used with GSEA. The gene sets implicated by this analysis showed a strong correlation with expression patterns in the brain.
Our analysis of differentially expressed genes in the blood of macaques undergoing cerebral ischemia revealed that, like the brain, the majority of transcripts in the blood is up‐regulated. When samples are pooled between monkeys, and all post‐occlusion samples are pooled, 651 genes have a high level of differential expression of these 513 are up‐regulated. Notable among these differentially expressed genes are four S100 genes: S100A8, S100A12, S100P, and S100A9. S100 are calcium‐binding proteins from glial cells.
Gene sets implicated in cerebral ischemia in the brain and the blood
Brain
Gene set enrichment analysis of brain expression data revealed several gene sets involved in DNA repair and apoptosis that are up‐regulated in the ischemic brain samples. The top five enriched gene sets were TNF‐α signaling via NF‐κB, apoptosis, p53 pathway, hypoxia, and UV response up. Some of these pathways have been described as related to cerebral ischemia in previous studies.
Blood
Gene set enrichment analysis of the differential expression results from the blood revealed pathways involved in signaling, hypoxia, and inflammatory response. The top five genes sets that enriched ischemic blood samples were TNF‐α signaling via NF‐κB, hypoxia, hedgehog signaling, inflammatory response, and angiogenesis.
Interestingly, two of the most enriched gene sets in brain are also enriched in the blood: TNF‐α signaling via NF‐κB and hypoxia response (Table 2). Both of these pathways fit well into the model of ischemia response. Thirty‐seven out of the 108 genes in the TNF‐α signaling via NF‐κB gene set have core enrichment in the blood (NES = 1.89), and 42 out of 118 genes in the hypoxia response gene set have core enrichment (NES = 1.90). The gene set TNF‐α signaling via NF‐κB appears to be the pathway most involved in ischemia, with over 50% of the set having core enrichment in brain and over 30% in the blood. Furthermore, the interactions of genes products analysis are highly connected within each other specifically ATF3 and IL6 (Fig. 5).
Common expression between brain and blood
In order to compare brain and blood differential expression all genes were sorted by fold change and every possible hypergeometric overlap was tested, which found that there was a significant overlap of both up‐ and down‐regulated genes in the gene lists. Our analysis of the most significant hypergeometric overlap of up‐ and down‐regulated genes revealed that the intersections contain 493 and 2156 genes, respectively (Fig. 6A and B).
An additional, more stringent analysis of blood expression data was also performed where pre‐ and post‐occlusion samples in each monkey were compared separately, and only the time point resulting in the largest fold change was kept. These results were merged with the brain expression data and genes were filtered for high differential expression in both brain (fold change ≥ 2) and blood (fold change ≥ 1.5). Nine genes were identified as highly differentially expressed in both the brain and blood (Table 1). All nine of these genes appeared in the most significant hypergeometric overlap described above.
The top nine genes are HSPA1B, PTGS2, BAG3, ADM, TM4SF1, DUSP1, HMOX1, LDLR‐like, and G0S2: (1) HSPA1B (Hsp70): Numerous studies have identified heat shock proteins as highly up‐regulated in ischemia.26, 28, 29, 30 The human ortholog of this gene is well characterized as a stress induced gene that stabilizes proteins against aggregation, and is involved in the ubiquitin–proteasome pathway; (2) DUSP1, dual‐specific phosphatase, has been implicated in cerebral ischemia.31 Their protein products are able to inactivate MAPK proteins. Furthermore, DUSP1 was also identified in an ischemic versus control human study10; (3) Prostaglandin‐Endoperoxide Synthase 2 (PTGS2/COX2) is an enzyme that plays a role in prostaglandin biosynthesis. This gene is known to become up‐regulated during inflammation, and is a target of aspirin32, 33; (4) Adrenomedullin (ADM) gene expression levels in the blood have been shown to be associated with the severity of ischemic stroke34 in human. Liu et al. suggests that ADM expression levels in peripheral blood leukocytes could indicate the severity of tissue damage. Furthermore, ADM was also identified in an ischemic versus control study10; (5) Heme oxygenase 1 (HMOX1) is a member of the heat shock family of proteins we have discussed previously. It is believed to be part of the cellular defense system for oxidative stress‐mediated injury, like stroke.35 Additionally, previous studies have found HMOX1 to be up‐regulated in the brain after cerebral ischemia36; (6) The apoptotic gene BAG3 was also significantly up‐regulated in ischemic brain tissue. An expression study of cerebral ischemia in rats also reported this gene as highly up‐regulated28; (7) Transmembrane‐4 L‐six family member‐1 (TM4SF1) is a small plasma membrane glycoprotein that regulates cell motility and proliferation. No effect is known, to our knowledge in stroke, ischemia or hypoxia but TM4SF1 regulates apoptosis, cell cycle and ROS metabolism in human cancer cells37; (8) Hypoxia‐inducible protein G0/G1 switch gene 2 (G0s2) is a positive regulator of oxidative phosphorylation and protects cells by preserving ATP production, even under hypoxic conditions38; (9) LDL receptor blockade reduces mortality in a mouse model of ischemic stroke. Furthermore up‐regulation of low‐density lipoprotein receptor expression in the ischemic core and the peri‐ischemic area after transient MCA occlusion in rats.39
While the nine overlap genes are of interest, some of the genes that are differentially expressed in the blood but not in the brain may also be of interest as stroke biomarkers.
Conclusions
Our data showed that there is a common transcriptomic signature between the ischemic brain and the blood and support the development of blood transcriptomics as a tool for biopsy transcriptome expression profiling to characterize patients with ischemic stroke in order to develop a companion biomarker for the assessment of neuroprotection drugs or even for delayed thrombectomy after 6 h onset25 in patients with ischemic stroke. Although it is known that plasma extracellular noncoding RNAs (ex‐RNAs) are a class of circulating RNA molecules that directly modulate networks of gene expression in target tissues,40 it was quite unexpected to identify common profile of RNA coding sequence in the brain and blood.
Author Contributions
LR contributed to the analysis of the data and drafted a significant portion of the final manuscript and figures. MLQ, CO, ER participated to the acquisition and analysis of data, CF contributed to the analysis of data. GLG contributed to the acquisition and analysis of data, EG contributed to the analysis of data and drafted a significant portion of the final manuscript and figures. ST contributed to the conception and design of the study, the acquisition and analysis of data, and drafted a significant portion of the manuscript and figures.
Conflict of Interest
A patent has been filed after this work.
Supporting information
Table S1. Top 50 differentially expressed genes in the brain (pooled analysis: S3 + S2).
Table S2. Top 50 differentially expressed genes in the blood (pooled analysis).
Table S3. Physiological parameters of operated animals.
Acknowledgments
The authors thank Denis Vivien (Department of Physiopathology and Imaging of Neurological Disorders, INSERM U1237, University Caen Normandie, GIP Cyceron) for welcoming Marie‐Lise Quillé, Estelle Rousselet and Serge Timsit in their laboratory to perform animal stroke experiments. This work was supported by grants from Association Gaëtan Saleun and by ANR (Research French National Agency) Strokinin project: ANR‐10‐BIOT‐005.
Funding Information
This work was supported by grants from Association Gaëtan Saleun and by ANR (Research French National Agency) Strokinin project: ANR‐10‐BIOT‐005.
Funding Statement
This work was funded by Agence Nationale de la Recherche grant ANR-10-BIOT-005.; Association Gaëtan Saleun grant .
References
- 1. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol 2017;18:123–131. [DOI] [PubMed] [Google Scholar]
- 2. Kim YJ, Gallien S, van Oostrum J, Domon B. Targeted proteomics strategy applied to biomarker evaluation. Proteomics Clin Appl 2013;7:739–747. [DOI] [PubMed] [Google Scholar]
- 3. Rothstein L, Jickling GC. Ischemic stroke biomarkers in blood. Biomark Med 2013;7:37–47. [DOI] [PubMed] [Google Scholar]
- 4. Cox‐Limpens KE, Gavilanes AW, Zimmermann LJ, Vles JS. Endogenous brain protection: what the cerebral transcriptome teaches us. Brain Res 2014;1564:85–100. [DOI] [PubMed] [Google Scholar]
- 5. Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD‐95 inhibitor in the gyrencephalic primate brain. Nature 2012;483:213–217. [DOI] [PubMed] [Google Scholar]
- 6. Cook DJ, Tymianski M. Nonhuman primate models of stroke for translational neuroprotection research. Neurotherapeutics 2012;9:371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swyngedouw NE, Jickling GC. RNA as a stroke biomarker. Fut Neurol 2017;12:71–78. [Google Scholar]
- 8. Barr TL, Conley Y, Ding J, et al. Genomic biomarkers and cellular pathways of ischemic stroke by RNA gene expression profiling. Neurology 2010;75:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang Y, Xu H, Du X, et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J Cereb Blood Flow Metab 2006;26:1089–1102. [DOI] [PubMed] [Google Scholar]
- 10. Moore DF, Li H, Jeffries N, et al. Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischemic stroke: a pilot investigation. Circulation 2005;111:212–221. [DOI] [PubMed] [Google Scholar]
- 11. Stamova B, Xu H, Jickling G, et al. Gene expression profiling of blood for the prediction of ischemic stroke. Stroke 2010;41:2171–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jickling GC, Xu H, Stamova B, et al. Signatures of cardioembolic and large‐vessel ischemic stroke. Ann Neurol 2010;68:681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jickling GC, Stamova B, Ander BP, et al. Profiles of lacunar and nonlacunar stroke. Ann Neurol 2011;70:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jickling GC, Stamova B, Ander BP, et al. Prediction of cardioembolic, arterial, and lacunar causes of cryptogenic stroke by gene expression and infarct location. Stroke 2012;43:2036–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jickling GC, Zhan X, Stamova B, et al. Ischemic transient neurological events identified by immune response to cerebral ischemia. Stroke 2012;43:1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khaitovich P, Muetzel B, She X, et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Res 2004;14:1462–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gauberti M, Obiang P, Guedin P, et al. Thrombotic stroke in the anesthetized monkey (Macaca mulatta): characterization by MRI–a pilot study. Cerebrovasc Dis (Basel, Switzerland) 2012;33:329–339. [DOI] [PubMed] [Google Scholar]
- 18. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol 2004;3:1–25. [DOI] [PubMed] [Google Scholar]
- 19. Smyth GK. limma: linear models for microarray data In: Gentleman RCVJ, Huber W, Irizarry RA, Dudoit S, eds. Bioinformatics and computational biology solutions using R and bioconductor. New York, NY: Springer, 2005. [Google Scholar]
- 20. Street SL, Kyes RC, Grant R, Ferguson B. Single nucleotide polymorphisms (SNPs) are highly conserved in rhesus (Macaca mulatta) and cynomolgus (Macaca fascicularis) macaques. BMC Genom 2007;8:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plaisier SB, Taschereau R, Wong JA, Graeber TG. Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene‐expression signatures. Nucleic Acids Res 2010;38:e169‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szklarczyk D, Franceschini A, Wyder S, et al. v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic acids Res 2015;43(Database issue):D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Albers GW, Marks MP, Kemp S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med 2018;378:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nogueira RG, Jadhav AP, Haussen DC, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med 2018;378:11–21. [DOI] [PubMed] [Google Scholar]
- 26. Buttner F, Cordes C, Gerlach F, et al. Genomic response of the rat brain to global ischemia and reperfusion. Brain Res 2009;1252:1–14. [DOI] [PubMed] [Google Scholar]
- 27. Hori M, Nakamachi T, Rakwal R, et al. Unraveling the ischemic brain transcriptome in a permanent middle cerebral artery occlusion mouse model by DNA microarray analysis. Dis Model Mech 2012;5:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt‐Kastner R, Zhang B, Belayev L, et al. DNA microarray analysis of cortical gene expression during early recirculation after focal brain ischemia in rat. Brain Res Mol Brain Res 2002;108:81–93. [DOI] [PubMed] [Google Scholar]
- 29. Kawahara N, Wang Y, Mukasa A, et al. Genome‐wide gene expression analysis for induced ischemic tolerance and delayed neuronal death following transient global ischemia in rats. J Cereb Blood Flow Metab 2004;24:212–223. [DOI] [PubMed] [Google Scholar]
- 30. Tang Y, Lu A, Aronow BJ, et al. Genomic responses of the brain to ischemic stroke, intracerebral haemorrhage, kainate seizures, hypoglycemia, and hypoxia. Eur J Neurosci 2002;15:1937–1952. [DOI] [PubMed] [Google Scholar]
- 31. Wang L, Zhou C, Wang Z, et al. Dynamic variation of genes profiles and pathways in the hippocampus of ischemic mice: a genomic study. Brain Res 2011;1372:13–21. [DOI] [PubMed] [Google Scholar]
- 32. Tohgi H, Konno S, Tamura K, et al. Effects of low‐to‐high doses of aspirin on platelet aggregability and metabolites of thromboxane A2 and prostacyclin. Stroke 1992;23:1400–1403. [DOI] [PubMed] [Google Scholar]
- 33. Eikelboom JW, Hirsh J, Weitz JI, et al. Aspirin‐resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002;105:1650–1655. [DOI] [PubMed] [Google Scholar]
- 34. Liu J, Yan J, Greer JM, et al. Correlation of adrenomedullin gene expression in peripheral blood leukocytes with severity of ischemic stroke. Int J Neurosci 2014;124:271–280. [DOI] [PubMed] [Google Scholar]
- 35. Chen K, Maines MD. Nitric oxide induces heme oxygenase‐1 via mitogen‐activated protein kinases ERK and p38. Cell Mol Biol 2000;46:609–617. [PubMed] [Google Scholar]
- 36. Zhao Z, Lu Z, Sun X, et al. Global transcriptomic profiling of cortex and striatum: cerebral injury after ischemia/reperfusion in a mouse model. J Stroke Cerebrovasc 2017;26:1622–1634. [DOI] [PubMed] [Google Scholar]
- 37. Lu XC, Williams AJ, Yao C, et al. Microarray analysis of acute and delayed gene expression profile in rats after focal ischemic brain injury and reperfusion. J Neurosci Res 2004;77:843–857. [DOI] [PubMed] [Google Scholar]
- 38. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009;40:e331–e339. [DOI] [PubMed] [Google Scholar]
- 39. Zhang W, Potrovita I, Tarabin V, et al. Neuronal activation of NF‐kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab 2005;25:30–40. [DOI] [PubMed] [Google Scholar]
- 40. Mick E, Shah R, Tanriverdi K, et al. Stroke and circulating extracellular RNAs. Stroke 2017;48:828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Top 50 differentially expressed genes in the brain (pooled analysis: S3 + S2).
Table S2. Top 50 differentially expressed genes in the blood (pooled analysis).
Table S3. Physiological parameters of operated animals.