

Abstract
Objective
We identified a novel de novo KCNT1 variant in a patient with early‐infantile epileptic encephalopathy (EIEE) and status dystonicus, a life‐threatening movement disorder. We determined the functional consequences of this variant on the encoded KNa1.1 channel to investigate the molecular mechanisms responsible for this disorder.
Methods
A retrospective case review of the proband is presented. We performed manual and automated electrophysiologic analyses of the KCNT1‐L437F variant expressed heterologously in Chinese hamster ovary (CHO) cells in the presence of channel activators/blockers.
Results
The KCNT1‐L437F variant, identified in a patient with refractory EIEE and status dystonicus, confers a gain‐of‐function channel phenotype characterized by instantaneous, voltage‐dependent activation. Channel openers do not further increase L437F channel function, suggesting maximal activation, whereas channel blockers similarly block wild‐type and variant channels. We further demonstrated that KCNT1 current can be measured on a high‐throughput automated electrophysiology platform with potential value for future screening of novel and repurposed pharmacotherapies.
Interpretation
A novel pathogenic variant in KCNT1 associated with early‐onset, medication‐refractory epilepsy and dystonia causes gain‐of‐function with rapid activation kinetics. Our findings extend the genotype–phenotype relationships of KCNT1 variants to include severe dystonia.
Introduction
Pathogenic variants in KCNT1, the gene encoding the sodium‐activated potassium channel KNa1.1, are associated with intractable epilepsy, most prominently epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). EIMFS represents an epileptic encephalopathy distinguished electroencephalographically by nearly continuous seizures that “migrate” between regions, and clinically by seizure onset coincident with developmental arrest or regression that is resistant to conventional anticonvulsants.1, 2 Although ADNFLE typically begins in adolescence and is restricted both by nocturnal seizure predominance and by regionally restricted seizure onset, it is similarly intractable and comorbid with cognitive impairment when attributed to KCNT1 pathogenic variants.3, 4
As awareness of KCNT1 pathogenicity has spread, the phenotype broadened to include individuals with other early‐onset epilepsies such as Ohtahara and West syndromes,5, 6 other focal epilepsies,7 and leukoencephalopathies.8, 9 More recently, non‐epileptic movement disorders have been described including choreiform movements and generalized dystonia,10 consistent with robust KCNT1 expression in the striatum, thalamus, substantia nigra, and cerebellum.11, 12
To date, the majority of pathogenic variants localize within two of the six transmembrane domains (S5 and S6) and the C‐terminal regulator of potassium domains (RCK1 and RCK2) of the KNa1.1 channel. With the exception of p.Phe932Ile, all pathogenic variants functionally annotated thus far confer a convergent KNa1.1 gain‐of‐function demonstrated as elevated potassium current amplitudes by in vitro electrophysiologic assays.10, 13 Yet, the mechanism by which current density is increased appears more variable. Multiple mechanisms including altered PKC‐dependent phosphorylation favoring the open channel conductance state,14 and altered cooperativity between channel subunits leading to relatively less time spent in subconductance states15 have been suggested. In a neuron, upon intracellular sodium binding and membrane depolarization, high conductance KNa1.1 channels open, contributing to action potential frequency adaptation and afterhyperpolarization potentials.16 In this report, we build upon reported phenotypes in KCNT1‐associated epilepsy (i.e., EIMFS or ADNFLE) to include status dystonicus, a life‐threatening movement disorder. We undertook manual patch‐clamp recording to define the impact of a novel KCNT1 variant identified in our patient on channel function in a mammalian heterologous expression system, and demonstrated the utility of high‐efficiency channel expression in an automated patch‐clamp system for reproducibly testing the effects of anticonvulsant drugs.
Methods
Study subjects
Deidentified patient information was provided by treating physicians for inclusion in this report. Informed consent for release of medical information was obtained from the proband and his family per institutional policies. A review by the Lurie Children’s Hospital International Review Board deemed the heterologous expression studies as exempt, nonhuman subject research using deidentified patient data.
Heterologous KCNT1 expression
A recombinant human KCNT1 cDNA matching the long isoform (Refseq sequence NM_020822.2) was synthesized (Atum, Newark, CA) then subcloned into a pCMV‐IRES‐eGFP reporter plasmid described previously.17 The L437F (c.1309C>T) and R428Q (c.1283G>A) variants were introduced using QuikChange II site‐directed mutagenesis and the complete open reading frames of final constructs were Sanger sequenced to verify the mutation and exclude polymerase or cloning errors.
CHO‐K1 cells (ATCC, Manassas, VA), chosen as the heterologous expression because of minimal endogenous potassium conductance, were maintained at passages 5–15 in DMEM/F12, penicillin/streptomycin, and 10% FBS, prior to transient transfection with Fugene using 1 µg of endotoxin‐free plasmid DNA (Clontech, Mountain View, CA). The cells were maintained at <70% confluence and selected for manual patch‐clamp recording on the basis of moderate GFP expression after a 48–72 h incubation period at 37°C. Inducible stable cell lines expressing either WT or variant (L437F) KCNT1 were generated by dual transfection of CHO‐K1 cells with pCMV‐rtTA and ptetO‐KCNT1‐IRES‐eGFP plasmids using the piggyBac transposase system as described previously.18 The cells were selected in 5 µg/mL puromycin 48hrs after Fugene transfection and serially diluted to obtain individual clones. In the presence of 2 µg/mL doxycycline for 48 h, cell lines selected for further study demonstrated robust expression of eGFP and of KCNT1, as measured by automated patch‐clamp recording of KNa1.1 channel currents and by flow cytometry for eGFP positivity.
Manual patch‐clamp electrophysiology
All whole‐cell recordings were performed at room temperature. External solution contained (in mmol/L) 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES. Internal solution contained (in mmol/L) 10 NaCl, 50 KCl, 60 KMeSO4, 1 MgCl2, 10 EGTA, 10 HEPES, 0.5 Na‐GTP, 0.5 Mg‐ATP, 1 NAD+ (free acid) titrated to a pH of 7.2 and osmolarity of 300 mOsm/kg. Whole‐cell currents were acquired with an Axopatch Multiclamp 700B amplifier at 5 kHz and filtered at 1 kHz without leak subtraction. Fast and slow capacitive transients were compensated. Whole‐cell currents were measured from a holding potential of −80 mV with 2000 msec voltage steps to +30 mV in 10 mV increments. The instantaneous current (peak within the first 50 msec) and quasi steady‐state current (peak within the last 50 msec) were quantified as a ratio and presented as median ± SEM of at least 10 individual cells. Quinidine (anhydrous; Sigma) was maintained as a 1 mg/mL stock solution dissolved in DMSO, with final solvent concentration of 0.1% or less. Liquid junction potential was estimated as −8 mV based on previous measurements and not corrected.
Automated patch‐clamp electrophysiology
Automated planar‐array patch‐clamp recording was performed with a SyncroPatch 768 PE system (Nanion, Germany) as previously described.19 External solution was identical to that used for manual patch‐clamp recording; internal solution contained (in mmol/L) 40 NaCl, 60 KF, 20 KCl, 1 MgCl2, 10 EGTA, 20 HEPES, 0.5 Na‐GTP, 1 NAD+ (free acid) titrated to a pH of 7.2 and osmolarity of 300 mOsm/kg. Pulse generation was performed with PatchControl software (Nanion), and whole‐cell currents were acquired at 5 kHz and filtered at 1 kHz. Currents were not leak subtracted. Fast and slow capacitances were compensated using internal algorithms within PatchControl software. Whole‐cell currents were measured from a holding potential of −80 mV and elicited with depolarizing steps (2000 msec) from −80 to +60 mV (10 mV steps). The cells were included for analysis if the whole‐cell capacitance measured at −80 mV was less than 100 pF, the holding current was not more negative than −500 pA, the seal resistance was greater than 0.5 GΩ, and the access resistance was less than 200 MΩ. Data are presented as mean ± SEM, with statistical significance calculated using nonparametric Wilcoxon rank sum or Kruskal–Wallis tests of significance, as indicated.
Results
Clinical case description
The proband was born to a G2P1 mother with a prenatal course complicated by hyperemesis gravidarum and cholelithiasis requiring cholecystectomy. Vaginal delivery at 39 weeks was uncomplicated, with birth weight, height, and head circumference between the 25th‐50th percentile. Secondary cleft palate with micrognathia was noted at birth in the absence of other clinically apparent dysmorphisms. At 5 weeks, the proband developed focal clonic seizures described as variable limb jerking and horizontal nystagmus. Interictal EEG at that time was notable for rhythmic, sharply contoured delta with embedded, multifocal spikes with shifting laterality and variable evolution; during sleep, a discontinuous record with burst suppression emerged (Fig. 1A–C). Brain MRI was normal for age, whereas MRI of the spine revealed central cord dilation and hydromyelia of the lower thoracolumbar spine (images not available). Extensive metabolic work‐up was performed (Table 1), but was not diagnostic. Comparative genomic hybridization microarray and an infantile epilepsy panel including sequencing and deletion/duplication analysis of 38 genes (GeneDx, assayed August 2012) were negative. The proband was initially treated with phenobarbital, but daily seizures persisted and then evolved in semiology to asymmetric tonic seizures with a vibratory component. Levetiracetam, topiramate, dexamethasone, and clobazam were all trialed without reduction in seizure frequency. Following gastrostomy tube placement, he was started on the ketogenic diet and escalated to a ratio of 4:1 (protein/fat: carbohydrate) without improvement.
Figure 1.
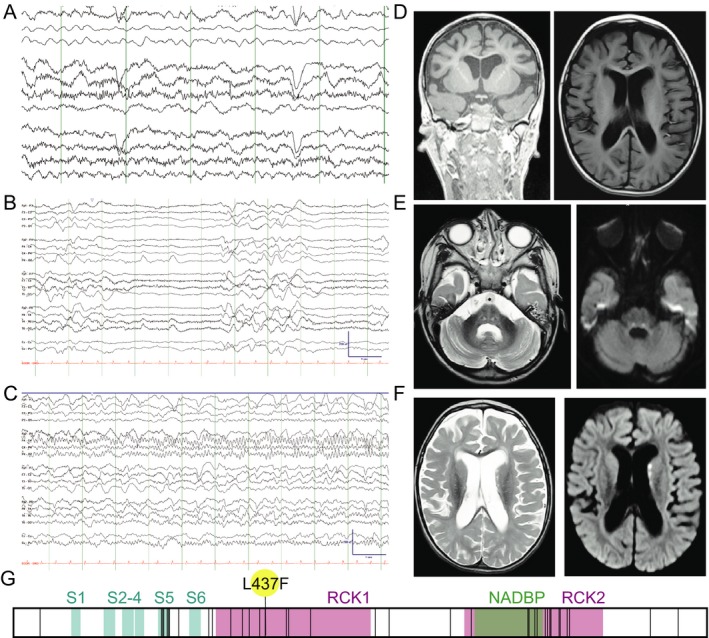
A proband with a de novo KCNT1‐L437F variant. (A–C) EEG at age 4 years old. (A) Awake cerebral electrical activity was characterized by a moderately disorganized background without a discernible posterior dominant rhythm. There was a moderate excess of delta activity diffusely. There were occasional small‐amplitude, multifocal spike wave discharges. (B) Deeper stages of sleep were marked by bursts of diffuse slowing alternating with epochs of severe (~10 μV) diffuse attenuation of voltage lasting 2–6 sec, giving the record a discontinuous appearance. (C) Example of an electroclinical focal seizure. On the video, the semiology was fairly stereotyped and characterized by a combination of upward and rightward eye deviation, eye blinking, change in breathing, chin quivering, jaw movements, and limb jerking. At the offset, the patient often coughed or sighed. Duration ~1–2 min. Electrographically, the onset was fairly stereotyped with focal rhythmic alpha activity in the right parietal region that typically spread (shown here) to the right posterior temporal region and then the remainder of the right hemisphere and then the left hemisphere. Offset was gradual and followed by diffuse background slowing. (D) Baseline MRI was notable for hydrocephalus ex vacuo. (E and F) MRI obtained at time of status dystonicus is notable for bilateral restricted diffusion in the basal ganglia, corticospinal, and pontine tracts. (G) Schematic of the KCNT1 coding sequence (from NM_020822.2) where all variants reported pathogenic in HGMD (6/24/19) are denoted by vertical lines superimposed on the transmembrane (S1–S6), regulator of potassium conductance (RCK), and NAD binding protein (NADBP) domains, with the proband variant (L437F) flagged.
Table 1.
Metabolic work‐up.
| Serum amino acids, CSF amino acids |
| Urine organic acids |
| Serum lactate, CSF lactate |
| CSF pyruvate |
| CSF glucose, protein |
| Serum ammonia |
| CSF neurotransmitters, neopterin, tetrahydrobiopterin (GCH1 sequencing performed due to low CSF tetrahydrobiopterin) |
| Acylcarnitine profile |
| Lysosomal enzyme screen |
| Cortisol, IGF‐1, IGBBP‐1, Prolactin, GH |
KCNT1‐associated epilepsy with status dystonicus.
At age 15 months, he developed opisthotonic posturing during a presumed viral upper respiratory infection, which progressed to status dystonicus that did not improve with baclofen, oral diazepam, trihexyphenidyl, or carbidopa/levodopa treatments. Nearly continuous posturing and autonomic instability with associated respiratory insufficiency ultimately responded to a continuous midazolam drip. Video EEG revealed no ictal correlate despite repeated studies while the patient remained dystonic. MRI at this time was notable for generalized volume loss and ventriculomegaly (Fig. 1D) as well as symmetric restricted diffusion of the pontine tracts and T2 FLAIR hyperintensities in the bilateral head of the caudate and thalamus (Fig. 1E and F). Cerebrospinal fluid studies demonstrated normal protein, glucose, cell count and differential, and CSF lactate. His muscle tone improved following slow benzodiazepine taper, botulinum toxin injections, and subsequent inpatient rehabilitation for intensive physical and occupational therapy. No subsequent episodes of dystonia or chorea have occurred. At age 3 years, he developed tonic seizures associated with breath holding and variable bradycardia. Interictal EEG demonstrated a suppression‐burst pattern with multifocal epileptiform discharges, with intermittent epochs consistent with 4–5 Hz posterior dominant rhythm during wakefulness. His neurodevelopment plateaued with the onset of seizures and is limited to intermittent visual fixing and tracking; he has never rolled, sat, or babbled. He has over time acquired the ability to lift his head when prone, to attend to familiar voices and a family dog, and respond with a social smile. He is fed by gastrostomy‐tube only and does not have purposeful use of his hands. At age 8 years, seizure frequency is relatively unchanged, with multiple persistent focal tonic seizures per day, and likely additional electroclinical events with subtle behavioral correlates, as demonstrated on prior EEGs. His current medical regimen includes oxcarbazepine, added most recently and ostensibly the most effective for decreasing seizure cluster frequency, clobazam, and the ketogenic diet. Multiple echocardiograms obtained during periods of critical respiratory illnesses have all been normal.
Whole‐exome sequencing performed at age 5 years revealed a novel, de novo KCNT1 variant c.1309C>T (p.L437F), classified as likely pathogenic by ACMG criteria.20 The KCNT1‐L437F variant is not present in ExAC or gnomAD databases, suggesting it is not a rare population variant. The encoded leucine residue is highly conserved across species including C. elegans, Drosophila, and common house mouse. The variant is in close proximity to other recurrent pathogenic variants in the first regulator of potassium conductance (RCK1) domain within the C‐terminus, a key domain for sodium‐triggered exposure of the channel pore to the intracellular environment,21 and has a deleterious impact on secondary structure predicted by some in silico algorithms (e.g., MutationTaster, Grantham score of 22) but not others (e.g., predicted benign in Polyphen‐2).
KCNT1‐L437F expression yields a KNa1.1 gain‐of‐function channel phenotype
Whole‐cell patch‐clamp recording in voltage clamp mode from CHO cells expressing wild‐type (WT) KCNT1 revealed an outward current at depolarizing potentials with a near‐instantaneous component and a slowly‐activating, non‐inactivating component with a reversal potential (n = 15, median: −75.8 ± 0.85 mV; Fig. 2) near the calculated K+ equilibrium potential (E K = −80 mV), in agreement with known characteristics of KNa1.1 channels.11, 22 The current‐voltage relationship exhibited strong outward rectification most pronounced at depolarized potentials, was ~60% blocked by 100 μmol/L quinidine (cf. IC50 = 89 µmol/L in HEK cells23), and had near‐complete block by 1 mmol/L quinidine. By comparison, untransfected CHO cells had a maximal peak current amplitude of <200 pA at +60 mV (n = 9, median: 172 ± 12 pA). Taken together, this kinetic and pharmacologic profile is consistent with the reported KCNT1‐encoded KNa1.1 channel.
Figure 2.
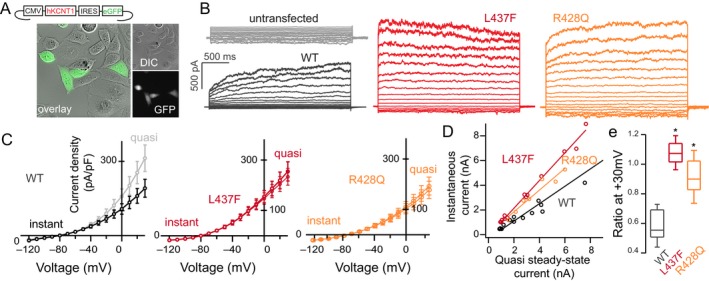
Whole‐cell patch‐clamp recording of CHO cells expressing WT and mutant human KNa1.1 (A) Transfected cells selected for recording were selected by GFP expression and showed no change in morphology or cell health. (B) In comparison to untransfected cells, KCNT1‐expressing CHO cells exhibit a voltage‐dependent potassium current with relatively slow activation kinetics (black) as compared to the L437F variant (red) or the R428Q variant (orange), (C) evident in I–V curves drawn at initial depolarization (instantaneous current) and at 2s (quasi steady‐state). (D) This kinetic change is quantified by instantaneous:steady‐state ratio at a suprathreshold potential of +30 mV (n = 8–14 cells/group, P < 0.05, Kruskal–Wallis H‐test, (E). No significant difference in current density between WT and L437F or R428Q was noted (P > 0.1, Mann–Whitney U test).
By contrast, cells expressing KCNT1‐L437F exhibited an outward potassium conductance with nearly instantaneous activation at depolarized potentials, best quantified by an instantaneous to steady‐state current ratio that was significantly greater than WT channels (WT: n = 10, median ratio: 0.57 ± 0.02; L437F: n = 14, median: 1.02 ± 0.03; P < 0.001, Mann–Whitney U test). There was no significant difference in the current density between the mutant and WT channels (P > 0.1, Mann–Whitney U test). As the majority of KCNT1 variants in the literature have been studied functionally in Xenopus oocytes,10, 15, 24 we studied for comparison a previously reported recurrent KCNT1 variant (p.R428Q) expressed in CHO cells. The cells transfected with KCNT1‐R428Q also exhibited a greater instantaneous to steady‐state current ratio compared to WT channels (n = 13, median ratio: 0.91 ± 0.03, P < 0.05, Mann–Whitney U test). Taken together, the conductance generated by KCNT1‐L437F expression with instantaneous activation compared to that of the WT channel with comparatively slow activation kinetics is consistent with a gain‐of‐function channel phenotype.
Automated patch‐clamp recording of CHO cells expressing KCNT1‐L437F
To further investigate the properties of KCNT1‐L437F, we optimized a high‐throughput, automated 384‐well planar patch‐clamp electrophysiology platform to record currents carried by wild‐type and mutant KNa1.1 channels as well as to perform pharmacological studies. Given that cells are sampled on this assay in an unbiased fashion due to robotic plating within wells of a recording chip, consistent channel expression and current density were essential. However, transient expression by electroporation resulted in 50% or less of cells expressing KCNT1 48–72 h later, despite extensive optimization of culture and transfection conditions. Similarly, stable expression of a bicistronic pCMV‐KCNT1‐IRES‐EGFP construct yielded cell lines in which KNa1.1 current was present in less than 50% of cells and the proportion of KCNT1‐expressing cells fell during serial passaging despite constant antibiotic selection suggesting channel‐mediated toxicity. To avoid potential KCNT1‐associated cell toxicity, we created a stable cell line with tetracycline‐inducible bicistronic KCNT1‐IRES‐EGFP, facilitating more consistent and persistent channel expression. The current‐voltage relationships and activation kinetics of WT KCNT1‐expressing cells compared with KCNT1‐L437F cells were not significantly different between manual and automated patch clamp experiments (Fig. 3).
Figure 3.
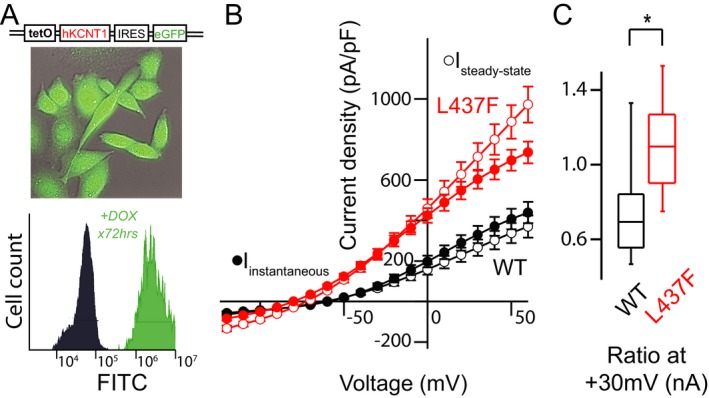
Automated planar whole‐cell patch clamp recording of stable, inducible KCNT1‐expressing CHO cells recapitulate altered KNa1.1 channel activation and allow for pharmacologic screening. (A) GFP‐expressing, doxycycline‐induced CHO cells exhibit a near‐homogeneous shift in population fluorescence 72 h post‐induction verified by flow cytometry. (B and C) I–V curves drawn at initial depolarization (instantaneous current) demonstrate a voltage and time‐independent effect. This kinetic change is quantified by instantaneous:steady‐state ratio at a suprathreshold potential of +30 mV (n = 56–169 cells/group, P < 0.05, Kruskal–Wallis H‐test).
We next tested the impact of bithionol, a KNa1.1 channel opener,23 on both WT and KCNT1‐L437F expressing cells (Fig. 4). At +60 mV in the presence of 5 µmol/L bithionol, WT KCNT1‐expressing cells exhibited a 144% greater current density (baseline: n = 50, mean I steady‐state = 439.9 pA/pF ± 53.2; bithionol: mean = 634.7 pA/pF ± 40.4), whereas current density of KCNT1‐L437F cells was not changed significantly by the compound (baseline: n = 42, mean I steady‐state = 735.8 pA/pF ± 53.5; bithionol: mean = 678.8 pA/pF ± 49.9, P > 0.1). The activated current carried by both WT and L437F variant channels in the presence of bithionol were similarly blocked at all potentials by 125 µmol/L quinidine (WT: 87.2% block, mean I steady‐state = 80.9 pA/pF ± 11.5; L437F: 86.68%, mean I steady‐state = 90.4 pA/pF, ±13.8).
Figure 4.
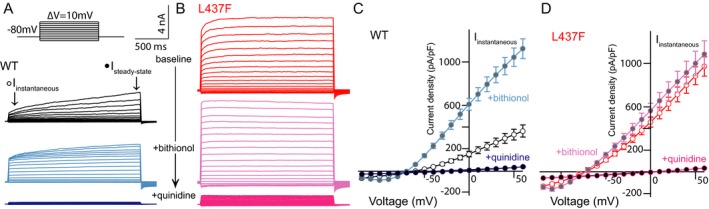
KCNT1‐L437F‐expressing CHO cells are insensitive to bithionol activation yet blocked by quinidine. (A–D) Voltage‐dependent outward current in WT expressing cells measured in whole‐cell voltage clamp mode (black) is increased by application of the KCNT1 activator bithionol (blue) and blocked by quinidine; in comparison, outward current measured in cells expressing the L437F variant (red) is not significantly changed by bithionol application, yet is also blocked by quinidine.
We additionally compared the differential activation between WT and variant channels using loxapine, an antipsychotic drug previously identified as a selective activator of KNa1.1 channels.25 Upon addition of 5 µmol/L loxapine, we observed no change in current amplitude in cells expressing KCNT1‐L437F compared to a robust activation of current in treated WT cells (L437F: mean % change = 2.5% ± 17.8; WT: mean %change = 525.2% ± 64.1). These findings suggest that KCNT1‐L437F is near‐maximally activated at baseline. In addition to quinidine, we also examined the effects of bepridil, which has been described as a potent KNa1.1 channel blocker.23 In the presence of 10 µmol/L bepridil, current amplitude in cells expressing either WT or KCNT1‐L437F were diminished (WT: mean %change = –50.0% ± 8.76; L437F: mean %change = –57.4% ± 9.5) to a similar extent as in the presence of 100 μmol/L quinidine.
The availability of automated patch‐clamp electrophysiology for interrogation of genetic ion channel variants is conducive to identification of precisely effective anticonvulsants, including not only FDA‐approved anticonvulsants but also other repurposed drugs and novel rationally designed compounds. As an initial screen, we tested quinidine, a class I antiarrhythmic used off‐label in EIMFS with variable efficacy,26 against a panel of anticonvulsants at concentrations consistent with peak therapeutic serum levels in patients. The drugs selected included those known to target sodium and calcium channels with variable specificity, as well as the conventional potassium channel blocker 4‐aminopyridine (4‐AP) (Fig. 5). Consistent with literature supporting limited pharmacologic blockers for KNa1.1 channels and medication‐refractory seizures in patients with KCNT1‐associated epilepsy, the five anticonvulsants tested had no significant consistent effect on current density (n = 58–64/drug, Wilcoxon signed rank test, P > 0.05 for all). In contrast, currents in 4‐AP and quinidine block the KCNT1‐L437F conductance to a limited but significant extent (4‐AP: mean %change = −89.5% ± 7.5; quinidine: mean %change = −78% ± 5.5). Given the implications of quinidine as a novel anticonvulsant for EIMFS, a dose‐response curve was performed on parallel cell samples, with estimated 50% block of the channel at 66 µmol/L.
Figure 5.
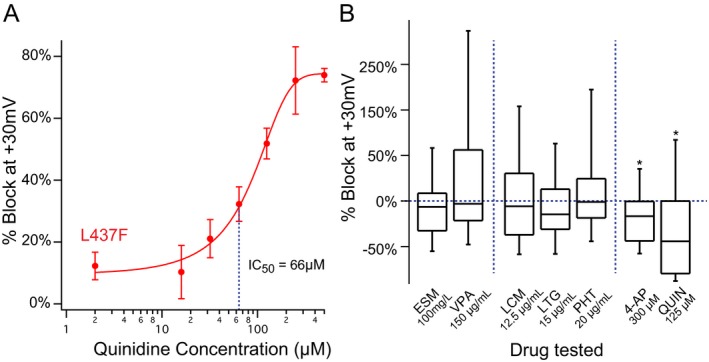
KCNT1‐L437F channels are blocked by quinidine but not by other conventional anticonvulsants. (A) Automated patch‐clamp recording of KCNT1‐L437F expressing cells demonstrated a dose‐response to quinidine with an IC50 unchanged from WT CHO cells. (B) Screening of conventional anticonvulsants at maximum therapeutic serum concentrations including ethosuximide (ESM), valproic acid (VPA), lacosamide (LCM), lamotrigine (LTG), phenytoin (PHT) compared to a traditional potassium channel blocker (4‐AP) and quinidine displays a lack of channel block, consistent with prior reports of seizures in KCNT1‐associated epilepsy refractory to conventional anticonvulsants.
Discussion
Missense mutations in KCNT1 are implicated in 40% of cases of EIMFS.27 In this report, we describe a male patient with a novel de novo pathogenic KCNT1 variant presenting with unusual clinical features. While early infantile epileptic encephalopathy is within the spectrum of epilepsies thus far ascribed to KCNT1 variants,10 our patient’s presentation expands the phenotypic spectrum of comorbid movement disorders to include status dystonicus, a rare but life‐threatening neurologic emergency.28 Whereas movement disorders comorbid with EIMFS have been included as part of the clinical phenotype, to our knowledge, KCNT1 mutations have not been associated with status dystonicus. As in our patient, acute infection is a noted trigger which may contribute to development of status dystonicus in individuals with an underlying predisposition.29 In a report of 31 patients at a tertiary care center in Australia with pathogenic KCNT1 variants presenting with early‐onset epilepsy, seven exhibited movement disorders including focal dystonia, generalized dystonia, and choreoathetosis.10 KCNT1 expression is well‐documented in both the striatum and substantia nigra,11, 12 consistent with case series of patients with focal brain lesions leading to dystonia30 and with a clinical manifestation suggestive of impaired dopamine signaling. Similar to other ion channelopathies underlying early‐onset epileptic encephalopathies with coincident movement disorders (e.g., KCNQ2, SCN8A), the expression pattern of the involved channel and careful clinical observation are likely to continue to expand the phenotypic spectrum.31, 32
The patient we describe in this report developed an epileptic encephalopathy over time characterized by intractable, multifocal electroclinical seizures of variable semiologies with prominent dysautonomia, which is representative of other patients with KCNT1‐assoociated epilepsy with multiple seizure types and poor response to classical anticonvulsants. Given the relatively small number of patients whose genotypes and medication trials have been published, it is not yet possible to preemptively gauge medication responsiveness by location of a pathogenic variant. Our patient has not yet been trialed on quinidine due to ongoing medical comorbidities increasing the risk of drug‐drug interactions and cardiac arrhythmias. Although initial epilepsy gene panel testing five years ago was negative for pathogenic variants, subsequent whole exome sequencing identified a de novo likely pathogenic variant in KCNT1, c.1390C>T (p.L437F). Given the availability of clinical whole exome sequencing and the exponential rate of discovery of epilepsy genes identified in epileptic encephalopathies,33 expanding the gamut of diagnostic testing performed for our patient as it became available was essential in ultimately identifying an etiologic diagnosis. KCNT1 was subsequently added to the same gene panel performed for our patient one year later, reiterating the need for timely updates in genetic testing for patients who lack a specific diagnosis.
Similar to ~50% of pathogenic KCNT1 variants, the L437F variant resides within one of the regulator of potassium conductance (RCK) domains in the C‐terminus of the channel. Crystallography studies have suggested that the RCK1 and RCK2 domains form a gating ring on the cytoplasmic interface between the channel and the cytosol, requiring sodium binding to alter the confirmation of the ring and mechanically alter the shape of the electrostatic pore between transmembrane domains, thus allowing passage of potassium.21, 34 Although a sodium binding site has been localized to RCK2,35 the conformational shift suggested by structural analyses of the channel in the presence and absence of sodium suggest an all‐or‐none phenomenon requiring coordinated movement of the S5, RCK1, and RCK2 domains within which the overwhelming majority of pathogenic variants have been localized. Many of the studies to date investigating the functional consequences of KCNT1 variants have used Xenopus oocytes to describe relatively higher current density in pathogenic variants conferring a gain‐of‐function channel phenotype.13, 14, 15, 36 Several studies have probed the properties of rodent KCNT1 in heterologous expression systems,15, 22, 37, 38, 39 but to our knowledge only one has investigated the human KCNT1 channel, also in an inducible cell system, but without quantification of the activation kinetics.25 This report supports parallel mechanistic findings in oocytes and mammalian systems for the KCNT1‐L437F variant.
Our study leveraged reported, available pharmacologic tools including KNa1.1 activators (bithionol and loxapine) as well as blockers (quinidine and bepridil) to begin to address the mechanism of gain‐of‐function.23, 25 Bithionol, an anti‐helminthic drug that additionally activates BK channels, is understood mechanistically to be an allosteric modulator of KNa1.1 through inhibition of soluble adenylyl cyclase.40 While KNa1.1 is thought to be ATP‐insensitive as it lacks the ATP binding domain of its family member KNa2.1 (encoded by KCNT2), it is sensitive to divalent cation concentration,40 which also changes with inhibition of adenylyl cyclase. Importantly, our study demonstrated that the KCNT1‐L437F pathogenic variant is not activated by either compound with presumably different mechanisms of action, whereas current conducted by the wild‐type channel is dramatically increased, suggesting the L437F channel is already maximally conducting, with a high open probability. Comparatively, both quinidine and bepridil exerted a similar degree of block on WT and L437F channels, arguing against either conformational changes in the folded channel causing failed binding of activators or expression of compensatory ion channels with overlapping current profiles. While our study supports altered channel gating, perhaps as a function of disturbed sodium binding, our ability to extrapolate the findings to the human brain are limited by the use of transient overexpression in a non‐neuronal cell. To better understand the mechanisms of pathogenesis underlying KCNT1‐associated epilepsy, additional experiments of pathogenic variants expressed under the native promoter, in relevant neuronal subpopulations, and throughout development will be needed.
Authors’ Contributions
The study was conceived, all experiments were performed, and the paper was written by TSG. Generation of the inducible, stable plasmid construct was performed with assistance from CT. Automated patch clamp recordings were obtained with assistance from CV. The proband reported was treated clinically by JJM. The study was overseen and paper was edited by ALG.
Conflict of Interest
Gertler, Thompson, and Vanoye declare no conflicts of interest with the work described herein. Millichap reports personal fees from American Academy of Neurology, personal fees from Up‐To‐Date, personal fees from BMJ Best Practice, personal fees from Invitae, grants and personal fees from UCB Pharma, grants and personal fees from Mallinkrodt, personal fees from Esai, personal fees from Xenon, personal fees from Biomarin, personal fees from Ionis, personal fees from Greenwich, personal fees from Sunovion, outside the submitted work. George reports personal fees from Amgen, Inc., grants from Praxis Precision Medicines, Inc., personal fees from Otsuka Pharmaceutical Co., grants from Merck & Co., Inc., outside the submitted work.
Acknowledgments
The authors acknowledge support from R25NS070695 (TSG), KL2TR001424 (TSG) and NS108874 (ALG).
Funding Information
The authors acknowledge support from R25NS070695 (TSG), KL2TR001424 (TSG) and NS108874 (ALG).
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grants NS108874 and R25NS070695; National Center for Advancing Translational Sciences grant KL2TR001424.
References
- 1. Caraballo RH, Fontana E, Darra F, et al. Migrating focal seizures in infancy: analysis of the electroclinical patterns in 17 patients. J Child Neurol 2008;23:497–506. [DOI] [PubMed] [Google Scholar]
- 2. Coppola G. Malignant migrating partial seizures in infancy: an epilepsy syndrome of unknown etiology. Epilepsia 2009;50(Suppl 5):49–51. [DOI] [PubMed] [Google Scholar]
- 3. Steinlein OK. Genetic heterogeneity in familial nocturnal frontal lobe epilepsy. Prog Brain Res 2014;213:1–15. [DOI] [PubMed] [Google Scholar]
- 4. Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet 2016;53:217–225. [DOI] [PubMed] [Google Scholar]
- 5. Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole‐genome sequencing in severe early‐onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 2014;23:3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukuoka M, Kuki I, Kawawaki H, et al. Quinidine therapy for West syndrome with KCNTI mutation: a case report. Brain Dev 2017;39:80–83. [DOI] [PubMed] [Google Scholar]
- 7. Hansen N, Widman G, Hattingen E, et al. Mesial temporal lobe epilepsy associated with KCNT1 mutation. Seizure 2017;45:181–183. [DOI] [PubMed] [Google Scholar]
- 8. Arai‐Ichinoi N, Uematsu M, Sato R, et al. Genetic heterogeneity in 26 infants with a hypomyelinating leukodystrophy. Hum Genet 2016;135:89–98. [DOI] [PubMed] [Google Scholar]
- 9. Vanderver A, Simons C, Schmidt JL, et al. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol 2014;50:112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McTague A, Nair U, Malhotra S, et al. Clinical and molecular characterization of KCNT1‐related severe early‐onset epilepsy. Neurology 2018;90:e55–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol 2002;454:241–254. [DOI] [PubMed] [Google Scholar]
- 12. Rizzi S, Knaus HG, Schwarzer C. Differential distribution of the sodium‐activated potassium channels slick and slack in mouse brain. J Comp Neurol 2016;524:2093–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dilena R, DiFrancesco JC, Soldovieri MV, et al. Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain‐of‐function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 2018;15:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barcia G, Fleming MR, Deligniere A, et al. De novo gain‐of‐function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim GE, Kronengold J, Barcia G, et al. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. 2014;9:1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Budelli G, Hage TA, Wei A, et al. Na+‐activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat Neurosci 2009;12:745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vanoye CG, Welch RC, Daniels MA, et al. Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE subunits. J Gen Physiol 2009;134:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson MH, Coates CJ, George AL Jr. PiggyBac transposon‐mediated gene transfer in human cells. Mol Ther 2007;15:139–145. [DOI] [PubMed] [Google Scholar]
- 19. Vanoye CG, Desai RR, Fabre KL, et al. High‐throughput functional evaluation of KCNQ1 Decrypts variants of unknown significance. Circ Genom Precis Med 2018;11:e002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hite RK, Yuan P, Li Z, et al. Cryo‐electron microscopy structure of the Slo2.2 Na(+)‐activated K(+) channel. Nature 2015;527:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joiner WJ, Tang MD, Wang LY, et al. Formation of intermediate‐conductance calcium‐activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci 1998;1:462–469. [DOI] [PubMed] [Google Scholar]
- 23. Yang B, Gribkoff VK, Pan J, et al. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology 2006;51:896–906. [DOI] [PubMed] [Google Scholar]
- 24. Tang QY, Zhang FF, Xu J, et al. Epilepsy‐related slack channel mutants lead to channel over‐activity by two different mechanisms. Cell Rep 2016;14:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Biton B, Sethuramanujam S, Picchione KE, et al. The antipsychotic drug loxapine is an opener of the sodium‐activated potassium channel Slack (Slo2.2). J Pharmacol Exp Ther 2012;340:706–715. [DOI] [PubMed] [Google Scholar]
- 26. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium‐gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012;44:1188–1190. [DOI] [PubMed] [Google Scholar]
- 28. Allen NM, Lin JP, Lynch T, King MD. Status dystonicus: a practice guide. Dev Med Child Neurol 2014;56:105–112. [DOI] [PubMed] [Google Scholar]
- 29. Manji H, Howard RS, Miller DH, et al. Status dystonicus: the syndrome and its management. Brain 1998;121(Pt 2):243–252. [DOI] [PubMed] [Google Scholar]
- 30. Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994;117(Pt 4):859–876. [DOI] [PubMed] [Google Scholar]
- 31. Dhamija R, Goodkin HP, Bailey R, et al. A case of KCNQ2‐associated movement disorder triggered by fever. J Child Neurol 2017;32:1123–1124. [DOI] [PubMed] [Google Scholar]
- 32. Singh R, Jayapal S, Goyal S, et al. Early‐onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure 2015;26:69–71. [DOI] [PubMed] [Google Scholar]
- 33. Oyrer J, Maljevic S, Scheffer IE, et al. Ion channels in genetic epilepsy: from genes and mechanisms to disease‐targeted therapies. Pharmacol Rev 2018;70:142–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hite RK, MacKinnon R. Structural titration of Slo2.2, a Na(+)‐dependent K(+) channel. Cell 2017;168:390–399.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Z, Rosenhouse‐Dantsker A, Tang QY, et al. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J Neurosci 2010;30:7554–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bearden D, Strong A, Ehnot J, et al. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol 2014;76:457–461. [DOI] [PubMed] [Google Scholar]
- 37. Brown MR, Kronengold J, Gazula VR, et al. Amino‐termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J Physiol 2008;586:5161–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fleming MR, Brown MR, Kronengold J, et al. Stimulation of slack K(+) channels alters mass at the plasma membrane by triggering dissociation of a phosphatase‐regulatory complex. Cell Rep 2016;16:2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y, Brown MR, Hyland C, et al. Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci 2012;32:15318–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kleinboelting S, Ramos‐Espiritu L, Buck H, et al. Bithionol potently inhibits human soluble adenylyl cyclase through binding to the allosteric activator site. J Biol Chem 2016;291:9776–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]