

Abstract
Objective
To identify a convergent, multitarget proliferation characteristic for astrocytoma transformation that could be targeted for therapy discovery.
Methods
Using an integrated functional genomics approach, we prioritized networks associated with astrocytoma progression using the following criteria: differential co‐expression between grade II and grade III IDH1‐mutated and 1p/19q euploid astrocytomas, preferential enrichment for genetic risk to cancer, association with patient survival and sample‐level genomic features. Drugs targeting the identified multitarget network characteristic for astrocytoma transformation were computationally predicted using drug transcriptional perturbation data and validated using primary human astrocytoma cells.
Results
A single network, M2, consisting of 177 genes, was associated with glioma progression on the basis of the above criteria. Functionally, M2 encoded physically interacting proteins regulating cell cycle processes and analysis of genome‐wide gene‐regulatory interactions using mutual information and DNA–protein interactions revealed the known regulators of cell cycle processes FoxM1, B‐Myb, and E2F2 as key regulators of M2. These results suggest functional disruption of M2 via gene mutation or altered expression as a convergent pathway regulating astrocytoma transformation. By considering M2 as a multitarget drug target regulating astrocytoma transformation, we identified several drugs that are predicted to restore M2 expression in anaplastic astrocytoma toward its low‐grade profile and of these, we validated the known antiproliferative drug resveratrol as down‐regulating multiple nodes of M2 including at nanomolar concentrations achievable in human cerebrospinal fluid by oral dosing.
Interpretation
Our results identify M2 as a multitarget network characteristic for astrocytoma progression and encourage M2‐based drug screening to identify new compounds for preventing glioma transformation.
Background
Traditionally, drugs have been designed with the aim of targeting a single biological entity, usually a protein, with high selectivity to avoid unwanted “off‐target” effects which are perceived generally as being associated with an increased risk of side effects.1 Consequently, a major focus of recent cancer drug development has been the discovery of drugs targeting single pathogenic variants or molecular entities.2 While this approach can be effective in situations where a pervasive driver underlies a particular cancer diagnosis, such as BCR‐ABL1 fusion gene in chronic myeloid leukaemia,3 the majority of cancers including gliomas represents a complex spectrum of diseases characterized by hundreds of molecular aberrations with different cancers exhibiting varied combinations of driver genes, which can further vary between patients and even within a single tumor.4 In this context, the effectiveness of targeting a single target may be limited.5 Despite the genetic heterogeneity of cancer, the phenotype of uninhibited cellular proliferation is shared by essentially all tumors, suggesting common convergent multitarget oncogenic pathways that could themselves be targeted as a novel therapeutic discovery strategy. Moreover, in contrast to perceived wisdom, multitarget drugs where the selectivity is for a certain group of “targets” rather than a single entity can lead to both increased efficacy and reduced side effects.6 For the prevention of transformation of low‐grade gliomas to ultimately fatal high‐grade tumors, where continuous drug treatment may be required for years or even decades, a reduced side effect profile is a prerequisite to delivering clinically implementable new therapeutics.
In this study, we aimed to identify a multitarget pathway that could be targeted for the discovery of novel therapeutics for the prevention or delay in transformation of non‐pediatric diffuse low‐grade astrocytomas (LGAs). Classified as grade II by the revised 4th edition of the World Health Organisation (WHO) classification of brain tumors, adult LGAs are one of the most common causes of cancer‐related morbidity and death in patients younger than fifty years old.7 Surgical resection is often not achievable due to extensive brain infiltration that goes far beyond the tumor epicenter.8 LGAs typically carry mutations in the isocitrate dehydrogenase 1 and 2 genes (IDH1, IDH2), TP53 and in the ATRX and are 1p/19q euploid.9 Almost invariable transformation of LGA into aggressive anaplastic astrocytoma (AA) and to secondary glioblastoma (GBM) is ultimately fatal. The timescale for transformation is unpredictable and the mechanisms causing them to become aggressive are poorly characterized.10
To identify a convergent proliferation characteristic underlying transformation of LGAs, we utilized an integrated systems‐level functional genomics framework which posits that multiple diverse biological perturbations (genetic, epigenetic) carrying risk for a given disorder may converge mechanistically on a common pathway.11 Specifically, network analyses represent a systems‐level approach for capturing the convergent role of heterogeneous perturbations in complex diseases12, 13, 14 such that tissue‐specific clusters of transcriptionally co‐regulated genes (“modules”) represent candidate regulators and drivers of disease states.12, 15 To identify transcriptional modules underpinning LGA transformation, we made use of publicly available IDH1‐mutant 1p/19q euploid LGA and AA gene expression data from The Cancer Genome Atlas (TCGA).16 We focused on IDH1‐mutant 1p/19q euploid astrocytomas, as these two molecular markers define a transcriptionally and epigenetically distinct subclass of gliomas.17, 18 Multilevel integrative analyses were then applied to determine the relationship between individual modules and LGA transformation, including differential co‐expression, integration with genetic risk data and integration with patient‐level genomic features and clinical survival. Reverse engineering analyses of genome‐wide gene–gene interactions and DNA–protein interactions were used to identify key regulatory drivers of network gene expression.19 Candidate drugs targeting the identified multitarget LGA transformation characteristic were computationally prioritized by integration with gene expression profiles from human cancer cell lines perturbed by small molecules (“drug perturbation data”), including Food and Drug Administration (FDA) approved drugs.20 Candidate drug predictions were validated using primary human anaplastic astrocytoma cells. Our analyses presented below outline a novel paradigm for elucidating a convergent pathway for astrocytoma progression and provide a proof‐of‐principle for multitarget drug screening as a novel strategy for prevention of transformation for low‐grade gliomas.
Materials and Methods
Glioma cell culture and treatment
Primary Astrocytoma WHO grade III cultures (TB98, TB62 and TB58) were established from fresh tumors and maintained in DMEM/F12 (1:1) supplemented with 10% fetal bovine serum (Complete medium, CM). Primary (passage 5) cell lines were treated with 20 µmol/L, 10 nmol/L or 2 nmol/L resveratrol (Sigma) or dimethyl sulfoxide (DMSO) (Sigma) and incubated for 24 h before extracting total RNA using the RNasey Mini kit according to the manufacturer’s instructions (QIAGEN). In order to keep the proportion of the solvent in medium low (below 1%), for the 20 μmol/L treatment experiment 2 mmol/L resveratrol working solution was prepared in DMSO due to resveratrol’s limited solubility in water (3 mg/mL, 131.44 μmol/L).
The effect of resveratrol on cell proliferation was determined at various time points using the sulforodamine B (SRB) assay (Sigma). For the long‐term exposure, resveratrol (2 nmol/L or 10 μmol/L) or DMSO was added to the cell culture twice per week over a period of 60 days. The cells were counted in the end of the treatment period.
RNA sequencing
Libraries for RNA sequencing (RNA‐seq) were prepared using the TruSeq Stranded Total RNA kit. The library was then sequenced on Illumina HiSeq4000 using 75‐bp paired‐end reads. The reads were mapped to the hg38 using the Spliced Transcripts Alignment to a Reference (STAR) software (version 2.5.0a).21 Quantification of mapped reads was carried out using HTSeq (version 1.6.1p1).22
TCGA low‐grade glioma data
Clinical data, somatic mutation annotation files and SNP array data as well as raw and normalized RNA‐seq read counts for low‐grade glioma samples were acquired from TCGA data portal (https://tcga-data.nci.nih.gov). For RNA‐seq data, fragments per million reads mapped (FPKM) was calculated for all genes.23 The genes with very low expression were removed and the gene expression was corrected for batch effects.24 We selected 49 grade II and 70 grade III IDH1‐mutated and 1p/19q euploid astrocytomas for the analysis. Clinical covariates used in this study are listed in Supplementary Table S1.
Differential gene co‐expression and differential gene expression
Differentially co‐expressed genes between IDH1 mut LGA and AA were calculated using DiffCoEx25 using the upper‐quartile normalized, batch‐corrected log2‐transfomed expression data. The statistical significance of co‐expression changes was determined using a permutation of intramodular correlation dispersion.25 To compare correlation of module genes between conditions, Wilcoxon rank‐sum test was used to determine the significance of the difference between average absolute correlation of the module genes. All differential gene expression analysis was performed using DESeq2.26 The batch and grade were added as factors in the design matrix for AA and LGA. Expression data of TB98 cells treated with 2 nmol/L and 10 nmol/L resveratrol were adjusted for surrogate variables.27, 28
Functional enrichment analysis
The gene ontology (GO) enrichment analysis was carried out using MSigDB v6.2 web‐based gene set investigation tool whereas the pathway enrichments were performed using WebGestalt.29 Enrichment ratio (r) representing the proportion of genes overlapping with genes in a GO/pathway category (k) compared to an expected value of k (ke). The expected value of k was calculated as number of module genes (n) divided by total number of genes in the reference gene set (m) multiplied by number of genes in the GO/pathway category (j), therefore ke = (n/m) * j and r = k/ke.25 In the WebGestalt analysis, background of 13,284 genes expressed in AA and LGA was used as reference, whereas in MSigDB analysis, all known human gene symbols (m = 45,956) were considered.
Module preservation
Expression data for additional glioma subtypes, other epithelial cancers and healthy tissues were downloaded from various sources and processed as described in Supplementary Table S6. Preservation of the differentially co‐expressed modules in other datasets was determined by calculating the Z summary preservation score.30 For Z summary scores > 10, module can be interpreted as preserved while Z summary scores < 2 suggest no preservation. Z summary scores between 2 and 10 indicate weak to moderate module preservation.
Cox proportional‐hazards model
To investigate the effect of module M2 activity on survival, Cox multivariable regression model was applied to AA cohort using the coxph function from the survival R‐package (number of events = 19, censored = 51). In the LGA cohort, number of events was not sufficient for the analysis (number of events = 5, censored = 44).
Genomic lesions in TCGA low‐grade gliomas
To identify significant mutations in LGA and AA, MutSigCV tool version 1.3 was applied. As a modification to the algorithm, instead of gene expression values averaged over 91 cancer cell lines Cancer Cell Line Encyclopaedia, the average gene expression in IDH1 mut and 1p/19q euploid astrocytomas was used.31 For the identification of arm‐level and focal chromosomal aberrations, the GISTIC 2.0 was employed. The confidence level for identifying focal regions containing a driver was set to 0.9 and the threshold for a copy‐number variation (CNV) occurrence was kept default. Genomic alterations (focal and arm‐level) were visualized using the ComplexHeatmap R‐package.32
Association between module gene expression and CNVs in regions harboring cancer genes
Oncogenes and tumor suppressor genes (TSG) were defined according to the COSMIC v76 list of genes with mutations causally implicated in cancer (Cancer Gene Census) (http://cancer.sanger.ac.uk/census/).33 For each oncogene and TSG, the association between somatic CNVs overlapping this oncogene or TSG and the average expression of M1 and M2 was calculated using a linear model. Additionally, we calculated the difference in the relative frequency of events in a given oncogene or TSG between LGA and AA. The oncogene or TSG alteration event was determined using GISTIC2.0 output, where values >1 were considered as amplification and values <−1 as deletions of the gene. The sum of alteration events per oncogene or TSG in the samples of given histopathological grade was divided by the number of samples in the cohort. Relative incidence of alterations was calculated as the difference between cohort‐adjusted sum of alterations in an oncogene or TSG. Values over 0 indicate that the cancer gene was preferentially altered in AA.
Cancer predisposition genes
Cancer predisposition gene lists were retrieved from the literature. Genes in which germline mutations increase the risk of developing cancer (n = 114) were acquired from Supplementary Table S1 in Rahman33 and from the Catalogue Of Somatic Mutations In Cancer (COSMIC) v76 Cancer Gene Census catalogue (n = 84) (http://cancer.sanger.ac.uk/census). 30 Additionally, a set of cancer‐associated genes harboring rare germline truncation variants across 12 tumor types (n = 249) was attained from Supplementary Data 2 in Lu et al.34 The statistical significance of overrepresentation of cancer predisposition and cancer‐associated genes was calculated using Fisher’s Exact test (FET) and the odds ratios (ORs) were aggregated into a summary estimate using the fixed effects model in the metafor R‐package.35
Transcription factor binding site enrichment
To identify transcription factor (TF) binding sites (TFBS) overrepresented in the promoters of M2 module genes, two databases were used. WebGestalt29 was accessed online at http://www.webgestalt.org. In parallel, the TRANSFAC® database release 2017.1. was queried, as experimental evidence of binding and regulation can be obtained in addition to TF – regulatory site binding predictions. We employed the “FMatch” analysis to identify overrepresented TFs in the gene set list of interest. Best promoters supported with binding site defined −500 to +200 bp from the transcription start site were set as parameters.
Cell division cycle gene sets
Full list of human cell division cycle genes was downloaded from GO database AmiGO v1.836 accession GO:0007049 (http://amigo.geneontology.org/amigo) (n = 1,768). The rest of the cell cycle gene sets were obtained from the Molecular Signature Database (MSigDB v6.2) (http://software.broadinstitute.org/gsea/msigdb/index.jsp).
Master regulator analysis
To infer TF–target interactions, Accurate Reconstruction of Cellular Networks (ARACNe) was applied.37 ARACNe‐AP (ARACNe with Adaptive Partitioning) tool (version 1.4) was run as a command‐line java executable described by Lachmann et al.38 Batch‐corrected FPKM values for both, LGA and AA along with genes defined as TFs in FANTOM 5 project39 were used as input. First, the significance threshold for mutual information was calculated (αMI = 0.21 at P‐value < 10− 8) and thereafter ARACNe was ran in 100 bootstraps to generate a consensus network robust to expression outliers (edge significance P‐value < 0.05 with Bonferroni correction).40 The ARACNe‐derived network was converted into a regulon by determining the direction for each ARACNe‐inferred TF–target pair using the Spearman’s correlation. Positively correlated TF and its predicted target expression is interpreted as positive TF mode, while TFs' expression is negatively correlated with its inferred targets is considered a negative regulator of the gene expression.41 Master regulators (MRs), which are up‐regulated in one condition and its positively regulated targets are enriched for overexpressed genes and negatively regulated targets for underexpressed genes, are termed activated MRs while for repressed MRs behave in the opposite manner. Most strongly influenced targets (highly ranked members of gene sets) were extracted for each MR in leading edge analysis. After extracting the leading edges of gene set enrichment for all the significant MRs, we performed a one‐tailed FET to calculate the significance of the overlap between the activated and repressed MR target gene sets and the differentially co‐expressed modules M1 and M2. The ssmarina package R‐functions (version 2) implemented in the MR analysis were downloaded from http://dx.doi.org/10.6084/m9.figshare.785718.
Literature‐based discovery of FoxM1 and B‐Myb targets
FoxM1 and B‐Myb binding data were collected from the literature. Chromatin immunoprecipitation (ChIP) data for FoxM1 binding peaks were retrieved from the Supplementary Table S1 in Wiseman et al.42 Supplementary Table S1 in Chen et al.43 and Supplementary Table S8 in Fisher et al.44. ChIP peaks for B‐Myb were obtained from deposited Supplementary Table S2 in Sadasivam et al.45 and processed ENCODE B‐Myb binding data Supplementary Table S8 in Fisher et al.44 We considered gene a target if it appeared in two‐thirds of FoxM1 binding peak lists and in 2/2 of B‐Myb binding studies.
E2F2 ChIP‐seq data analysis
Raw ChIP‐seq data were downloaded for Gene Expression Omnibus (GEO) database (accession number GSM1208606 for E2F2 and GSM1208813 for IgG control).46 The reads were aligned to human genome NCBI GRCh38 using the bowtie aligner version 1.0.0 and pre‐built index file sourced from http://bowtie-bio.sourceforge.net/manual.shtml#what-is-bowtie on 3/07/2017. Reads with more than one reportable alignment were omitted from further analysis. ChIP‐seq quality was assessed by applying Phantompeakqualtools 46 version 1.1 and the quality metrics of experiments satisfied the recommendations by ENCODE and modENCODE consortia.47 Peaks were called using the Macs2 software version 2.1.1., with False Discovery Rate ( FDR) cut‐off of 0.01 to call significant regions.48 Called peaks annotated using the annotatePeak function from the ChIPseeker R‐package.49 Peaks between 500 bp upstream to 200 downstream from the transcription start sites were retained for further analysis.
Connectivity Map analysis
Connectivity Map (CMap) is a public database containing genome‐wide expression profiles for three human cancer cell lines (MCF7, PC3, HL60), 6 hours after treatment with different concentrations of ~1300 small chemical compounds. Raw .CEL files and pre‐ranked data are available for download at https://portals.broadinstitute.org/cmap/. Expression was profiled using one of Human Affymetrix U133A platforms (HT_HG‐U133A, HT_HG‐U133A_EA, HG‐U133A). Raw .CEL files were processed from probe to gene‐level using custom (CDF) probe annotations to GENCODE gene ids.50, 51 Background correction and normalization of expression were performed using RMA and variance stabilization normalization (vsn) methods as implemented in the affy R‐package.52, 53 To conduct differential expression analysis, assays with less than two control or drug treatment samples were excluded. For ease of interpretation, GENCODE gene IDs were converted HUGO gene IDs. Differential expression was quantified using the limma package,54 while accounting for batch effects within the model. To perform module enrichments for genes whose expression is modulated by drug treatment, we first looked for drugs that modulate at least five genes at 10% FDR in the dataset of interest. Subsequently, significance of the overlap of module genes with the list of genes up or down‐regulated by the drug was calculated using one‐tailed FET. P‐values of enrichment were corrected for multiple testing using Benjamini–Hochberg FDR procedure.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed using GSEA Java desktop application by the Broad Institute (version 2.2.4). The significance of module M2 gene set enrichments in the extremities of ranked list with values ordered by –log10(P‐value) × log2(fold change) was assessed by permuting the rank list 10,000 times. The ranked list was ordered from the most up‐regulated to the most down‐regulated genes so that positive normalized enrichment score would indicate overrepresentation of the gene set among the most down‐regulated genes. GSEA of genes up‐ and down‐regulated by resveratrol in different concentrations, the Hallmark Gene Set from MSigDB were used.55
Protein–protein interactions
The protein‐protein interactions (PPIs) were acquired from the STRING database v10.5.56 Interaction sources selected were text‐mining, experiments, and databases. The minimum required interaction score was set to 0.4.
Kinase inhibition data
Kinase Profiling Inhibitor Database (http://www.kinase-screen.mrc.ac.uk) accessed 02/11/2018. The database was queried for resveratrol.
Results
Gene network and differential co‐expression analysis in astrocytomas
Transcriptional changes can be related to a disease state via two paradigms, the analysis of gene covariation (differential gene co‐expression) and gene expression levels (differential gene expression).57 In order to identify sets of genes differentially covarying in the progression of LGA, we selected supratentorial IDH1 mutant and 1p/19q euploid adult astrocytoma samples from the TCGA database (Supplementary Table S1). The cohort included 49 LGA and 70 AA samples (Supplementary Fig. S1). We used DiffCoEx algorithm25 to identify set of genes with differential co‐expression between the tumor grades (see Methods), yielding two modules of significantly differentially co‐expressed genes: M1 (n genes = 272) (P‐value < 0.001) and M2 (n genes = 177) (P‐value < 0.001) (Fig. 1A) (Supplementary Figure S2) (Supplementary Table S2). Genes without differential co‐expression cluster assignment were grouped in module M0 (n genes = 12,835). The average correlation of M1 genes was significantly higher in LGAs (P‐value < 10− 16, Mann–Whitney U‐test) while the average correlation of module genes for M2 was higher in AAs (P‐value < 10− 16, Mann–Whitney U‐test) (Fig. 1B).
Figure 1.
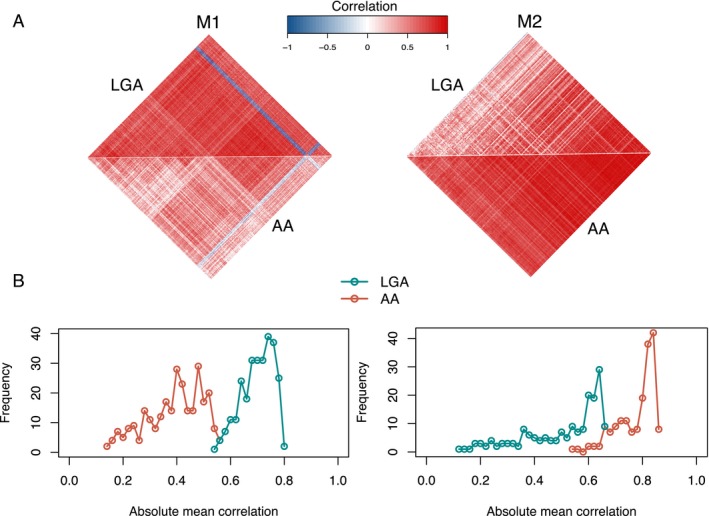
Identification of differential co‐expression modules M1 and M2. (A) Heatmaps showing the strength of correlation between module genes in low‐grade (LGA) and high‐grade (AA) astrocytomas. (B) Frequency plots of absolute mean correlation of module M1 (right panel) and M2 (left panel) genes in LGA and AA samples. The absolute mean correlation of M1 was significantly higher in LGA samples (P‐value < 10–16, Wilcoxon rank‐sum test), and the absolute mean correlation of M2 higher in AAs (P‐value < 10–16, Wilcoxon rank‐sum test).
We then investigated whether modules differentially co‐expressed between LGA and AA were also differentially expressed. Out of 272 genes in M1, 18 genes (6.6%) were significantly (FDR < 5%) differentially expressed and down‐regulated in AA, whereas 160 out of 177 genes (90%) in module M2 were up‐regulated in AA (FDR < 5%) (Supplementary Fig. S3) (Supplementary Table S3). The average expression of module M2 genes was significantly higher in AAs (P‐value = 1.32 × 10− 5, Mann–Whitney U‐test), while the average expression of M1 did not vary significantly between LGAs and AAs (P‐value = 0.60) (Supplementary Fig. S4).
As co‐expressed genes may share related biological functions,58, 59 we investigated whether the two differentially co‐expressed modules were enriched for genes relating to specific biological pathways or GO terms using web‐based tools – WebGestalt29 and MSigDB.60 Module M1 genes were enriched for GO terms related to neuronal constituents such as “neuron part” (Enrichment ratio (r) = 11.62, FDR = 1.89 × 10− 63) and “synapse” (r = 14.57, FDR = 3.10 × 10− 52), while M2 was highly enriched for cell division genes such as GO term “cell cycle” (r = 16.82, FDR = 3.83 × 10− 160) (Fig. 2A and 2) (Supplementary Tables S4 and S5), suggesting a functional distinction between these two modules. In keeping with their functional specialization from the analysis of GO terms, M1 and M2 were highly enriched for biological pathways such as “neuronal” system (M1, r = 5.46, FDR = 2.98 × 10− 6) and “cell cycle” (M2, r = 17.98, FDR = 1.18 × 10− 68), respectively (Supplementary Fig. S5).
Figure 2.
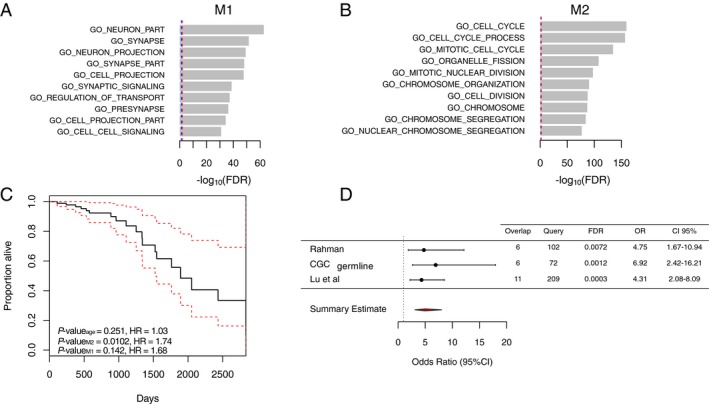
Characterization of modules M1 and M2. (A) Bar‐plot showing the functional enrichment of module M1 genes. The top enriched genes encode for genes annotated by GO as components of a neuron. (B) Bar‐plot of module M2 functional enrichment. The genes with cell cycle GO term were most significantly enriched. The x‐axis represents the significance of overrepresentation of biological pathway genes in modules calculated by the hypergeometric test. Blue‐dashed line represents FDR = 0.05, and red‐dashed line represents FDR = 0.01. (C) Estimated survival function for the Cox regression of time to death in AA cohort. Factors analyzed were the mean expression of module M2 and M1 in the AA and age at diagnosis. The dashed lines show point‐wise 95% confidence intervals around the survival function. Number of events = 19, censored = 55. (D) Forest plot showing the enrichment of M2 genes among cancer predisposition genes and genes harboring rare germline variants in cancer. The gene sets tested were derived from publications by Rahman33 (n = 102), and Lu et al.34 (n = 209), as well as germline mutations from COSMIC Cancer Gene Consensus (CGC)61 (n = 72). The figure shows the OR representing that the module M2 is more likely to contain genes from these gene sets than expected by chance with corresponding 95% confidence intervals. Overlap, number of query genes overlapping the module M2; Query, number of genes in each query list; OR, odds ratio; CI, confidence intervals.
Next, to provide evidence for the biological validity of modules M1 and M2, we undertook a series of comparative network analyses assessing the conservation of co‐expression between module genes in unrelated gene expression datasets. We explored module preservation in other malignancies such as oligodendroglioma, IDH1‐wild type astrocytoma, GBM, colon adenocarcinoma, liver hepatocellular carcinoma, lung adenocarcinoma, as well as in healthy tissue with proliferative potential (colon, lung, liver), and normal cerebral cortex tissue (Supplementary Table S6). We used the module composite preservation statistic Z summary score, where Z summary scores >10 indicate strong module preservation, Z summary scores < 2 suggest no preservation and Z summary scores between 2 and 10 indicate weak to moderate module preservation.30 Since module M1 genes were on average more highly co‐expressed in LGA and M2 genes more strongly co‐expressed in AA, the reference datasets to test module preservation were set to LGA for M1 and AA for M2. Taking this Z summary score approach, we found that co‐expression of M1 was preserved only in neural tissue, in keeping with its functional specificity (“neuronal processes”) (Table 1), while module M2 (“cell cycle”) was highly preserved in proliferating tissues, including different types of cancers as well as renewing healthy tissue.
Table 1.
Preservation of differential co‐expression networks
| Tissue Origin | Histopathology | Data source | Tissue | Z summary score M1 | Z summary score M2 |
|---|---|---|---|---|---|
| Neural | Malignant | TCGA | WHO grade III IDH1wt astrocytoma | 26.8 | 46.5 |
| Neural | Malignant | TCGA | WHO grade II IDH1mut 1p/19q codeleted oligodendroglioma | 63.3 | 48.8 |
| Neural | Malignant | TCGA | WHO grade III IDH1mut 1p/19q codeleted oligodendroglioma | 16.4 | 18.9 |
| Neural | Malignant | TCGA | Glioblastoma | 23.0 | 67.1 |
| Neural | Malignant | GEO | Glioblastoma (GSE77530) | 12.8 | 29.9 |
| Neural | Malignant | GEO | Glioblastoma (non‐enhancing) (GSE59612) | 30.0 | 18.9 |
| Neural | Malignant | GEO | Glioblastoma (enhancing) (GSE59612) | 3.7 | 45.9 |
| Nonneural | Malignant | TCGA | Colon adenocarcinoma | 0.8 | 33.8 |
| Nonneural | Malignant | TCGA | Liver hepatocellular carcinoma | 1.4 | 58.8 |
| Nonneural | Malignant | TCGA | Lung adenocarcinoma | 2.0 | 101.9 |
| Nonneural | Nonmalignant | GTEx | Colon | 1.1 | 15.0 |
| Nonneural | Nonmalignant | GTEx | Lung | 0.5 | 16.6 |
| Nonneural | Nonmalignant | GTEx | Liver | 0.1 | 6.9 |
| Neural | Nonmalignant | GTEx | Cerebral cortex | 11.8 | –1.3 |
Preservation of modules M1 and M2 in neural and non‐neural malignant and non‐cancerous tissues assessed by the composite network preservation statistic Z summary.30 Z summary scores >10 represent strong evidence for preservation, Z summary < 2 is interpreted as no preservation and Z summary 2–10 suggests weak to moderate preservation. LGA expression was used as the reference set to investigate M1 preservation and AA to explore module M2 preservation in other datasets.
To further assess the biological validity of module M2, we investigated the physical interactions between the protein products of M2 genes. Out of the 177 genes in module M2, 133 (75%) were identified as cell cycle genes according to GO biological process annotation36 (FDR = 1.18 × 10− 68). Selecting sources of PPI from the STRING database56 for the total 1,768 cell cycle genes defined by the GO biological process annotation (GO:0007049, n = 1,768), we found that the most highly connected cell cycle genes were significantly (P‐value < 10− 4) overrepresented in module M2 (Supplementary Fig. S6A and B). These results highlight that the genes constituting M2 represent central hubs of the overall set of genes governing the cell cycle and point toward an important influence of M2 on this biological process.
M2 gene network’s relevance to astrocytoma
The above results suggest that M2 network represents a set of co‐regulated genes capturing the known relationship between cell cycle regulation and progression of LGA to AA.19 To investigate this further, we assessed the relationship between M2 genes and known cancer predisposition genes; the identification of disease predisposition genes (i.e., genes which when impacted by germline mutation increase the lifetime risk of a disease) has been instrumental to the discovery of causal pathways.57
For module M2, we observed a significant (OR = 4.75, 95% CI [1.67–10.94], FDR = 0.007) overrepresentation of bona fide cancer predisposition genes annotated by Rahman,33 for cancer genes listed in the COSMIC Cancer Gene Census catalogue61 (OR = 6.92, 95% CI [2.42–16.21], FDR = 0.001) and a significant overrepresentation of rare germline truncation variants identified in 249 cancer‐associated genes across 4,034 cancer cases from TCGA34 (OR = 4.31, 95% CI [2.08–8.09], FDR = 0.0003) (Fig. 2D). The biological functions of the cancer predisposition genes overlapping M2 genes were related to cell division and DNA repair (Supplementary Table S7), therefore the enrichment of these genes in module M2 is influenced by overrepresentation of cell cycle genes among module M2 genes. These results highlight that M2 is significantly enriched for cancer predisposing genes in excess of random expectation suggesting M2 captures molecular processes associated with the development and maintenance of cancer.
Next, we explored the relationship between M2 expression in the human glioma tissue sample and patient survival. Survival analysis among the AA cohort (n = 70, number of events = 19) using the Cox proportional‐hazards regression model62 revealed that AA patient survival was slightly but significantly dependent on average expression of module M2 (Hazard Ratio (HR) = 1.74, 95% CI [1.14–2.64], P‐value = 0.01), but not average expression of module M1 (HR = 1.68, 95% CI [0.83–3.369], P‐value = 0.142) or age at diagnosis (HR = 1.03, 95% CI [0.98–1.08], P‐value = 0.25) (Fig. 2C), suggesting an association between M2 expression and tumor progression.
Finally, we explored the relationship between M2 expression in LGA and AA, and patient‐level genetic features. Among the TCGA cohort of IDH1 mut LGA and AA, we observed that AA samples had on average more single nucleotide variants and INDEL mutations per sample than LGAs (Supplementary Fig. S7A). However, only the TP53 (n mut = 134, FDR < 10− 13), ATRX (n mut = 96, FDR < 10− 13) and Mucin‐4 (MUC4) (n mut = 17, FDR = 4.80 × 10− 7) genes carried significantly more somatic mutations than expected considering the average background mutation frequency of the genes in these samples (see Methods) (Supplementary Table S8). Somatic mutations in other genes were not frequent in this collection of samples with the large proportion of mutated genes observed only once or seldom in more than 3% of the glioma samples (Supplementary Fig. S7B). In contrast, we observed recurrent somatic CNVs in multiple samples including copy number loss of well‐known TSG CDKN2A at 9p21.3 (n del = 45/119), RB1 at 13q14.3 (n del = 40/119) and PTEN at 10q23.31 (n del = 22/119) (Supplementary Fig. S8). We next explored the effect of copy number change (i.e., gene dosage) on the genes presenting the alteration. We focused on the known oncogenes and TSG obtained from the COSMIC list of genes with somatic mutations causally implicated in cancer (Cancer Gene Census).61 Out of 484 oncogenes and TSG from the COSMIC Cancer Gene Census database, 338 (69.8%) were affected by CNVs in our sample set (based on the GISTIC analysis, see Methods). As not all CNVs induce aberrant activity of their gene products, we selected these oncogenes and TSG‐altering CNVs, whose ploidy influences expression of that gene (functional copy number alterations, FCNAs).63 Among the 338 oncogenes and TSGs altered by CNVs, 156 genes (46.2%) were considered influenced by FCNAs, that is, their gene dosage had a significant (FDR < 0.05) effect on the expression of the affected gene. The 338 oncogenes and TSG as well as 156 oncogenes and TSG affected by FCNA were not significantly (P‐value > 0.05) over‐ or underrepresented among module M1 and M2 genes. Only 5 of 338 oncogenes and TSG (OR = 1.12, P‐value = 0.81) and 2 of 156 oncogenes and TSG affected by FCNV (OR = 0.96, P‐value = 1.00) were found in module M2 and 2 of 338 oncogenes and TSG (OR = 0.28, P‐value = 0.05) and none of the 156 oncogenes and TSG affected by FCNV (OR = 0, P‐value = 0.08) in module M1. We observed, however, that the gene dosage of 61 of 338 (18.1%) oncogenes and TSG and 36 of 156 (23.1%) oncogenes and TSG affected by FCNA were significantly (FDR < 0.05) associated with an increase in the average expression level of module M2 across the patient cohort (n = 119) (Supplementary Table S9). In contrast, M1 expression was not associated with gene dosage of any oncogenes or TSG. The change in average M2 expression in relation to FCNA in oncogene or TSG dosage was strongest (FDR < 0.0003) for loss of cancer‐related genes located on chromosome arm 10q (VTI1A, TCF7L2, WDR11 and PTEN) a structural genetic variant previously associated with poor prognosis in LGAs and AAs and occurring more frequently in AAs in this cohort (Fig. 3).64, 65, 66 Additionally, strong associations were found between PDGFRA amplification (FDR < 0.001) and CIC deletion (FDR < 0.001). The COSMIC Cancer Gene Census gene list does not list WDR11 as a cancer gene, but the gene was included in the analysis since its disruption in glioma has been previously reported.67, 68
Figure 3.
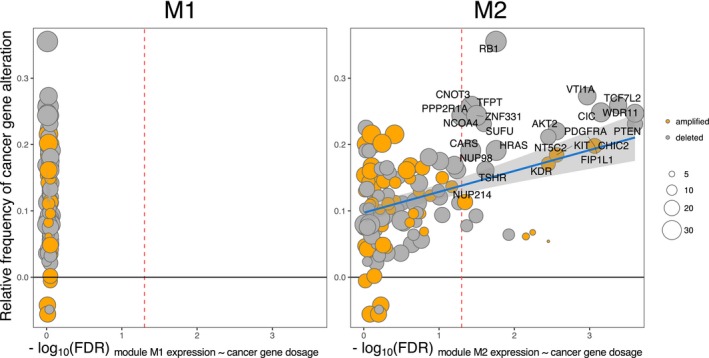
Association of module expression with dosage of oncogenes and tumor suppressor genes. Average module M2 expression is significantly associated with gene dosage of several functionally aberrant oncogenes and TSG (cancer genes) (‐log10(FDR) > 1.30, red dashed line) (right panel), whereas average module M1 expression is not significantly associated with gene dosage changes in any oncogenes and TSG (left panel). M2 module is most significantly associated with deletion of PTEN, TCF7L2, WDR11, CIC and amplification of PDGFRA, FIPL1, CHIC2. The stronger association between module genes and alterations in oncogenes and TSG more frequent in AA is highlighted by linear model fit represented by blue line with 95% confidence intervals in gray shading. On the y‐axis: difference of sample size‐adjusted number of CNV events in a given cancer gene. Relative frequency >0 represents gene dosage alteration events more often found in AA whereas relative frequency <0 is assigned to gene dosage alteration events more frequent in LGA. The horizontal gray line marks the relative frequency = 0. On the x‐axis: negative logarithm of the FDR of difference in average module expression between samples where a given oncogene or TSG is altered and samples where the gene is intact. The oncogenes or TSG having significant associations (FDR < 0.05) with module expression and are preferentially altered in AA (ratio > 0.1) are labelled. Vertical dashed red line represents log10(FDR) = 1.3. The data point size corresponds to the combined number of alteration events in the cancer gene (LGA + AA).
In summary, these integrative systems‐level analyses reveal that module M2 is differentially co‐expressed between IDH1 mut LGA and AA, overexpressed in AA compared to LGA, enriched for known cancer predisposition genes, functionally enriched for genes related to the cell cycle, and has a pattern of expression in human glioma samples associated with both patient survival and genetic aberrations in chromosomal regions harboring known oncogenes or TSG. These results suggest M2 as a candidate network associated with astrocytoma progression. We therefore set out to investigate the transcriptional regulation of M2 in more detail.
Transcriptional regulation of M2 gene network
We first investigated whether the gene co‐expression might result from the regulatory control of a common TF; this may be reflected by enrichment for TFBS sequences in a set of co‐expressed genes.69 Analysis for enrichment of TFBS among M2 genes using the web‐based gene set enrichment analysis tools – WebGestalt70 and the TRANSFAC®71 database revealed an overrepresentation of TFBS in M2 for the E2F‐family TFs (WebGestalt, FDR = 3.42 × 10− 17) (TRANSFAC®, P‐value = 2.16 × 10− 4). The E2F‐family of TFs has an established direct role in cell cycle control and loss of function in E2F‐binding suppressor protein Rb leads to uncontrolled cell proliferation.72, 73 Additionally, we observed enriched TFBS among M2 genes for CUX1 (Cut Like Homeobox 1), Myb‐like factors and NF‐Y (Nuclear Factor Y) TF family, which have been previously associated with the regulation of cell division74, 75, 76 (Supplementary Table S10).
These TFBS enrichment analyses provided an independent line of evidence to support the validity of M2 as a gene co‐regulatory network which is functionally related to cell cycle regulation. However, since sequence specificities of TFBS motifs have yet to be established for more than half of bona fide TFs,77 enrichments for TFBS alone are unlikely to provide a complete picture of the regulatory control of gene co‐expression networks. Factors like cell lineage‐specific chromatin conformation, promoter methylation, and utilization of alternative promoters may all contribute to the complexity of transcriptional regulation.78 With this in mind, we employed the ARACNe through Adaptive Partitioning inference of mutual information (ARACNe‐AP)37 to derive a global IDH1 mut and 1p/19q euploid astrocytoma‐specific transcriptional regulatory network (“regulome”). In addition, this approach allowed us to investigate the relationship of this empirically derived tissue‐specific regulome and module M2 (and M1) expression. ARACNe uses mutual information to infer statistical dependency between gene expression profiles of TFs and their target genes. Significant mutual information between a given TF and a gene’s expression is interpreted as a functional relationship between the two.79 We applied the ARACNe‐AP algorithm to LGA and AA gene expression profiles combined and obtained a regulome with 1,293 regulators, 13,260 targets and 171,429 interactions (see Methods). In the second step, we used the Master Regulator Inference Algorithm (MaRInA)52,53 to prioritize TFs influencing the transcriptional programs associated with progressing glioma. The MaRInA method makes use of differentially expressed genes between two conditions (i.e., between LGA and AA) and the directionality of the ARACNe‐inferred TF targets in the regulon. If the transition of the phenotype between LGA and AA is prompted by activation or repression of specific TFs, then their positively and negatively regulated target genes would be among the most differentially expressed. Using this strategy, we identified 23 candidate MRs of low‐grade astrocytoma progression (P‐value < 0.01), among which 16 were activated and seven repressed in the anaplastic astrocytoma state (Fig. 4).
Figure 4.
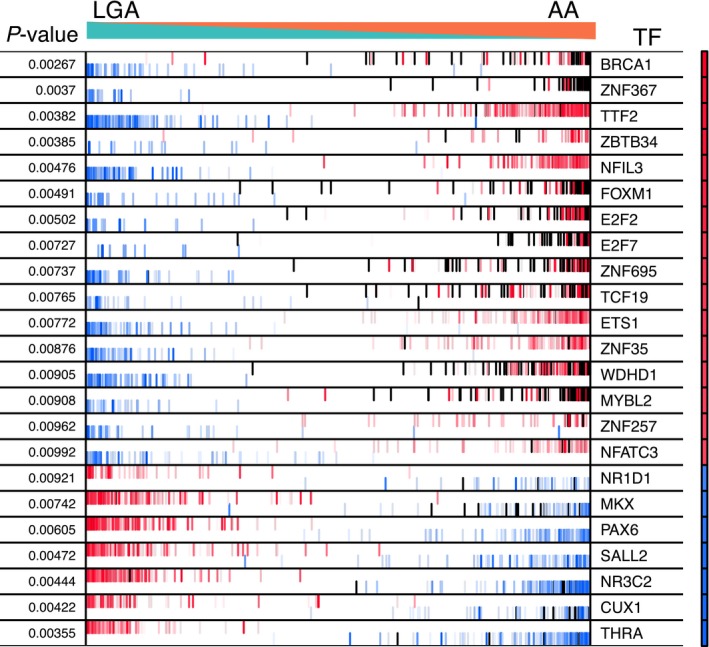
Significant MRs determined by MaRInA analysis for the IDH1 mut astrocytoma‐specific transcriptional signature (P‐value < 0.01). Each row of the plot corresponds to the MaRInA results obtained for the respective MR. The narrow heatmap column on the right illustrates the predicted MR activity, with red indicating an increase of activity in AA samples (positive NES) and blue indicating a decrease (negative NES). The x‐axis represents the AA‐specific gene expression signature, rank‐ordered from the most down‐regulated to most up‐regulated gene in AA, compared to LGA. The red and blue bar code lines correspond to the ARACNe‐predicted target genes of a given MR. Positively regulated target genes are shown in red and negatively regulated target genes are shown in blue. Module M2 genes are noted as black lines. Increased activity of an MR is implied by the enrichment of its positive targets among the overexpressed genes in the AA signature and of its negative targets among the underexpressed genes in the AA signature. Conversely, decreased MR activity is indicated by the enrichment of its positive targets among the underexpressed genes in the AA signature and of its negative targets among the overexpressed genes in the AA signature. AA, anaplastic astrocytoma, LGA, low‐grade astrocytoma, NES, normalized enrichment score, TF, transcription factor.
Next, we integrated the information about differential TF activity and differential co‐expression in LGA and AA (Supplementary Fig. S9). We found significant enrichment of module M2 genes among the overexpressed genes of the activated MR targets (FET, P‐value = 1.4 × 10− 198, one‐tailed) and the underexpressed genes of repressed MR targets (FET, P‐value = 9.3 × 10− 35, one‐tailed) (Supplementary Table S11). Module M1 genes were not significantly overrepresented in any of the MR‐regulated gene sets. To prioritize the MRs with the greatest influence on M2 expression, we ranked these TFs based on the significance of overlap of their individual regulons with the M2 module (see Methods). We found that cumulatively the top three TFs (FOXM1, MYBL2, E2F2) are predicted to influence the expression of 149 of 177 (84%) of M2 network genes (Table 2). The TFs FoxM1 and B‐Myb are known to cooperate during the G2 to M transition of the cell cycle and are up‐regulated in several cancers, including gliomas.19, 45, 80 Supporting the regulation of M2 network by FOXM1, MYBL2, and E2F2, we found that binding targets of FoxM1 detected by ChIP‐seq analysis (coded by FOXM1), B‐Myb (coded by MYBL2) and E2F2 were also highly significantly enriched among M2 module genes (FDR = 6.06 × 10− 34, FDR = 1.74 × 10− 92 and FDR = 1.10 × 10− 21, respectively) (Supplementary Table S12). A considerable proportion of ChIP‐seq detected FoxM1, B‐Myb, and E2F2 targets in the M2 module had been predicted to be positively regulated by these TFs in the MaRInA analysis (Fig. 5A–C) (Supplementary Fig. S10). The large overlap between the predictions of MaRInA approach and experimentally detected target gene sets for the FoxM1 and B‐Myb in M2, supports a role for these TFs and their functional relationship to M2 in orchestrating aberrant cell cycle gene expression (Fig. 5D).44, 81 Compared to a list of cell cycle genes and cell cycle phase‐specific gene signatures, the strongest enrichment of ChIP‐seq detected targets of FoxM1 and B‐Myb was among the M2 genes (Supplementary Fig. S11), suggesting that M2 captures the downstream targets of these MRs.
Table 2.
Transcription factor regulons with strongest overlap with M2 module genes.
| TF | # regulon genes | # overlap with M2 | FET FDR | # unique added genes | Cumulative contribution | % M2 network coverage | |
|---|---|---|---|---|---|---|---|
| 1. | FOXM1 | 156 | 124 | 3.69 × 10–25 | 124 | 124 | 70 |
| 2. | MYBL2 | 174 | 126 | 6.43 × 10–220 | 15 | 139 | 79 |
| 3. | E2F2 | 167 | 121 | 8.49 × 10–210 | 10 | 149 | 84 |
| 4. | ZNF367 | 141 | 115 | 5.12 × 10–209 | 9 | 158 | 89 |
| 5. | E2F7 | 138 | 108 | 7.66 × 10–191 | 4 | 162 | 92 |
| 6. | ZNF695 | 153 | 105 | 3.35 × 10–174 | 2 | 164 | 93 |
| 7. | WDHD1 | 174 | 103 | 4.80 × 10–160 | 3 | 167 | 94 |
| 8. | TCF19 | 134 | 85 | 3.01 × 10–133 | 1 | 168 | 95 |
| 9. | BRCA1 | 82 | 53 | 2.16 × 10–81 | 1 | 169 | 95 |
| 10. | MKX | 86 | 26 | 2.89 × 10–28 | 0 | 169 | 95 |
| 11. | NR3C2 | 135 | 22 | 1.30 × 10–17 | 0 | 169 | 95 |
| 12. | TTF2 | 189 | 24 | 1.40 × 10–16 | 1 | 170 | 96 |
| 13. | CUX1 | 51 | 10 | 3.27 × 10–09 | 0 | 170 | 96 |
| 14. | THRA | 122 | 12 | 2.35 × 10–07 | 0 | 170 | 96 |
| 15. | ZNF257 | 20 | 4 | 3.75 × 10–04 | 0 | 170 | 96 |
| 16. | NR1D1 | 27 | 4 | 1.26 × 10–03 | 0 | 170 | 96 |
| 17. | NFATC3 | 40 | 4 | 5.75 × 10–03 | 0 | 170 | 96 |
| 18. | ZNF35 | 56 | 4 | 1.96 × 10–02 | 0 | 170 | 96 |
The TFs are ranked by significance of overlap of regulon genes with the M2 module genes (FDR corrected P‐value < 0.05). The cumulative contribution of TF regulon genes to the M2 module. Taken together, the genes belonging to regulons of the MR cover 96% (170/177) of module M2 genes. The number of unique genes each MR contributes in addition to the higher ranking MRs is shown in the # unique added genes column. Collectively the top 5 TF regulons include 92% (162/177) genes of the M2 module.
Figure 5.
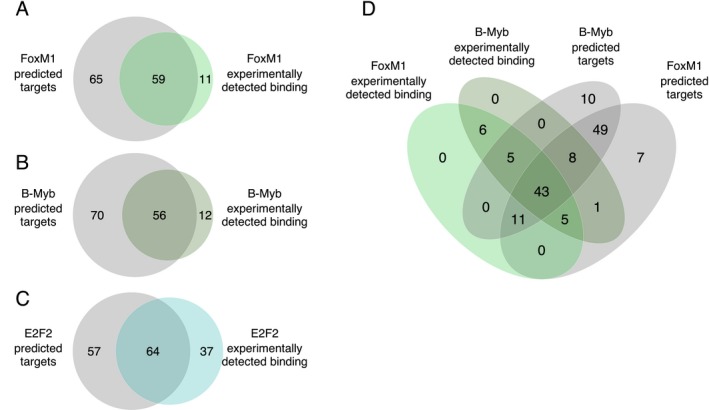
Concordance between the predicted and validated TF targets in the M2 module. (A–C) Pairwise comparison of positively regulated TF target predictions by ARACNe‐MaRInA algorithm and experimentally detected bindings in ChIP‐seq analysis. (D) Overlap between up‐regulated FoxM1 and B‐Myb targets predicted by ARACNe‐MaRInA analysis and respective promoter bindings identified in a ChIP‐sequencing experiments.
In silico prediction of compounds targeting the M2 gene network
The above results highlight module M2 as a gene co‐regulatory network capturing convergent cellular process active in AA. To translate this insight to therapeutic discovery, we sought to identify compounds with the ability to repress M2 overexpression in AA as a potential means of inhibiting the growth of AA and, by extension, delaying transformation of LGA to AA.14 To this purpose, we used publicly available data reporting changes in gene expression in human cancer cells following exposure to ~1300 bioactive compounds (CMap).14, 82 We aimed to identify compounds that are able to induce a cellular transcriptional response (i.e., a gene expression profile) that is anticorrelated to the coordinated transcriptional program M2 underpinning AA. To identify drugs predicted to down‐regulate module M2, we assessed the significance of overlap between M2 module genes and genes significantly down‐regulated by any given compound (Supplementary Table S13). Taking this approach, we found that genes down‐regulated in response to the known anticancer drug etoposide had the most significant overlap with M2 module genes (FET, FDR = 4.17 × 10− 83, one‐tailed), while those down‐regulated by methotrexate had the fifth most significant overlap with M2 (FET, FDR = 1.28 × 10− 51, one‐tailed) (Table 3). Etoposide is a topoisomerase inhibitor and its efficacy in improving survival in high‐grade glioma patients has been shown in a large scale meta‐analysis,83 while adjuvant methotrexate was suggested to prolong event‐free survival in pediatric high‐grade glioma in Phase II clinical trial.84 In addition to drugs with known efficacy in glioma, several drugs not previously implicated in the treatment of glioma were predicted to down‐regulate M2 including the naturally occurring stilbenoid resveratrol, which was the second most efficient drug predicted to down‐regulate M2 (resveratrol 10 µmol/L, FET FDR = 2.44 × 10− 63, one‐tailed; resveratrol 17.6 µmol/L, FDR = 1.90 × 10− 51, one‐tailed; resveratrol 50 µmol/L, FDR = 1.15 × 10− 28, one‐tailed). The effect of resveratrol on M2 network expression provides a novel insight to the mechanism underlying its reported antineoplastic effects.85, 86, 87
Table 3.
Connectivity Map hits.
| Compound | FDR (FET) | Cell line | Concentration (µM) | Treated | Controls | |
|---|---|---|---|---|---|---|
| 1. | Etoposide | 1.26 × 10–80 | MCF7 | 6.8 | 2 | 12 |
| 2. | Resveratrol | 7.38 × 10–61 | MCF7 | 10.0 | 2 | 12 |
| 3. | Monobenzone | 3.66 × 10–60 | MCF7 | 20.0 | 2 | 12 |
| 4. | Thioridazine | 8.15 × 10–52 | PC3 | 10.0 | 3 | 13 |
| 5. | Methotrexate | 1.28 × 10–51 | HL60 | 8.8 | 2 | 12 |
| 6. | 15‐delta prostaglandin J2 | 2.82 × 10–51 | MCF7 | 10.0 | 5 | 30 |
| 7. | Resveratrol | 5.65 × 10–49 | MCF7 | 17.6 | 2 | 12 |
| 8. | Trifluridine | 1.56 × 10–47 | MCF7 | 13.6 | 2 | 11 |
| 9. | Pyrvinium | 4.65 × 10–47 | MCF7 | 3.4 | 2 | 11 |
| 10. | 0173570‐0000 | 4.15 × 10–46 | PC3 | 10.0 | 2 | 11 |
Top 10 compounds significantly down‐regulating module M2 genes according to the CMap dataset determined by Fisher’s Exact Test (FET). Resveratrol was represented among the top 10 twice.
To explore the mechanism by which resveratrol might act upon M2 network, we investigated the direct PPI between MRs (FoxM1, B‐Myb, E2F2) and the rest of the M2 gene products. We reasoned that proteins directly binding to the MRs may have greater influence on their activity than proteins with no known interaction with the MRs. We identified 26 proteins that bind one or all three of the MRs, among which several protein kinases (Fig. 6A). We queried the Kinase Profiling Database (see Methods) to identify kinases strongly inhibited by resveratrol in low concentrations. We identified MELK as the only kinase, which activity was reduced over 50% by exposure to resveratrol concentrations as low as 1 μmol/L (Fig. 6B). Serine‐threonine kinase MELK has been shown to phosphorylate and activate FoxM1 in a PLK1‐dependent manner.88 We therefore propose MELK inhibition and subsequent dephosphorylation of FoxM1 as one of the potential mechanisms by which resveratrol could exert its effect on module M2.
Figure 6.
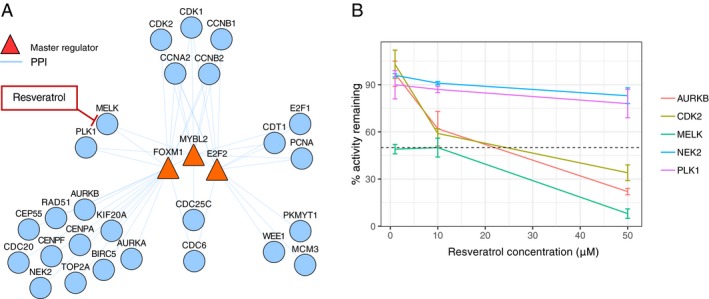
FOXM1 and MYBL2 are bound by serine‐threonine kinase MELK, which is inhibited by resveratrol. (A) Network of M2 MRs and the M2 genes found to bind them directly via PPI. Serine‐threonine kinases MELK and PLK1 bind to both FOXM1 and MYBL2 as well as are regulated by these TFs. (B) Inhibition of kinases in module M2 with direct PPI interaction with module M2 regulators FOXM1, MYBL2 or E2F2 by 1 µmol/L, 10 µmol/L, and 50 µmol/L resveratrol according to the Kinase Profiling Inhibitor Database. PPI, protein–protein interaction.
Resveratrol treatment of human primary anaplastic astrocytoma cell cultures
To validate the in silico predictions of drugs targeting M2 network from the analysis of drug perturbation data (above), we chose to study the of effect of resveratrol on M2 module genes in human low‐passage primary anaplastic astrocytoma cell culture as resveratrol has not previously been implicated in prevention of glioma progression. We chose first to study resveratrol at 20 µmol/L dissolved in DMSO to match the concentrations and solvent used in the CMap database. We used low‐passage IDH1 mutant human tumor‐derived TB98 cells and exposed them to 20 µmol/L resveratrol or DMSO or left the cells untreated in triplicates for 24 h in vitro. Genome‐wide transcriptional changes in response to exposure were captured using RNA‐seq. Resveratrol treatment had a widespread effect on gene expression at 20 µmol/L concentration with 10,622 protein coding genes being significantly (FDR < 0.05) differentially expressed (Supplementary Table S14). In agreement with the CMap data, the genes showing more robust down‐regulation (FDR < 0.01, log2FC < −1) in resveratrol‐treated cells were enriched for module M2 genes (FET FDR = 1.12 × 10− 41, GSEA P‐value < 10− 5) (Fig. 7A–C).
Figure 7.
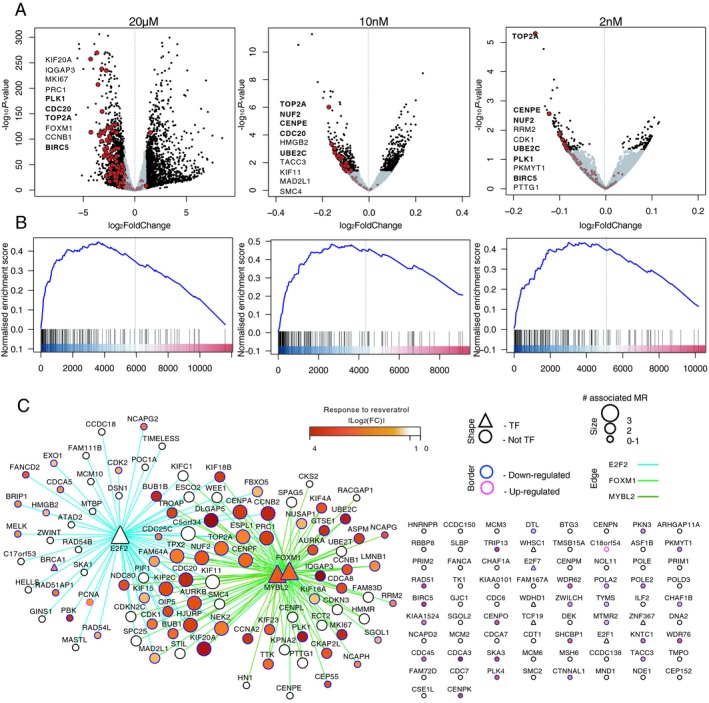
Down‐regulation of module M2 genes by resveratrol. (A) Volcano plots showing the genes changing in response to resveratrol treatment in different concentrations (n = 3, TB98 cells). Genes with P‐value < 0.05 (10 nmol/L and 2 nmol/L treatment) or changed more than twofold (20 µmol/L treatment) are in black, module M2 genes are marked in red. Top 10 down‐regulated module M2 genes are labelled (P‐value ranked). M2 genes names among the top 10 down‐regulated genes in at least two assays are written in bold. (B) Gene Set Enrichment Analysis (GSEA) plots showing enrichment of module M2 genes among differentially expressed genes between DMSO and resveratrol treatment. Genes were ranked based on log10(fold change) × P‐value from the most up‐regulated to most down‐regulated genes. Positive normalized enrichment score indicates enrichment among the down‐regulated genes. (C) M2 network depicted as consensus TF – target interactions for MRs FOXM1, MYBL2, and E2F2. Differential expression in response to 20 µmol/L resveratrol treatment is shown in yellow to red (absolute log2 fold change value of gene expression between resveratrol and DMSO‐treated primary astrocytoma‐derived cells). Targets of FOXM1 and MYBL2 were strongly down‐regulated after 24 h treatment with 20 µmol/L resveratrol (TB98 cells). Network edges represent an interaction between MR and its target as inferred by MaRInA detected by ChIP‐seq (intersect). Edge colors are assigned to individual MR–target interactions. All TFs in the network are noted as triangles. The size of the node represents the number of MR–target interactions for a given gene and the border color symbolizes up‐ or down‐regulation in response to resveratrol.
This analysis confirmed that M2 genes are substantially and significantly down‐regulated as a result of resveratrol exposure. However, the concentration used in this experiment (20 µmol/L) is not achievable in human subjects due it being rapidly metabolised by the liver.89 In contrast, resveratrol concentrations of 2 nmol/L and 10 nmol/L have been shown to be achievable in human cerebrospinal fluid (CSF) by oral dosing.90 We therefore explored the transcriptional signature of TB98 cells following exposure to 2 nmol/L and 10 nmol/L resveratrol. Analysis of the change in expression of M2 using GSEA revealed that M2 remains highly significantly enriched (P‐value < 10− 5) for genes down‐regulated at both 2 nmol/L and 10 nmol/L resveratrol concentrations (Fig. 7A–B) (Supplementary Fig. S12A,B; Supplementary Tables S15, S16). In particular, genes with expression peaks during G2 to M phase of the cell cycle, such as TOP2A and PLK1 were consistently down‐regulated after resveratrol exposure, including at nanomolar concentrations. Consistent with resveratrol acting on G2 to M progression, using the hallmark gene set collection from the Molecular Signature Database,91 we further confirmed that genes encoding G2M progression, E2F targets and MYC targets were consistently and significantly (FDR < 0.1) enriched among the genes down‐regulated after 24 h exposure with 20 µmol/L, 10 nmol/L and 2 nmol/L resveratrol (Table 4; Supplementary Table S17).
Table 4.
Gene sets significantly (FDR < 0.1) enriched among genes down‐regulated by resveratrol in at least two experiments.
| Hallmark Gene Set | 20 µmol/L | 10 nmol/L | 2 nmol/L |
|---|---|---|---|
| G2M checkpoint | ✓ | ✓ | ✓ |
| E2F targets | ✓ | ✓ | ✓ |
| MYC targets | ✓ | ✓ | ✓ |
| Mitotic spindle | ✓ | ✓ |
These above results confirmed that resveratrol is capable of inducing a transcriptional response in genes related to cell cycle progression in primary anaplastic astrocytoma cell culture, including at nanomolar concentrations. We therefore investigated the phenotypic effect of resveratrol exposure on cell growth in vitro in two low‐passage IDH1 mut AA‐derived cell lines (TB98, TB58) and IDH1 wt AA cell line (TB62). After 8 days, the wells treated with 20 µmol/L resveratrol had significantly (P‐value < 0.05) reduced number of cells than the DMSO‐treated wells indicated by colorimetric cell proliferation assay (Fig. 8A). The experiment was replicated in TB98‐derived cells with an added harvesting time‐point of 4 days (Fig. 8B). Treatment with 20 µmol/L resveratrol consistently and significantly (P‐value < 0.0001, independent two sample t test) reduced the cell numbers in the TB98 cells. Similarly, we observed a significant (P‐value < 0.05, independent two sample t test) reduction in cell numbers after exposing TB98 cells to 2 nmol/L and 10 nmol/L resveratrol for 60 days, compared to DMSO control (Fig. 8C).
Figure 8.
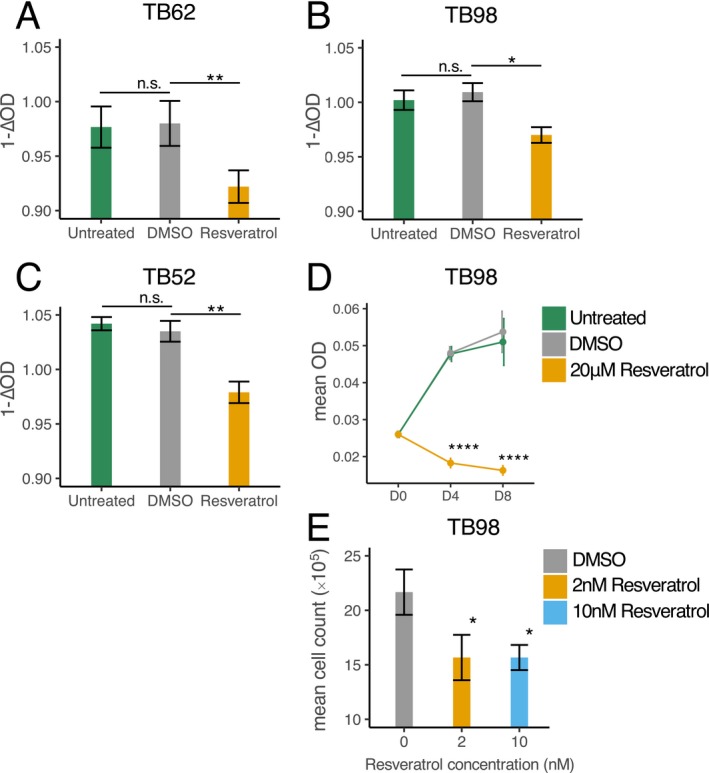
Resveratrol’s effect on cell numbers. (A–C) Optic density change (∆OD = ODday8 – ODday0) after treatment with 20 µmol/L resveratrol in three different cell lines was significantly greater in comparison to the control (DMSO), which indicates reduced number of cells (n = 3). (D) Exposure to 20 µmol/L resveratrol significantly reduced the cell numbers (reflected by OD measurement) after 4 and 8 days in TB98 cells (n = 3) in comparison to DMSO. (E) Cells treated with 2 nmol/L and 10 nmol/L resveratrol over a period of 60 days were significantly fewer than DMSO‐treated cells (n = 3). P‐value < 0.05*; P‐value < 0.01**; P‐value < 0.001***, P‐value < 0.0001**** (independent two sample Student’s t test).
Discussion
Progression of relatively slowly growing grade II diffuse astrocytomas to an ultimately lethal higher grade gliomas is, at large, inevitable.92 While aggressive chemotherapy followed by radiotherapy may improve survival of low‐grade glioma patients,93, 94 a challenge remains to identify treatment strategies that improve survival of patients with low‐grade gliomas without impacting quality of life or neurocognitive function. Despite recent success in single‐target cancer therapies focusing on unique “driver” genetic aberrations, this has not translated into clinical benefit in gliomas.95 The research presented here was driven by the view that tumorigenesis and anaplastic transformation of gliomas is likely to be determined by perturbed biological processes that converge on a common, signal‐independent proliferation characteristic that could be targeted for therapy discovery.
In this study, we took advantage of publicly available glioma data from TCGA database. In order to reduce inter‐patient variability, a paired study design, that is, analysis of tumor samples from the same patient before and after progression would be desirable. However, currently available paired studies19, 96 lack a sufficient sample size to perform differential co‐expression analysis. Using the TCGA dataset, we uncovered two modules of differentially co‐expressed genes between low and high‐grade IDH1‐mutated and 1p/19q euploid astrocytomas (M1 and M2). Functionally, the modules are enriched for neuronal and cell cycle genes, respectively. M2 had higher intramodular gene correlation in AA compared to LGA and the average expression of M2 module was significantly increased in samples with loss of chromosome 10q ‐ an event previously identified as prognostically unfavorable in 1p/19q euploid low‐grade gliomas.64 In addition to loss of TSG on 10q such as PTEN, M2 expression was higher in individuals with oncogene PDGFRA locus amplification and deletion of TSG CIC.
For module M2, we also observed an overrepresentation of cancer predisposition genes including for genes functionally related to DNA repair and chromosomal segregation, which are central to tumor development. In contrast to M2, M1 had stronger co‐expression in LGA compared to AA, was not enriched for cancer predisposition genes and was functionally enriched for neuronal processes. With grade II astrocytomas containing less tumor cells, it is plausible that the emergence of the M1 network differential co‐expression signature is due to the samples containing variable amounts of normal brain parenchyma and lack of association between M1 expression and genomic cancer events as well as conservation of its co‐expression in neural tissue further supports this line of reasoning. Collectively, these integrative analyses point to an association between module M2 and astrocytoma progression. We therefore posited that targeting expression could represent a novel therapeutic approach to delaying glioma progression.
Using a reverse engineering approach, we first attempted to identify MRs of M2 as candidate drug targets. Utilizing gene–gene and DNA–protein interaction data, we predicted E2F2, MYBL2, and FOXM1 as candidate MRs of M2 expression. Consistent with this, E2F‐family and Myb‐like TFBS were overrepresented in module M2 gene promoters, but not of FoxM1 most likely because FoxM1 acts on cell cycle regulation via B‐Myb‐MuVB complex rather than its canonical binding motif.43, 45 E2F2 is one of the E2F‐family TFs responsible for the regulation of early cell cycle genes.97 The E2F‐family TFs govern the transition from G1 to S phase of the cell cycle, and are strongly inhibited by Retinoblastoma Protein (coded by the RB1), which was altered in ~30% in this IDH1‐mutant and 1p/19q euploid astrocytoma cohort. Additionally, E2F2, MYBL2, and FOXM1 are consistently up‐regulated in other TP53‐mutated tumors, and FoxM1‐ and E2F2‐mediated cell division cycle has been previously linked to progression of IDH1‐mutated gliomas and atypical meningiomas.19, 98, 99 While collectively these systems‐level analyses highlight M2 as a gene‐regulatory network governed by FoxM1, B‐Myb, and E2F2 that captures the context relevant targets of these TFs better than existing gene signatures, neither E2F2, B‐Myb or FoxM1 represents easily “druggable” targets. We therefore took an alternative approach to “drugging” the M2 network as a whole using a strategy that leverages gene expression changes that occur in neoplastic cells in response to drug exposures.14, 82 Using module M2 as a “multitarget” gene network, we computationally screened for drugs that induce a cellular transcriptional response that shifts M2 expression from its profile in anaplastic astrocytoma in a low‐grade (i.e., down‐regulated) direction. Taking this approach, we identified the health food supplement resveratrol among the known anticancer drugs etoposide and methotrexate as candidate antiproliferative agents acting via a transcriptional effect on M2 network genes.
Resveratrol (3,4',5‐trihydroxy‐trans‐stilbene) is a natural polyphenol synthesized by seed producing plants such as grapevines, in response to stress or fungal attack, and is currently marketed as a health food supplement.100 Among the processes thought to be targeted by resveratrol are cell cycle progression, apoptosis, and angiogenesis.101 The anticancer properties of resveratrol have been shown in the clinical setting – it reduces cell proliferation in colorectal cancer patients and increases apoptosis in hepatic metastasis of colorectal cancer.102, 103 However, despite peak plasma concentrations of resveratrol ranging from 117.0 ng/mL (0.51 µmol/L) to 538.8 ng/mL (2.36 µmol/L) upon administration of 1 g or 5 g of resveratrol, respectively,104 and evidence from animal105, 106 and human90 studies that resveratrol can penetrate the blood–brain barrier, the low nanomolar concentrations of resveratrol in human CSF in individuals taking up to 2 × 1 g of resveratrol daily90 have to date precluded its consideration as an antineoplastic agent for low‐grade gliomas.
Advancing on these studies, here we found that resveratrol had an effect on transcription of M2‐related cell cycle division genes even at nanomolar concentrations (2 nmol/L and 10 nmol/L), providing a novel insight in the antiproliferative mechanism of resveratrol unrelated to its known binding partners at µM concentrations. Additionally, we observed that FoxM1 and B‐Myb are strongly down‐regulated by resveratrol, suggesting that the late cell cycle genes are most affected, and that genes which peak in expression during G2 to M phase of the cell cycle, such as TOP2A and PLK1, are consistently down‐regulated after resveratrol treatment, including at nanomolar concentrations. Further support for a resveratrol effect on the second phase of the cell cycle can also be found from previous studies showing the attenuation of the G2 to M progression in response to resveratrol in vitro.107, 108, 109 The results presented here suggest therefore that resveratrol down‐regulates the M2 gene‐regulatory network governing G2 to M progression. Resveratrol is known to bind a wide spectrum of protein targets, many of which play a key role in cell cycle regulation, such as DNA polymerase α/β and mitogen‐activated protein kinases ERK1 and JNK1. However, the inhibition of given targets occurs in the concentrations exceeding what is achievable in humans.110, 111 Currently, the highest known affinity of resveratrol is toward quinone reductase NQO2 (K d = 35 nmol/L). 110, 112 Inhibition of NQO2 has been shown to attenuate NF‐κB activity, which promotes tumor cell proliferation.113, 114 Down‐regulation of module M2 genes and the subsequent decrease in cell division could therefore be mediated by reduced NF‐κB signaling. We also propose a potentially novel mechanism, by which the cellular effects of resveratrol may be mediated via inhibiting a serine‐threonine kinase MELK, which is responsible for activation of M2 MR FoxM1.88 Further studies investigating MELK inhibition and concomitant reduction in FoxM1 phosphorylation upon treatment with nanomolar concentrations of resveratrol are therefore warranted.
Besides confirming the feasibility and effectiveness of network‐based multitarget drug discovery for delaying time to progression for low‐grade gliomas, the well‐established safety and high tolerability of resveratrol make resveratrol itself an ideal candidate for directly repurposing to patients with inoperable low‐grade gliomas. While resveratrol’s potential antineoplastic effect is likely not unique to IDH1‐mutated 1p/19q euploid astrocytomas, this drug would be of particular interest in these tumors due to their less aggressive clinical course compared to IDH1wt astrocytomas, and hence the undesirable use strong chemotherapeutic drugs in this clinical context.
Author Contributions
MRJ, EP, and LL conceived and designed the study and acquired funds. MRJ, EP, LL, PKS, KS, OJLR, SRL and ADD performed data analysis, and developed and implemented the methodology. Data generation, curation and/or laboratory experiments were performed by SCC, AB, JS, NS, QZ, KO, MW and FR. MRJ, EP and LL wrote the manuscript and all authors contributed, read, and approved the final version.
Ethics Approval
Approval for the patient tumor‐derived cell cultures was obtained from the institutional ethics review committee and Imperial College Healthcare Tissue Bank (ICHTB) [project no. R13061, ICHTB Human Tissue Authority (HTA) license 12275]. All procedures were conducted in accordance with the Declaration of Helsinki.
Competing Interests
The authors declare that they have no competing interests.
Supporting information
Figure S1. Sample selection diagram.
Figure S2. Significance of differential expression.
Figure S3. Differential expression analysis between AA and LGA.
Figure S4. Average expression of module M1 and M2 genes in LGA and AA gliomas.
Figure S5. Pathway enrichments results of module M1 and M2 from Web‐Gestalt Pathway Commons analysis.
Figure S6. Cell cycle genes with high connectivity are significantly enriched for the M2 genes.
Figure S7. Frequency of somatic mutations (base substitutions and small insertions/deletions) in IDH1‐mutated 1p/19q euploid astrocytomas.
Figure S8. Heatmap and frequencies of copy‐number alterations across the IDH1‐mutated and 1p/19 euploid astrocytoma cohort.
Figure S9. Workflow to determine the source of regulatory influence on DiffCoEx modules M1 and M2.
Figure S10. Overall concordance between the predicted and validated TF of FoxM1, B‐Myb and E2F2.
Figure S11. Overlap of ChIP‐seq targets of FoxM1, B‐Myb and E2F2 with gene signatures.
Figure S12. M2 network depicted as consensus transcription factor (TF) – target interactions for master regulators (MRs) FOXM1, MYBL2 and E2F2.
Table S1. Sample information about TCGA grade II and grade III astrocytoma samples used in this analysis.
Table S2. List of genes belonging to differential co‐expression modules M1 and M2, as well as genes not differentially co‐expressed between grade II and grade III IDH1‐mutated anaplastic astrocytomas (module M0).
Table S3. Differential expression of genes between TCGA IDH1‐mutated AA and LGA.
Table S4. Top 10 significant enrichments or Gene Ontology Biological Process in module M1.
Table S5. Top 10 significant enrichments or Gene Ontology Biological Process in module M2.
Table S6. Information about the datasets used for calculating preservation of differential co‐expression modules M1 and M2.
Table S7. Biological function of cancer predisposition genes in module M2.
Table S8. Genes significantly mutated in AA and LGA ‐ MutSig analysis results.
Table S9. Gene dosage of cancer genes (COSMIC database + WDR11) in IDH1‐mutated AA and LGA and gene expression and correlation between cancer gene dosage and module M1 and M2 expression.
Table S10. Transcription factor binding site enrichment results using WebGestalt and TRANSFAC databases.
Table S11. Fisher's exact test results of Master Regulators (MR) targets over‐representation in modules M1 and M2, list of MRs and their target genes and functional enrichment of activated and repressed MR target genes.
Table S12. Lists of ChIP‐seq detected targets of B‐Myb, FoxM1 and E2F2.
Table S13. Significance of over‐representation of genes up‐ or down‐regulated by a given compound in Connectivity Map database among module M2 genes.
Table S14. Genes up‐ and down‐regulated after 24 h 20 µmol/L resveratrol treatment.
Table S15. Genes up‐ and down‐regulated after 24 h 10 nmol/L resveratrol treatment.
Table S16. Genes up‐ and down‐regulated after 24 h 2 nmol/L resveratrol treatment.
Table S17. Gene Set Enrichment analysis of gene expression changes after 24 h resveratrol treatment in concentrations 20 µmol/L, 10 nmol/L and 2 nmol/L.
Acknowledgments
This study was supported by Imperial College NIHR Biomedical Research Centre (BRC) Scheme, the Brain Tumour Research Campaign (BTRC), Brain Tumour Research (BTR) Centres of Excellence and Imperial College London President’s PhD Scholarship Programme. The Imperial BRC Genomics Facility has provided resources and support that have contributed to the research results reported within this paper. The authors thank Simona Parinello and Lucy Brooks (University College London, UK) for their assistance with in vitro experiments and valuable discussions.
Funding Information
This study was supported by Imperial College NIHR Biomedical Research Centre (BRC) Scheme, the Brain Tumour Research Campaign (BTRC), Brain Tumour Research (BTR) Centres of Excellence and Imperial College London President’s PhD Scholarship Programme. The Imperial BRC Genomics Facility has provided resources and support that have contributed to the research results reported within this paper.
Funding Statement
This work was funded by Imperial College NIHR Biomedical Research Centre grant ; Brain Tumour Research Campaign grant ; Brain Tumour Research Centres of Excellence grant ; Imperial College London grant ; University College London grant .
Contributor Information
Liisi Laaniste, Email: l.laaniste15@imperial.ac.uk.
Enrico Petretto, Email: enrico.petretto@duke-nus.edu.sg.
Michael R. Johnson, Email: m.johnson@imperial.ac.uk.
Data Availability Statement
Gene expression, somatic mutations, and CNV data in grade II and III gliomas were acquired from TCGA and are publicly available at Genomic Data Commons Data Portal: (https://portal.gdc.cancer.gov). Datasets used for module preservation analysis are also publicly available in TCGA and Gene Expression Omnibus (see Supplementary Table S6). ChIP‐seq data of E2F2 binding have been previously published and are available at GEO under accession GSE49402. RNA‐seq data of astrocytoma cell line TB98 response to 20 µmol/L, 10 nmol/L and 2 nmol/L resveratrol is available in GEO under accession GSE117620.
References
- 1. Ramsay RR, Popovic‐Nikolic MR, Nikolic K, et al. A perspective on multi‐target drug discovery and design for complex diseases. Clin Transl Med 2018;7:3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendelsohn J. Personalizing oncology: perspectives and prospects. J Clin Oncol 2013;31:1904–1911. [DOI] [PubMed] [Google Scholar]
- 3. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2012;105:2640–2653. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 5. Malkki H. Trial Watch: glioblastoma vaccine therapy disappointment in Phase III trial. Nat Rev Neurol 2016;12:190. [DOI] [PubMed] [Google Scholar]
- 6. Brunschweiger A, Hall J. A decade of the human genome sequence‐how does the medicinal chemist benefit? ChemMedChem 2012;7:194–203. [DOI] [PubMed] [Google Scholar]
- 7. Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:1–18. [DOI] [PubMed] [Google Scholar]
- 8. Sahm F, Capper D, Jeibmann A, et al. Addressing diffuse glioma as a systemic brain disease with single‐cell analysis. Arch Neurol 2012;69:523–526. [DOI] [PubMed] [Google Scholar]
- 9. TCGA . Integrative genomic analysis of diffuse lower‐grade gliomas. NEMJ 2015;372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sepúlveda‐Sánchez JM, Muñoz Langa J, Arráez M, et al. SEOM clinical guideline of diagnosis and management of low‐grade glioma (2017). Clin Transl Oncol 2018;20:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willsey AJ, Morris MT, Wang S, et al. The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 2018;174:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson MR, Behmoaras J, Bottolo L, et al. Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat Commun 2015;6:6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Srivastava PK, van Eyll J, Godard P, et al. A systems‐level framework for drug discovery identifies Csf1R as an anti‐epileptic drug target. Nat Commun 2018;9:3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delahaye‐Duriez A, Srivastava P, Shkura K, et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol 2016;17:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson M, Shkura K, Langley S, et al. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat Neurosci 2015;19:1–10. [DOI] [PubMed] [Google Scholar]
- 16. Grossman RL, Heath AP, Ferretti V, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med 2016;375:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 2015;18:458. [DOI] [PubMed] [Google Scholar]
- 18. TCGA . Integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med 2015;372:2481-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bai H, Harmancı AS, Erson‐Omay EZ, et al. Integrated genomic characterization of IDH1‐mutant glioma malignant progression. Nat Genet 2015;48:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene‐expression signatures to connect small molecules, genes, and disease. Science (80‐). 2006;313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 21. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anders S, Pyl PT, Huber W. HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oshlack A, Wakefield M. Transcript length bias in RNA‐seq data confounds systems biology. Biol Direct 2009;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2017. [Google Scholar]
- 25. Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics 2010;11:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li S, Labaj PP, Zumbo P, et al. Detecting and correcting systematic variation in large‐scale RNA sequencing data. Nat Biotechnol 2014;32:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics 2012;28:882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005;33:W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hammerman PS, Lawrence MS, Voet D, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 33. Rahman N. Realizing the promise of cancer predisposition genes. Nature 2014;505:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu C, Xie M, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun 2015;6:10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Viechtbauer W. Conducting meta‐analyses in R with the metafor package. J Stat Softw 2010;36:1–48. [Google Scholar]
- 36. Carbon S, Ireland A, Mungall CJ, et al. AmiGO: online access to ontology and annotation data. Bioinformatics 2009;25:288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Margolin AA, Nemenman I, Basso K, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006;7:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lachmann A, Giorgi FM, Lopez G, Califano A. ARACNe‐AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 2016;32:2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abugessaisa I, Shimoji H, Sahin S, et al. FANTOM5 transcriptome catalog of cellular states based on Semantic MediaWiki. Database 2016;2016:baw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Margolin AA, Wang K, Lim WK, et al. Reverse engineering cellular networks. Nat Protoc 2006;1:662–671. [DOI] [PubMed] [Google Scholar]
- 41. Aytes A, Mitrofanova A, Lefebvre C, et al. Cross-species analysis of genome-wide regulatory networks identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 2014:25:638–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wiseman EF, Chen X, Han N, et al. Deregulation of the FOXM1 target gene network and its coregulatory partners in oesophageal adenocarcinoma. Mol Cancer 2015;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen X, Muller GA, Quaas M, et al. The forkhead transcription factor FOXM1 controls cell cycle‐dependent gene expression through an atypical chromatin binding mechanism. Mol Cell Biol 2012;33:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB‐FOXM1 and RB‐E2F target gene analyses identifies cell cycle networks. Nucleic Acids Res 2016;44:6070–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B‐Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012;26:474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marinov GK, Kundaje A, Park PJ, Wold BJ. Large‐scale quality analysis of published chip‐seq data. G3 2014;4:209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landt SG, Marinov GK, Kundaje A, et al. ChIP‐seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012;1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu G, Wang L‐G, He Q‐Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 50. Lu X, Zhang X. The effect of GeneChip gene definitions on the microarray study of cancers. BioEssays 2006;28:739–746. [DOI] [PubMed] [Google Scholar]
- 51. Dai M, Wang P, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 2005;3:e175–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Irizarry RA, Bolstad BM, Collin F, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003;31:15e–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huber W, Von Heydebreck A, Ultmann H, et al. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002;18:96–104. [DOI] [PubMed] [Google Scholar]
- 54. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments*. Stat Appl Genet Mol Biol 2004;3:1–25. [DOI] [PubMed] [Google Scholar]
- 55. Liberzon A, Birger C, Ghandi M, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2016;417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Franceschini A, Szklarczyk D, Frankild S, et al. 1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013;41:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rotival M, Petretto E. Leveraging gene co‐expression networks to pinpoint the regulation of complex traits and disease, with a focus on cardiovascular traits. Brief Funct Genomics 2014;13:66–78. [DOI] [PubMed] [Google Scholar]
- 58. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA 1998;95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carlson M, Zhang B, Fang Z, et al. Gene connectivity, function, and sequence conservation: predictions from modular yeast co‐expression networks. BMC Genom 2006;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Futreal P, Coin L, Marshall L, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cox DR. Regression models and life‐tables. J R Stat Soc Ser B 1972;34:187–220. [Google Scholar]
- 63. Chen JC, Alvarez MJ, Talos F, et al. Identification of causal genetic drivers of human disease through systems‐level analysis of regulatory networks. Cell 2014;159:402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Thuijl HF, Scheinin I, Sie D, et al. Spatial and temporal evolution of distal 10q deletion, a prognostically unfavorable event in diffuse low‐grade gliomas. Genome Biol 2014;15:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Houillier C, Mokhtari K, Carpentier C, et al. Chromosome 9p and 10q losses predict unfavorable outcome in low‐grade gliomas. Neuro Oncol 2010;12:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dehais C, Laigle‐donadey F, Marie Y, et al. Prognostic stratification of patients with anaplastic gliomas according to genetic profile. Cancer 2006;107:1891–1897. [DOI] [PubMed] [Google Scholar]
- 67. Chernova OB, Hunyadi A, Malaj E, et al. A novel member of the WD‐repeat gene family, WDR11, maps to the 10q26 region and is disrupted by a chromosome translocation in human glioblastoma cells. Oncogene 2001;20:5378–5392. [DOI] [PubMed] [Google Scholar]
- 68. Katoh M, Katoh M. FGFR2 and WDR11 are neighboring oncogene and tumor suppressor gene on human chromosome 10q26. Int J Oncol 2003;22:1155–1159. [PubMed] [Google Scholar]
- 69. Beyer A, Workman C, Hollunder J, et al. Integrated assessment and prediction of transcription factor binding. PLoS Comput Biol 2006;2:0615–0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang J, Duncan D, Shi Z, Zhang B. WEB‐based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013;41:W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matys V, Kel‐Margoulis OV, Fricke E, et al. TRANSFAC(R) and its module TRANSCompel(R): transcriptional gene regulation in eukaryotes. Nucl Acids Res 2006;34(suppl_1):D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen H‐Z, Tsai S‐Y, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009;9:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chellappan SP, Hiebert S, Mudryj M, et al. The E2F transcription factor is a cellular target for the RB protein. Cell 1991;65:1053–1061. [DOI] [PubMed] [Google Scholar]
- 74. Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene 1999;18:3017–3033. [DOI] [PubMed] [Google Scholar]
- 75. Ly LL, Yoshida H, Yamaguchi M. Nuclear transcription factor Y and its roles in cellular processes related to human disease. Am J Cancer Res 2013;3:339–346. [PMC free article] [PubMed] [Google Scholar]
- 76. Seguin L, Liot C, Mzali R, et al. CUX1 and E2F1 regulate coordinated expression of the mitotic complex genes Ect2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol 2009;29:570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Balwierz PJ, Pachkov M, Arnold P, et al. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res 2014;24:869–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 2012;13:233–245. [DOI] [PubMed] [Google Scholar]
- 79. Basso K, Margolin AA, Stolovitzky G, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet 2005;37:382–390. [DOI] [PubMed] [Google Scholar]
- 80. Liu M, Dai B, Kang S‐H, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–3602. [DOI] [PubMed] [Google Scholar]
- 81. Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle‐dependent gene expression. Nat Rev Cancer 2013;13:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chandran V, Coppola G, Nawabi H, et al. A systems‐level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 2016;89:956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leonard A, Wolff JE. Etoposide improves survival in high‐grade glioma: a meta‐analysis. Anticancer Res 2013;33:3307–3316. [PubMed] [Google Scholar]
- 84. Wolff JE, Kortmann RD, Wolff B, et al. High dose methotrexate for pediatric high grade glioma ‐ Results of the HIT‐GBM‐D Pilot study. J Neurooncol 2011;102:433–442. [DOI] [PubMed] [Google Scholar]
- 85. Clément MV, Hirpara JL, Chawdhury SH, Pervaiz S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling‐dependent apoptosis in human tumor cells. Blood 1998;92:996–1002. [PubMed] [Google Scholar]
- 86. Lin HY, Tang HY, Keating T, et al. Resveratrol is pro‐apoptotic and thyroid hormone is anti‐apoptotic in glioma cells: both actions are integrin and ERK mediated. Carcinogenesis 2008;29:62–69. [DOI] [PubMed] [Google Scholar]
- 87. Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006;5:493–506. [DOI] [PubMed] [Google Scholar]
- 88. Joshi K, Banasavadi‐Siddegowda Y, Mo X, et al. MELK‐dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells 2013;31:1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gambini J, Inglés M, Olaso G, et al. Properties of resveratrol: in vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid Med Cell Longev 2015;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Turner RS, Thomas RG, Craft S, et al. A randomized, double‐blind, placebo‐controlled trial of resveratrol for Alzheimer disease. Neurology 2015;85:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The molecular signatures database hallmark gene set collection. Cell Syst 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ohgaki H, Kleihues P. Population‐based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 2005;64:479–489. [DOI] [PubMed] [Google Scholar]
- 93. Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus procarbazine, CCNU, and vincristine in low‐grade glioma. N Engl J Med 2016;374:1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Le Rhun E, Taillibert S, Chamberlain MC. Current management of adult diffuse infiltrative low grade gliomas. Curr. Neurol. Neurosci. Rep. 2016;16:15. [DOI] [PubMed] [Google Scholar]
- 95. Staedtke V, Bai R‐Y, Laterra J. Investigational new drugs for brain cancer. Expert Opin Investig Drugs 2016;25:937–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mazor T, Pankov A, Brett E, et al. Article DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors article DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain. Cancer Cell 2015;28:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bertoli C, Skotheim JM, de Bruin R. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Parikh N, Hilsenbeck S, Creighton CJ, et al. Effects of TP53 mutational status on gene expression patterns across 10 human cancer types. J Pathol 2014;232:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Harmancı AS, Youngblood MW, Clark VE, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 2017;8:14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Frémont L. Biological effects of resveratrol. Antioxid. Redox Signal. 2001;3:1041–1064. [DOI] [PubMed] [Google Scholar]
- 101. Varoni EM, Lo Faro AF, Sharifi‐Rad J, Iriti M. Anticancer molecular mechanisms of resveratrol. Front Nutr 2016;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Patel KR, Brown VA, Jones D, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res 2010;70:7392–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Howells LM, Berry DP, Elliott PJ, et al. Phase I randomized, double‐blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases ‐ Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev Res 2011;4:1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Boocock DJ, Faust G, Patel KR, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 2007;16:1246–1252. [DOI] [PubMed] [Google Scholar]
- 105. Vitrac X, Desmoulière A, Brouillaud B, et al. Distribution of [14C]‐trans‐resveratrol, a cancer chemopreventive polyphenol, in mouse tissues after oral administration. Life Sci 2003;72:2219–2233. [DOI] [PubMed] [Google Scholar]
- 106. Sale S, Verschoyle R, Boocock D, et al. Pharmacokinetics in mice and growth‐inhibitory properties of the putative cancer chemopreventive agent resveratrol and the synthetic analogue trans 3,4,5,4’ ‐tetramethoxystilbene. Br J Cancer 2004;90:736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Filippi‐Chiela EC, Villodre ES, Zamin LL, Lenz G. Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PLoS ONE 2011;6:e20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yu X‐D, Yang J, Zhang W‐L, Liu D‐X. Resveratrol inhibits oral squamous cell carcinoma through induction of apoptosis and G2/M phase cell cycle arrest. Tumor Biol 2016;37:2871–2877. [DOI] [PubMed] [Google Scholar]
- 109. Wang G, Guo X, Chen H, et al. A resveratrol analog, phoyunbene B, induces G2/M cell cycle arrest and apoptosis in HepG2 liver cancer cells. Bioorg Med Chem Lett 2012;22:2114–2118. [DOI] [PubMed] [Google Scholar]
- 110. Britton RG, Kovoor C, Brown K. Direct molecular targets of resveratrol: identifying key interactions to unlock complex mechanisms. Ann N Y Acad. Sci 2015;1348:124–133. [DOI] [PubMed] [Google Scholar]
- 111. Pirola L, Fröjdö S. Resveratrol: one molecule, many targets. IUBMB Life 2008;60:323–332. [DOI] [PubMed] [Google Scholar]
- 112. Buryanovskyy L, Fu Y, Boyd M, et al. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004;43:11417–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nolan KA, Dunstan MS, Caraher MC, et al. In silico screening reveals structurally diverse, nanomolar inhibitors of NQO2 that are functionally active in cells and can modulate NF‐ B signaling. Mol Cancer Ther 2012;11:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Xia Y, Shen S, Verma IM. NF‐ kB, an Active player in human cancers. Cancer Immunol Res 2014;2:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lefebvre C, Rajbhandari P, Alvarez MJ, et al. A human B‐cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol 2010;6:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yan J, Enge M, Whitington T, et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 2013;154:801–813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sample selection diagram.
Figure S2. Significance of differential expression.
Figure S3. Differential expression analysis between AA and LGA.
Figure S4. Average expression of module M1 and M2 genes in LGA and AA gliomas.
Figure S5. Pathway enrichments results of module M1 and M2 from Web‐Gestalt Pathway Commons analysis.
Figure S6. Cell cycle genes with high connectivity are significantly enriched for the M2 genes.
Figure S7. Frequency of somatic mutations (base substitutions and small insertions/deletions) in IDH1‐mutated 1p/19q euploid astrocytomas.
Figure S8. Heatmap and frequencies of copy‐number alterations across the IDH1‐mutated and 1p/19 euploid astrocytoma cohort.
Figure S9. Workflow to determine the source of regulatory influence on DiffCoEx modules M1 and M2.
Figure S10. Overall concordance between the predicted and validated TF of FoxM1, B‐Myb and E2F2.
Figure S11. Overlap of ChIP‐seq targets of FoxM1, B‐Myb and E2F2 with gene signatures.
Figure S12. M2 network depicted as consensus transcription factor (TF) – target interactions for master regulators (MRs) FOXM1, MYBL2 and E2F2.
Table S1. Sample information about TCGA grade II and grade III astrocytoma samples used in this analysis.
Table S2. List of genes belonging to differential co‐expression modules M1 and M2, as well as genes not differentially co‐expressed between grade II and grade III IDH1‐mutated anaplastic astrocytomas (module M0).
Table S3. Differential expression of genes between TCGA IDH1‐mutated AA and LGA.
Table S4. Top 10 significant enrichments or Gene Ontology Biological Process in module M1.
Table S5. Top 10 significant enrichments or Gene Ontology Biological Process in module M2.
Table S6. Information about the datasets used for calculating preservation of differential co‐expression modules M1 and M2.
Table S7. Biological function of cancer predisposition genes in module M2.
Table S8. Genes significantly mutated in AA and LGA ‐ MutSig analysis results.
Table S9. Gene dosage of cancer genes (COSMIC database + WDR11) in IDH1‐mutated AA and LGA and gene expression and correlation between cancer gene dosage and module M1 and M2 expression.
Table S10. Transcription factor binding site enrichment results using WebGestalt and TRANSFAC databases.
Table S11. Fisher's exact test results of Master Regulators (MR) targets over‐representation in modules M1 and M2, list of MRs and their target genes and functional enrichment of activated and repressed MR target genes.
Table S12. Lists of ChIP‐seq detected targets of B‐Myb, FoxM1 and E2F2.
Table S13. Significance of over‐representation of genes up‐ or down‐regulated by a given compound in Connectivity Map database among module M2 genes.
Table S14. Genes up‐ and down‐regulated after 24 h 20 µmol/L resveratrol treatment.
Table S15. Genes up‐ and down‐regulated after 24 h 10 nmol/L resveratrol treatment.
Table S16. Genes up‐ and down‐regulated after 24 h 2 nmol/L resveratrol treatment.
Table S17. Gene Set Enrichment analysis of gene expression changes after 24 h resveratrol treatment in concentrations 20 µmol/L, 10 nmol/L and 2 nmol/L.
Data Availability Statement
Gene expression, somatic mutations, and CNV data in grade II and III gliomas were acquired from TCGA and are publicly available at Genomic Data Commons Data Portal: (https://portal.gdc.cancer.gov). Datasets used for module preservation analysis are also publicly available in TCGA and Gene Expression Omnibus (see Supplementary Table S6). ChIP‐seq data of E2F2 binding have been previously published and are available at GEO under accession GSE49402. RNA‐seq data of astrocytoma cell line TB98 response to 20 µmol/L, 10 nmol/L and 2 nmol/L resveratrol is available in GEO under accession GSE117620.