

Abstract
Objective
To determine the association between plasma insulin‐like growth factor binding protein 2 (IGFBP‐2) and cognitive outcomes.
Methods
We measured plasma IGFBP‐2 levels in 1596 (53% women, mean age 68.7 [SD 5.7] years) dementia‐free Framingham Offspring cohort participants between 1998 and 2001. Multivariable Cox proportional hazards models related plasma IGFBP‐2 to subsequent risk of incident dementia and Alzheimer’s disease. MRI brain measures and cognitive performance were included as secondary outcomes.
Results
During a median follow‐up of 11.8 (Q1, Q3: 7.1, 13.3) years, 131 participants developed incident dementia, of whom 98 were diagnosed with Alzheimer’s disease. The highest tertile of IGFBP‐2, compared to the lowest tertile, was associated with an increased risk of incident all‐cause dementia (hazard ratio [HR] 2.89, 95% CI 1.63–5.13) and Alzheimer’s disease (HR 3.63, 95% CI 1.76–7.50) in multivariable analysis. Higher circulating IGFBP2 levels were also cross‐sectionally associated with poorer performance on tests of abstract reasoning but not with MRI‐based outcomes. After adding plasma IGFBP‐2 levels to a conventional dementia prediction model, 32% of individuals with dementia were correctly assigned a higher predicted risk, while 8% of individuals without dementia were correctly assigned a lower predicted risk (overall net reclassification improvement index, 0.40, 95% CI 0.22–0.59).
Interpretation
Elevated circulating IGFBP‐2 levels were associated with an increased risk of both all‐cause dementia and Alzheimer’s disease. Addition of IGFBP2 plasma levels to a model of traditional risk factors significantly improved dementia risk classification. Manipulation of insulin‐like growth factor signaling via IGFBP‐2 may be a promising therapeutic target for dementia.
Background
Identifying novel biomarkers for dementia and cognitive decline can inform our understanding of the complex biological pathways underpinning the development of dementia, improve our ability to detect disease at an earlier stage, and identify future potential therapeutic targets for investigation.1 There is growing evidence to suggest that metabolic dysfunction and insulin resistance in the brain play an important role in the development of dementia.2, 3, 4 The insulin‐like growth factor (IGF) signaling system is known to play a role in neuroregeneration, neuronal survival and proliferation, and cerebral metabolic function.5, 6 Circulating IGF‐I and IGF‐II are typically bound to one of six known IIGF‐binding proteins. While IGFBP‐3 is the most prominent IGF‐binding protein in the circulation, insulin‐like growth factor binding protein 2 (IGFBP‐2) is the most predominant IGF‐binding protein in the brain, and thus was selected as the focus of this study. Elevated levels of IGF‐binding proteins, particularly IGFBP‐2, are thought to reduce IGF‐I and IGF‐II bioavailability in the brain and impair IGF signaling, thereby inhibiting the neuroprotective effects of IGF‐I and IGF‐II.7, 8, 9 Elevated plasma and cerebrospinal fluid (CSF) IGFBP‐2 levels have previously been cross‐sectionally associated with hippocampal atrophy and Alzheimer's disease (AD) in small studies.6, 7, 10, 11, 12, 13, 14, 15, 16
It is unknown, however, if plasma IGFBP‐2 levels are predictive of adverse cognitive outcomes, including risk of developing dementia, over long‐term follow‐up. Given the growing interest in dementia therapeutics targeting dysregulated insulin metabolism and IGF signaling,17, 18 IGFBP‐2 could be a useful circulating biomarker for predicting dementia risk, a surrogate marker for future clinical trials and a potential therapeutic target. In this study, we aimed to determine the association between plasma IGFBP‐2 levels and structural magnetic resonance imaging (MRI) brain measures, cognitive performance, and incident dementia and AD dementia, in a large, prospective cohort of cognitively healthy adults.
Methods
Study sample
The Framingham Heart Study (FHS) Offspring cohort is a large community‐based cohort that has been followed for more than 40 years (instituted between 1971 and 1975) for the development of vascular risk factors, decline in cognitive performance, stroke, and dementia.19 Every 4 years, participants attend a follow‐up examination. In this study, we included participants in the Offspring cohort aged 60 years or older who attended examination cycle 7 (1998–2001), had plasma IGFBP‐2 measured at this exam, and were free of a diagnosis of dementia with data available on dementia status on follow‐up. The study protocols and consent forms were approved by the institutional review board at Boston University Medical Center (H‐35519, H‐22765) and all participants provided written informed consent. At cohort inception in 1971, 5124 participants were recruited into the FHS Offspring cohort, of whom 3539 were alive and attended examination 7. Of these, 3219 were free of dementia with available data on dementia status during follow‐up, 1603 were aged 60 years or older, and 1596 had plasma IGFBP‐2 levels measured, resulting in a cohort sample size of 1596 participants for the primary outcome analysis (Fig. 1).
Figure 1.
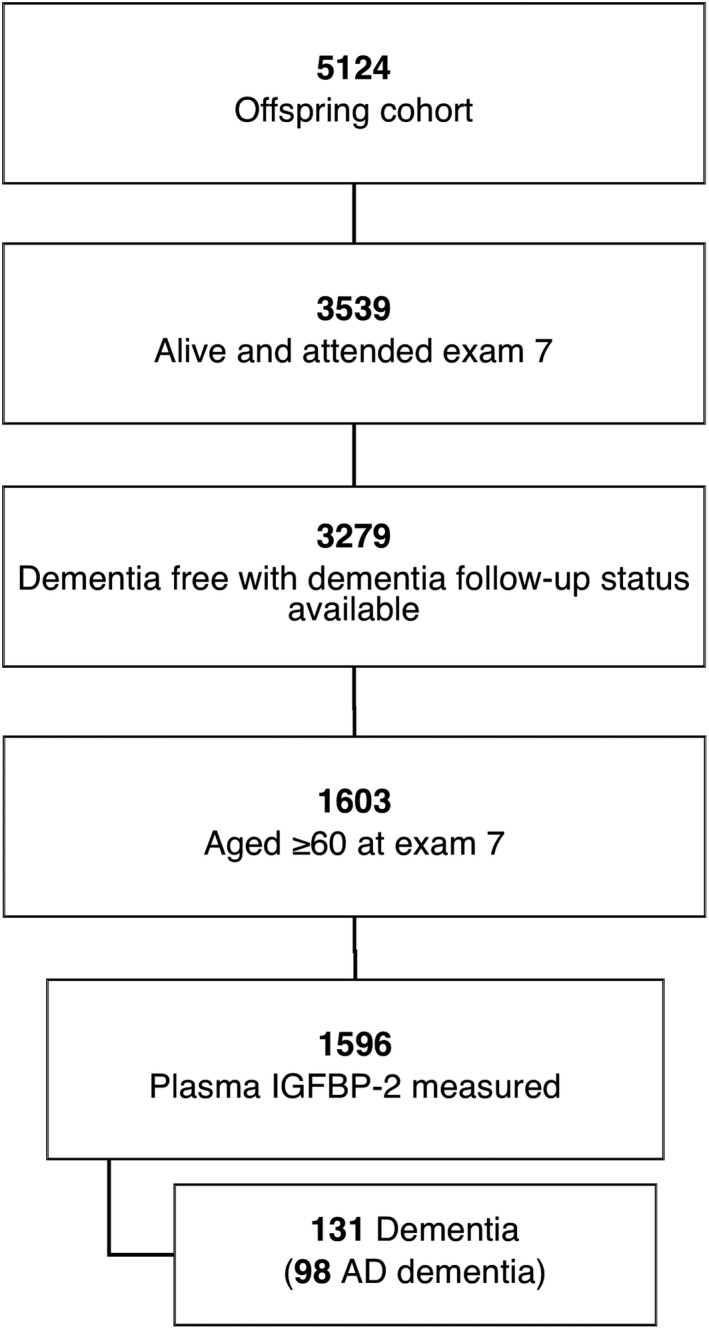
Flow of cohort participants
Outcome measures
The primary outcome measure was incident all‐cause dementia developing after examination cycle 7 up to December 2014. In the FHS Offspring cohort, all participants are systematically screened for the development of dementia via the Mini‐Mental State Examination (MMSE) and annual health status updates starting from examination 5. In addition, all participants were invited to complete a brain MRI and neuropsychological testing starting after each of examinations 7 through 9. If any concern for cognitive impairment is raised (e.g. by a participant, family member, or study physician) or if the MMSE score is five points lower than the participant’s previous highest recorded score, three points lower than the preceding examination, or below the education‐based cutoff, in‐depth neuropsychological testing is performed.20 The Diagnostic and Statistical Manual of Mental Disorders (4th edition) criteria were used to diagnose dementia, requiring impairment in memory and at least one other domain of cognitive function, in addition to documented functional disability. The final diagnosis of dementia was based on a committee review of neurological examination records, neuropsychological assessments, neuroimaging results, outpatient/inpatient/nursing home records, family interviews, and, where available, autopsy results. The committee included at least one neuropsychologist and one neurologist. AD dementia was also a primary outcome measure. A diagnosis of AD was based on the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association for definite, probable, or possible Alzheimer’s disease.21
We included structural MRI brain measures as secondary measures (n = 2074) according to the following methods. Removal of nonbrain tissues: Using an atlas‐based method, the skull is removed. Structural MRI brain images are nonlinearly registered via a cubic B‐spline deformation to a minimal deformation template synthetic brain image. A template‐based iterative method is used to correct for any field inhomogeneity bias. Gray matter, white matter, and CSF measurement: an expectation‐maximization algorithm is employed that iteratively refines its segmentation estimates to produce outputs that are most consistent with the input intensities from the native‐space T1 images along with a model of image smoothness. The segmentation yielded by these appearance models is refined using a Markov random field model and an adaptive prior model. Total brain volume (TBV) was defined as supratentorial brain volume as a percentage of the intracranial volume determined via coronal sections. White matter hyperintensity (WMH) volume is performed on a combination of FLAIR and 3D T1 images using a modified Bayesian probability structure based on a previously published method of histogram fitting and adjusting for intracranial volume. Prior probability maps for WMH were created from more than 700 individuals with semiautomatic detection of WMH followed by manual editing. Likelihood estimates of the native image are calculated through histogram segmentation and thresholding. All segmentation is initially performed in standard space resulting in probability likelihood values of WMH at each voxel in the white matter. These probabilities are then thresholded at 3.5 SD above the mean to create a binary WMH mask. Further segmentation is based on a modified Bayesian approach that combines image likelihood estimates, spatial priors, and tissue class constraints. The segmented WMH masks are then back‐transformed on to native space for tissue volume calculation. Volumes are log‐transformed to normalize population variance. The automatic hippocampal segmentation method employs a standard atlas‐based diffeomorphic approach with the minor modification of label refinement. We further modified this approach to include the European Alzheimer's Disease Consortium‐Alzheimer's Disease Neuroimaging Initiative (EADC‐ADNI) harmonized hippocampal masks using the following approach: (1) Subject image preprocessing with extraction of intracranial cavity, nonuniformity correction, tissue classification as discussed above; (2) Atlas registration of all EADC‐ADNI hippocampal masks to each subject; (3) Atlas fusion; and (4) intensity‐based label refinement. Hippocampal volume was adjusted for intracranial volume. The presence of MRI covert brain infarcts (CBIs) was determined from the size, location, and imaging characteristics of the lesions. The image analysis system allowed for superimposition of the subtraction image, the proton density image, and the T2‐weighted image at three times magnified view to assist in interpretation of lesion characteristics. Signal void, best seen on the T2‐weighted image, was interpreted to indicate a vessel. Only lesions 3 mm or larger qualified for consideration as cerebral infarcts. Kappa values for agreement among the three raters range from 0.73 to 0.90.22, 23 We also included peak width of skeletonized mean diffusivity (PSMD) and fractional anisotropy (FA) in those participants who had an MRI brain performed at exam 9 ([n = 896], as these measures were not included prior to exam 9), according to previously published methods.24 Imaging data were centrally processed and analyzed by operators blinded to all participant characteristics including cognitive performance on neuropsychological testing.
We included performance on neuropsychological tests at examination 7 as additional secondary outcomes, to determine if IGFBP‐2 levels were cross‐sectionally associated with deficits in specific cognitive domains. Specific tests included the visual reproductions delayed recall test, Similarities test, logical memory delayed recall, Trail Making Test (Parts B and A), and Hooper visual organization test (HVOT). Trail Making Test scores were inverse transformed and higher scores for individual test components indicated superior performance. More in‐depth details on neuropsychological testing in the Framingham Offspring cohort have been published previously.25
Laboratory measurements of IGFBP2
We measured IGFBP‐2 plasma levels as part of the Systems Approach to Biomarker Research in Cardiovascular Disease (SABRe CVD) Initiative.26 Blood samples were obtained at the baseline visit (examination cycle 7), drawn in the early morning from participants who had been fasting and were lying supine for 10 min. Samples were immediately centrifuged and stored at − 80°C until the time of assay performance. A modified ELISA sandwich approach was employed, multiplexed on a Luminex xMAP platform (Sigma‐Aldrich, St. Louis, MO). The contract lab (Sigma‐Aldrich) conducted multiplex assay development using protocols developed by Luminex which incorporated key steps to measure sensitivity, specificity, precision, accuracy, and linearity. Singleton assays were initially developed and subsequently combined into multiplex panels as part of the SABRe CVD initiative. Further details of assay methods have previously been published.26 The lower detection limit of IGFBP2 was 5970 pg/mL and the upper detection limit was 5.11 × 107 pg/mL. The intra‐assay coefficient of variation ranged from 2.8% to 6.0%. The inter‐assay coefficient of variation ranged from 8.7% to 10.2%
Covariates
We adjusted for baseline demographic and clinical covariates (measured at examination 7) associated with an increased risk of dementia including age, sex, education (self‐reported with the following categories: no high school degree, high school degree but no college degree, college degree or higher), systolic blood pressure, use of antihypertensive medication, apolipoprotein E4 (ApoE4) carrier status, prevalent diabetes mellitus, prevalent cardiovascular disease (CVD included peripheral vascular disease; coronary heart disease [including coronary insufficiency, angina, myocardial infarction]; and cerebrovascular disease [including transient ischemic attack and stroke]; and congestive heart failure), and body mass index (BMI), as BMI is known to impact the IGF system and dietary restriction has been associated with declining IGF‐1 levels and increased serum IGFBP‐2 levels.27
Statistical analysis
IGFBP‐2 values were natural logarithmically transformed and standardized to normalize the distribution and facilitate comparisons. We also calculated tertiles of plasma IGFBP‐2 comparing tertiles two and three with tertile one. We used multivariable‐adjusted Cox proportional hazards models to evaluate the association between plasma IGFBP‐2 (natural logarithmically transformed values and tertiles) and risk of incident all‐cause dementia and AD dementia. We followed participants from the baseline examination (exam 7) to the time of the incident event (for those with incident dementia), time to death, or time the participant was last confirmed to be dementia‐free (for those without incident events) up to December 2014. The proportional hazards assumption was upheld. Results were reported as hazard ratio (HR) with 95% confidence interval (CI). In model 1, we adjusted for age and sex. In model 2, we adjusted for age, sex, education, systolic blood pressure/use of antihypertensive medication, ApoE4 carrier status, prevalent diabetes mellitus, and prevalent CVD. In model 3, we additionally adjusted for BMI.
Logistic and linear regression models were used to cross‐sectionally relate plasma IGFBP2 levels to MRI‐based structural brain measures (including CBI, WMHV, TBV, HV, PSMD, and FA) and neuropsychological test performance, adjusting for age, age squared, sex, education (neurocognitive outcomes only), time from blood draw to MRI brain or neuropsychological testing, systolic blood pressure, use of antihypertensive medication, prevalent diabetes mellitus, prevalent cardiovascular disease, and BMI.
We assessed for interactions according to ApoE4 carrier status, age (age < median 68 years vs. ≥median of 68 years), sex, and BMI (<30 vs. ≥30) for the primary outcome measures. We also completed a sensitivity analyses excluding participants with a history of stroke at baseline (n = 1554). For the primary outcomes, we determined model discrimination (using the C‐statistic28 and integrated discrimination improvement [IDI] index29, 30) and the net reclassification improvement [NRI]29, 31 following addition of IGFBP‐2 to the conventional models. P < 0.05 was considered significant. We conducted all analyses using SAS statistical software, v9.4 (SAS Institute Inc., Cary, NC). Results were significant at P < 0.05 for the main analyses and at P < 0.10 for interactions.
Results
A total of 1596 participants were included in the primary analysis for incident dementia. The median (Q1, Q3) follow‐up was 11.8 (7.1, 13.3) years, during which 131 participants were diagnosed with dementia of whom 98 were diagnosed with AD (including 93 with AD dementia and five with both AD dementia and vascular dementia), four with vascular dementia, and 29 with other forms of dementia. The mean age of the cohort was 69 years (SD 6) and 53% of participants were women. The interquartile range for plasma IGFBP‐2 levels was 5.3 × 106 to 8.2 × 106 pg/mL in tertile one, 1.1–1.4 × 107 in tertile two, and 1.8–2.6 × 107 in tertile three. Participants with higher IGFBP‐2 levels tended to be older, have a lower BMI, and were less likely to have diabetes mellitus. Baseline characteristics are shown in Table 1.
Table 1.
Baseline characteristics
| Variable | Overall (n = 1596) |
IGFBP‐2 T1 (n = 531) |
IGFBP‐2 T2 (n = 533) |
IGFBP‐2 T3 (n = 532) |
|---|---|---|---|---|
| No. (%) | ||||
| Age, year, mean (SD) | 68.7 (5.7) | 66.7 (4.8) | 68.4 (5.5) | 71.0 (5.9) |
| Women | 841 (52.7) | 289 (54.4) | 265 (49.7) | 287 (54.0) |
| Systolic blood pressure, mmHg, mean (SD) | 132.4 (19.2) | 132.3 (17.3) | 132.0 (19.6) | 132.9 (20.6) |
| BMI, kg/m2, median (Q1, Q3) | 28.1 (24.7, 30.9) | 29.5 (26.7, 32.8) | 27.5 (25.0, 30.8) | 25.7 (23.2, 28.3) |
| IGFBP‐2, pg/mL, median (Q1, Q3) | 1.2 × 107 (8.2 × 106, 1.8 × 107) | 6.7 × 106 (5.3 × 106, 8.2 × 106 | 1.2 × 107 (1.1 × 107, 1.4 × 107 | 2.1 × 107 (1.8 × 107, 2.6 × 107) |
| Education | ||||
| No high school degree | 101 (6.5) | 29 (5.7) | 28 (5.4) | 44 (8.5) |
| High school degree | 525 (33.9) | 182 (35.6) | 168 (32.3) | 175 (33.8) |
| Some years of college | 450 (29.0) | 145 (28.3) | 151 (29.0) | 154 (29.7) |
| College degree | 475 (30.6) | 156 (30.5) | 174 (33.4) | 145 (28.0) |
| Antihypertensive medication | 700 (43.9) | 240 (45.3) | 230 (43.2) | 230 (43.2) |
| Current smoker | 135 (8.5) | 27 (5.1) | 47 (8.8) | 61 (11.5) |
| Prevalent diabetes mellitus | 262 (16.4) | 114 (21.5) | 84 (15.8) | 64 (12.0) |
| ApoE4 allele | 353 (22.4) | 120 (23.1) | 122 (23.0) | 111 (21.2) |
| Prevalent CVD | 302 (18.9) | 73 (13.8) | 97 (18.2) | 132 (24.8) |
| Atrial fibrillation | 94 (5.9) | 23 (4.3) | 37 (6.9) | 34 (6.4) |
| Stroke | 42 (2.6) | 10 (1.9) | 10 (1.9) | 22 (4.1) |
Baseline demographic and clinical characteristics were defined at examination 7. IGFBP‐2, insulin‐like growth factor binding protein‐2; SD, standard deviation; BMI, body mass index; CVD, cardiovascular disease; APOE E4, apolipoprotein E4 allele.
IGFBP‐2 and dementia
On multivariable analysis adjusting for age and sex, elevated plasma IGFBP‐2 was associated with an increased risk of dementia (HR 1.48, 95% CI 1.21–1.82) and AD dementia (HR 1.53, 95% CI 1.21–1.94) per each standard deviation unit (SDU) increment in natural log‐transformed biomarker value. There was minimal attenuation of the association when education and vascular risk factors were added to the models. On fully adjusted multivariable analysis (accounting for age, sex, education, SBP, use of antihypertensive medications, prevalent diabetes mellitus, prevalent CVD, BMI, and ApoE4 carrier status), IGFBP‐2 remained associated with an increased risk of dementia (HR 1.56, 95% CI 1.25–1.95) and AD dementia (HR 1.56, 95% CI 1.21–2.02). There was an apparent dose–response relationship between plasma IGFBP2 levels and risk of both dementia and AD dementia. Compared to the lowest tertile, the middle and top tertiles of plasma IGFBP‐2 were associated with an increased risk of both dementia (HR 1.87, 95% CI 1.07–3.27, and HR 2.89, 95% CI 1.63–5.13, respectively) and AD dementia (HR 2.68, 95% CI 1.33–5.40 and HR 3.63, 95% CI 1.76–7.50, respectively) (Table 2, Figs. 2, 3). There was no significant interaction between IGFBP‐2 and age (P = 0.62), sex (P = 0.43), BMI (P = 0.77), or ApoE4 carrier status (P = 0.39) on the risk of dementia. Sensitivity analyses excluding participants with prior stroke yielded similar results (Table 3).
Table 2.
IGFBP‐2 and risk of incident dementia and AD dementia
| Model | Dementia | Alzheimer’s disease | |||
|---|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | ||
| Per SDU increase | 1 | 1.48 (1.21–1.82) | 0.0002 | 1.53 (1.21–1.94) | 0.0004 |
| 2 | 1.57 (1.27–1.93) | <0.001 | 1.58 (1.24–2.01) | 0.0002 | |
| 3 | 1.56 (1.25–1.95) | <0.001 | 1.56 (1.21–2.02) | 0.0006 | |
| Tertile 1 | 1 | Reference | Reference | ||
| Tertile 2 | 1.71 (0.99–2.97) | 0.056 | 2.44 (1.23–4.85) | 0.01 | |
| Tertile 3 | 2.50 (1.48–4.23) | 0.0006 | 3.17 (1.62–6.19) | 0.0007 | |
| Tertile 1 | 2 | Reference | Reference | ||
| Tertile 2 | 1.87 (1.07–3.26) | 0.03 | 2.71 (1.36–5.42) | 0.005 | |
| Tertile 3 | 2.90 (1.68–5.00) | 0.0001 | 3.72 (1.86–7.42) | 0.0002 | |
| Tertile 1 | 3 | Reference | Reference | ||
| Tertile 2 | 1.87 (1.07–3.27) | 0.029 | 2.68 (1.33–5.40) | 0.006 | |
| Tertile 3 | 2.89 (1.63–5.13) | 0.0003 | 3.63 (1.76–7.50) | 0.0005 | |
Model 1: adjusted for age and sex. Model 2: adjusted for model 1 + education, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, prevalent diabetes mellitus, and ApoE4 carrier status. Model 3: adjusted for model 2 + BMI. IGFBP2 was natural logarithmically transformed and standardized. IGFBP‐2, insulin‐like growth factor binding protein‐2; SDU, standard deviation unit; HR, hazard ratio; CI, confidence interval.
Figure 2.
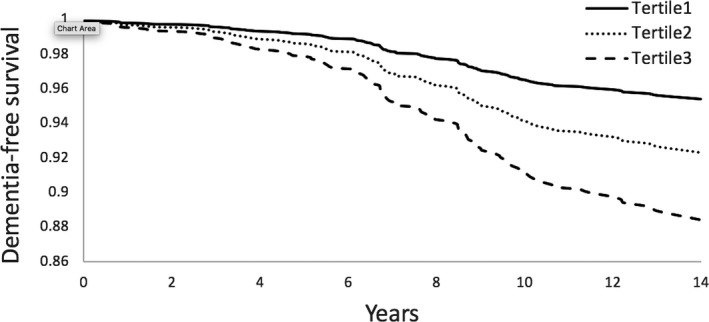
IGFBP‐2 and time to dementia
Figure 3.
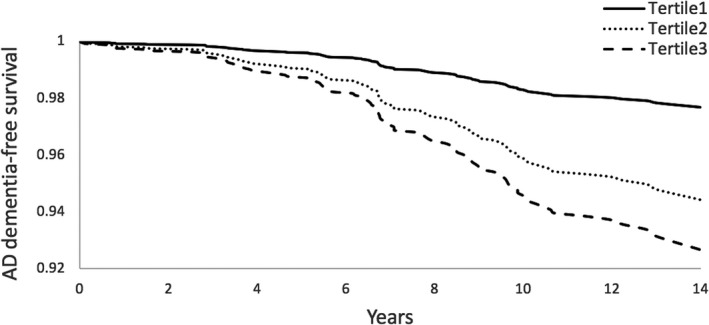
IGFBP‐2 and time to AD dementia
Table 3.
IGFBP‐2 and risk of incident dementia and AD dementia, excluding those with prior stroke
| Model | Dementia | Alzheimer’s disease | |||
|---|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | ||
| Per SDU increase | 1 | 1.58 (1.28–1.95) | <0.0001 | 1.65 (1.29–2.11) | <0.0001 |
| 2 | 1.72 (1.38–2.15) | <0.0001 | 1.77 (1.37–2.29) | <0.0001 | |
| 3 | 1.70 (1.35–2.14) | <0.0001 | 1.74 (1.33–2.27) | <0.0001 | |
| Tertile 2 | 1 | 1.90 (1.06–3.43) | 0.03 | 2.79 (1.32–5.89) | 0.007 |
| Tertile 3 | 2.87 (1.63–5.05) | 0.0003 | 3.83 (1.85–7.93) | 0.0003 | |
| Tertile 2 | 2 | 1.97 (1.09–3.58) | 0.03 | 2.89 (1.36–6.15) | 0.006 |
| Tertile 3 | 3.25 (1.81–5.83) | <0.0001 | 4.37 (2.06–9.27) | 0.0001 | |
| Tertile 2 | 3 | 1.94 (1.06–3.53) | 0.04 | 2.81 (1.31–6.02) | 0.008 |
| Tertile 3 | 3.12 (1.69–5.75) | 0.0003 | 4.13 (1.89–9.01) | 0.0004 | |
Model 1: adjusted for age and sex. Model 2: adjusted for model 1 + education, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, prevalent diabetes mellitus, and ApoE4 carrier status. Model 3: adjusted for model 2 + BMI. IGFBP2 was natural logarithmically transformed and standardized. Reference for tertiles is the lowest tertile. IGFBP‐2, insulin‐like growth factor binding protein‐2; SDU, standard deviation unit; HR, hazard ratio; CI, confidence interval.
IGFBP‐2 and secondary outcomes
On adjusted analysis (adjusted for age, age squared, sex, education, time from blood draw to neuropsychological testing, systolic blood pressure, use of antihypertensive medication, prevalent diabetes mellitus, prevalent cardiovascular disease, and BMI), higher plasma IGFBP‐2 levels were cross‐sectionally associated with poorer performance on tests of abstract reasoning (Similarities test [β ± SE − 0.499 ± 0.18, P = 0.006]), when comparing the highest tertile with the lowest tertile. Results were consistent when modeling IGFBP‐2 as a continuous variable (Table 4). Plasma IGFBP‐2 levels were not cross‐sectionally associated with performance on other neuropsychological tests or with MRI markers of small vessel disease or neurodegeneration (Table 5).
Table 4.
IGFBP‐2 and neuropsychological test performance
| Similarities (n correct) | Visual reproductions (n correct after delay) | Logical memory (n correct after delay) | Trail making B‐A (min)* | Hooper visual organization test* | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Β ± SE | P‐value | β ± SE | P‐value | β ± SE | P‐value | β ± SE | P‐value | β ± SE | P‐value | |
| Per SDU increase** | −0.266 ± 0.07 | 0.0005 | −0.044 ± 0.07 | 0.57 | −0.055 ± 0.08 | 0.57 | −0.0088 ± 0.005 | 0.11 | −0.011 ± 0.01 | 0.48 |
| T2 versus T1 | −0.200 ± 0.16 | 0.23 | −0.044 ± 0.16 | 0.80 | −0.077 ± 0.18 | 0.70 | −0.011 ± 0.01 | 0.24 | 0.033 ± 0.03 | 0.28 |
| T3 versus T1 | −0.499 ± 0.18 | 0.006 | −0.0044 ± 0.17 | 0.98 | −0.066 ± 0.19 | 0.76 | −0.022 ± 0.01 | 0.18 | −0.011 ± 0.03 | 0.72 |
Model adjusted for age, age squared, sex, education, time from blood draw to neuropsychological testing, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, prevalent diabetes mellitus, and BMI. IGFBP‐2, insulin‐like growth factor binding protein‐2; SDU, standard deviation unit; SE, standard error.
Natural log‐transformed to restore normality (higher scores indicate better performance).
Natural log‐transformed and standardized.
Table 5.
IGFBP‐2 and MRI measures of brain injury
| TBV (%×100) | Hippocampal volume (%) | WMHV* (%) | Covert brain infarcts (n) | PSMD* | Mean FA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β ± SE | P‐value | β ± SE | P‐value | β ± SE | P‐value | OR (95% CI) | P‐value | β ± SE | P‐value | β ± SE | P‐value | |
| Per SDU increase** | 0.022 ± 0.05 | 0.73 | −0.0022 ± 0.001 | 0.08 | 0.011 ± 0.02 | 0.57 | 1.07 (0.90, 1.27) | 0.44 | −0.0022 ± 0.01 | 0.82 | 0.0011 ± 0.001 | 0.29 |
| T2 versus T1 | −0.177 ± 0.11 | 0.11 | −0.00022 ± 0.003 | 0.92 | −0.044 ± 0.05 | 0.48 | 1.34 (0.92, 1.95) | 0.13 | 0.0066 ± 0.01 | 0.68 | 0.0011 ± 0.002 | 0.59 |
| T3 versus T1 | 0.099 ± 0.12 | 0.44 | −0.0044 ± 0.003 | 0.18 | 0.011 ± 0.05 | 0.82 | 0.88 (0.57–1.36) | 0.57 | −0.022 ± 0.02 | 0.18 | 0.0022 ± 0.002 | 0.13 |
PSMD and FA were measured in stroke and dementia‐free participants who had MRI brain completed at exam 9 (n = 896). IGFBP‐2, insulin‐like growth factor binding protein‐2; SDU, standard deviation unit; SE, standard error; TBV, total brain volume; WMHV, White matter hyperintensity volume; PSMD, peak width of skeletonized mean diffusivity; FA, fractional anisotropy. Model adjusted for age, age squared, sex, education, time from blood draw to MRI brain, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, prevalent diabetes mellitus, and BMI.
Natural log‐transformed.
Natural log‐transformed and standardized.
Risk prediction for dementia
In a model of conventional risk factors for dementia (age, sex, education, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, and ApoE4 carrier status), the C‐statistic was 0.80 (95% CI 0.77–0.84), with minimal change following the addition of IGFBP‐2 to the model. Addition of IGFBP‐2 to the conventional model, however, resulted in a relative IDI of 15% (95% CI 7–23%) compared to the base model. Furthermore, on addition of IGFBP‐2 to the conventional model, 32% of individuals with dementia were correctly assigned a higher predicted risk, while 8% of individuals without dementia were correctly assigned a lower predicted risk (overall NRI, 0.40, 95% CI 0.22–0.59) (Table 6).
Table 6.
Model discrimination and risk reclassification
| Dementia | Alzheimer’s | |||||||
|---|---|---|---|---|---|---|---|---|
| C‐statistic (95% CI) | Relative IDI (95% CI) | Overall NRI (95% CI) |
NRI, events NRI, nonevents* |
C‐statistic (95% CI) | Relative IDI (95% CI) | Overall NRI (95% CI) |
NRI, events NRI, nonevents* |
|
| Model 2 | 0.81 (0.77–0.85) | – | – | – | 0.85 (0.82–0.88) | – | – | – |
| Model 2 + I GFBP‐2 | 0.82 (0.79–0.86) | 0.15 (0.07–0.23) | 0.40 (0.22–0.59) |
0.32 0.08 |
0.87 (0.84–0.90) | 0.13 (0.05–0.22) | 0.42 (0.20–0.65) |
0.34 0.08 |
Model 2: adjusted for age, sex, education, systolic blood pressure, use of antihypertensive medication, prevalent cardiovascular disease, prevalent diabetes mellitus, and ApoE4 carrier status. IGFBP‐2 was natural logarithmically transformed and standardized. IDI, integrated discrimination improvement; NRI, net reclassification improvement.
Proportion of events correctly reclassified. Proportion of nonevents correctly reclassified.
Discussion
We found that elevated plasma IGFBP‐2 was associated with an increased risk of incident all‐cause and AD dementia as well as with poorer performance on tests of abstract reasoning. Addition of plasma IGFBP‐2 to a model of traditional risk factors for dementia resulted in improved dementia risk prediction.
Elevated plasma IGFBP‐2 was associated with an increased risk of incident all‐cause dementia as well as AD dementia. This relationship followed a dose–response pattern, with an increasing risk of dementia and AD dementia per increasing tertile of plasma IGFBP‐2. Previous cross‐sectional studies have noted elevated CSF levels of IGFBP‐2 in patients with AD,6, 10 as well as elevated levels of IGFBP‐2 in the plasma of patients with AD.6, 12, 13, 14, 16 Our results extend these findings by demonstrating a longitudinal association between plasma IGFBP‐2 levels measured at a median age of 68 years and subsequent risk of developing dementia over long‐term follow‐up. Unlike one prior study,6 we found no attenuation in this association after accounting for age, sex, education, or BMI.
Interestingly, we found that the association between IGFBP‐2 was stronger for AD dementia than all‐cause dementia. Adjusting for vascular risk factors, including diabetes mellitus, on multivariable analysis did not appreciably attenuate the association. In addition, we observed no cross‐sectional association between plasma IGFBP‐2 levels and measures of vascular brain injury, including WMHV or the presence of CBI. Indeed plasma IGFBP‐2 levels have been shown to be inversely associated with plasma triglycerides32 and risk of diabetes33 and in our cohort, participants in the higher tertiles of IGFBP2, compared to the bottom tertile, had a lower prevalence of diabetes mellitus. Thus, vascular pathways are unlikely to account for the association we observed.
IGFs have been reported to protect against hippocampal injury in the presence of ischemia and neurotoxins, including B‐amyloid,9, 34, 35 while elevated IGF‐binding proteins are thought to inhibit these neuroprotective effects.7, 8, 9 It has been suggested that ApoE4 may modify the association between impaired IGF signaling and dementia.36 In our study, we found a similar prevalence of the ApoE4 allele across tertiles of IGFBP‐2 levels. Furthermore, after adjusting for ApoE4 carrier status, the association between IGFBP‐2 and dementia was not materially altered and we observed no evidence of effect modification according to ApoE4 carrier status, consistent with a prior study.16 While elevated plasma IGFBP‐2 levels have been cross‐sectionally associated with smaller hippocampal volumes, this was only noted in individuals with a CSF biomarker profile inconsistent with AD (i.e. CSF Aß‐422> 1.92 pg/mL).11 In another study, elevated CSF IGFBP‐2 was associated with increased CSF Aß‐42.16 Thus, ApoE4‐mediated pathways do not appear to account for the observed association between elevated plasma IGFBP‐2 and risk of dementia.
Elevated IGFBP‐2 has also been linked to the development of tau pathology.37 Both increased plasma and CSF IGFBP‐2 levels have been associated with elevated CSF tau levels,6, 7, 15, 37 which are likely mediated through impaired IGF signaling resulting in increased tau phosphorylation and reduced clearance of ß‐amyloid.18, 38, 39 IGFBP‐2 has been shown to have differential cortical expression in transgenic mouse models of tauopathy compared to wild‐type mice, further implicating a potential role for IGFBP‐2 and subsequent impaired IGF signaling in the development of tauopathy and neuronal death.37
We did not find an association between plasma IGFBP‐2 levels and MRI structural brain measures, including hippocampal volume. There is limited published data on the relationship between IGFBP‐2 and structural brain measures. As mentioned, elevated plasma IGFBP‐2 levels have been linked with lower hippocampal volumes,11 while in transgenic mouse models, overexpression of IGFBP‐2 was noted to be associated with lower total brain weight, as well as reduced hippocampal, prefrontal cortical, and cerebellar weights.40, 41 Baseline CSF IGFBP‐2 levels have also been longitudinally associated with atrophy in nonhippocampal regions including the parahippocampus, entorhinal cortex as well as inferior and superior temporal lobes.37
We observed a cross‐sectional association between elevated plasma IGFBP‐2 levels and poorer performance on tests of abstract reasoning. There was no association with tests of other cognitive domains including visual memory, logical memory, visuospatial skills, or other executive function. One prior study found an association between elevated plasma IGFBP‐2 levels and poorer performance on tests of episodic memory but not executive function.11 Our analysis was cross‐sectional so we are unable to comment on the possibility of a longitudinal association between IGFBP‐2 levels and change in performance in other cognitive domains.
Circulating IGFBP‐2 offers promise as a candidate biomarker for predicting risk of dementia. On addition of IGFBP‐2 to a conventional demographic and vascular risk factor model, the relative IDI index was 15% compared to the base model, suggesting that IGFBP‐2 adds incremental value beyond that of traditional risk factors. Moreover, when we added plasma IGFBP‐2 to the conventional model, 32% of individuals with dementia were correctly assigned a higher predicted risk while 8% of individuals without dementia were correctly assigned a lower predicted risk, indicating improved dementia risk classification.
There is increasing interest in manipulating insulin sensitivity and IGF signaling in the brain to help target cognitive decline and dementia. Evidence to date supports a role for insulin, and growth hormone‐releasing hormone in improving cognitive performance in patients with mild cognitive impairment and AD dementia,42, 43, 44, 45, 46 while administration of IGF‐I and IGF‐II in murine models has been related to improved cognitive performance and neurological function as well as reduced ß‐amyloid burden in the brain.47, 48, 49, 50 Blocking IGFBP‐2 may be an attractive therapeutic target for dementia prevention by enhancing IGF signaling in the brain. Plasma IGFBP‐2 may also be useful as a surrogate outcome in phase II trials of future dementia therapeutics targeting IGF signaling or for selecting suitable patients for inclusion in phase III trials of such therapies.
The unique strengths of our study include our relatively large sample size compared to prior studies to date, the high proportion of individuals with measured IGFBP‐2 levels, the availability of prospective, long‐term follow‐up data in a population confirmed to be dementia‐free at baseline, the rigorous approach to surveillance, and diagnosis of clinically confirmed dementia and AD (dementia diagnosis was confirmed based on clinical assessments, medical record review by a behavioral neurologist, and rigorous application of standardized criteria), and the detailed assessment and follow‐up for development of vascular risk factors as well as intermediary cognitive and structural MRI brain measures. Limitations include the use of clinical criteria rather than biomarker‐based data (e.g. CSF Aß‐42 or amyloid positron emission tomography) for dementia diagnosis and subtyping. The Framingham Offspring cohort is predominantly Caucasian, which while allowing homogeneity, may limit the generalizability of our findings to more heterogeneous populations. Furthermore, we were unable to explore the association between CSF levels of IGFBP‐2 and cognitive outcomes. However, plasma biomarkers, compared to CSF biomarkers, offer the advantages of being less invasive and can be more easily incorporated into routine clinical practice. In addition, we did not have CSF tau levels available for a mediation analysis to further understand the association between elevated plasma IGFBP‐2 and dementia.
Conclusions
Elevated plasma levels of IGFBP‐2 were associated with an increased risk of all‐cause dementia, including AD dementia, in a community‐based population of cognitively healthy adults. Addition of IGFBP2 plasma levels to a model of traditional risk factors significantly improved dementia risk classification, suggesting that plasma IGFBP‐2 may be a useful biomarker for predicting dementia risk. Manipulating IGF‐signaling pathways via IGFBP‐2 may be a promising therapeutic target for dementia prevention.
Author Contributions
Study concept and design: McGrath, Seshadri, Himali, Levy, Vasan. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: McGrath. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Himali, Conner. Obtained funding: Seshadri, Levy, McGrath. Administrative, technical, or material support: Conner, Study supervision: Seshadri.
Conflict of Interest
None declared.
Funding Information
The Framingham Heart Study is supported by the National Heart, Lung, and Blood Institute (contract no. N01‐HC‐25195 and no. HHSN268201500001I). This research was supported by an Alzheimer’s Association Clinician Scientist Fellowship (AACSF‐18‐566570), NHLBI grants R01 HL60040 and R01 HL70100, grants from the National Institute on Aging (R01 AG054076, R01 AG049607, R01 AG033193, U01 AG049505, U01 AG052409), and grants from the National Institute of Neurological Disorders and Stroke (NS017950 and UH2 NS100605). Additional funding for the Systems Approach to Biomarker Research in Cardiovascular Disease (SABRe CVD) initiative was provided by the Division of Intramural Research, NHLBI, and the Population Sciences Branch, NHLBI. MPP is funded by a National Heart Foundation of Australia Future Leader Fellowship (102052). SCC is funded by the National Institute of General Medical Sciences (NIGMS) Interdisciplinary Training Grant for Biostatisticians (T32GM74905‐14). None of the funding entities had any role in the design and conduct of the study; collection, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication. The views and opinions offered in this manuscript are those of the authors and are not necessarily those of the funding agencies.
Funding Statement
This work was funded by National Institute on Aging grants R01 AG033193, R01 AG049607, R01 AG054076, U01 AG049505, and U01 AG052409; National Institute of Neurological Disorders and Stroke grants NS017950 and UH2 NS100605; National Heart, Lung, and Blood Institute grants HHSN268201500001I, N01-HC-25195, R01 HL60040, and R01 HL70100; Alzheimer’s Association Clinician Scientist Fellowship grant AACSF-18-566570; National Heart Foundation of Australia Future Leader Fellowship grant 102052; National Institute of General Medical Sciences (NIGMS) grant T32GM74905-14.
References
- 1. Molinuevo JL, Ayton S, Batrla R, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol 2018;136:821–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF‐1 resistance, IRS‐1 dysregulation, and cognitive decline. J Clin Invest 2012;122:1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bomfim TR, Forny‐Germano L, Sathler LB, et al. An anti‐diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease‐ associated Abeta oligomers. J Clin Invest 2012;122:1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bedse G, Di Domenico F, Serviddio G, Cassano T. Aberrant insulin signaling in Alzheimer's disease: current knowledge. Front Neurosci 2015;9:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez AM, Torres‐Aleman I. The many faces of insulin‐like peptide signalling in the brain. Nat Rev Neurosci 2012;13:225–239. [DOI] [PubMed] [Google Scholar]
- 6. Hertze J, Nagga K, Minthon L, Hansson O. Changes in cerebrospinal fluid and blood plasma levels of IGF‐II and its binding proteins in Alzheimer's disease: an observational study. BMC Neurol 2014;14:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aberg D, Johansson P, Isgaard J, et al. Increased cerebrospinal fluid level of insulin‐like growth factor‐II in male patients with Alzheimer's disease. J Alzheimers Dis 2015;48:637–646. [DOI] [PubMed] [Google Scholar]
- 8. Hoeflich A, Wu M, Mohan S, et al. Overexpression of insulin‐like growth factor‐binding protein‐2 in transgenic mice reduces postnatal body weight gain. Endocrinology 1999;140:5488–5496. [DOI] [PubMed] [Google Scholar]
- 9. Mackay KB, Loddick SA, Naeve GS, et al. Neuroprotective effects of insulin‐like growth factor‐binding protein ligand inhibitors in vitro and in vivo. J Cereb Blood Flow Metab 2003;23:1160–1167. [DOI] [PubMed] [Google Scholar]
- 10. Tham A, Nordberg A, Grissom FE, et al. Insulin‐like growth factors and insulin‐like growth factor binding proteins in cerebrospinal fluid and serum of patients with dementia of the Alzheimer type. J Neural Transm Park Dis Dement Sect 1993;5:165–176. [DOI] [PubMed] [Google Scholar]
- 11. Lane EM, Hohman TJ, Jefferson AL. Insulin‐like growth factor binding protein‐2 interactions with Alzheimer's disease biomarkers. Brain Imaging Behav 2016;11:1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salehi Z, Mashayekhi F, Naji M. Insulin like growth factor‐1 and insulin like growth factor binding proteins in the cerebrospinal fluid and serum from patients with Alzheimer's disease. BioFactors 2008;33:99–106. [DOI] [PubMed] [Google Scholar]
- 13. Doecke JD, Laws SM, Faux NG, et al. Blood‐based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol 2012;69:1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu WT, Holtzman DM, Fagan AM, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 2012;79:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toledo JB, Da X, Bhatt P, et al. Relationship between plasma analytes and SPARE‐AD defined brain atrophy patterns in ADNI. PLoS ONE 2013;8:e55531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLimans KE, Webb JL, Anantharam V, et al. Peripheral versus central index of metabolic dysfunction and associations with clinical and pathological outcomes in Alzheimer's disease. J Alzheimers Dis 2017;60:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benedict C, Grillo CA. Insulin resistance as a therapeutic target in the treatment of Alzheimer's disease: a state‐of‐the‐art review. Front Neurosci 2018;12:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res 2012;9:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feinleib M, Kannel WB, Garrison RJ, et al. The framingham offspring study. Design and preliminary data. Prev Med 1975;4:518–525. [DOI] [PubMed] [Google Scholar]
- 20. Seshadri S, Wolf P, Beiser A, et al. Lifetime risk of dementia and Alzheimer's disease the impact of mortality on risk estimates in the Framingham study. Neurology 1997;49:1498–1504. [DOI] [PubMed] [Google Scholar]
- 21. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 22. Massaro JM, D'Agostino RB Sr, Sullivan LM, et al. Managing and analysing data from a large‐scale study on Framingham Offspring relating brain structure to cognitive function. Stat Med 2004;23:351–367. [DOI] [PubMed] [Google Scholar]
- 23. DeCarli C, Massaro J, Harvey D, et al. Measures of brain morphology and infarction in the framingham heart study: establishing what is normal. Neurobiol Aging 2005;26:491–510. [DOI] [PubMed] [Google Scholar]
- 24. Echouffo‐Tcheugui JB, Conner SC, Himali JJ, et al. Circulating cortisol and cognitive and structural brain measures: the Framingham Heart Study. Neurology 2018;91:e1961–e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Au R, Seshadri S, Wolf PA, et al. New norms for a new generation: cognitive performance in the framingham offspring cohort. Exp Aging Res 2004;30:333–358. [DOI] [PubMed] [Google Scholar]
- 26. Ho JE, Lyass A, Courchesne P, et al. Protein biomarkers of cardiovascular disease and mortality in the community. J Am Heart Assoc 2018;7:pii:e008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thissen JP, Underwood LE, Ketelslegers JM. Regulation of insulin‐like growth factor‐I in starvation and injury. Nutr Rev 1999;57:167–176. [DOI] [PubMed] [Google Scholar]
- 28. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982;143:29–36. [DOI] [PubMed] [Google Scholar]
- 29. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr. Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med 2008;27:157–172; discussion 207–212. [DOI] [PubMed] [Google Scholar]
- 30. Kerr KF, McClelland RL, Brown ER, Lumley T. Evaluating the incremental value of new biomarkers with integrated discrimination improvement. Am J Epidemiol. 2011;174:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pencina MJ, D'Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med 2011;30:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carter S, Li Z, Lemieux I, et al. Circulating IGFBP‐2 levels are incrementally linked to correlates of the metabolic syndrome and independently associated with VLDL triglycerides. Atherosclerosis 2014;237:645–651. [DOI] [PubMed] [Google Scholar]
- 33. Wittenbecher C, Ouni M, Kuxhaus O, et al. Insulin‐like growth factor binding protein 2 (IGFBP‐2) and the risk of developing type 2 diabetes. Diabetes 2019;68:188–197. [DOI] [PubMed] [Google Scholar]
- 34. Doré S, Kar S, Quirion R. Insulin‐like growth factor I protects and rescues hippocampal neurons against β‐amyloid‐ and human amylin‐induced toxicity. Proc Natl Acad Sci USA 1997;94:4772–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei W, Wang X, Kusiak JW. Signaling events in amyloid β‐peptide‐induced neuronal death and insulin‐like growth factor I protection. J Biol Chem 2002;277:17649–17656. [DOI] [PubMed] [Google Scholar]
- 36. Galle SA, van der Spek A, Drent ML, et al. Revisiting the role of insulin‐like growth factor‐I receptor stimulating activity and the apolipoprotein E in Alzheimer’s disease. Front Aging Neurosci 2019;11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonham LW, Geier EG, Steele NZR, et al. Insulin‐like growth factor binding protein 2 is associated with biomarkers of Alzheimer's disease pathology and shows differential expression in transgenic mice. Front Neurosci 2018;12:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate‐2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci 2003;23:7084–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watanabe K, Uemura K, Asada M, et al. The participation of insulin‐like growth factor‐binding protein 3 released by astrocytes in the pathology of Alzheimer's disease. Mol Brain 2015;8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoeflich A, Lahm H, Nedbal S, et al. Growth inhibition in giant growth hormone transgenic mice by overexpression of insulin‐like growth factor‐binding protein‐2. Endocrinology 2001;142:1889–1898. [DOI] [PubMed] [Google Scholar]
- 41. Schindler N, Mayer J, Saenger S, et al. Phenotype analysis of male transgenic mice overexpressing mutant IGFBP‐2 lacking the Cardin‐Weintraub sequence motif: reduced expression of synaptic markers and myelin basic protein in the brain and a lower degree of anxiety‐like behaviour. Growth Horm IGF Res 2017;33:1–8. [DOI] [PubMed] [Google Scholar]
- 42. Claxton A, Baker LD, Hanson A, et al. Long‐acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early‐stage Alzheimer's disease dementia. J Alzheimers Dis 2015;44:897–906. [DOI] [PubMed] [Google Scholar]
- 43. Reger MA, Watson GS, Green PS, et al. Intranasal insulin improves cognition and modulates beta‐amyloid in early AD. Neurology 2008;70:440–448. [DOI] [PubMed] [Google Scholar]
- 44. Craft S, Claxton A, Baker LD, et al. Effects of regular and long‐acting insulin on cognition and Alzheimer's disease biomarkers: a pilot clinical trial. J Alzheimers Dis 2017;57:1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 2012;69:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baker LD, Barsness SM, Borson S, et al. Effects of growth hormone–releasing hormone on cognitive function in adults with mild cognitive impairment and healthy older adults: results of a controlled trial. Arch Neurol 2012;69:1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lupien SB, Bluhm EJ, Ishii DN. Systemic insulin‐like growth factor‐I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. J Neurosci Res 2003;74:512–523. [DOI] [PubMed] [Google Scholar]
- 48. Liu XF, Fawcett JR, Thorne RG, et al. Intranasal administration of insulin‐like growth factor‐I bypasses the blood‐brain barrier and protects against focal cerebral ischemic damage. J Neurol Sci 2001;187:91–97. [DOI] [PubMed] [Google Scholar]
- 49. Chen DY, Stern SA, Garcia‐Osta A, et al. A critical role for IGF‐II in memory consolidation and enhancement. Nature 2011;469:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carro E, Trejo JL, Gomez‐Isla T, et al. Serum insulin‐like growth factor I regulates brain amyloid‐β levels. Nat Med 2002;8:1390. [DOI] [PubMed] [Google Scholar]