

Plasmonic nanostructures supporting strong spatially confined field enhancement are developed to probe single-molecule SERS.
Abstract
Tailored metal nanoclusters have been actively developed to manipulate light at the subwavelength scale for nanophotonic applications. Nevertheless, precise arrangement of molecules in a hot spot with fixed numbers and positions remains challenging. Here, we show that DNA origami metamolecules with Fano resonances (DMFR) can precisely localize single dye molecules and produce quantified surface-enhanced Raman scattering (SERS) responses. To enable tailored plasmonic permutations, we develop a general and programmable method for anchoring a set of large gold nanoparticles (L-AuNPs) on prescribed n-tuple docking sites of super-origami DNA frameworks. A tetrameric nanocluster with four spatially organized 80-nm L-AuNPs exhibits peak-and-dip Fano characteristics. The drastic enhancement at the wavelength of the Fano minimum allows the collection of prominent SERS spectrum for even a single dye molecule. We expect that DMFR provides physical insights into single-molecule SERS and opens new opportunities for developing plasmonic nanodevices for ultrasensitive sensing, nanocircuits, and nanophotonic lasers.
INTRODUCTION
Metallic nanostructures that support surface plasmons are of great interest because of their ability to manipulate light at the nanoscale (1, 2). In particular, metal nanoclusters with spatially coupled nanoparticles, known as metamolecules (3), resemble molecules with spatially coupled atoms and display optical properties that make them attractive as potential metamaterials. These properties include nanocircuits (4), plasmonic sensors (5), and subwavelength waveguides (6, 7). Both theoretical and experimental studies have verified that the strong field localization at hot spots of plasmonic nanostructures can result in drastic spectroscopic enhancement approaching the single-molecule regime (8–11). However, direct quantification of single molecules within hot spots remains difficult due to the challenges in simultaneous nanometer-precise control of the geometry of metallic nanoparticle ensembles and the number and position of single molecules localized in the hot spot (12, 13).
During the past decades, top-down lithography (14, 15) and bottom-up self-assembly (16–18) approaches have made remarkable progress in fabricating complex plasmonic nanostructures with high precision. Nevertheless, interfacing these metal nanostructures with chemical and biological molecules of interest arouses considerable research efforts. Self-assembled DNA nanostructures, especially DNA origami, provide a highly programmable approach for designing precise nanopatterns with nanoscale addressability for organizing molecules and nanoparticles (19–22). For example, DNA origami–supported nanoantennas have proven effective to plasmonically enhance the emission of a fluorophore or a Raman dye placed to the proximity of metal nanoparticles (23–27). The use of DNA origami nanostructures is thus expected to open a new door for fabricating plasmonic nanodevices with spatial organization and high complexity.
Here, we report a general strategy for precisely organizing large gold nanoparticles (L-AuNPs) into plasmonic metamolecules with super-origami DNA frameworks. Because pronounced Fano resonances are usually achieved with >80-nm L-AuNPs (28), we designed DNA super-origami with n-tuple docking sites, which allowed the formation of rhombic tetrameric nanoclusters of 80-nm AuNPs. By exploiting the very strong electromagnetic field localized in hot spots at the wavelength of the Fano minimum (29–31), we developed a platform for quantizing surface-enhanced Raman scattering (SERS) of single dye molecules localized in the hot spot of a DNA origami metamolecule with Fano resonances (DMFR).
RESULTS
Design of super-origami DNA frameworks with n-tuple anchoring sites
In an attempt to tailor plasmonic permutations, we used super-origami as templates (see fig. S1) for anchoring L-AuNPs on prescribed n-tuple docking sites. In mathematics, an n-tuple is an ordered list of n elements. First, we constructed three kinds of super-origami templates with capture strands at special positions (Fig. 1A and fig. S2A). These rhombus- and trapezoid-shaped super-origami nanostructures were well formed (Fig. 1B), and the counted yields were 87.9 and 86.5%, respectively (see fig. S2B). Next, a set of L-AuNPs with two different diameters, S = {x, y} (50- and 80-nm L-AuNPs are as x and y, respectively), was anchored site-specifically on purified super-origami templates via DNA hybridization. By choosing unique anchoring strands, the rhombus template with inner edge anchors (RI) could be used for building 2-tuples over the set S, resulting in total permutations of 22 = 4 (Fig. 1C). Similarly, 3-tuples with the permutations of 23 = 8 (Fig. 1D) and 4-tuples with the permutations of 24 = 16 (Fig. 1E) were built from the trapezoid template with inner edge anchors (TI) and the rhombus template with outer corner anchors (RO), respectively. Scanning electron microscopy (SEM) images showed that L-AuNPs were quantitatively anchored on super-origami templates, with geometry covering all the permutations of n-tuples. We note that, due to the symmetry of the structure and the randomness of adsorption to the glass substrate, several n-tuple structures appear to be similar under SEM characterization. Agarose gel electrophoresis revealed that the target metamolecules appeared as distinct bands compared with free L-AuNPs and by-products, enabling high-yield isolation (fig. S3A). The yield of 80-nm L-AuNP dimer tuple (y, y) was as high as 86% (fig. S3B), while for other metamolecules such as heterostructures [e.g., tuple (x, y)] and tetrameric structures [e.g., tuple (y, y, y, y)], the yields were both higher than 60% (see fig. S4). The high-yield formation of tailored L-AuNP plasmonic permutations arises from several reasons. First, DNA origami in nature is a perfect structure to realize nanometer precision. Second, super-origami templates provide larger platforms to accommodate L-AuNPs. Third, abundant capture strands immobilized on an appropriately sized area (12 for outer corner and 18 for inner edge) enable cooperative binding of an L-AuNP to the correct position.
Fig. 1. Design principle and SEM characterization of super-origami DNA nanostructures with n-tuples.
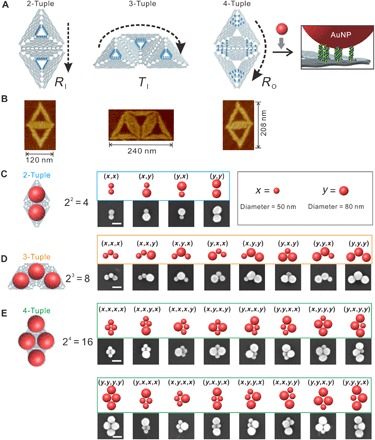
(A) Oligomeric super-origami templates for the construction of AuNP n-tuples. Arrows indicate the directions. (B) Atomic force microscope (AFM) characterization of DNA super-origami. (C to E) SEM characterizations of AuNPs n-tuples. Scale bars, 100 nm.
Plasmonics of tetrameric Fano metamolecules
Next, we used an 80-nm AuNP tetrameric cluster [tuple (y, y, y, y)] to study the structure-correlated optical and plasmonic properties of individual tetramers (Fig. 2A). Previous studies established that L-AuNPs exhibit intense absorption and scattering cross sections (32). Figure 2B displays the finite-difference time-domain (FDTD) calculations for estimating both the size of the hot spot region and the |E|4 enhancement. The electric field in the hot spot region (the center green box) was calculated to be 90 times stronger than the field of the incident light. We then immobilized these tetrameric metamolecules on an indium tin oxide (ITO) glass substrate, the tetrameric morphology of which was confirmed with SEM (Fig. 2C). To correlate individual metamolecule morphology with its optical and plasmonic properties, we developed a colocalization method by using a laser-etched nine-square grid as the reference on glass (see fig. S5). The scattering and Raman spectra of the metamolecule were then characterized by polarization-dependent dark-field microscopy (DFM) (Fig. 2D) and Raman spectroscopy (Fig. 2E).
Fig. 2. Correlative SEM, DFM, and Raman characterization of tetrameric metamolecules.
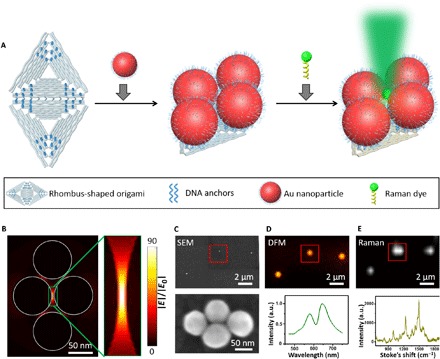
(A) Schematic illustration of the fabrication procedure. L-AuNPs (80 nm) and dyes could be immobilized site-specifically on a rhombus-shaped super-origami through DNA hybridizations. (B) FDTD calculations for an 80-nm L-AuNP tetramer cluster. A hot spot is present in the green box. (C to E) Correlative SEM characterization (C), DFM characterization (D), and Raman mapping (E) of an 80-nm L-AuNP tetramer cluster. Six ROX (carboxy-X-rhodamine) molecules were placed in the hot spot shown in (B). a.u., arbitrary units.
Next, the plasmonic properties of individual tetramers were characterized by SEM-DFM correlative imaging. The metamolecules were immobilized on an ITO glass substrate in air and imaged with an inverted DFM (Fig. 3A). As shown in Fig. 3B, a selected area with two well-formed tetramers was colocalized with SEM and DFM. The polarization-dependent scattering spectra with incident light polarized from 0° to 90° were collected on one identified structure (Fig. 3C). Apparently, at 0° orientation, a single peak was observed from the spectrum. By gradually changing the incident light orientation to 45°, a narrow dip emerged near 645 nm. This dip became pronounced at 60° and reached its minimized value at 90°. This narrow and asymmetric dip near 645 nm was an evidence of a typical Fano resonance, which arises from interference between a superradiant “bright” mode and a subradiant “dark” mode (28, 33). A similar trend in incident light orientation–dependent spectra evolution was obtained from calculations using the finite element simulation software COMSOL (Fig. 3D).
Fig. 3. DFM-SEM correlative characterization of the plasmonic properties of a tetrameric metamolecule.
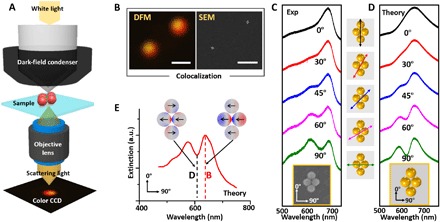
(A) Schematic of the DFM setup for measuring the scattering spectra of a single 80-nm L-AuNP tetrameric metamolecule. (B) Colocalized DFM and SEM images. Scale bars, 1 μm. (C and D) SEM image and scattering spectra (the experimental and theoretic) of the tetrameric metamolecule at different polarization angles of incident light. The orientation angles of the incident light relative to the cluster are shown in the middle column. (E) Theoretic extinction spectrum and surface charge distribution plot of the tetrameric metamolecule when the polarization angle of the incident light was 90°.
The simulation of the surface charge distributions provided insights into the Fano metamolecule’s bright and dark modes (Fig. 3E). The charge density plot of the bright mode at its peak of 650 nm showed that the charge distributions in each L-AuNP were oriented in the same direction, resulting in strong scattering due to the constructive interference of their radiated fields. The charge density plot at the dark mode peak frequency at 615 nm showed that the charge distributions in individual L-AuNPs were oriented in different directions, indicating that the bright mode is suppressed and energy is stored in the dark mode. We note that the experimental and calculated Fano minimums differed slightly, which were possibly caused by the DNA coating and origami substrate (34).
To further establish the direct relationship between the observed DMFR and the tetrameric metamolecules, we examined the scattering spectra of several other types of metamolecules: a homodimer [tuple (y, y)], a heterodimer [tuple (x, y)], a homotrimer [tuple (y, y, y)], and a homotetramer [tuple (x, x, x, x)]. Experimental and calculated spectra matched very well for these metamolecules, confirming the validity of tailoring their structure-correlated plasmonic properties (see figs. S6 and S7). For example, simulations on the electromagnetic radiation revealed that the homodimer had a transverse mode and a longitudinal mode, which was in coincidence with the experimental data (see fig. S6). By increasing incident light polarization from 0° to 90°, we found that the transverse mode weakened gradually, while the longitudinal mode increased. Nevertheless, these spectra containing a bright electric dipole peak did not exhibit Fano minima that strongly overlap with these modes. The scattering intensities of tetramers and trimers of different sizes were also compared (see fig. S8), which were consistent with the theoretical predictions that the scattering signal of the 50-nm AuNP metamolecules was much weaker.
Fano-like resonances support single-molecule SERS
Having substantiated DMFR from the tetrameric metamolecule, we next explored the potential of using metamolecules for SERS analysis. First, DNA minor groove–binding dye SYBR Green I was used to study the structure-correlated Raman properties (Fig. 4A) (35). A wavelength incident light of 633 nm was selected because it sits in the Fano minimum (see fig. S9). We expect that the metamolecule emits minimal light in this region, which shows minimal energy depletion of the plasmon oscillations (31). By intercalating SYBR Green I on surface-bound DNA on L-AuNPs and DNA origami template, we measured the Raman enhancement from tetrameric metamolecules with the SEM-Raman colocalization method (Fig. 4B). To illustrate the role of DMFR in Raman enhancement, we deliberately selected an area with two tetramers: one well-formed symmetric tetramer (i) and the other distorted asymmetric tetramer (ii) (Fig. 4C). We observed strong distinctions in brightness and color for these two different metamolecules from the real-color photograph (Fig. 4B). We performed FDTD calculations on the electric field of tetramer (i) and tetramer (ii); the strongest SERS enhancement was expected from molecules that were located precisely at the hot spot. FDTD simulations revealed that the integrity of the symmetric electric field in the Fano metamolecule (i) was broken in the distorted metamolecule (ii) (Fig. 4D). Nonpolarized scattering spectra of the two metamolecules measured by DFM (Fig. 4E) showed an obvious Fano minimum at ~633 nm for tetramer (i), but not for tetramer (ii). The corresponding SERS spectra further established that tetramer (i) exhibited pronounced Raman fingerprint bands at 1239, 1340, and 1452 cm−1 (Fig. 4F). Thus, we establish that the Fano-like resonances in tetramer (i) result in high electric field enhancement for SERS.
Fig. 4. Characterization and SERS spectra of tetrameric metamolecules.
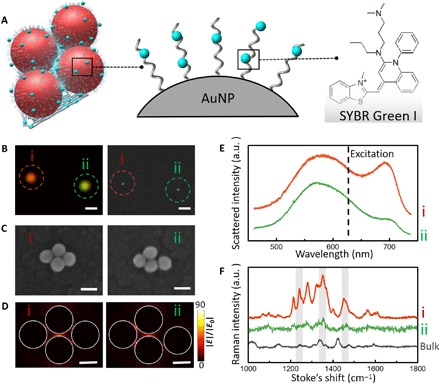
(A) Schematic of the tetrameric metamolecule that is incorporated with Raman dye. (B) Real-color photograph and the corresponding SEM images of the two individual tetramers (i and ii). Scale bars, 1 μm. (C) High-magnification SEM images reveal the difference between two tetramers. Scale bars, 100 nm. (D) FDTD calculations for two tetramer clusters. Scale bars, 50 nm. (E) Nonpolarized experimentally scattering spectra of the two individual tetramers. (F) Raman spectra of individual tetramers with intercalated SYBR Green I molecules (spectra i and ii) and the highly concentrated bulk solution (black curve) of SYBR Green I. All measurements were performed with a 633-nm excitation laser (10-s exposure).
Having established the association of DMFR with SERS, we further quantitatively studied metamolecules at the single-molecule level. A ROX (carboxy-X-rhodamine) molecule was selected as the Raman dye, which is non-intercalative with DNA strands. ROX molecules with a certain number were deliberately anchored in the hot spot region of the tetrameric cluster (Fig. 5A). By hybridizing ROX-tagged DNA strands on the corresponding sites of the rhombus-shaped DNA origami, we could precisely control the number and position of ROX. By comparing the molecule size of ROX with the size of the hot spot of the Fano tetramer, we expected that up to six ROX molecules could be accommodated (Fig. 5A). By exploiting the nanoaddressability of DNA origami, we prescribed fixed numbers of ROX in the hot spot region (Fig. 5B). We found that the SERS intensity quantitatively increased along with the number of ROX but saturated when the number reached six (Fig. 5, C and D, and fig. S10), characteristic of a quantized phenomenon. Notably, the Raman signal of even a single ROX was readily detectable. We next calculated enhancement factor (EF) values for individual ROX embedded in the metamolecules (Fig. 5E). The EF values were generally on the order of 108 for metamolecules with 1 to 12 ROX molecules. A single ROX leads to the broadest distribution of EF values, which suggested that the position variation of ROX arises from the local random perturbations of the DNA strands.
Fig. 5. Quantized single-molecule SERS.
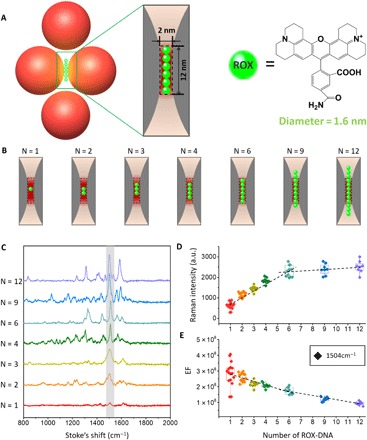
(A) Schematic of the tetrameric metamolecules with accurate number of Raman dye ROX molecules in the hot spot. The diameter of ROX is ~1.6 nm, while the diameter of double-stranded DNA is 2 nm. (B) Schematic of the hot spot region with different numbers of ROX (N = 1, 2, 3, 4, 6, 9, 12). According to the calculated size of hot spot and the diameter of the ROX, six ROX can fill in the hot spot region. (C) SERS spectra taken from seven individual tetramers with different numbers of ROX. (D) Quantized SERS responses as measured by the intensity plot at 1504 cm−1 along with the increase of the number of ROX per particle (N = 12, red, 1 ROX; N = 14, orange, 2 ROX; N = 9, claybank, 3 ROX; N = 9, green, 4 ROX; N = 11, light blue, 6 ROX; N = 8, dark blue, 9 ROX; N = 8, purple, 12 ROX). (E) Measured EFs at 1504 cm−1. All measurements for EF calculations were performed with a 633-nm excitation laser (10-s exposure).
DISCUSSION
In summary, we demonstrate the potential of super-origami DNA frameworks as a general method for fabricating plasmonic nanostructures. The successful construction of metamolecules with DMFR allows quantitative analysis of Raman enhancement localized in the hot spot, which provides direct physical evidence for the single-molecule SERS. Thus, these super-origami–templated metamolecules with strong plasmonic enhancement provide an ideal platform for single-molecule biophysical studies and ultrasensitive sensing. Given the flexibility in the origami construction and designable affinity for various targets, we envision that this strategy could be extended to versatile applications in nanoelectronics, nanophotonics, and biosensing.
MATERIALS AND METHODS
Materials
All unmodified staple strands were purchased from Invitrogen (China) and used as received. All thiol-functionalized DNA strands and ROX-functionalized DNA strands were purchased from TAKARA (Dalian, China) and used as received. M13mp18 single-stranded DNA was purchased from New England Biolabs. Chemicals were purchased from Sinopharm and Sigma-Aldrich. Colloidal solutions of 50- and 80-nm AuNPs were purchased from BBI Solutions.
Preparation of DNA-functionalized L-AuNPs
Before use, colloidal solutions of 50- and 80-nm AuNPs were subjected to centrifugation to concentrate 10 times (50-nm AuNPs, 7000 rpm for 10 min; 80-nm AuNPs, 4000 rpm for 10 min). Concentrated colloidal solution (800 μl) of 80-nm AuNPs was mixed with freshly dissolved thiol-modified DNA (100 μM) in a 1:50,000 ratio (1:10,000 for 50-nm AuNPs) in Milli-Q water, and the mixture was incubated for 2 hours at room temperature (300 rpm). Then, 100 μl of phosphate buffer (PB) [100 mM (pH 7.4)] was added to the mixture. After 30 min, we added 10 μl of NaCl solution (2 M) every 20 min for four times and then 20 μl of NaCl solution (2 M) every 30 min for three times. The NaCl concentration was gradually increased to ensure the full coverage of L-AuNPs with thiolated DNA. The final concentration of NaCl was 200 mM, and the mixture was incubated at room temperature (300 rpm) overnight. The AuNP-DNA conjugates were purified by 0.5% agarose gel electrophoresis [running buffer, 0.5× tris-borate-EDTA (TBE); loading buffer, 50% sucrose; 1 hour at a constant 100 V]. Desired bands were cut out, and thiolated DNA–modified AuNP clusters were extracted from the gel using a protocol given by Bellot et al. (36). Freshly prepared, fully covered 50- and 80-nm AuNPs did not precipitate in the 0.6× TAE-Mg2+ buffer [24 mM tris, 12 mM acetic acid, 1.2 mM EDTA, and 7.5 mM magnesium acetate (pH 8.0)]. This high-salt resistance property of fully covered L-AuNPs makes it possible to assemble metamolecules on a DNA origami template.
Formation of DNA super-origami structures
As mentioned above, we first assembled the simple triangle DNA origami monomers named triangle A and triangle C, respectively. Each kind of simple triangle DNA origami monomer was prepared according to Liu et al. (20). A molar ratio of 1:10 between the long M13 scaffold and each required staple strand (“extended staples,” selected “anchoring staples,” and the rest staple strands) was used, and DNA origami was assembled in 1× TAE-Mg2+ buffer [40 mM tris, 20 mM acetic acid, 2 mM EDTA, and 12.5 mM magnesium acetate (pH 8.0)] by the reported annealing program (95°C for 3 min, 95° to 15°C, 0.1°C/10 s) (21). The triangle DNA origami monomers were subsequently purified four times with Microcon centrifugal filtration devices (100-kDa molecular weight cutoff filters, Millipore) to remove the excess staple strands. The concentration of each purified DNA origami monomer was estimated from the optical absorbance at 260 nm. Then, the two kinds of purified DNA origami triangular monomers were mixed in a 1:1 ratio and annealed from 45° to 15°C at a speed of 0.1°C/min. The trapezoid-shaped super-origami was prepared in the same way.
Self-assembly of metamolecule using super-origami templates
The DNA-modified AuNP solution was added to DNA origami solution (purified) in 0.6× TAE-Mg2+ buffer with a ratio of 2:1 for the 80-nm AuNP dimer [tuple (y, y)], 3:1 for the 80-nm AuNP trimer [tuple (y, y, y)], and 4:1 for the 80-nm AuNP tetramer [tuple (y, y, y, y)]. The mixtures were annealed from 45° to 15°C at a speed of 0.1°C/min to promote hybridization of the DNA on the L-AuNPs with the complementary capture strands on the DNA origami. Successful formation of desired structures (28 permutations of n-tuples) and separation from other by-products are accomplished by gel electrophoresis in a 0.5% agarose gel run at 100 V and maintained at 4°C by a surrounding ice bath. Desired bands were cut out, and DNA origami–templated AuNP clusters were extracted from the gel using a protocol given by Bellot et al. (36).
Characterization of the plasmonic properties of metamolecules using SEM-DFM correlative imaging
The ITO glasses with a special marker on the surface were immersed in the piranha solution (H2SO4:H2O2 = 3:1) for 5 min followed by rinsing with copious amounts of ultrapure water. The cleaned ITO glasses were dried by N2 and then treated with oxygen plasma to make the surface hydrophilic [Harrick Plasma PDC-32G cleaner for 1 min at high radio frequency (RF) level]. The purified metamolecule sample (10 μl) was left to adsorb on the surface of ITO glass for 10 min. Then, the ITO glasses were washed with ultrapure water and dried with N2 immediately.
The sample was first scanned using SEM (LEO 1530 VP, Zeiss). We found the desired tetrameric metamolecule near the laser marker under SEM image. Then, we relocated this desired tetrameric metamolecule under the DFM. We use an inverted microscope (Olympus IX71, Japan) equipped with a dark-field condenser [0.8 < numerical aperture (NA) < 0.95] and a 60× or 40× objective lens (NA = 0.8). The sample slides were immobilized on a platform, and a 100-W halogen lamp provided a white light source to excite the tetrameric metamolecule to generate plasmon resonance scattering light. The scattered light was collected with a true-color digital camera (Olympus DP70, Japan) to generate the dark-field color image and was also split with a monochromator (Acton SP2300i, Princeton Instruments, USA), which was equipped with a grating (grating density, 300 lines/mm; blazed wavelength, 500 nm) and recorded with a spectrograph charge-coupled device (CCD) (Cascade 512B, Roper Scientific, Princeton Instruments, USA) to obtain the scattering spectra. The scattering spectra were integrated as 10 s for the 80-nm AuNP tetramer [tuple (y, y, y, y)]; 20 s for homotrimer [tuple (y, y, y)], heterodimer [tuple (x, y)], and homodimer [tuple (y, y)]; and 30 s for the 50-nm AuNP trimer [tuple (x, x, x)]. The spectrum of an individual tetrameric metamolecule was corrected by subtracting the background spectrum taken from the adjacent regions without the metamolecule.
Raman measurements and calculation of the EFs
We acquired the single-point Raman spectrum with an XPLORA (HORIBA, Jobin Yvon, France) Raman microscope system. Raman mapping was performed in air using the 633-nm laser and a 60× S Plan Fluor air objective (NA, 0.7; Nikon). The laser power after the objective was measured to be 10.7 mW, and the laser power we used was reduced to 50% to protect the Raman dye. Raman mapping was performed at a step of 1.5 μm × 1.5 μm, and the acquisition time was 10 s per point. To obtain the pure Raman signal, the combined background of Au and SYBR Green I was subtracted from the spectrum.
We performed the reference Raman measurements of SYBR Green I in bulk solution at 2000× concentration. The corresponding concentration of commercial SYBR Green I was assumed to be 3.92 mM according to Zipper et al. (35). This dye can bind to DNA strands nonspecifically and can therefore randomly distribute on the whole surface of the tetrameric metamolecule (37). The SERS EF was calculated by comparing the signals measured from a single tetrameric metamolecule with the intensity of the Raman signal from the bulk solution. The EF was estimated from the excitation power P, the Raman intensity I, and the amount of molecules N contributing to the signal for the bulk SYBR Green I solution and for the tetrameric metamolecules, respectively
ISERS is the peak intensity of SYBR Green I at 1340 cm−1 in the tetrameric metamolecules, and Ibulk is the peak intensity of SYBR Green I in the solution. Pbulk is the same with PSERS. Nbulk is the estimated number of SYBR Green I molecules in the volume of the laser spot in the bulk measurement (~1.8 × 107). It has been reported that the DNA molecule has a 13-nm2 footprint for the 80-nm L-AuNPs (38). Therefore, NSERS was calculated, as the SYBR Green I molecules were absorbed on the surface of the L-AuNPs in the plasmonic hot spot (~6). The size of the plasmonic hot spot was estimated according to FDTD calculation results from fig. S9.
COMSOL simulations
Finite-element method (COMSOL) was used to simulate the scattering spectra of the metamolecule and to map the surface charge density distribution plots. All calculations were performed in an effective embedding medium with a dielectric constant of ε = 2.4025 (or n = 1.55) (39) to match the experiment. The optical constants of gold were taken from Johnson and Christy (40). S-polarized incident light with oblique incident angle of 30° to the normal of the substrate plane was chosen strictly compared with the experimental results. The cluster geometries used in the simulations were based on the cluster SEM images, from which the gold nanoparticle dimensions could be measured with high precision. However, the interparticle gaps were difficult to resolve because SEM yields projection images; if there is any offset of the nanoparticle in the vertical direction, the transmission electron microscopy (TEM) image cluster appears fused. To fit the calculated spectra to experiment, we simulated clusters with different interparticle gaps. We found that the 3-nm gap gives good agreement between theory and experiment for the 80-nm AuNP homodimer [tuple (y, y)] structure, and the 2-nm gap for the tetrameric metamolecule [tuple (y, y, y, y)].
FDTD simulations and data processing
FDTD simulations of tetrameric metamolecules (particle diameter, 80 nm; gap size, 2 nm) were carried out using Lumerical FDTD solutions (Lumerical Solutions, Canada). Optical constant data for gold were taken from Johnson and Christy (40). Linear polarized light was injected with a total-field/scattered-field source. Convergence was reached for a mesh size of 0.4 nm around the plasmonic particles. The follow-up data processing is constructed using MATLAB.
Supplementary Material
Acknowledgments
Funding: We appreciate financial support from the National Key R&D Program of China (2016YFA0201200), the National Science Foundation of China (21675167, 21722310, 21834007, and 21873071), the Key Research Program of Frontier Sciences (QYZDJ-SSW-SLH031), the Open Large Infrastructure Research of CAS, the LU Jiaxi International Team of the Chinese Academy of Sciences, and K. C. Wong Foundation of Shanghai Jiao Tong University. Author contributions: C.F. and H.L. conceived the project. W.F. and S.J. designed and performed the experiments and contributed to the simulation. J.C., Liqian Wang, Q.L., X.Z., Lihua Wang, and Lianhui Wang contributed to characterizations and data analysis. X.D. and N.L. contributed to the simulation. W.F., H.L., and C.F. wrote the manuscript. All authors read and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaau4506/DC1
Fig. S1. Schematic drawings of DNA origami template.
Fig. S2. Super-origami templates.
Fig. S3. Characterization of 80-80 nm AuNP metamolecules.
Fig. S4. SEM images of 50-80 nm and 80-80-80-80 nm AuNP metamolecules.
Fig. S5. Schematic representation of SEM-DFM-Raman correlative imaging for plasmonic property investigations.
Fig. S6. Characterization of the plasmonic properties of 80-nm homodimer [tuple (y, y)] using the “DFM-SEM correlative imaging.”
Fig. S7. Characterization of the plasmonic properties of three metamolecules using the “DFM-SEM correlative imaging.”
Fig. S8. The absolute scattering spectra of two homotetramers and two homotrimers.
Fig. S9. FDTD calculations of the electromagnetic field (E) at mid-height of the tetrameric metamolecules.
Fig. S10. SERS spectra taken from individual tetrameric metamolecules with different numbers of ROX.
Supplementary Appendix
REFERENCES AND NOTES
- 1.Barnes W. L., Dereux A., Ebbesen T. W., Surface plasmon subwavelength optics. Nature 424, 824–830 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Noginov M. A., Zhu G., Belgrave A. M., Bakker R., Shalaev V. M., Narimanov E. E., Stout S., Herz E., Suteewong T., Wiesner U., Demonstration of a spaser-based nanolaser. Nature 460, 1110–1112 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Smith D. R., Pendry J. B., Wiltshire M. C., Metamaterials and negative refractive index. Science 305, 788–792 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Engheta N., Circuits with light at nanoscales: Optical nanocircuits inspired by metamaterials. Science 317, 1698–1702 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Liu N., Tang M. L., Hentschel M., Giessen H., Alivisatos A. P., Nanoantenna-enhanced gas sensing in a single tailored nanofocus. Nat. Mater. 10, 631–636 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Schuller J. A., Barnard E. S., Cai W., Jun Y. C., White J. S., Brongersma M. L., Plasmonics for extreme light concentration and manipulation. Nat. Mater. 9, 193–204 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Roller E.-M., Besteiro L. V., Pupp C., Khorashad L. K., Govorov A. O., Liedl T., Hot spot-mediated non-dissipative and ultrafast plasmon passage. Nat. Phys. 13, 761–765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kneipp K., Wang Y., Kneipp H., Perelman L. T., Itzkan I., Dasari R., Feld M. S., Single molecule detection using surface-enhanced Raman scattering (SERS). Phys. Rev. Lett. 78, 1667–1670 (1997). [Google Scholar]
- 9.Nie S., Emory S. R., Probing single molecules and single nanoparticles by surface-enhanced Raman scattering. Science 275, 1102–1106 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Lim D. K., Jeon K. S., Kim H. M., Nam J. M., Suh Y. D., Nanogap-engineerable Raman-active nanodumbbells for single-molecule detection. Nat. Mater. 9, 60–67 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Lim D. K., Jeon K. S., Hwang J. H., Kim H., Kwon S., Suh Y. D., Nam J. M., Highly uniform and reproducible surface-enhanced Raman scattering from DNA-tailorable nanoparticles with 1-nm interior gap. Nat. Nanotechnol. 6, 452–460 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Lassiter J. B., Sobhani H., Fan J. A., Kundu J., Capasso F., Nordlander P., Halas N. J., Fano resonances in plasmonic nanoclusters: Geometrical and chemical tunability. Nano Lett. 10, 3184–3189 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Pilo-Pais M., Watson A., Demers S., LaBean T. H., Finkelstein G., Surface-enhanced Raman scattering plasmonic enhancement using DNA origami-based complex metallic nanostructures. Nano Lett. 14, 2099–2104 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Hentschel M., Saliba M., Vogelgesang R., Giessen H., Alivisatos A. P., Liu N., Transition from isolated to collective modes in plasmonic oligomers. Nano Lett. 10, 2721–2726 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Duan H., Fernández-Domínguez A. I., Bosman M., Maier S. A., Yang J. K., Nanoplasmonics: Classical down to the nanometer scale. Nano Lett. 12, 1683–1689 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Sharma J., Chhabra R., Cheng A., Brownell J., Liu Y., Yan H., Control of self-assembly of DNA tubules through integration of gold nanoparticles. Science 323, 112–116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen X., Song C., Wang J., Shi D., Wang Z., Liu N., Ding B., Rolling up gold nanoparticle-dressed DNA origami into three-dimensional plasmonic chiral nanostructures. J. Am. Chem. Soc. 134, 146–149 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Jones M. R., Seeman N. C., Mirkin C. A., Programmable materials and the nature of the DNA bond. Science 347, 1260901 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Rothemund P. W., Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Liu W., Halverson J., Tian Y., Tkachenko A. V., Gang O., Self-organized architectures from assorted DNA-framed nanoparticles. Nat. Chem. 8, 867–873 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Zhang H., Chao J., Pan D., Liu H., Qiang Y., Liu K., Cui C., Chen J., Huang Q., Hu J., Wang L., Huang W., Shi Y., Fan C., DNA origami-based shape IDs for single-molecule nanomechanical genotyping. Nat. Commun. 8, 14738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X., Zhang F., Jing X., Pan M., Liu P., Li W., Zhu B., Li J., Chen H., Wang L., Lin J., Liu Y., Zhao D., Yan H., Fan C., Complex silica composite nanomaterials templated with DNA origami. Nature 559, 593–598 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Acuna G. P., Möller F. M., Holzmeister P., Beater S., Lalkens B., Tinnefeld P., Fluorescence enhancement at docking sites of DNA-directed self-assembled nanoantennas. Science 338, 506–510 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Prinz J., Heck C., Ellerik L., Merk V., Bald I., DNA origami based Au-Ag-core-shell nanoparticle dimers with single-molecule SERS sensitivity. Nanoscale 8, 5612–5620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simoncelli S., Roller E.-M., Urban P., Schreiber R., Turberfield A. J., Liedl T., Lohmüller T., Quantitative single-molecule surface-enhanced Raman scattering by optothermal tuning of DNA origami-assembled plasmonic nanoantennas. ACS Nano 10, 9809–9815 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Tanwar S., Haldar K. K., Sen T., DNA origami directed Au nanostar dimers for single-molecule surface-enhanced Raman scattering. J. Am. Chem. Soc. 139, 17639–17648 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Zhan P., Wen T., Wang Z. G., He Y., Shi J., Wang T., Liu X., Lu G., Ding B., DNA origami directed assembly of gold bowtie nanoantennas for single-molecule surface-enhanced Raman scattering. Angew. Chem. Int. Ed. 57, 2846–2850 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Fan J. A., Bao K., Wu C., Bao J., Bardhan R., Halas N. J., Manoharan V. N., Shvets G., Nordlander P., Capasso F., Fano-like interference in self-assembled plasmonic quadrumer clusters. Nano Lett. 10, 4680–4685 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Ye J., Wen F., Sobhani H., Lassiter J. B., Van Dorpe P., Nordlander P., Halas N. J., Plasmonic nanoclusters: Near field properties of the Fano resonance interrogated with SERS. Nano Lett. 12, 1660–1667 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Zhen Y. R., Neumann O., Day J. K., Nordlander P., Halas N. J., Coherent anti-stokes Raman scattering with single-molecule sensitivity using a plasmonic Fano resonance. Nat. Commun. 5, 4424 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Stockman M. I., Dark-hot resonances. Nature 467, 541–542 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Vietz C., Lalkens B., Acuna G. P., Tinnefeld P., Functionalizing large nanoparticles for small gaps in dimer nanoantennas. New J. Phys. 18, 045012 (2016). [Google Scholar]
- 33.Fan J. A., Wu C., Bao K., Bao J., Bardhan R., Halas N. J., Manoharan V. N., Nordlander P., Shvets G., Capasso F., Self-assembled plasmonic nanoparticle clusters. Science 328, 1135–1138 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Thacker V. V., Herrmann L. O., Sigle D. O., Zhang T., Liedl T., Baumberg J. J., Keyser U. F., DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commun. 5, 3448 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Zipper H., Brunner H., Bernhagen J., Vitzthum F., Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucleic Acids Res. 32, e103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellot G., McClintock M. A., Lin C., Shih W. M., Recovery of intact DNA nanostructures after agarose gel-based separation. Nat. Methods 8, 192–194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhler P., Roller E.-M., Schreiber R., Liedl T., Lohmüller T., Feldmann J., Plasmonic DNA-origami nanoantennas for surface-enhanced Raman spectroscopy. Nano Lett. 14, 2914–2919 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Hurst S. J., Lytton-Jean A. K. R., Mirkin C. A., Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 78, 8313–8318 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inagaki T., Hamm R. N., Arakawa E. T., Painter L. R., Optical and dielectric properties of DNA in the extreme ultraviolet. J. Chem. Phys. 61, 4246–4250 (1974). [Google Scholar]
- 40.Johnson P. B., Christy R. W., Optical constants of the noble metals. Phys. Rev. B 6, 4370–4379 (1972). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaau4506/DC1
Fig. S1. Schematic drawings of DNA origami template.
Fig. S2. Super-origami templates.
Fig. S3. Characterization of 80-80 nm AuNP metamolecules.
Fig. S4. SEM images of 50-80 nm and 80-80-80-80 nm AuNP metamolecules.
Fig. S5. Schematic representation of SEM-DFM-Raman correlative imaging for plasmonic property investigations.
Fig. S6. Characterization of the plasmonic properties of 80-nm homodimer [tuple (y, y)] using the “DFM-SEM correlative imaging.”
Fig. S7. Characterization of the plasmonic properties of three metamolecules using the “DFM-SEM correlative imaging.”
Fig. S8. The absolute scattering spectra of two homotetramers and two homotrimers.
Fig. S9. FDTD calculations of the electromagnetic field (E) at mid-height of the tetrameric metamolecules.
Fig. S10. SERS spectra taken from individual tetrameric metamolecules with different numbers of ROX.
Supplementary Appendix