

Abstract
Exposure of cells to a dose of ionizing radiation as low as 5 mGy can induce changes in radiation sensitivity expressed by cells exposed to subsequent higher doses at later times. This is referred to as an adaptive effect. We describe a unique survivin-associated adaptive response in which increased radiation resistance or sensitization of cells can be induced by exposure to 5 mGy or to the reactive oxygen species (ROS) generating drug Emodin (1,3,8-trihydroxy-6-methylanthraquinone), a naturally occurring anthraquinone. The purpose of this study was to determine the role of ROS generating processes in affecting both the intracellular localization of the inhibitor of apoptosis protein survivin and its subsequent effect on radiation response in the presence or absence of the anti-oxidant N-acetyl-L-cysteine (NAC). Experiments were performed using two well characterized murine sarcomas: SA-NH p53 wild-type (WT) and FSa p53 mutant (Mut), grown either in culture or as solid tumors in the right hind legs of C3H mice. Doses of 5 mGy or 50 μM Emodin were used to induce changes in the response of these tumor cells to higher radiation exposures using a multi-dosing paradigm. Effects on radiation sensitivity were determined for SA-NH and FSa cells as a function of survivin translocation either to the cytoplasm or nucleus in the presence or absence of 10 mM NAC treatment. In vitro survival assays (2 Gy per fraction, two once daily fractions) and tumor growth delay (TGD) (5 Gy per fraction, five once daily fractions) studies were performed. Intracellular localization of survivin was determined by enzyme-linked immunosorbent assay (ELISA) and correlated to survival response and treatment conditions. 2 Gy alone had no effect on intracellular translocation of survivin. When preceded 15 min earlier by 5 mGy or Emodin exposures, survivin became elevated in the cytoplasm of p53 WT SA-NH as compared to the nuclei of p53 Mut FSa cells. SA-NH cells transfected with p53 small interfering RNA (siRNA), in contrast, responded similarly to p53 Mut FSa cells by becoming more radiation sensitive if exposed to 5 mGy prior to each 2 Gy irradiation. In contrast to their respective responses to five once daily 5 Gy fractions, SA-NH tumors were protected by 5 mGy exposures administered 15 min prior to each daily 5 Gy dose as evidenced by a more rapid growth (1.9 day decrease in TGD, P=0.032), while FSa tumors were sensitized, growing at a much slower rate (4.5 day increase in TGD, P < 0.001). Exposure of SA-NH and FSa tumor cells to 10 mM NAC inhibited the ability of 5 mGy and Emodin to induce intracellular translocation of survivin and the corresponding altered adaptive survival response. The survivin-associated adaptive response can be induced following a multi-dosing scheme in which very low radiation doses are followed shortly thereafter by higher doses consistent with a standard image guided radiotherapy protocol that is currently widely used in the treatment of cancer. While induced by exposure to ROS generating stresses, the ultimate expression of changes in radiation response is dependent upon the bi-functionality of the tumor associated protein survivin and its intracellular translocation.
Keywords: Survivin, Emodin, Adaptive response, Reactive oxygen species, p53, Radiation response
1. Introduction
Exposure of cells to a very low radiation dose of 5 mGy prior to irradiation with higher doses can result in an altered radiation response that is generally characterized by an increase over the expected level of survival. Originally described as occurring in human lymphocytes [1], this phenomenon has been extensively investigated in a wide range of both normal and malignant cell systems [2–5]. With the development and implementation of ionizing radiation-based imaging such as computerized axial tomography (CAT) for routine use in image guided radiotherapy (IGRT), the potential exists that induction of these responses in malignant cells by very low imaging level doses may affect treatment outcomes [6]. Recently we reported on a novel adaptive response mediated by the inhibitor of apoptosis protein survivin that appears to be induced only following a multi-fraction irradiation scheme [7]. The focus of that study was in assessing the role of elevated survivin levels induced by exposure to doses ranging from 5 to 100 mGy on enhancing the radiation resistance of p53 wild-type (WT) murine and human tumor cells to exposures of 2 Gy. Survivin is found primarily in malignant cells and is a bifunctional protein. It can exhibit different properties depending upon its intracellular location. If located in the cytoplasm, survivin can enhance cellular resistance to stress-inducing agents and increase survival through its ability to inhibit apoptosis as was demonstrated in p53 WT RKO36 human colon carcinoma cells, SA-NH murine sarcoma cells and transformed mouse embryo fibroblast cells [7]. Nuclear localization of survivin, in contrast, is not cytoprotective but rather can enhance apoptosis and is associated with regulation of cellular proliferation and division [8–11]. Transport of survivin from the nucleus to the cytoplasm is highly regulated and occurs through the nuclear pore complex. This is facilitated through the association of survivin with the redox sensitive chromosome region maintenance 1 nuclear export protein (CRM-1, exportin 1 or XPO1) and the Ras-related nuclear guanosine 5′-triphosphate binding protein (RanGTP) complex [11–13]. Its transport into the nucleus from the cytoplasm, however, is not through an active transport process requiring a nuclear import protein complex, but rather is by simple diffusion [14–16]. Nuclear survivin, in turn, has been reported to be susceptible to accelerated proteosomal degradation resulting in a loss of its cytoprotective properties [17].
In this study we expand our initial observations regarding the survivin-associated adaptive response to investigate the effects of exposure of murine p53 mutant (Mut) or p53 WT tumor cells to a very low dose of 5 mGy or to the ROS generating anthraquinone Emodin (1,3,8-trihydroxy-6-methylanthraquinone). Emodin was chosen for study because it is a known redox effector capable of acting as a pro-oxidant and generating ROS. As an effective electron acceptor it can interact intracellularly with molecular oxygen to generate superoxide anion [18]. Emodin is a naturally occurring agent found in the roots and barks of certain plants, molds, lichens, and Chinese herbs [19]. It has been used therapeutically to generate reactive oxygen species (ROS) in several tumor systems and potentiate the anticancer effects of various chemotherapeutic drugs [20,21]. The focus of this investigation is to characterize the effects of two different ROS generating processes on changes in intracellular survivin localization and the accompanying change in radiation response. Both radiation and Emodin protocols will be utilized in a multi-dose treatment scheme. The SA-NH murine tumor model, capable of both in vitro and in vivo growth, has been extensively investigated in our laboratory regarding the role of manganese superoxide dismutase (SOD2) in mediating a classical very low radiation dose-induced radio-adaptive response [4] and was also used to originally identify and characterize survivin’s role in a novel adaptive response expressed in a multi dosing paradigm more reflective of those used in standard radiotherapy treatment protocols [7]. Since the functional loss of wild-type p53 has been associated in a number of different cell lines with not only reduced expression of survivin [15,16,22,23], but also with downstream ROS production and redox regulation [23–25] we have expanded our investigation to include a p53 Mut FSa tumor model (murine fibrosarcoma cells with a substitution of methionine for valine at position 169) that has been extensively studied in our laboratory [26–28] along with SA-NH cells transfected with p53 small interfering RNA (siRNA) [29]. Tumor cells were evaluated under in vitro conditions utilizing a split dose radiation protocol of 2 Gy per fraction separated by 24 h with or without an additional 5 mGy or 50 μM Emodin dose administered 15 min prior in the presence or absence of 10 mM NAC. SA-NH and FSa were also evaluated and compared following a five fraction irradiation scheme of 5 Gy per fraction each separated by 24 h using a tumor growth delay assay.
2. Materials and methods
2.1. Ethics statement
All animal studies, housing and experiments were carried out with The University of Chicago Animal Care and Use Committee (IACUC) approval, ACUP number 70,672. The University of Chicago has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (A3523–01) and conducts its reviews in accordance with United States Public Health Service (USPHS) regulations and applicable federal and local laws.
2.2. Cells and culture conditions
SA-NH cells derived from a spontaneously arisen SA-NH murine sarcoma tumor and FSa cells from a methylcholanthrene-induced C3H female mouse fibrosarcoma and adapted for in vitro growth were supplied by Dr. Luka Milas, the University of Texas MD Anderson Cancer Institute. Both cell lines were routinely checked for mycoplasma contamination and were used at passages 5–10. SA-NH cells were cultured in McCoy’s 5A medium and FSa cells in RPMI 1640 110 (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA), penicillin (100 units/ml) and streptomycin (100 μg/ml) (Invitrogen Life Technologies) as described in detail elsewhere [29]. All cell cultures were maintained at 37 °C in a humidified environment containing 5% CO2. For all cell survival experiments, cells were grown to confluence and maintained in confluence for three days. One day prior to all experiments the cells were refed with fresh medium. Under these culture conditions SA-NH cells had a plating efficiency (PE) of 80%, while FSa cells had a PE of 15%.
2.3. Drugs
N-acetyl-L-cysteine (NAC) and Emodin were obtained from Sigma-Aldrich, St. Louis, MO. NAC was dissolved in phosphate-buffered saline immediately prior to use at a concentration of 100 mM and sterilized using a 0.2 µm syringe filter. Emodin was dissolved in DMSO at a concentration of 100 mM. Cells were treated with 10 mM NAC for 30 min and/or 50 μM Emodin for 15 min.
2.4. Irradiation conditions
For in vitro studies tumor cells were irradiated with very low doses ranging from 5 to 100 mGy 15 min before exposure to a 2 Gy dose and then 24 h later. In vivo irradiations of SA-NH and FSa tumors were performed on tumor-bearing mice that were placed in lead-shielded cylindrical clear-plastic holders. Their unshielded tumor-bearing right hind legs were secured in place with surgical tape during irradiation. All irradiations were performed using a Philips RT250 x-ray generator with a 1 mm copper filter operating at 250 kVp and 15 mA. Dosimetry for the mGy and Gy dose exposures were performed by a board certified member of the Medical Physics faculty in the Department of Radiation and Cellular Oncology at The University of Chicago. Dosimetry procedures involved the use of TLD’s placed at various locations in the radiation field to calculate the dose rate under various exposure conditions. For the 2 Gy in vitro and 5 Gy in vivo exposures, the dose rate was 1.33 Gy/min; for 100 mGy and 20 mGy exposures the dose rate was 0.3 Gy/min; and for 5 mGy exposures, the dose rate was 0.05 Gy/min. Exposures were administered daily for 2 consecutive days for all in vitro studies and 5 consecutive days in all tumor growth delay experiments.
2.5. Cell survival assay
Immediately following each treatment protocol, cell survival was determined using an in vitro colony forming assay. Cells were counted, diluted, and known numbers seeded into 100-mm tissue culture dishes to allow the development of 100–200 colonies per dish. Colonies of SA-NH and FSa cells were fixed and stained 10 and 14 days later, respectively. Each experiment was repeated three times.
2.6. Enzyme-linked immunosorbent assay (ELISA)
Cytoplasmic and nuclear protein was prepared from SA-NH and FSa cells according to a method described in detail elsewhere [30]. Cell pellets were suspended in 400 μl ice-cold Buffer A (10 mM 4-(2-hy-droxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.9; 10 mM potassium chloride [KCl]; 0.1 mM ethylenediaminetetraacetic acid [EDTA]) containing 1 mM dithiothreitol (DTT) (Sigma-Aldrich, St. Louis, MO); 0.5 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich); 1 μg/ml leupeptin (Sigma-Aldrich) and 5 μg/ml aprotinin (Sigma-Al-drich) and allowed to swell on ice for 15 min. Fifty microliters of octylphenoxypolyethoxyethanol (IGEPAL CA-630) (Sigma-Aldrich) was added and the suspension vortexed on high for 30 s. Nuclei were pelleted by centrifugation at 25,000 × g for 1 min at 4 °C and the supernatant containing cytoplasmic protein transferred to a 1.5 ml tube. The nuclei were re-suspended in 50 μl ice-cold Buffer B (20 mM HEPES, pH 7.9; 0.4 M NaCl; 1 mM EDTA; 25% glycerol) containing the same protease inhibitors in Buffer A and incubated on ice for 15 min. The nuclei were pelleted by centrifugation at 25,000 × g for 5 min at 4 °C and the supernatant containing nuclear proteins transferred to a clean 1.5 ml tube. Protein was quantified by the Bradford method [31] and adjusted to 4 μg/μl. Survivin protein levels were determined using the Survivin BioAssay ELISA Kit (Mouse) from US Biological Life Sciences (Salem, MA) according to the manufacturer’s instructions. The averages of duplicate wells for standards and samples were calculated, a standard curve generated, and the concentration of survivin protein in each of the samples determined.
2.7. p53 siRNA transfection
SA-NH cells were grown to confluence and transfected with 100 nanoM p53 or negative control (NC) short interfering RNA (siRNA, OriGene Technologies, Rockville, MD) using Lipofectamine 2000 reagent (Invitrogen Life Technologies) as described elsewhere [10]. The sequence of the p53 siRNA was 5′-AGGAGUCA-CAGUCGGAUAUCAGCCT-3′, which bound to the beginning of the open reading frame in exon 2 of the p53 gene. siRNA oligomer and Lipofectamine 2000 were diluted in serum-free media and incubated for 5 min at room temperature. The siRNA oligomer and Lipofectamine 2000 were then combined and incubated for 20 min. Growth medium was aspirated from the dishes and cells were washed with phosphate-buffered saline (PBS, Invitrogen Life Technologies) at 37 °C. siRNA Lipofectamine 2000 complexes were added to the dishes and incubated for 24 h with cells under their normal growth conditions. The medium was then aspirated, the cells washed with PBS at 37 °C and fresh complete medium at 37 °C added to the dishes.
2.8. Western blotting
Total proteins were prepared from both SA-NH and FSa cells using a method described elsewhere [10]. Cells were washed with cold PBS, harvested on ice, and transferred to a 15 ml tube (total protein) and pelleted at 200 × g for 5 min at 4 °C. The total protein cell pellet was resuspended in 350 μl 50 mM potassium phosphate buffer, pH 7.8 and sonicated on ice three times for 15 s each. Each protein fraction was quantified by the Bradford method and adjusted to 2 μg/μl with potassium phosphate buffer. Phosphorylated survivin, CRM-1 (Santa Cruz Biotechnology, Santa Cruz, CA), and α-Tubulin (Cell Signaling Technology, Danvers, MA) protein levels were assessed using the WesternBreeze Chemiluminescent Western Blotting Immunodetection System (Invitrogen Life Technologies). Protein (10 μg) was electrophoresed on a 12% Tris Gyl-cine gel and transferred onto polyvinylidene fluoride (PVDF) membranes. The blots were blocked for 1 h with 10 ml blocking solution, washed twice with 20 ml ddH2O for 5 min each, and then incubated with primary antibody (1:1000 dilution of rabbit anti-phosphorylated survivin; 1:1000 dilution of rabbit anti-CRM-1; 1:1000 dilution of rabbit anti-α-Tubulin) for 1 h at room temperature. The blots were then washed 4× for 5 min each with antibody wash solution and incubated with goat anti-rabbit immunoglobulin G (IgG) conjugated with alkaline phosphatase for 30 min at room temperature. The blots were again washed 4 × for 5 min each with antibody wash solution followed by three 2 min washes with ddH2O. The protein bands were visualized by applying 2.5 ml chemiluminescent substrate to the membranes for 5 min. Membranes were exposed to BioMax XAR film (Kodak, Rochester, NY).
2.9. Animals and tumor models
Female C3H mice, 8–11 weeks old (Harlan Laboratories, Indianapolis, IN), were housed in a specific pathogen free barrier facility at The University of Chicago. Experiments were carried out with The University of Chicago Animal Care and Use Committee (IACUC) approval, ACUP number 70672. The University of Chicago has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (A3523–01) and conducts its reviews in accordance with United States Public Health Service (USPHS) regulations and applicable federal and local laws. For tumor growth delay experiments, mice were housed up to five per cage under standard conditions (12 h light and 12 h dark at 48% relative humidity and a constant temperature of 22 °C). The right hind legs of mice were injected subcutaneously with either 5 × 106 SA-NH or 3 × 106 FSa cells in a volume of 0.1 ml and were monitored daily. Total numbers of mice in each experimental group from two separate experiments ranged from 12 to 17 animals. Experiments were initiated when tumors reached approximately 250 mm3 in volume for all tumor growth delay studies.
2.10. Tumor growth delay measurements
Treatment and measurements began when tumors grew to a minimum volume of 250 mm3 as measured in three dimensions (length along the right hind leg, width at its widest point, and the height of the tumor measured from the femur to the highest point of the tumor). Tumor-bearing mice in the control group were loaded into the animal holding chamber with their hind legs taped to the chamber for the same amount of time that the tumor-bearing mice in the irradiation groups were secured. Animals in all experimental groups were sacrificed when tumors reached an approximate volume of 1000 mm3.
2.11. Statistical analysis
Means and standard errors were calculated for all data points from at least three independent experiments. Pairwise comparisons of survivin protein concentrations and cell survival between each of the experimental conditions were performed using a Student’s two-tailed t-test (SigmaPlot software 11.0, SPSS, Chicago, IL). Mean tumor volumes over time were compared among groups using repeated measures analysis of covariance (ANOVA) assuming a quadratic model and accounting for separate variances in each group. Post-hoc pairwise comparisons among groups were done using the Tukey-Kramer method [32]. Absolute tumor growth delay values, defined as the time in days for the tumors in the treated groups to grow from 250 mm3 to 1000 mm3 minus the time in days for untreated control tumors to grow across this same size range, were also determined [29].
3. Results
3.1. The effect of a very low radiation dose (5 mGy) administered in the presence or absence of 10 mM NAC on SA-NH and FSa cell survival and associated intracellular translocation of survivin in a split dose paradigm
The SA-NH murine sarcoma which is p53 WT expresses a prosurvival adaptive response that is mediated by survivin and characterized by an increase in radiation resistance [7,33]. Presented in Fig. 1 is a comparison between the radiation responses of SA-NH p53 WT sarcoma and p53 Mut FSa fibrosarcoma cells following their exposure, either in the presence or absence of 10 mM NAC for 30 min, to 5 mGy 15 min prior to each of two 2 Gy doses separated by 24 h. Exposure to 5 mGy induced an altered radiation response, e.g. an adaptive response, in both SA-NH and FSa cells. Survival increased about 20% (P<0.001) over that observed in SA-NH cells exposed to only two 2 Gy doses. In contrast, under similar radiation conditions FSa cells were sensitized by the 5 mGy doses as evidenced by a 15% decrease in surviving fraction as compared to FSa cells exposed to only two 2 Gy doses (P=0.003). Exposure of either SA-NH or FSa cells to the anti-oxidant NAC resulted in the abrogation of their respective adaptive responses (P = 0.572 and P = 0.841, respectively).
Fig. 1.

Demonstration of very low radiation dose induced adaptive responses in two mouse tumor cell lines differing in p53 mutational status. P values were determined by comparing the survival of cells following two 2 Gy doses with those exposed to two 2 Gy doses along with the 5 mGy doses using a Student’s two-tailed t-test with values ≤0.05 identified as significant. Exposure to 5 mGy induced a significant elevation in SA-NH p53 WT (P=0.001) and reduction in FSa p53 Mut (P<0.003) cell survival. The presence of NAC during the 5 mGy irradiation inhibited these effects in both SA-NH (P=0.572) and FSa (P=0.841). Each experiment was repeated three times and error bars represent the standard error of the mean (SEM).
Intracellular survivin localization as a function of treatment was measured by ELISA in SA-NH and FSa cells (see Fig. 2). Survivin protein was measured in cytoplasmic and nuclear fractions 24 h following the first 2 Gy exposure. No significant differences in the distribution of survivin between the cytoplasmic and nuclear fractions were observed in either SA-NH or FSa cells under non-irradiated control conditions or if exposed to a dose of 2 Gy. Following exposure of cells to 5 mGy 15 min prior to irradiation with 2 Gy, a robust but differential translocation of survivin occurred between the cytoplasmic and nuclear fractions of SA-NH and FSa cells at 24 h. Survivin concentration became elevated in the cytoplasm of SA-NH cells, while FSa cells exhibited an elevation of survivin in their nuclear fraction. Ratios of cytoplasmic to nuclear survivin for each of these cell lines are presented in Table 1 for comparison.
Fig. 2.

Survivin protein in cytoplasmic and nuclear fractions measured by ELISA as a function of treatment protocol in SA-NH and FSa tumor cells. P values were determined by comparing the protein content in the unirradiated control cytoplasmic (C) and nuclear (N) fractions with their corresponding groups from the three treatment protocols: 2 Gy only (SA-NH, P = 0.416 and 0.686, C and N, respectively and FSa, P = 0.814 and 0.469, C and N, respectively; 5 mGy+ 2 Gy ( SA-NH, P = 0.020 and 0.133, C and N, respectively; FSa, P = 0.597 and 0.009, C and N, respectively; and 5 mGy+NAC +2 Gy (SA-NH, P = 0.773 and 0.700, C and N, respectively; FSa, P = 0.016 and 0.996, C and N, respectively. Comparisons were performed using a Student’s two-tailed t-test with P values 0.05 identi ed as signi cant. Each experiment was repeated three times and error bars represent the SEM.
Table 1.
Ratio of cytoplasmic/nuclear survivin levels.
| Control | 2 Gy – 24 h | 5 mGy – 15 min – 2 Gy – 24 h | 10 mM NAC – 30 min – 5mGy – 15 min – 2 Gy – 24 h | 50 μM Emodin – 15 min – 2 Gy– 24 h | 10 mM NAC – 30 min – 50 μM Emodin – 15 min – 2 Gy – 24 h | |
|---|---|---|---|---|---|---|
| SA-NH (p53 WT) | 1.12 | 0.92 | 2.94 | 0.98 | 2.10 | 0.98 |
| FSa (p53 Mut) | 1.20 | 0.88 | 0.22 | 0.95 | 0.56 | 0.97 |
3.2. The effect of Emodin administered in the presence or absence of 10 mM NAC on SA-NH and FSa cell survival and associated intracellular translocation of survivin in a split dose paradigm
The anthraquinone Emodin is a multifunctional drug that has been reported to be an effective ROS generator capable of altering cellular redox status [18–21]. To further assess the role of ROS in the initiation and subsequent expression of the disparate adaptive responses observed for SA-NH and FSa cells following exposure to 5 mGy prior to their irradiation with 2 Gy, the experimental protocol was altered to replace the 5 mGy irradiations with exposures of cells to 50 μM Emodin. As shown in Fig. 3, exposure of SA-NH and FSa cells to Emodin 15 min prior to each 2 Gy dose resulted in adaptive responses similar to those observed following exposure of cells to 5 mGy (see Fig. 2). SA-NH cells expressed an enhanced survival response (P=0.002), while FSa cells were significantly sensitized (P<0.001). The presence of NAC completely inhibited these Emodin-induced protective (SA-NH) and sensitization (FSa) responses (P=0.546 and P=0.649, respectively). Intracellular translocation of survivin following Emodin treatment mirrored that observed following exposure of cells to 5 mGy. The Emodin-induced protective effect expressed in SA-NH cells was accompanied by elevated cytoplasmic and reduced nuclear survivin levels, while its sensitizing effect in FSa correlated with a buildup of nuclear survivin (see Fig. 4). Pre-treatment of cells with NAC inhibited the Emodin-induced translocation of survivin in both cell lines.
Fig. 3.

Demonstration of Emodin induced adaptive responses in SA-NH and FSa tumor cell lines. P values were determined by comparing the survival of cells following two 2 Gy doses with those exposed to two 2 Gy doses along with Emodin using a Student’s two-tailed t-test with values ≤0.05 identified as significant. Exposure to 50 μM Emodin induced a significant elevation in SA-NH (P=0.002) and reduction in FSa (P <0.001) cell survival. The presence of NAC during the Emodin treatment inhibited these effects in both SA-NH (P = 0.546) and FSa (P = 0.649). Each experiment was repeated three times and error bars represent the SEM.
Fig. 4.

Survivin protein in cytoplasmic and nuclear fractions measured by ELISA as a function of Emodin treatment protocol in SA-NH and FSa tumor cell lines. P values were determined by comparing the protein content in the unirradiated control cytoplasmic (C) and nuclear (N) fractions with their corresponding groups from the three treatment protocols: 2 Gy only (SA-NH, P = 0.407 and 0.045, C and N, respectively and FSa, P = 0.377 and 0.173, C and N, respectively; Emodin + 2 Gy ( SA-NH, P = 0.012 and 0.004, C and N, respectively; FSa, P = 0.186 and 0.002, C and N, respectively; and Emodin+ NAC +2 Gy (SA-NH, P = 0.827 and 0.192, C and N, respectively; FSa, P = 0.186 and 0.122, C and N, respectively. Comparisons were performed using a Student’s two-tailed t-test with P values ≤0.05 identi ed as signi cant. Each experiment was repeated three times and error bars represent the SEM.
3.3. p53, phosphorylated survivin and the nuclear transport protein CRM-1
Redox effectors have been reported to be regulated by p53 [24]. Because the effects of exposure to both 5 mGy and Emodin on altering radiation response in p53 WT SA-NH and p53 Mut FSa cells are associated with ROS generating processes that result in such different outcomes, it was of interest to determine what effect, if any, inhibition of p53 in WT SA-NH cells might have on altering the expression of the adaptive response. SA-NH were transfected with p53 siRNA, negative control (NC) siRNA, or mock transfected (MT). As shown in Fig. 5, NC siRNA transfection had no effect on the expression of the pro-survival adaptive response, while transfection with p53 siRNA resulted in a significant sensitization of SA-NH cells to the additional 5 mGy exposures (P= 0.003). Western blotting was performed on proteins from both SA-NH and FSa cells. As described in Fig. 6, the nuclear transport protein CRM-1 involved in survivin export from the nucleus to the cytoplasm was elevated in SA-NH as compared to FSa cells when measured 24 h following the first and just prior to the second radiation exposures. Total phosphorylated survivin levels, however, appeared to be similar between both cell systems at that time.
Fig. 5.
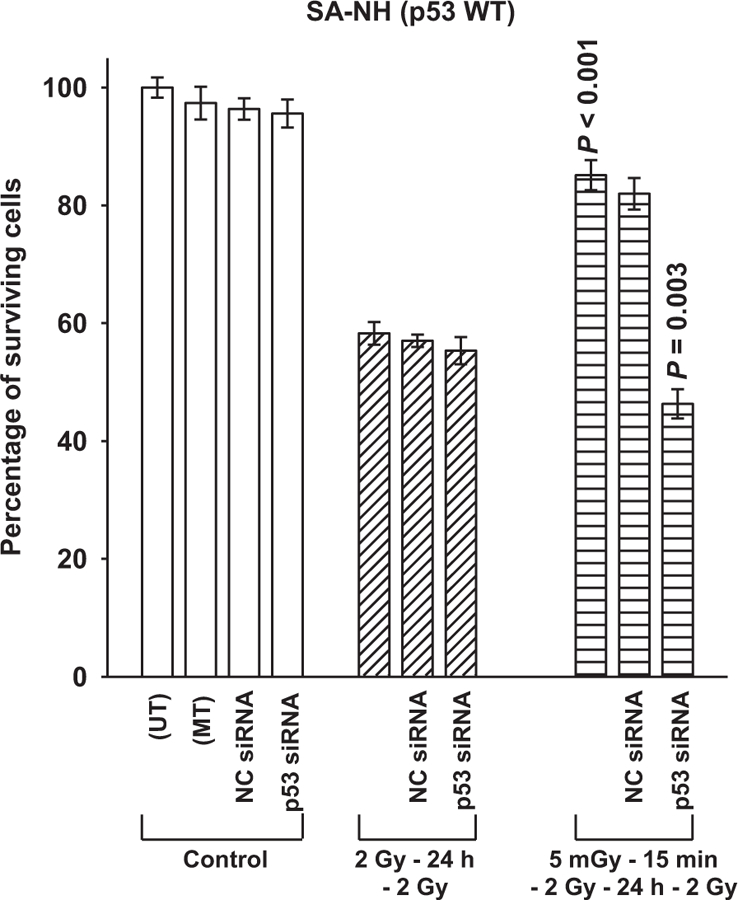
The effect of p53 siRNA transfection on the expression of the survivin-associated adaptive response in SA-NH tumor cells. The cell survival of untransformed (UT) and mock transformed (MT) SA-NH control cells were contrasted to negative control (NC) and p53 siRNA transfected cells following two 2 Gy doses separated by 24 h and 5 mGy administered 15 min prior to each of two 2 Gy doses. P values were determined by comparing the surviving fractions of the two 2 Gy treatment with that of the 5 mGy+ 2 Gy exposures × 2 of nontransfected and p53 siRNA transfected SA-NH. 5 mGy induced a cytoprotective adaptive response in nontransfected cells (P < 0.001) and a sensitizing response in p53 siRNA transfected SA-NH cells (P = 0.003). Experiments were repeated three times and error bars represent the SEM.
Fig. 6.
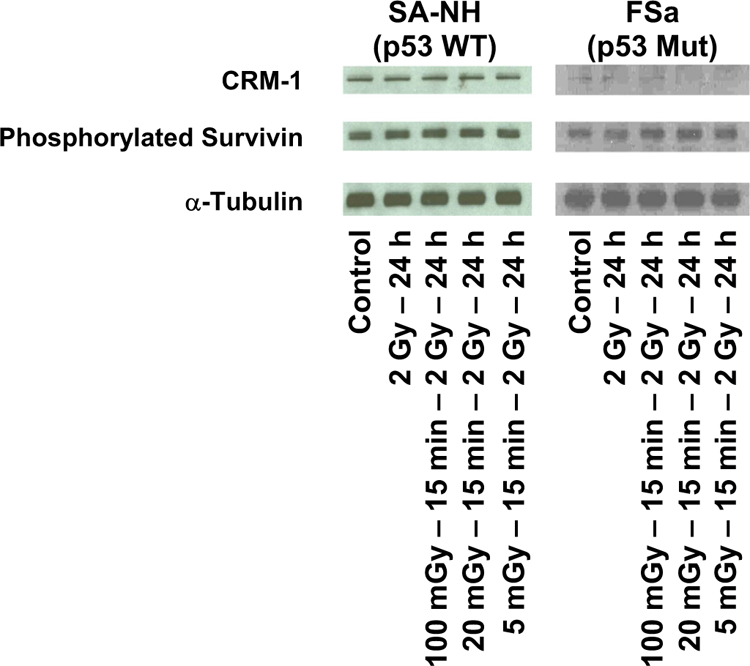
CRM-1 and phosphorylated survivin protein levels measured by Western Blotting as a function of radiation treatment in SA-NH and FSa tumor cells. Representative blots from three experiments.
3.4. Effect of adaptive responses on tumor growth delay
To assess the potential magnitude of the survivin-associated adaptive response following a multiple dosing protocol under in vivo conditions, we performed tumor growth delay studies on SA-NH and FSa tumors that were grown in the right hind legs of C3H mice. Because mouse tumors are relatively aggressive and require substantial radiation dosing to observe effects before animals either die or are required to be euthanized, the two radiation protocols that were used involved exposing mice to five 5 Gy doses each separated by 24 h, with or without the addition of 5 mGy exposures administered 15 min prior to each therapeutic dose. When tumors reached a volume of 250 mm3 irradiations were initiated. After tumors grew to a volume of 1000 mm3, animals were removed from the study and sacrificed. As demonstrated in Fig. 7(A), 5 mGy delivered before each 5 Gy dose to SA-NH tumors induced elevated radiation resistance as manifested by a significant reduction in tumor growth delay of 1.9 days compared to that for tumors irradiated only with 5 Gy doses (P < 0.001 for 5 mGy+5 Gy compared to 5 Gy only). Under these same conditions, FSa tumors had an increase in tumor growth delay of about 4.5 days, reflecting an increased radiation sensitivity induced by 5 mGy (see Fig. 7B). Absolute growth delay values are presented for comparison in Table 2.
Fig. 7.

Tumor growth delay studies demonstrating the effects of the very low radiation dose induced adaptive responses in SA-NH and FSa tumors. Each tumor type is compared to their respective unirradiated control tumor growth curve. Irradiations, designated by (↓), were performed after tumors reached a volume of approximately 250 mm3 and tumor growth was monitored daily until they reached a volume of about 1000 mm3. Mean growth times and SEM were determined. Mean tumor volumes over time were compared among groups using repeated measures analysis of covariance (ANOVA) assuming a quadratic model and accounting for separate variances in each group. A post-hoc pairwise comparison among groups was performed using the Tukey-Kramer method.
Table 2.
Effect of very low dose radiation on tumor growth in C3H mice.
| Tumor | p53 Status | Time in days for tumor to grow from 300 mm3 to 1000 mm3 in volume |
Absolute growth delaya,b,c | P value (t-test) | ||
|---|---|---|---|---|---|---|
| Untreated | 5 Gy × 5 Days | 5 mGy – 15 min – 5 Gy × 5 Days | ||||
| SA-NH | WT | 3.6 ± 70.3 | 7.2 ± 70.9 | 5.3 ± 70.4 | 3.6a | <0.001 |
| 1.7b | =0.001 | |||||
| −1.9c | =0.032 | |||||
| FSa | Mutant | 4.1 ± 70.6 | 7.4 ± 70.8 | 11.9 ± 70.7 | 3.3a | =0.005 |
| 7.8b | <0.001 | |||||
| 4.5c | <0.001 | |||||
Time in days for tumors exposed to 5 Gy for 5 consecutive days to grow from 300 mm3 to 1000 mm3 minus the time in days for untreated tumors to grow from 300 mm3 to 1000 mm3.
Time in days for tumors exposed to 5 mGy – 15 min – 5 Gy for 5 consecutive days to grow from 300 mm3 to 1000 mm3 minus the time in days for untreated tumors to grow from 300 mm3 to 1000 mm3.
Time in days for tumors exposed to 5 mGy – 15 min – 5 Gy for 5 consecutive days to grow from 300 mm3 to 1000 mm3 minus the time in days for tumors exposed to 5 Gy for 5 consecutive days to grow from 300 mm3 to 1000 mm3
4. Discussion
Exposure to a very low dose of a stress-inducing agent such as ionizing radiation results in the increased tolerance of the exposed organism to a subsequent much larger dose. This is referred to as an adaptive response. Most studies on adaptive responses have focused on the use of a single exposure to a very low dose of ionizing radiation followed at a later time by a much larger radiation dose [1–5]. Underlying this response are redox driven processes that involve the generation and amplification of ROS-mediated signals and the activation of redox sensitive proteins such as tumor necrosis factor α (TNFα), nuclear factor kappa B (NFκB), and SOD2 [3–5]. The discovery of the survivin-associated adaptive response represents a new expression of this redox driven phenomenon, which as a result of its bifunctional properties dependent upon its intracellular localization, can be manifested as conferring either enhanced radiation resistance or sensitization [7,33].
Two ROS generating agents, very low dose ionizing radiation and the drug Emodin [18–21], were found to be effective in altering cellular radiation sensitivity to higher radiation doses in tumor cells differing in their p53 mutational status and in inducing the translocation of survivin into the subcellular compartments consistent with these changes in radiation sensitivity (see Figs. 1–4). The tumor suppressor protein p53 is a redox responsive transcription factor that among other things can regulate cellular ROS generation [24,25]. It has been reported that WT p53 is a potent suppressor of transforming growth factor-β (TGF-β)-induced nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) ROS production in lung and breast epithelial cells, while mutant p53 has been associated with enhanced NOX4 expression and elevated levels of ROS production [22]. Enhanced ROS production has also been correlated with a down regulation of survivin and its migration from the cytosol to the nucleus of cells, suggesting that mutation or inhibition of p53 could inhibit a radioprotective adaptive response and possibly even convert it into a radiation sensitization response [23]. Transfection of p53 WT SA-NH cells with p53 siRNA appeared to lead to just such a change in radiation response (see Fig. 5). The robust 5 mGy-induced radioadaptive protective response in the WT cells (P <0.001) was converted into a significant radiosensitization response (P = 0.003) following knockdown of WT p53.
The relative distribution of survivin between the cytoplasm and nucleus was similar and unchanged for both unirradiated control cells and cells exposed to only two 2 Gy doses in both tumor models. Translocation of survivin was observed to occur only when cells were exposed to either a 5 mGy dose or a 50 μM concentration of Emodin just prior to each high dose of radiation. The inhibitory effects of NAC on the induction of adaptive responses by 5 mGy reflect the involvement of redox sensitive processes in the translocation of survivin and the subsequent radio-adaptive response expressed. These results are best understood in the context of survivin’s bivariate properties which are known to be a function of its intracellular localization [8–12].
Survivin is a bifunctional protein whose mode of action is dependent upon its intracellular localization [11]. Survivin is a nuclear cytoplasmic shuttling protein that has an active nuclear exportation signal (NES) in its linker region but lacks a nuclear importation signal (NIP) [8–12]. Its movement into the nucleus is not through an active nuclear import process, but rather appears to occur through simple diffusion. Nuclear survivin is subject to accelerated proteosomal degradation, further diminishing any potential for a cytoprotective adaptive response [17]. This suggests that the basis for the observed bifunctional survivin-associated adaptive response induced by very low dose irradiation is dependent upon the underlying nuclear export process responsible for the cytoplasmic translocation of survivin, e.g., the CRM-1 nuclear export protein. Western blots presented in Fig. 6 show a fairly uniform distribution of total phosphorylated survivin under all treatment conditions and between both tumor cell systems. Total CRM-1, in contrast, appears to be uniform regardless of treatment but is observed to be relatively higher in SA-NH as compared to FSa cells. CRM-1 is known to be highly responsive to changes in oxidative stress, with increased ROS resulting in the inhibition of its nuclear transport activity [12,13]. Mut p53 has been associated with increased oxidative stress in human lung and breast epithelial cells [22]. The relationship between p53 status and intracellular ROS production may, therefore, play an important role in affecting CRM-1 activity and be a determining factor in the redox driven process that drives the survivin-associated adaptive response to be either a cyto-protecting or sensitizing phenomenon [24,25]. This is supported by the cytoplasmic to nuclear survivin concentration ratios that were calculated for SA-NH and FSa contained in Table 1. Cells exposed to 5 mGy or 50 μM Emodin resulted in a 2-to 3-fold elevation in cytoplasmic survivin in p53 WT SA-NH sarcoma cells as compared to a 60% or greater reduction in the p53 Mut FSa cell line. The resulting intracellular localization of survivin was measured 24 h after initial radiation or Emodin exposures because it represents the altered state of intracellular survivin distribution existing at the time of the subsequent treatment in a multi-dosing radiation treatment protocol delivered on a daily basis. Since exposure to the anti-oxidant NAC resulted in the inhibition of both 5 mGy and Emodin in inducing an intracellular translocation of survivin in both tumor lines independent of p53 status, it is reasonable to suggest that subsequent changes in the intracellular redox environment underlies both survivin-associated adaptive responses (see Table 1). A model describing the proposed survivin-associated pro-survival and sensitizing adaptive responses is presented in Fig. 8.
Fig. 8.

Model of the redox sensitive, survivin-associated adaptive response as a function of very low radiation dose-induced translocation of the bi-functional inhibitor of apoptosis protein survivin in tumor cells differing in p53 mutational status.
Since the survivin-associated adaptive response is only demonstrable in a multiple dosing protocol and changes in survival, as measured under in vitro conditions, range from 10% to 20%, it was not clear whether these differences represented the maximum effect that could be attained or whether such differences could be propagated with additional doses of radiation. To address this issue, tumor growth delay studies were performed in vivo using both tumor models in which five 5 Gy doses were administered 24 h apart with or without 5 mGy doses given 15 min earlier. Under these conditions, the addition of 5 mGy exposures to the treatment protocols resulted in the robust expression of both the radio-protecting and sensitizing adaptive responses. SA-NH tumors exhibited a significant reduction in tumor growth delay of 1.9 days (P=0.032) (see Fig. 7A), while FSa tumors experienced an increase in growth delay of 4.5 days (P<0.001) (see Fig. 7B). The tumor growth parameters and associated statistical significance are included in Table 2 for comparison and demonstrate that the respective survivin-associated prosurvival and sensitization adaptive responses for these two tumors were highly significant and suggestive of being propagated with each dosing fraction in a multi-fraction exposure protocol.
The classical adaptive response paradigm most often described involves exposure of cells to a single very low dose of ionizing radiation prior to a second exposure to a much larger dose at a later time [3–5]. The kinetics of increased intracellular SOD2 induced by the very low radiation dose generally correlated with the increased magnitude of cell survival measured as a function of the time following when the higher challenge dose was administered. Other factors that were identified as important in this process included an intact and functioning TNFα signaling pathway, activation of NFκB and enhanced SOD2 gene expression [3–5]. It was expected that these factors would also be involved if an adaptive response were expressed in a multiple dosing paradigm. However, when very low radiation doses were administered shortly before or following exposure to high doses delivered over 24 h intervals, neither TNFα nor SOD2 were found to be involved [7]. Rather, this novel adaptive response was found to be mediated through the action of the tumor specific protein survivin. The uniqueness of the survivin-mediated adaptive response is the potential to exert either a pro-survival or a sensitization phenomenon depending upon the intracellular localization and the associated bifunctional properties of the survivin protein. That this process is redox driven is not unexpected given that the major molecular factors identified as being involved are known to be susceptible to changes in oxidative stress. These include p53, CRM-1, and the bifunctional inhibitor of apoptosis protein survivin. Since survivin is not found in differentiated normal tissues, but rather in malignant tissues, a better understanding of its properties and their dependence upon oxidative stress offers the potential for developing novel methods for enhancing tumor response to therapies while minimizing the probabilities of adversely enhancing tumor resistance.
Acknowledgments
Grant Support
This work was supported in part by the DOE Low Dose Program/Project Grant DE-413 SC0001271 awarded to Drs. Grdina, Li and Woloschak. Dr. Grdina was also supported by NIH NCI R01-CA132998 and Dr. Weichselbaum by NIH NCI R01-CA111423.
Footnotes
Disclosure of potential conflicts of interest
Drs. Grdina and Murley are minority equity partners in Pinnacle Oncology LLC regarding the potential novel uses of amifostine. Dr. Weichselbaum is a consultant to Reflexion, a radiotherapy company.
References
- [1].Wolff S, Afzal V, Wiencke JK, Olivieri G, Michaeli A, Human lymphocytes exposed to low doses of ionizing radiations become refractory to high doses of radiation as well as to chemical mutagens that induce double strand breaks in DNA, Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med 53 (1) (1988) 39–47. [DOI] [PubMed] [Google Scholar]
- [2].Bonner WM, Low-dose radiation: thresholds, bystander effects, and adaptive responses, PNAS USA 100 (9) (2003) 4973–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Murley JS, Baker KL, Miller RC, Darga TE, Weichselbaum RR, Grdina DJ, SOD2-mediated adaptive responses induced by low-dose ionizing radiation via TNF signaling and amifostine, Free Radic. Biol. Med 51 (10) (2011) 1918–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grdina DJ, Murley JS, Kataoka Y, Baker KL, Kunnavakkam R, Coleman MC, et al. , Amifostine induces antioxidant enzymatic activities in normal tissues and a transplantable tumor that can affect radiation response, Int. J. Radiat. Oncol. Biol. Phys 73 (3) (2009) 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grdina DJ, Murley JS, Miller RC, Mauceri HJ, Sutton HG, Thirman MJ, et al. , A manganese superoxide dismutase (SOD2)-mediated adaptive response, Radiat. Res 179 (2013) 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dixon RG, Ogden K, Optimizing dose in computed tomographic procedures, Tech. Vasc. Interv. Radiol 13 (3) (2010) 172–175. [DOI] [PubMed] [Google Scholar]
- [7].Grdina DJ, Murley JS, Miller RC, Mauceri HJ, Sutton HG, Li JJ, et al. , A survivin-associated adaptive response in radiation therapy, Cancer Res 73 (14) (2013) 4418–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li F, Yang J, Ramnath N, Javle MM, Tan D, Nuclear or cytoplasmic expression of survivin: What is the significance? Int. J. Cancer (2005) 509–512. [DOI] [PMC free article] [PubMed]
- [9].Stauber RH, Mann W, Knauer SK, Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential, Cancer Res 67 (13) (2007), 5999–02.. [DOI] [PubMed] [Google Scholar]
- [10].Knauer SK, Mann W, Stauber RH, Survivin’s dual role: an export’s view, Cell Cycle 6 (5) (2007) 518–521. [DOI] [PubMed] [Google Scholar]
- [11].Johnson ME, Howerth EW, Survivin: a bifunctional inhibitor of apoptosis protein, Vet. Pathol 41 (2004), 599–07. [DOI] [PubMed] [Google Scholar]
- [12].Crampton N, Kodiha M, Shrivastava S, Umar R, Stochaj U, Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1, Mol. Biol. Cell 20 (2009) 5106–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kodiha M, Stochaj U, Nuclear transport: a switch for oxidative stress-signaling circuit? J. Signal Transd 2012 (2012) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chan KS, Wong CH, Huang YF, Li HY, Survivin withdrawal by nuclear export failure as a physiological switch to commit cells to apoptosis, Cell Death Dis 1 (e57) (2010) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nguyen KT, Holloway MP, Altura RA, The CRM1 nuclear export protein in normal development and disease, Int. J. Biochem. Mol. Biol 3 (2) (2012) 137–151. [PMC free article] [PubMed] [Google Scholar]
- [16].Becskei A, Mattaj IW, The strategy for coupling the RanGTP gradient to nuclear protein export, PNAS 100 (4) (2003) 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Connell CM, Colnaghi R, Wheatly SP, Nuclear survivin has reduced stability and is not cytoprotective, J. Biol. Chem 283 (6) (2008) 3289–3296. [DOI] [PubMed] [Google Scholar]
- [18].Rahimipour S, Bilkis I, Peron V, Gescheidt G, Barbosa F, Mazur Y, et al. , Generation of free radicals by emodic acid and its [d-Lys6]GnRH-conjugate, Photochem. Photobiol 74 (2) (2001) 226–236. [DOI] [PubMed] [Google Scholar]
- [19].Srinivas G, Babykutty S, Satbiadevan PP, Srinivas P, Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent, Med. Res. Rev 27 (5) (2007) 591–608. [DOI] [PubMed] [Google Scholar]
- [20].Wang W, Sun Y, Li X, Li H, Chjen Y, Tian Y, et al. , Emodin potentiates the anticancer effect of cisplatin on gallbladder cancer cells through the generation of reactive oxygen species and the inhibition of survivin expression, Oncol. Rep 26 (2011) 1143–1148. [DOI] [PubMed] [Google Scholar]
- [21].Huang XZ, Wang J, Huang C, Chen YY, Shi GY, Hu QS, et al. , Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells, Cancer Biol. Ther 7 (3) (2008) 1–8. [DOI] [PubMed] [Google Scholar]
- [22].Boudreau HE, Casterline BW, Burke DJ, Leto TL, Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-β-mediated migration of human lung and breast epithelial cells, Br. J. Cancer 110 (2014) 2569–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pervin S, Tran L, Urman R, Braga M, Parveen M, Li SA, et al. , Oxidative stress specifically downregulates survivin to promote breast tumour formation, Br. J. Cancer 108 (2013) 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maillet A, Pervaiz S, Redox regulation of p53, redox effectors regulated by p53: a subtle balance, Antioxid. Redox Signal 16 (11) (2012) 1285–1294. [DOI] [PubMed] [Google Scholar]
- [25].Liu B, Chen Y, Clair DK ROS and p53: Versatile partnership, Free Radic. Biol. Med 44 (8) (2008) 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Grdina D, Hunter N, Kataoka Y, Murley JS, Milas L, Chemopreventive Doses of amifostine Confer No cytoprotection to Tumor Nodules growing in the Lungs of Mice treated with Cyclophosphamide, Semin. Oncol 26 (Suppl. 7) (1999) S22–S27. [PubMed] [Google Scholar]
- [27].Halpern HJ, Yu C, Peric M, Barth ED, Karczmar GS, River JN, Grdina DJ, Teicher BA, Measurement of differences in pO2 in response to perfluorocarbon/carbogen in FSa and NFSa murine fibrosarcomas with low frequency electron paramagnetic resonance oximetry, Radiat. Res 145 (5) (1996) 610–618. [PubMed] [Google Scholar]
- [28].Halpern HJ, Yu C, Peric M, Barth E, Grdina DJ, Teicher BA, Oxymetry deep in tissues with low frequency electron paramagnetic resonance, Proc. Natl. Acad. Sci. USA 91 (26) (1994) 13047–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schimming R, Mason KA, Hunter N, Weil M, Kishi K, Milas L, Lack of correlation between mitotic arrest or apoptosis and antitumor effect of docetaxel, Cancer Chemother. Pharm 43 (1999) 165–172. [DOI] [PubMed] [Google Scholar]
- [30].Murley JS, Kataoka Y, Weydert CJ, Oberley LW, Grdina DJ, Delayed cytoprotection after enhancement of Sod2 (MnSOD) gene expression in SA-NH mouse sarcoma cells exposed to WR-1065, the active metabolite of amifostine, Radiat. Res 158 (2002) 101–109. [DOI] [PubMed] [Google Scholar]
- [31].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding, Anal. Biochem 72 (1978) 248–254. [DOI] [PubMed] [Google Scholar]
- [32].Kramer CY, Extension of multiple range tests to group means with unequal numbers of replications, Biometrics 12 (1956) 309–310. [Google Scholar]
- [33].Grdina DJ, Murley JS, Miller RC, Woloschak GE, Li JJ, NFκB and survivin-mediated radio-adaptive response, Radiat. Res 183 (2015) 391–397. [DOI] [PubMed] [Google Scholar]