

Abstract
Poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) acts as a DNA damage sensor. It recognizes DNA damage and facilitates DNA repair by recruiting DNA repair machinery to damage sites. Recent studies reported that PARP-1 also plays an important role in DNA replication by recognizing the unligated Okazaki fragments and controlling the speed of fork elongation. On the other hand, emerging evidence reveals that excessive activation of PARP-1 causes chromatin DNA fragmentation and triggers an intrinsic PARP-1-dependent cell death program designated parthanatos, which can be blocked by genetic deletion or pharmacological inhibition of PARP-1. Therefore, PARP-1 plays an essential role in maintaining genomic stability by either facilitating DNA repair/replication or triggering DNA fragmentation to kill cells. A group of structure-specific nucleases is crucial for executing DNA incision and fragmentation following PARP-1 activation. In this review, we will discuss how PARP-1 coordinates with its associated nucleases to maintain genomic integrity and control the decision of cell life and death.
Keywords: PARP-1, nuclease, DNA damage, cell death, DNA replication/repair
1. Introduction
Poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) is the most extensively studied nuclear enzyme of the PARP superfamily [1–3]. It functions as a DNA damage sensor [1–3]. Upon DNA damage, PARP-1 utilizes NAD+ as the substrate and catalyzes the addition of mono-ADP-ribose or PAR to different acceptor proteins, including PARP-1 itself, which is the earliest event in response to DNA damage [1–3]. As a consequence, this event leads to the recruitment of DNA repair proteins and nucleases to damage sites, thereby facilitating DNA damage repair [4–10]. Recently, it was also shown that PARP-1 recognizes the unligated Okazaki fragments and promotes repair during DNA replication [11], indicating its important role in DNA replication. Therefore, inhibition of PARP-1 may sensitize cancer cells to death by interfering with DNA repair and replication. Indeed, PARP inhibitors have been approved by the FDA for the treatment of patients with homologous recombination (HR)-deficient ovarian and breast cancers [12–14].
On the other hand, excessive activation of PARP-1 causes large chromatin DNA fragmentation and triggers an intrinsic PARP-1-dependent cell death program designated parthanatos, which occurs in a variety of organ systems and is widely involved in different neurologic and non-neurologic diseases [15–17]. Pharmacological inhibition or genetic deletion of PARP-1 is profoundly protective against such cellular injury in models of human diseases [15–19]. Therefore, PARP-1 plays an essential role in maintaining genomic stability by either facilitating DNA repair/replication or triggering DNA fragmentation to kill cells (Figure 1). A group of structure-specific nucleases is required for the incision of DNA in multiple PARP-1-evoked DNA repair /damage pathways including single-stranded break (SSB) repair, double-stranded break (DSB) repair, and DNA replication process as well as parthanatos in response to DNA damage [4–10, 15]. In this review, we will summarize the progresses how PARP-1 coordinates with its associated nucleases to maintain genomic integrity and control cell fate.
Figure 1. PARP-1 cooperates with nucleases and controls genomic stability.
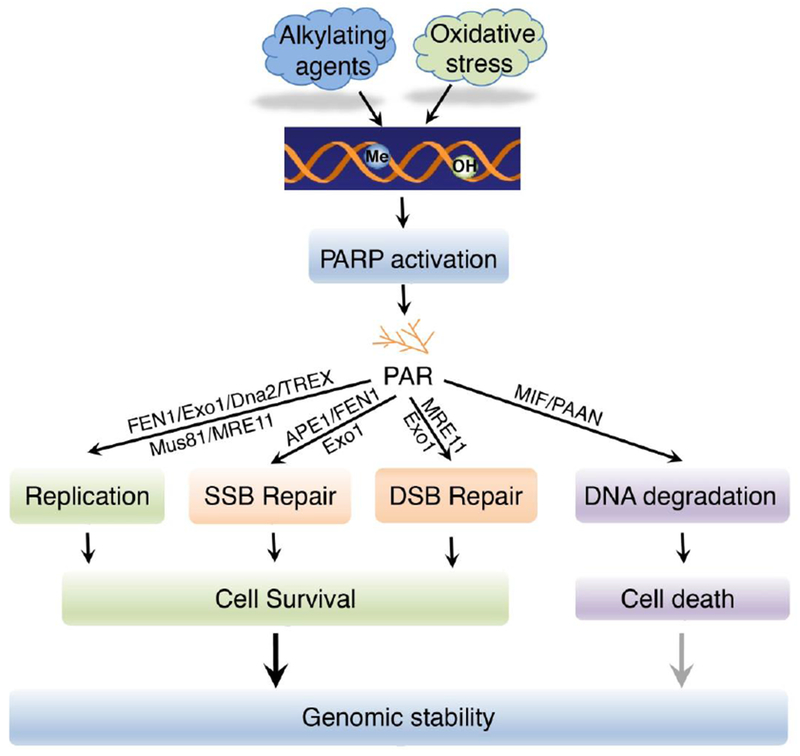
Upon DNA damage induced by oxidative stress or alkylating agents abundant in the environment, PARP-1 is activated to synthesize PAR. As a consequence, specific nucleases are recruited to DNA damage sites to either facilitate DNA damage repair/replication, or trigger DNA degradation leading to cell death. Me, methylation; OH, hydroxylation.
2. Role of PARP-1 in DNA replication and DNA damage repair
DNA replication is an essential process for maintaining genomic integrity. Emerging evidence shows that PARP-1 plays a role in DNA replication and genomic maintenance. Previously, it has been shown that the synthesis of PAR, the bioproduct of PARP-1 activation, is detected at sites of DNA replication fork stalling following DNA damage induced by replication inhibition and recruits meiotic recombination 11 homolog 1 (MRE11) to resect the fork and promote DNA replication restart [5]. Recently, it was found that endogenous PAR is detected at sites of DNA replication during S phase, which is mainly caused by unligated Okazaki fragments [11]. Suppression of Okazaki fragment formation decreases PARP-1 activity. Therefore, PARP-1 serves as a sensor of unligated Okazaki fragments to facilitate a XRCC1-dependent repair during DNA replication [11]. In line with this, another recent study showed that PARP-1 activation acts as a sensor of replication stress and controls the velocity of replication forks [20]. Inhibition of PARP increases the speed of fork elongation without causing fork stalling, which might reduce the fidelity of DNA polymerases and increase DNA damage and genomic instability.
PARP-1 has been well characterized for its role in base excision repair (BER), following DNA base modifications by exogenous genotoxic agents, such as alkylating agents, which are the most common damage to form SSBs [21]. PARP-1 rapidly recognizes these breaks and mediates the recruitment of a scaffold protein XRCC1 and subsequent assembly with other core factors at break sites to process the repair [22]. Besides BER, PAPR-1 has also been suggested to play a role in nucleotide excision repair (NER) by poly(ADP-ribos)ylating (PARylating) NER proteins xeroderma pigmentosum complementation group A (XPA) and XPC [23–25]. Inhibition of PARP-1 may impair SSB repair and cause DSBs.
DNA DSB is often caused by either exogenous damaging agents or endogenous collapse of DNA replication forks. HR and non-homologous end joining (NHEJ) are two major pathways responsible for DSB repair. Pharmacological inhibition of PARP-1 has shown a promising therapeutic effect on BRCA1/2-deficient ovarian cancer [9, 10]. PARP inhibitors have also been approved to treat BRCA mutated metastatic breast and prostate cancers [12–14]. BRCA1/2 are essential proteins required for HR repair. Although increasing evidences have linked PARP-1 to DSB repair, how PARP-1 contributes to DSB repair remains obscure. Nevertheless, two types of models about PARP-1 involvement in DSB repair have been reported. The first model is that PARP-1 directly regulates DSB repair. Upon PARP-1 activation, PAR is synthesized and controls ataxia telangiectasia mutated (ATM) activation and the recruitment of HR proteins to DNA damage sites [1, 26]. PARP-1 deficiency or inhibition interferes with the recruitment of the MRN complex proteins MRE11 and NBS1 to DNA lesions [6]. The 5’-3’ exonuclease activity of MRE11 is essential to generate 3’ single-stranded DNA (ssDNA) overhangs during HR initiation [27]. PARP-1 also contributes to the recruitment of another important HR component BRCA1 to DNA damage sites [28]. BRCA1 forms a heterodimer with BRAD1 and the BRCT domain of BRAD 1 can mediate the recmitment of BRCA1 to DNA lesions through binding with PAR [28]. Poly(ADP-ribos)ylation (PARylation) of BRCA1 stabilizes the BRCA1-RAP80 complex and PARP-1 has been proposed to limit HR process [1, 29]. Besides HR, PARP-1 has been shown to controls NHEJ by PARylating an important NHEJ factor DNA-dependent protein kinase catalytic subunit (DNA-PKcs) [30]. In the absence of Ku70 and Ku80, PARP-1 binds at DSB sites and recruits MRE11 to resect DNA. Then the resulting DNA ends join together by their microhomologue sequences and the gap is further sealed by polymerase Θ and ligase III, thereby promoting an error-prone alternative NHEJ (aNHEJ) repair for cell survival [1]. The second model is that PARP-1 indirectly impacts on DSB repair by controlling the SSB repair pathway, especially the BER. Inhibition of PARP-1 blocks the BER, and turns the SSBs into DSBs, thereby increasing HR stress in BRCA1/2-deficient cells, which at least partially explains the molecular mechanism underlying synthetic lethality of the PARP inhibitor in BRCA1/2-deficient cells [31].
Collectively, these findings suggest that PARP-1 plays a critical role in DNA replication and DNA damage repair to maintain genomic stability.
3. PARP-1 hyperactivation mediates a unique cell death program-parthanatos
As the other side of coin, excessive activation of PARP-1 leads to a unique cell death program designated parthanatos, which is a new type of programmed necrosis different from apoptosis and necrosis (Figure 2) [15–17]. Parthanatos has several key features, including nuclear shrinkage, chromatin condensation, and large DNA fragmentations ranged from 15 kb to 50 kb. Parthanatos occurs in many organ systems and is widely involved in the pathological processes of different neurologic and non-neurologic diseases, including ischemia-reperfusion injury after stroke and myocardial infarction, glutamate excitotoxicity, neurodegenerative diseases, inflammatory injury, reactive oxygen species (ROS)–induced injury [15–19]. Parthanatos specifically depends on PARP-1 hyperactivation and PAR accumulation. PARP-1 inhibitors or genetic deletion of PARP-1 completely block parthanatos, whereas caspase inhibitors fail to do so. PAR polymer has a very short half-life and is rapidly degraded by PAR glycohydrolase (PARG) [16]. It functions as a cell death signal and mediates parthanatos [32]. Following oxidative stress or brain injury, PARP-1 evokes PAR-mediated deadly crosstalk between the nucleus and mitochondria by triggering apoptosis-inducing factor (AIF) release from mitochondria (Figure 2). Then AIF recruits microphage migration inhibitory factor (MIF), a newly identified 3’ exonuclease, to the nucleus, where MIF cleaves the genomic DNA into large DNA fragments leading to cell death [15]. Genetic deletion of MIF, abolition of MIF nuclease activity, or disruption of MIF/AIF interaction prevents DNA degradation and cell death. These findings uncover a critical role of PARP-1 in cell death and may ultimately lead to new ways to protect against brain injury.
Figure 2. MIF is the executor for PARP-1 dependent cell death (parthanatos).
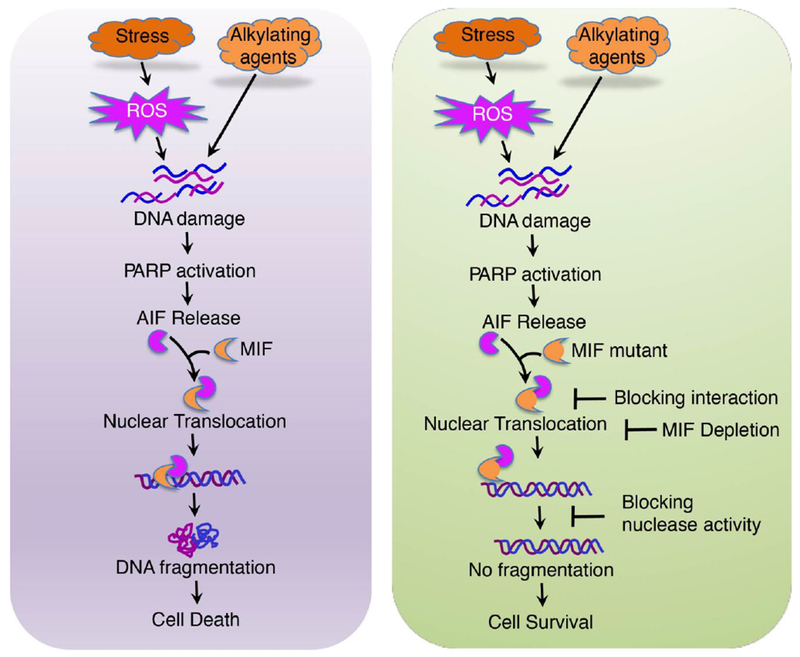
Stresses induce DNA damage, PARP1 activation, and subsequently PAR formation. PAR facilitates the release of AIF from mitochondria. AIF recruits MIF to the nucleus, where MIF cleaves genomic DNA into large DNA fragments and causes cell death. Interference with this cascade by blocking either the formation of the AIF-MIF complex or MIF nuclease activity prevents DNA fragmentation and promotes cell survival.
Besides its role in brain injury, the role of PARP-1 in alkylating agents-induced DNA damage and cell death has also been documented [33, 34]. N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG), which belongs to the mono- (SN1-) subtype alkylating agent, was first discovered in 1959 as an anti-cancer drug through a national cooperative cancer drug screen program [35]. It acts by adding alkyl groups to the O6 of guanine and O4 of thymine, thereby causing PARP-1 hyperactivation and eventually leading to cell death [33, 34]. PARP-1 inhibition blocks PAR synthesis and prevents MNNG-induced cell death [15]. MNNG is structurally similar to toxic compounds found in common chemotherapy drugs [33]. Its derivatives Lomustine and Carmustine are the currently most commonly used alkylating chemotherapy drugs [36]. Similarly, SN2-subtype alkylating agent methyl methanesulfonate (MMS) also activates PARP-1 to produce PAR and causes cell death, which can be robustly blocked by PARP-1 deletion both in vitro and in vivo [33, 34]. Although alkylating agents are the most commonly used chemotherapy drugs to kill cancer cells [33, 36–38], the importance of PARP-1 in alkylating agents-induced cancer cell death has not brought much attention yet. The precise mechanisms by which PARP-1 hyperactivation following alkylating agent treatment induces DNA degradation and cancer cell death still remain to be explored.
4. Nucleases and genomic stability
PARP-1 is widely involved in different DNA repair pathways, DNA replication and cell death, in response to DNA damage. The structure-specific nucleases are often required for DNA incision during these PARP-1-evoked processes to regulate genomic stability either by facilitating DNA repair or triggering DNA fragmentation. Multiple nucleases, including Flap endonuclease 1 (FEN1), DNA replication helicase/nuclease 2 (DNA2), MUS81-EME1, Apurinic/apyrimidinic endonuclease (APE1), 3’ repair exonuclease (TREX), MRE11, and exonuclease 1 (EXO1), have been previously identified for their critical role in the process of DNA replication, SSB repair, and DSB repair (Table 1). Recently, we discovered that MIF is a novel PARP-1 activity associated nuclease (PAAN) involved in DNA degradation during parthanatos [15]. The functions of these nucleases in PARP-1-mediated genomic integrity are summarized below.
Table 1.
Summary of PARP-1-associated nucleases in DNA repair, replication and cell death pathways.
| Nucleases | Polarity | Nuclease Activity | Functional Pathways | References |
|---|---|---|---|---|
| APE1 | 3’-5’ | AP site endonuclease; 3’ exonuclease | BER; DNA replication | [54, 55, 58] |
| FEN1 | 5’-3’ | 5’ flap endonuclease; 5’ exonuclease | BER; Okazaki fragment processing | [11, 39–41, 51] |
| DNA2 | 5’-3’ | 5’ flap endo/exonuclease; Resolve the long 5’ flap; | Long-patch BER; Maintenance of telomere integrity; Okazaki fragment processing; Stalled replication fork resolution | [42–49, 80] |
| Mus81 | 3’-5’ | 3’ flap endonuclease | Stalled replication fork resolution | [50, 81] |
| TREX1/2 | 3’-5’ | Aberrant ssDNA removal; 3’ exonuclease | DNA damage repair; Immune response; DNA replication proofreader | [59–62] |
| MRE11 | 3’-5’ | Blunt or recessed 3’ exonuclease | Stalled replication fork restart; telomere length maintenance; DSB repair | [5, 6, 27, 63] |
| EXO1 | 5’-3’ | 5’ exonuclease and 5’ flap endonuclease | DNA replication; MMR; BER; Translesion synthesis (TLS); HR | [64–67] |
| MIF | 3’-5’ | 3’ exonuclease; DNA degradation; | Parthanatos | [15] |
4.1. FEN1
FEN1 is a structure-specific nuclease with 5’ flap endonuclease and 5’ exonuclease activity. It plays an essential role in DNA replication as well as BER. During DNA replication, FEN1 is required for the removal of RNA-DNA primers in lagging-strand and the resulting nick is sealed by DNA ligase I, leading to the maturation of Okazaki fragments [39]. Recently, it was shown that perturbation of FEN1 increases the unligated Okazaki fragments, which causes PARP-1 activation and PAR accumulation in S phase and further activates XRCC-1-dependent repair during DNA replication [11]. The function of FEN1 to remove the 5’-flap structure has also been widely explored in BER. The long-patch BER includes more than one nucleotide incorporation through strand-displacement reaction, which activates PARP-1- and XRCC1-dependent repair pathway and requires FEN1 to resolve the 5’-flap intermediate [40]. The nuclease-deficient FEN1 mutant causes BER deficiency and genomic instability, and makes mice more susceptible to DNA alkylating agent methylnitrosourea-induced lung adenocarcinoma [41].
4.2. DNA 2
DNA2 possesses 5′-3′ helicase and endonuclease activities and plays an essential role in DNA replication and repair in both nucleus and mitochondrion [42]. It resolves the long 5’ flap during the process of Okazaki fragment maturation and long-patch BER [43]. When the 5’ flap length is more than 27 nt, the replication protein A (RPA) complex recruits DNA2 to incise the long flap into a short one suitable for FEN1 cleavage [44]. DNA replication stress often results in stalled or reversed replication fork. DNA2 can degrade the reversed forks and coordinate with the Werner syndrome ATP-dependent helicase (WRN) as well as the Bloom syndrome protein (BLM) to restart the arrested replication forks [45]. DNA2 also plays a role in maintenance of telomere integrity [46]. It cleaves G-quadruplex DNA in vitro. DNA2 deficiency displays a variety of genome instabilities and impairs telomere replication [46]. The small-molecule inhibitor of DNA2 and PARP inhibitor have a synergistic effect on sensitization of human cancer cells to camptothecin chemotherapy [47]. Thus, combined inhibition of PARP-1 and DNA2 has been considered as a potential strategy for inducing synthetic lethality [43, 47–49].
4.3. MUS81-EME1
MUS81-EME1 is another structure-dependent endonuclease involving in restart of the stalled DNA replication fork. It prefers to remove the branched DNA structure and provokes the DSBs under conditions of replication inhibition [50]. Lack of functional MUS81 makes cells more sensitive to DNA damaging agents and impairs DNA replication fork progression [51, 52]. Furthermore, MUS81 is also essential for maintaining normal DNA replication by reducing the frequency of the initiation events [53].
4.4. APE1
APE1, a primary apurinic/apyrimidinic endonuclease, plays an essential role in the BER pathway. DNA base modification and damage often cause SSBs, which can be rapidly detected by PARP-1. The latter recruits the scaffold protein XRCC1 and APE1 to damage sites to process the repair [22]. APE1 cleaves the phosphodiester backbone immediately adjacent to the 5’ to an AP site to generate 3’-hydroxyl and 5’-deoxyribose phosphate terminuses [54]. In the short-patch BER, the DNA nick incised by APE1 is sealed by DNA polymerase β and DNA ligase 3 [55]. In the long-patch BER, following APE1 incision, PCNA and DNA polymerase δ or β are recruited, which displaces 2 to 8 nucleotides from the damage sites and further elongate the nucleotides to seal the gap with the assistant of FEN1 to cleave the displaced nucleotides and DNA ligase I to incorporate de novo nucleotides [56, 57]. In vitro reconstituted BER system implies that APE1 contributes the rejoining efficiency and increases the fidelity of BER [54]. Besides its endonuclease activity, APE1 also functions as a 3’ to 5’ exonuclease to remove the 3’-mispaired termini, such as phosphoglycolate (3’-PG) caused by ROS [58].
4.5. TREX
TREX1 and TREX2 are the major 3’ exonucleases that cleave the mismatched DNA end. They serve as proofreaders to maintain the accuracy and fidelity of DNA replication. TREX1 predominantly locates in the cytoplasm and digests cytosolic ssDNA to prevent cytosolic DNA-induced immune response [59]. Upon DNA damage induced by genotoxic stress such as UV and γ irradiation, TREX1 translocates to the nucleus where it directly interacts with PARP-1 and contributes to DNA damage responses [60]. TREX1 deficiency causes persistent ssDNA accumulation in endoplasmic reticulum, impairs Gl/S transition during the cell cycle, and provokes ATM-dependent DNA damage checkpoint activation [61]. TREX2 is reported to interact with DNA polymerase δ and increases its accuracy to prevent mutagenesis in the cell [59, 62].
4.6. MRE11
MRE11 possesses endonuclease and 5’ exonuclease activities and is widely involved in DNA replication, telomere length maintenance, and DSB repair [5, 6, 27, 63]. Its 5’ exonuclease activity is essential to generate 3’ ssDNA overhangs during HR initiation [27]. MRE11 is one of the first nucleases recruited to DNA lesions, where it forms a MRN (Mre11-Rad50-Nbs1) complex with Nbs1 and Rad50 [6]. MRE11 physically interacts with PARP-1 via its PAR biding motif and PAPR-1 is required for the recruitment of MRE11 to DNA damage lesions. In addition, PARP-1 can be activated by stalled replication forks and further recruits MRE11 to resect the fork for initiation restart [5]. Interestingly, PARP-1 could protect the excessive degradation of nascent DNA caused by MRE11 in BRCA2-deficient cells. Thus, BRCA2-deficient cells exhibit more stalled forks degradation upon PAPR-1 inhibition. This phenomenon could partially explain the synthetic lethal effect of BRCA2 deficiency combined with PARP-1 inhibitors [63].
4.7. EXO1
EXO1 is a multifunctional nuclease with 5’ exonuclease activity and 5’ flap endonuclease activity and is involved in DNA replication, DNA mismatch repair, and DSB repair in the cell [64–67]. It has been shown that PARP-1 interacts with EXO1 and promotes its early recruitment to sites of DNA damage in cells upon laser micro irradiation. This recruitment process depends on PARP-1-induced PARylation at the N-terminal PIN domain of EXO1. Zhang F et al. also showed that BRCA2 is PARylated by PARP-1 and mediates the early recruitment of EXO1 for DNA end resection [68]. However, PAR binding inhibits EXO1’s nuclease activity. Deletion of EXO1 prevents PARP inhibitor-induced Rad51 accumulation in chromatin and switches HR repair to NHEJ repair pathway [69].
4.8. MIF/PAAN
MIF is a pleiotropic cytokine-like protein, which was originally identified by Bloom and Bennett as a protein secreted by T cells and inhibiting migration of macrophage [70, 71]. To date, it becomes clear that MIF is widely expressed in multiple cell types, including neurons, monocytes, macrophages, vascular smooth muscle cells, cardiomyocytes, and cancer cells, and plays an important role in inflammation, immune response, and tumor growth. Hypoxia-inducible factor induces the expression of MIF in hypoxic cells [72]. Increased expression of MIF has been observed in cancer patients and is correlated with poor clinical outcome in these patients [73]. Besides its known function as a non-classically secreted cytokines, MIF also possesses oxido-reductase and tautomerase activities in vitro, although its biological substrates and physiological relevance remain unknown. MIF tautomerase-null knock-in mice reveals that MIF-interacting protein rather than its tautomerase activity mediates MIF-dependent growth regulation [74]. Interestingly, MIF knockout mice have a longer life span than control mice [75].
We recently identified MIF as a novel Mg2+- and Ca2+-dependent nuclease [15]. It possesses 3’ exonuclease activity and has relatively higher (about 10-fold) binding affinity to the ssDNA with a stem-loop like structure than the double-stranded DNA in vitro. By screening DNA substrates either with different structures or different sequences using both in vitro nuclease assay and electrophoretic mobility shift assay, we discovered that MIF binds to the 5’ free unpaired region of ssDNA but cleaves it at the 3’ unpaired region (Figure 3), indicating that MIF recognizes the ssDNA substrate in a structure-dependent rather than a sequence-dependent manner. MIF binds to chromatin DNA and cleaves it into large DNA fragments in neurons following N-methyl-D-aspartate (NMDA) neurotoxicity or in cancer cells following a strong alkylating agent MNNG treatment, eventually leading to cell death [15]. MIF knockout or nuclease-deficient MIF mutant prevents large DNA fragmentations and protects from NMDA or MNNG-induced cell death (Figure 3). Since MIF is widely involved in inflammation, immune response, and cancer biology, further studies are required to study if MIF’s nuclease activity contributes to its effects on inflammation, immune response and tumor growth.
Figure 3. MIF is a novel Mg2+/Ca2+-dependent 3’ exonuclease.
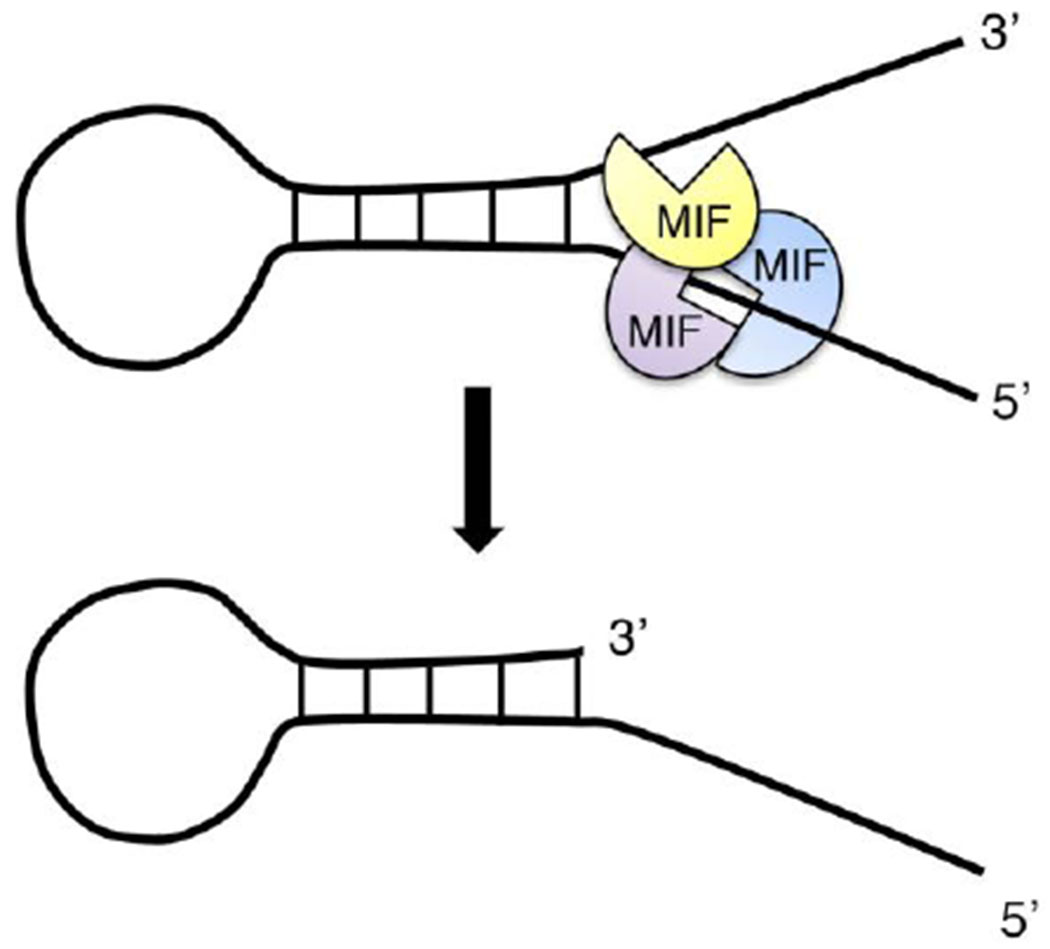
MIF trimer specifically recognizes the ssDNA with a stem loop structure as the substrate. It binds to the 5’ unpaired region of stem loop structured ssDNA, but selectively cleaves at its 3’ unpaired ssDNA.
5. Perspective
PARP-1 controls cell life and death in a context-dependent manner (Figure 4). In response to the mild DNA damage, PARP-1 activation facilitates DNA repair leading to cell survival [3, 4, 7–10, 76]. In this context, PARP inhibition may sensitize cancer cells to death by interfering with DNA repair. Indeed, the discovery of synthetic lethality of PARP inhibitors in BRCA1/2-deficient tumors has brought much attention and enthusiasm into the field of DNA repair and cancer therapy [9, 10]. Currently, four PARP inhibitors olaparib, rucaparib, niraparib and talazoparib have been approved by the FDA [12–14] and many others are being extensively tested in clinical trials either as a single agent or in combination with chemotherapy or radiotherapy approaches to treat different types of solid tumors. On the other hand, we and other groups have shown that PARP-1 mediates an intrinsic caspase-independent cell death program designated parthanatos in response to severe DNA damage [15–19, 77, 78]. In this context, inhibition of PARP-1 may prevent alkylating agent-induced cytotoxicity and increase chemoresistance. The precise mechanism by which PARP-1 regulates these antagonistic functions: DNA repair/cell survival versus DNA fragments/cell death, remains unclear. Interestingly, Tulin group recently showed that the interaction of N-terminal DNA binding domain of PARP-1 with DNA leads to a “hit and run activation of PARP-1”, which may initiate the DNA repair pathway [79]. In contrast, the interaction of the C-terminal domain of PARP-1 with histone H4 in the nucleosome leads to the long-term activation of PARP-1, which causes chromatin loosening. These studies indicate that different types of interaction with PARP-1 may contribute to distinct functions of PARP-1. Further studies are needed to explore PARP-1 interactors at its N- and C-terminal domains, which may help understand the regulation of the precise functions of PARP-1 under different DNA damage contexts.
Figure 4. The model of context-dependent functions of PARP-1 and nucleases in DNA repair and cell death.
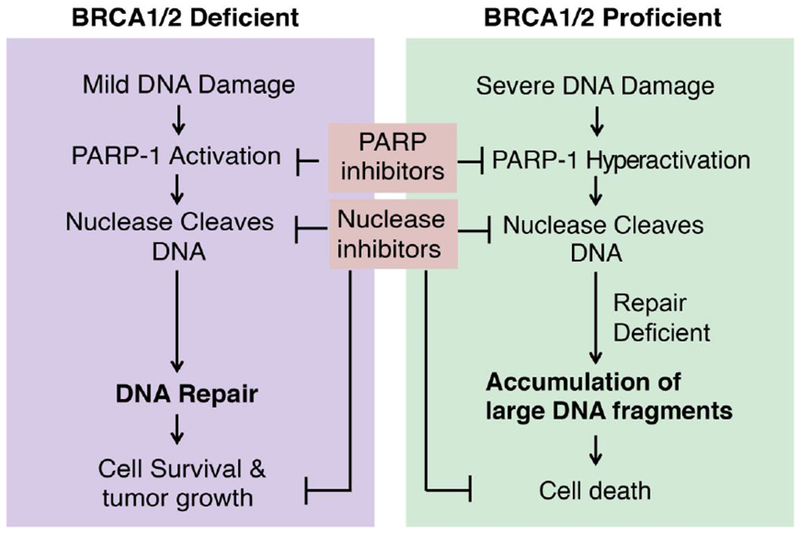
The role of PARP-1 is highly context-dependent in response to different types of DNA damage in different types of cells and tissues. In BRCA1/2-deficient cancer cells, PARP-1 plays a critical role in DNA damage repair. PARP-1 inhibitors combined with DNA damage chemotherapy drugs kill cancer cells. However, in BRCA1/2-proficient cells, DNA alkylating agents or oxidative stress may cause PARP-1 hyperactivation, PAR accumulation and DNA degradation leading to cell death. Inhibition of PARP-1 prevents cell death. A group of structure-specific nucleases is recruited by activated PARP-1 in response to DNA damage. Future studies are required to understand if these PARP-1-associated nucleases execute different functions under the different contexts.
Although it is clear that a group of structure-specific nucleases associates with PARP-1 activation and is crucial for maintaining genome integrity and controlling cell fate, several outstanding questions remain to be answered. For example, how does PARP-1 differentially coordinate with these nucleases under different DNA damage contexts? The polymerases ε and δ have been known to have the proofreading functions with its 3’ exonuclease activity. However, it is still unclear if PARP-1 recruits other structural-specific Ύ exonucleases to monitor the accuracy of DNA replication in real time and removes the misincorporated nucleotide while the polymerases copy DNA at a high speed. It is also important to know if the roles of these PARP-1-associated nucleases are context-dependent.
Overall, a comprehensive understanding of the crucial roles of PARP-1 and its associated nucleases in DNA repair and cell death is of paramount importance for the development of successful therapeutic regiments for the treatment of different cancers as well as prevention of normal tissue injury.
Acknowledgement:
This work was supported by grants from the National Institutes of Health (NIH, R00NS078049 and R35GM124693), Darrell K Royal Research Fund, Welch Foundation (I-1939), CPRIT (RP170671), TIBIR pilot Grant, the University of Texas (UT) Southwestern Medical Center Startup funds and UT Rising Stars to Y.W., and NIH (R01CA222393), CPRIT (RR140036), Susan G. Komen® (CCR16376227), Welch Foundation (I-1903), Mary Kay Foundation (08-19) to W.L..
Abbreviations:
- AIF:
Apoptosis-inducing factor
- APE1:
Apurinic/apyrimidinic endonuclease
- ATM:
Ataxia Telangiectasia Mutated
- BER:
Base excision repair
- DSB:
Double-stranded break
- EXO1:
Exonuclease 1
- FEN1:
Flap endonuclease 1
- HR:
Homologous recombination
- MIF:
Microphage migration inhibitory factor
- MRE11:
Meiotic Recombination 11 Homolog 1
- NHEJ:
Non-homologous end joining
- MNNG:
N-Methyl-N′-nitro-N-nitrosoguanidine
- NMDA:
N-methyl-D-aspartate
- PAAN:
PARP-1 activity associated nuclease
- PARP-1:
Poly (ADP-ribose) (PAR) polymerase-1
- PAR:
Poly (ADP-ribose)
- PARylation:
Poly (ADP-ribos)ylation
- Parthanatos:
PARP-1 dependent cell death
- ROS:
Reactive Oxygen Species
- SSB:
Single-stranded break
- TREX:
3’Repair Exonuclease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest: The authors declare that they have no conflict-of-interest.
References:
- 1.Ray Chaudhuri A and Nussenzweig A, The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol, 2017. 18(10): p. 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo X and Kraus WL, On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev, 2012. 26(5): p. 417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai P, Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol Cell, 2015. 58(6): p. 947–58. [DOI] [PubMed] [Google Scholar]
- 4.Beck C, et al. , Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res, 2014. 329(1): p. 18–25. [DOI] [PubMed] [Google Scholar]
- 5.Bryant HE, et al. , PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J, 2009. 28(17): p. 2601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haince JF, et al. , PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem, 2008. 283(2): p. 1197–208. [DOI] [PubMed] [Google Scholar]
- 7.Helleday T, Bryant HE, and Schultz N, Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle, 2005. 4(9): p. 1176–8. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, et al. , PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res, 2006. 34(21): p. 6170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant HE, et al. , Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 2005. 434(7035): p. 913–7. [DOI] [PubMed] [Google Scholar]
- 10.Farmer H, et al. , Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 2005. 434(7035): p. 917–21. [DOI] [PubMed] [Google Scholar]
- 11.Hanzlikova H, et al. , The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol Cell, 2018. 71(2): p. 319–331 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledermann J, et al. , Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med, 2012. 366(15): p. 1382–92. [DOI] [PubMed] [Google Scholar]
- 13.Mirza MR, et al. , Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med, 2016. 375(22): p. 2154–2164. [DOI] [PubMed] [Google Scholar]
- 14.Swisher EM, et al. , Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol, 2017. 18(1): p. 75–87. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, et al. , A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science, 2016. 354(6308). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Dawson VL, and Dawson TM, Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol, 2009. 218(2): p. 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, et al. , Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal, 2011. 4(167): p. ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacher P and Szabo C, Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol, 2008. 173(1): p. 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo C and Dawson VL, Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci, 1998. 19(7): p. 287–98. [DOI] [PubMed] [Google Scholar]
- 20.Maya-Mendoza A, et al. , High speed offork progression induces DNA replication stress and genomic instability. Nature, 2018. 559(7713): p. 279–284. [DOI] [PubMed] [Google Scholar]
- 21.Chen A, PARP inhibitors: its role in treatment of cancer. Chin J Cancer, 2011. 30(7): p. 463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldecott KW, et al. , XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res, 1996. 24(22): p. 4387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer JM, et al. , Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J, 2014. 281(16): p. 3625–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King BS, et al. , Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J Biol Chem, 2012. 287(47): p. 39824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maltseva EA, et al. , Poly(ADP-ribose) Polymerase 1 Modulates Interaction of the Nucleotide Excision Repair Factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J Biol Chem, 2015. 290(36): p. 21811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haince JF, et al. , Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novelpoly(ADP-ribose)-dependentpathway in the early response to DNA-damaging agents. J Biol Chem, 2007. 282(22): p. 16441–53. [DOI] [PubMed] [Google Scholar]
- 27.Williams RS, et al. , Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell, 2008. 135(1): p. 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M and Yu X, Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell, 2013. 23(5): p. 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, et al. , PARP1-drivenpoly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov, 2014. 4(12): p. 1430–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spagnolo L, et al. , Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res, 2012. 40(9): p. 4168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Andrea AD, Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst), 2018. 71: p. 172–176. [DOI] [PubMed] [Google Scholar]
- 32.Andrabi SA, et al. , Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A, 2006. 103(48): p. 18308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu D, Calvo JA, and Samson LD, Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer, 2012. 12(2): p. 104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calvo JA, et al. , Aag DNA glycosylasepromotes alkylation-induced tissue damage mediated by Parp1. PLoS Genet, 2013. 9(4): p. e1003413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nitrosoureas: Current Status and New Developments, ed. L.H B Prestayko Archie W., Crooke Stanley T. 1981: Academic Press. [Google Scholar]
- 36.Lawley PD and Thatcher CJ, Methylation of deoxyribonucleic acid in cultured mammalian cells by N-methyl-N’-nitro-N-nitrosoguanidine. The influence of cellular thiol concentrations on the extent of methylation and the 6-oxygen atom of guanine as a site of methylation. Biochem J, 1970. 116(4): p. 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maanen MJ, Smeets CJ, and Beijnen JH, Chemistry, pharmacology and pharmacokinetics of N,N’,N”-triethylenethiophosphoramide (ThioTEPA). Cancer Treat Rev, 2000. 26(4): p. 257–68. [DOI] [PubMed] [Google Scholar]
- 38.Lajous H, et al. , Rethinking Alkylating(-Like) Agents for Solid Tumor Management. Trends Pharmacol Sci, 2019. [DOI] [PubMed] [Google Scholar]
- 39.Balakrishnan L and Bambara RA, Flap endonuclease 1. Annu Rev Biochem, 2013. 82: p. 119–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klungland A and Lindahl T, Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J, 1997. 16(11): p. 3341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu H, et al. , Chemical-induced cancer incidence and underlying mechanisms in Fen1 mutant mice. Oncogene, 2011. 30(9): p. 1072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, et al. , hDNA2 nuclease/helicasepromotes centromeric DNA replication and genome stability. EMBO J, 2018. 37(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pawlowska E, Szczepanska J, and Blasiak J, DNA2-An Important Player in DNA Damage Response or Just Another DNA Maintenance Protein? Int J Mol Sci, 2017. 18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar S and Burgers PM, Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev, 2013. 27(3): p. 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sturzenegger A, et al. , DNA2 cooperates with the WRNandBLMRecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem, 2014. 289(39): p. 27314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin W, et al. , Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J, 2013. 32(10): p. 1425–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu W, et al. , A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine, 2016. 6: p. 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chai W, Zheng L, and Shen B, DNA2, a new player in telomere maintenance and tumor suppression. Cell Cycle, 2013. 12(13): p. 1985–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, et al. , Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis, 2017. 6(4): p. e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanada K, et al. , The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol, 2007. 14(11): p. 1096–104. [DOI] [PubMed] [Google Scholar]
- 51.Abraham J, et al. , Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J, 2003. 22(22): p. 6137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Regairaz M, et al. , Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J Cell Biol, 2011. 195(5): p. 739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu H, et al. , The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat Commun, 2015. 6: p. 6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou KM and Cheng YC, An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3’mispairedDNA. Nature, 2002. 415(6872): p. 655–9. [DOI] [PubMed] [Google Scholar]
- 55.Dianov G, Price A , and Lindahl T, Generation of single-nucleotide repair patches following excision of uracil residues from DNA. Mol Cell Biol, 1992. 12(4): p. 1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ranalli TA, Tom S, and Bambara RA, AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem, 2002. 277(44): p. 41715–24. [DOI] [PubMed] [Google Scholar]
- 57.Carter RJ and Parsons JL, Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol Cell Biol, 2016. 36(10): p. 1426–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitaker AM, Flynn TS, and Freudenthal BD, Molecular snapshots of APE1 proofreading mismatches and removing DNA damage. Nat Commun, 2018. 9(1): p. 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyazaki T, et al. , The 3’-5’DNA exonuclease TREX1 directly interacts with poly(ADP-ribose) polymerase-1 (PARP1) during the DNA damage response. J Biol Chem, 2014. 289(47): p. 32548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christmann M, et al. , Three prime exonuclease I (TREX1) is Fos/AP-1 regulated by genotoxic stress and protects against ultraviolet light and benzo(a)pyrene-induced DNA damage. Nucleic Acids Res, 2010. 38(19): p. 6418–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang YG, Lindahl T, and Barnes DE, Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell, 2007. 131(5): p. 873–86. [DOI] [PubMed] [Google Scholar]
- 62.Shevelev IV, Ramadan K, and Hubscher U, The TREX2 3’-->5’ exonuclease physically interacts with DNA polymerase delta and increases its accuracy. ScientificWorldJournal, 2002. 2: p. 275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ying S, Hamdy FC, and Helleday T, Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res, 2012. 72(11): p. 2814–21. [DOI] [PubMed] [Google Scholar]
- 64.Bolderson E, et al. , Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res, 2010. 38(6): p. 1821–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keijzers G, Bohr VA, and Rasmussen LJ, Human exonuclease 1 (EXO1) activity characterization and its function on flap structures. Biosci Rep, 2015. 35(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goellner EM, et al. , Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-bindingpartners. Nat Struct Mol Biol, 2018. 25(8): p. 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sertic S, et al. , Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol Cell, 2018. 70(1): p. 34–47 e4. [DOI] [PubMed] [Google Scholar]
- 68.Zhang F, et al. , Poly(ADP-Ribose) Mediates the BRCA2-Dependent Early DNA Damage Response. Cell Rep, 2015. 13(4): p. 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheruiyot A, et al. , Poly(ADP-ribose)-bindingpromotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amst), 2015. 35: p. 106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bloom BR and Bennett B, Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science, 1966. 153(3731): p. 80–2. [DOI] [PubMed] [Google Scholar]
- 71.Bloom J, Sun S, and Al-Abed Y, MIF, a controversial cytokine: a review of structural features, challenges, and opportunities for drug development. Expert Opin Ther Targets, 2016. 20(12): p. 1463–1475. [DOI] [PubMed] [Google Scholar]
- 72.Oda S, et al. , Macrophage migration inhibitory factor activates hypoxia-inducible factor in ap53-dependent manner. PLoS One, 2008. 3(5): p. e2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balogh KN, Templeton DJ , and Cross JV, Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLoS One, 2018. 13(6): p. e0197702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fingerle-Rowson G, et al. , A tautomerase-null macrophage migration-inhibitory factor (MIF) gene knock-in mouse model reveals that protein interactions and not enzymatic activity mediate MIF-dependent growth regulation. Mol Cell Biol, 2009. 29(7): p. 1922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harper JM, Wilkinson JE, and Miller RA, Macrophage migration inhibitory factor-knockout mice are long lived and respond to caloric restriction. FASEB J, 2010. 24(7): p. 2436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feng FY, et al. , Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol Cell, 2015. 58(6): p. 925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martire S, Mosca L, and d’Erme M, PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev, 2015. 146-148: p. 53–64. [DOI] [PubMed] [Google Scholar]
- 78.Virag L, et al. , Poly(ADP-ribose) signaling in cell death. Mol Aspects Med, 2013. 34(6): p. 1153–67. [DOI] [PubMed] [Google Scholar]
- 79.Thomas C, et al. , Hit and run versus long-term activation of PARP-1 by its different domains fine-tunes nuclear processes. Proc Natl Acad Sci U S A, 2019. 116(20): p. 9941–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karanja KK, et al. , DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle, 2012. 11(21): p. 3983–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amangyeld T, et al. , Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res, 2014. 42(9): p. 5846–62. [DOI] [PMC free article] [PubMed] [Google Scholar]