

Abstract
Objective
High-throughput gene expression profiling of tissue samples from patients with systemic sclerosis (SSc) has identified four “intrinsic” gene expression subsets: inflammatory, fibroproliferative, normal-like, and limited. Prior methods required agglomerative clustering of many samples. In order to classify individual patients in clinical trials or for diagnostic purposes, supervised methods that can assign single samples to molecular subsets are required. We introduce a novel machine learning classifier as a robust accurate intrinsic subset predictor.
Methods
Three independent gene expression cohorts were curated and merged to create a dataset covering 297 skin biopsies from 102 unique patients and controls to train a machine learning algorithm. We performed external validation using three independent SSc cohorts, including a gene expression dataset generated by an independent laboratory on a different microarray platform. In total, 427 skin biopsies from 213 individuals were analyzed in the training and testing cohorts.
Results
Repeated cross-fold validation identified consistent and discriminative markers using multinomial elastic net, performing with average classification accuracy of 88.1% with high sensitivity and specificity. In external validation, the classifier achieves average accuracy of 85.4%. Reanalyzing data from Assassi et al. study, we identified subsets of patients that represent the canonical inflammatory, fibroproliferative, and normal-like subsets.
Conclusion
We developed a highly accurate classifier for SSc molecular subsets for individual patient samples. The method can be used in SSc clinical trials to identify intrinsic subset on individual samples. Our method provides a robust data-driven approach to aid clinical decision-making and interpretation of heterogeneous molecular information from SSc patients.
Introduction
Systemic sclerosis (SSc) is a complex autoimmune, connective tissue disease characterized by skin fibrosis and internal organ dysfunction, vascular damage, and immunologic abnormalities. Patients are classified clinically according to the extent of skin involvement into limited cutaneous SSc (lcSSc) patients and diffuse cutaneous SSc (dcSSc) (1).
To further characterize disease heterogeneity and pathogenesis, transcriptomics has elucidated common biological processes in subsets of SSc patients using intrinsic gene expression analysis. Four intrinsic molecular subsets, identified through gene expression profiling in skin samples, are characterized by distinct molecular signatures and have been validated by multiple studies (2–5). Subset is consistent across the different skin biopsy sites within a single patient, regardless of clinically affected or unaffected status demonstrating the systemic nature of the disease (2–4, 6). These subsets have also been found across organ systems in analyses of multiple tissues (7, 8). The inflammatory subset is defined by enrichment in immune system response, inflammatory response, and vascular development (3). The fibroproliferative subset is characterized by increased expression of proliferative processes including cell cycle, mitosis, and chromosome segregation. The normal-like subset is composed of samples from SSc patients whose gene expression most closely resembles that of healthy controls. This subset has previously been characterized by fatty acid metabolism and lipid metabolism, though not consistently (5, 9). The limited subset consists exclusively of lcSSc patients and is the least characterized in terms of unique molecular signatures. Importantly, lcSSc patients can also be assigned to the inflammatory and normal-like subsets.
To date, there are no FDA-approved disease-modifying treatments for SSc (10). Although overall survival and treatment strategies for SSc are improving, the power in clinical trials is often compromised by patient heterogeneity. Following a clinical diagnosis of SSc, immunotherapeutic treatment regimens are often intense and exploratory; in addition to delayed relief, patients risk adverse side effects throughout this experimental approach. Only recently have clinical trials begun to consider molecular heterogeneity in the interpretation of outcomes, which may explain improvement in a subset of SSc patients and may identify patients that will improve naturally as part of their disease course (11). Thus, SSc is an ideal example of a disease in which outcomes may be improved by tools that will aid personalized medicine, especially in the context of molecular subsets.
The inflammatory intrinsic gene expression subset has been associated with response to immune modulating therapies. For example, Hinchcliff et al. showed that four out of seven patients treated with mycophenolate mofetil improved, and all four were classified as inflammatory at baseline (4). Additionally, four out of five improvers in a placebo-controlled study of abatacept were assigned to the inflammatory subset (12). Gordon et al. demonstrated that transitions between intrinsic subsets correlated strongly with clinical improvements in a randomized, double-blind, placebo-controlled trial of belimumab with mycophenolate mofetil background therapy (13). Specifically, movement from the inflammatory or fibroproliferative subsets to the normal-like subset strongly correlated with decreases in modified Rodnan Skin Score (MRSS). The use of genomic data and intrinsic subsets may help improve patient outcomes by identifying therapies with higher potential for success in each individual patient. Furthermore, longitudinal tracking of intrinsic subset assignment may provide insight to SSc pathogenesis and overall disease trajectory.
Landmark studies that first assigned intrinsic subsets in SSc used agglomerative methods including intrinsic gene analysis and unsupervised clustering algorithms to determine the number of intrinsic subsets and each sample’s membership to a subset (2–4, 14, 15). There are several limitations with these methods. First, intrinsic gene analysis requires paired samples from each individual (e.g. forearm and back skin biopsies). Paired skin samples are often not available in the setting of clinical trials. Second, the most “intrinsic” genes are agnostically derived from the samples in each dataset, and often the exact list differs between studies, although some genes are commonly found and biological processes are consistent (5). Third, unsupervised clustering algorithms rely on the assumption that at least two intrinsic subsets are represented in the dataset. This often requires a large number of samples (i.e. n>70) samples for all four intrinsic subsets to arise and be distinguishable in a dataset.
In order to classify patients in pilot clinical trials or for diagnostic purposes, supervised methods that can assign an individual sample to an intrinsic molecular subset are required. Here we have developed a method to assign single samples to intrinsic gene expression subsets using carefully curated and defined criteria using machine learning. The method uses a multinomial elastic net classifier and an optimized set of genes for assigning samples to intrinsic gene expression subset using objective molecular genomic data.
Methods
Training data set curation and preprocessing.
DNA microarray data (2–4) (described in Table 1) were collected with at least 80% probes passing filter, analyzed as log2 LOWESS-normalized Cy5/Cy3 ratios, and multiplied by −1 to convert them to log2(Cy3/Cy5) ratios. Each dataset was processed separately using the following pipeline from GenePattern (16): missing values were imputed using K-Nearest Neighbors with default settings, the CollapseDataset module was run using median collapse mode, and genes were median-centered. Datasets were combined using only genes present in all three sets. Arrays from morphea and eosinophilic fasciitis (EF) patients were excluded as well as any arrays which were not assigned to a subset in the original analyses. All gene expression data have previously been published on GEO.
Table 1.
Studies included in this analysis. Arrays were excluded if no subset classification was done in the original analysis or if the patient was diagnosed with morphea and/or EF.
| Dataset | GEO Accession | Samples | Platform | # Arrays Excluded | # Arrays Included | Purpose |
|---|---|---|---|---|---|---|
| Milano A, et al. (2008) | GSE9285 | 75 | Agilent (4×44k) | 13 | 62 | Training |
| Pendergrass S, et al. (2012) | GSE32413 | 89 | Agilent (4×44k) | 13 | 76 | Training |
| Hinchcliff M, et al. (2013) | GSE45485 | 165 | Agilent (4×44k) | 6 | 159 | Training |
| Chakravarty EF, et al. (2015) | GSE66321 | 16 | Agilent (8×60k) | 8 | 8 | Testing |
| Gordon J, et al. (2015) | GSE65405 | 12 | Agilent (8×60k) | 6 | 6 | Testing |
| Assassi S, et al. (2015) | GSE58095 | 102 | Illumina HT-12 v4 | 0 | 102 | Testing |
Classifier training and evaluation.
The KernSmooth (17), glmnet (18), random forest (19), and caret (20) packages implemented in R were used to train supervised classifiers. The support vector machine (SVM) was trained with a linear kernel. GLMnet and random forest were run with default parameters. Repeated cross-validation (10x, 3-fold) was used to train the model and simultaneously assess robustness. Accuracy metrics were measured across all repeated cross-validated folds.
External Validation.
We compared subset assignments made by our model to those reported in the studies described in Table 1. GSE65405 and GSE66321 were profiled using Agilent 8×60k array technology, an updated version of the platform used in the studies to train the classifier. We calculated concordance and Cohen’s kappa coefficient based on the intrinsic subset assignment determined by the classifier and compared to the intrinsic subset information from the original publication. GSE58095 was generated using Illumina HT-12 v4. We downloaded GSE58095 from NCBI Gene Expression Omnibus (GEO). GenePattern (16) was used to impute missing values in the dataset using K-nearest-neighbors algorithm and collapse probes to genes using a CHIP file for Illumina HT-12 v4.
SSc Subset Molecular Signatures.
Ranked genes with positive, non-zero coefficients in the final model for each molecular subtype were analyzed with g:Profiler using default g:SCS threshold (21). To further validate the gene signatures, we identified modules (groups of co-expressed genes) using weighted gene co-expression network analyses (WGCNA). We identified modules associated with molecular subsets using biweight midcorrelation with the bicor function in WGCNA R package (22). Modules were annotated with significant biological processes identified through g:Profiler. The entire workflow is shown on Figure 1.
Figure 1: Overall Study Schematic.
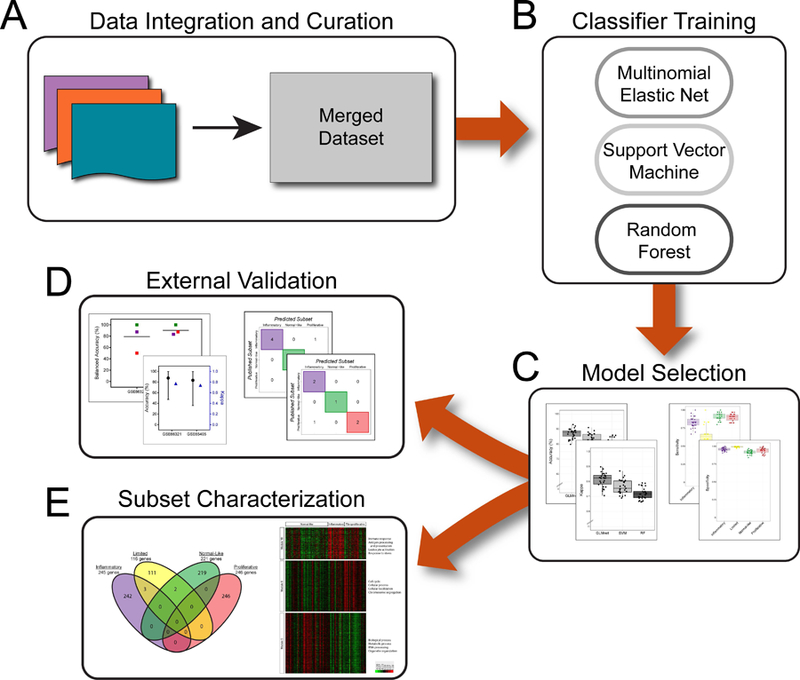
Gene expression data from three independent studies (A) were merged into a single dataset used to train a variety of machine learning classifiers (B). A final model was selected (C) and externally validated on other published gene expression data (D) and used to further characterize the intrinsic subsets (E).
Results
Dataset Curation.
Our goal for this study was to create the first validated classifier for the intrinsic molecular subsets of SSc using supervised machine learning algorithms. In order to train a broadly applicable classifier, we identified large gene expression datasets from three independent studies (Table 1) and developed a machine learning classifier using an optimized scheme (Figure 1). Many clinical studies are characterized by unique and specific inclusion criteria, and these criteria lead to limitations for generalization. While increasing the reliability of results, these criteria often result in a dataset of patients that do not represent the full spectrum of disease. By merging data from three studies, we are confident that our training dataset reflects a broad spectrum of SSc (Figure 2). We only included samples with definitive intrinsic subset labels, as determined in each respective original analysis (Table S2). Our final training dataset contained gene expression data for 11,430 genes across 297 microarrays from 102 unique patients. These arrays represent all four intrinsic gene expression subsets (Figure 2A, Figure S1), although the limited intrinsic subtype is somewhat underrepresented. The other three intrinsic subsets are well-balanced in the number of samples: 71 inflammatory, 102 fibroproliferative, and 107 normal-like. The 107 samples in the normal-like intrinsic subset represent both healthy controls (n=49) and SSc patients (n=58) that had a normal-like subset label. The patients in our cohort represent a diverse group based on age, sex, disease duration, and extent of skin involvement (Fig 2B, 2C; Table S1). We used guided Principal Component Analysis (gPCA) to determine if a significant batch effect existed as a result of combining three independent studies (23), and we did not see a significant study bias (p=0.993, gPCA) (Figure 2D).
Figure 2: Merge of Training Datasets.
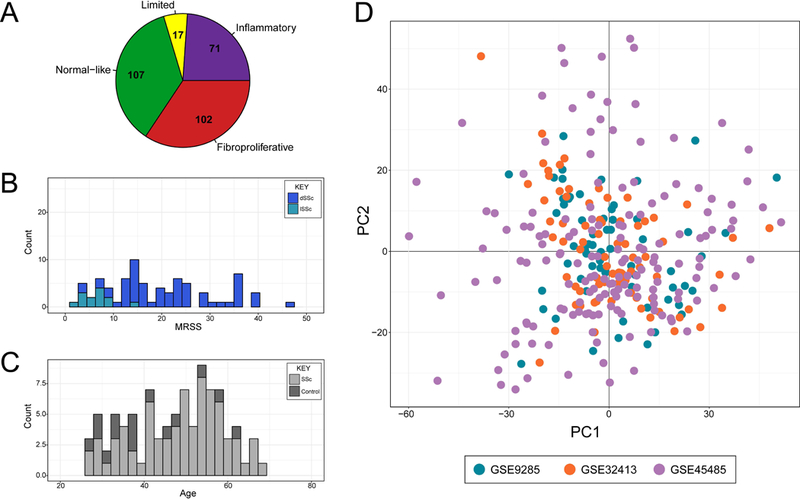
The merged dataset contains arrays from all four intrinsic subsets (A). The data are representative of a range in skin score severity as designated by MRSS (B) and a wide range of age for both SSc patients and controls (C). The first and second dimensions of a Principal Component Analysis are plotted and colored by dataset of origin (D).
Training Machine Learning Classifiers.
We trained supervised classifiers including multinomial elastic net (GLMnet), support vector machine (SVM) and random forest (RF), because they represent a popular and diverse set of machine learning algorithms. Initial evaluation of the classifiers was done using the performance over repeated cross-validation. We found that GLMnet outperforms SVM and RF in iterations of training in both average accuracy and kappa (Figure 3A, 3B). Therefore, we selected GLMnet as the primary classifier to further validate SSc intrinsic subsets using DNA microarray data. GLMnet displays high overall sensitivity and specificity for all intrinsic subsets (Figure 3C, 3D). Sensitivity and specificity for each intrinsic subset in the cross-validation of SVM and RF are shown in Figure S1. Specifically, GLMnet attains 83.3% sensitivity and 95.8% specificity for the inflammatory subset and 89.7% sensitivity and 94.1% specificity for the fibroproliferative subset. In contrast to the other subsets, the limited subset shows a greater range in classification sensitivity during training (Figure 3C). This is most likely due to fewer limited subset samples in the training set. Additionally, the limited subset has not been consistently associated with specific outcomes in SSc clinical trials and only represents a very small proportion of lcSSc patients, because lcSSc patients can also be classified as inflammatory or normal-like. Thus, the variable classification power for the limited subset is neither surprising nor of great importance. Interestingly, SVM is slightly more sensitive in detecting the limited intrinsic subset (Figure S1) than GLMnet and RF, but we ultimately selected GLMnet as the best model for DNA microarray data due to superior performance in the remaining three subsets. Notably, the SVM has slightly better performance on RNA-seq data using a small testing set (Table S7).
Figure 3: Model selection and validation.
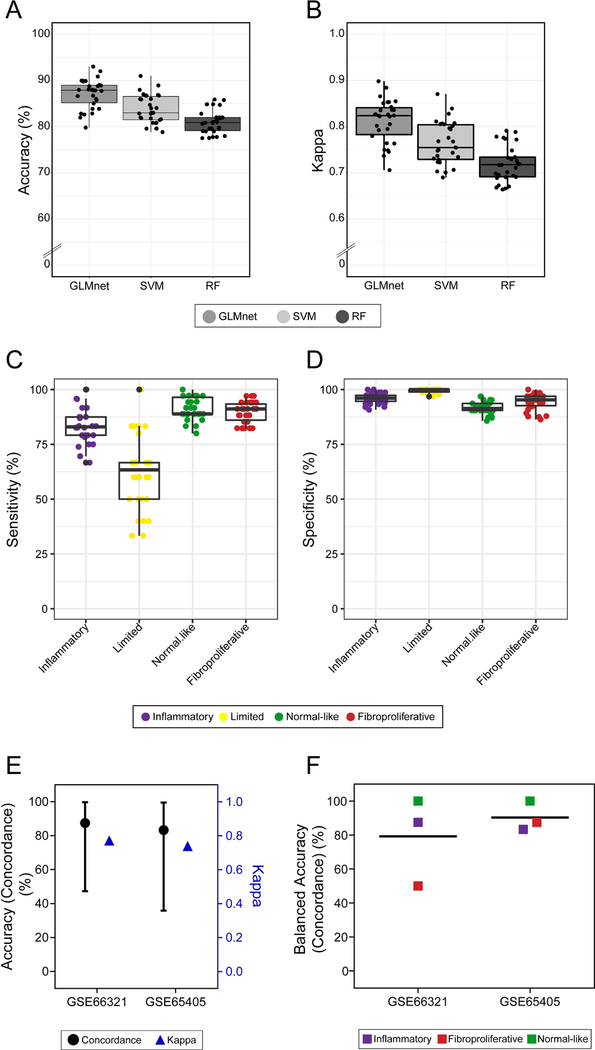
Accuracy (A) and Cohen’s Kappa (B) were calculated for each model during repeated cross-fold validation. Sensitivity (C) and specificity (D) for all four intrinsic SSc subsets are plotted from repeated cross-fold validation for the final model. In external validations, concordance of intrinsic subset assignment between the original publications and our analysis is plotted in black and Cohen’s Kappa in blue (E). Balanced accuracy (concordance) is shown for each of the intrinsic subsets represented in each dataset (F).
Characterizing Molecular Signatures.
GLMnet, through rigorous training, selects the most consistent and discriminative genes to assign intrinsic subsets across multiple cohorts. We identified those genes that were important for prediction of SSc intrinsic subsets by selecting genes with positive, non-zero coefficients from the final model (Table 2, Table S5). These gene lists were used to determine the significant biological processes for discriminating SSc molecular subsets. 245 genes were positively associated with prediction of the inflammatory subset and were annotated to Gene Ontology biological processes of immune system response, response to stress, and inflammatory response. Importantly, fibrotic processes such as angiogenesis, cell adhesion, and response to wounding are also up-regulated in the inflammatory subset, consistent with typical clinical presentations of early and active SSc. 246 genes were positively associated with prediction of the fibroproliferative subset. Functional terms including metabolic pathways and cellular process are up-regulated for the fibroproliferative subset. Although the fibroproliferative subset gene signature is not significantly enriched in proliferative processes, it still successfully identifies samples previously assigned to this subtype. In the normal-like subset, housekeeping processes such as electron transport chain and cellular respiration are highly expressed. These results validate earlier characterizations of each respective subset. We observed very few genes that overlapped gene lists for each molecular subset (Figure S3). This further implicates that the SSc molecular subsets represent distinct biological states.
Table 2.
Genes for profiling molecular pathways enriched in each intrinsic subset were selected using the ranked positive, non-zero coefficients in the final model.
| Subset | Summary of Significant Biological Processes (g:SCS corrected p< 0.05, g:Profiler) | Select Genes |
|---|---|---|
| Inflammatory | Response to stress, response to wounding, immune system process, inflammatory response, defense response, angiogenesis | CD33, CD52, CXCL2, CXCR4, CXCR3, CTGF, FN1, IL6, THBS1, COL11A1, COL8A2, VCAM1, SYK, SPHK1 |
| Fibroproliferative | Metabolic process, cellular metabolic process, ncRNA metabolic process, mitochondrial gene expression | CENPV, CXCL1, COMP, POLR1B, SPIN2B, MTOR, ALAD, TSFM, ELAC2 |
| Normal-like | Electron transport chain, cellular respiration | SP5, COX5B, NDUFV3, GPD2, ETFA |
| Limited | Actin filament depolymerization | FBLN1, STAT6, TRIM46, SPTBN1, GSN, VILL |
External Validation of SSc Molecular Subset Classifier.
In order to test the predictive accuracy of our classifier, we sought validation using additional published DNA microarray data from SSc skin samples with assigned molecular subsets (Table 1). Chakravarty et al. and Gordon et al. were small investigator-initiated clinical trials where intrinsic subset was determined by calculating Spearman correlations between each sample’s gene expression and the centroid of gene signature associated with each intrinsic subset from the Milano et al. study (2, 12, 24). Unfortunately, the subset labels in these publications do not represent a true gold standard for assessing classifier accuracy. Thus, for this study, we use concordance of samples being assigned to the same subset as a substitute measure of accuracy. We also report Cohen’s kappa coefficient as a robust measure of performance, which takes into account multiple classes and the possibility of agreement by chance.
Chakravarty et al. was an investigator-initiated pilot clinical trial of abatacept and contains gene expression data (GSE66321) with intrinsic subset assignments for eight SSc patients (12). Because it is not completely understood how abatacept therapy affects intrinsic subset assignment, we included only the baseline samples for each patient. Additionally, the accuracy metrics from baseline samples are the most relevant in the context of analyses for clinical trials and intrinsic subsets as potential diagnostic and/or prognostic biomarkers. No significant study bias existed between the original training dataset and the new data (p=0.989, gPCA); therefore, no batch correction measures were taken. We then assigned intrinsic subset labels to each baseline sample using GLMnet and compared these to the intrinsic subset labels reported in the original publication (Table S3, Figure S4A). We find that only one sample is classified differently with a change in subset from inflammatory to fibroproliferative, giving an overall concordance of 87.5% and kappa of 0.7714 (Figure 3E). The balanced accuracy for each class is very high for inflammatory (87.5%) and normal-like (100%) intrinsic subsets, and is lower for the fibroproliferative subset (50.0%) in this external validation because only one patient was classified as fibroproliferative (Fig 3F).
For a second validation dataset, Gordon et al. was an investigator-initiated clinical trial of nilotinib that contains gene expression data (GSE65405) and intrinsic subsets for six SSc patients (24). Again, there was no study bias between the testing and training data (p=1, gPCA). We find an overall concordance of 83.33% and kappa of 0.7391 (Figure 3E). Only one sample is classified differently from the original analysis, with a change in subset from fibroproliferative to inflammatory (Table S4, Figure S4B). There is high balanced accuracy among the three intrinsic subsets present in this dataset (inflammatory: 83.3%, fibroproliferative: 87.5%, normal-like: 100%) (Figure 3F). Overall, GLMnet performs consistently well, despite small sample sizes and unbalanced classes.
SSc Molecular Subsets from Assassi et al.
We further tested the predictive power of our algorithm by classifying samples with gene expression data generated by an independent lab using a different DNA microarray platform (25). This gene expression dataset (GSE58095) contains 102 samples from 97 individuals. The original study identified subsets of patients in this cohort, which they labeled as “keratin”, “fibro-inflammatory”, and “normal-like” using a different subsetting approach. GLMnet assigned intrinsic subsets to each of the 102 samples; 22 samples were classified as inflammatory, 27 samples were fibroproliferative, and 53 samples were normal-like. Of the 36 healthy control samples, 29 were correctly classified as normal-like, giving an accuracy of 80.6%. Additionally, this dataset contained paired early and late samples from five patients. For these longitudinal samples, three SSc patients were assigned to the same intrinsic subset at both time points and two SSc patients changed from inflammatory or fibroproliferative to normal-like (Table S6). This change to normal-like subset over time may represent natural disease process or response to therapy, but more samples and additional studies are required to understand this variation in subset assignment.
Because there were no gold-standard intrinsic subset assignments for the Assassi et al. cohort with which to compare the GLMnet classifier labels for a final validation of accuracy, we undertook an independent, data-driven procedure to infer the underlying structure of the raw data. In this analysis, we wanted to identify the major gene expression signatures associated with each intrinsic subset in order to evaluate agreement with the previous characterizations of the canonical intrinsic gene expression subsets. First, we used WGCNA to identify modules of co-expressed genes as previously described (5, 8, 22). Then, the GLMnet molecular classifier was used to assign each sample in the dataset to an intrinsic subset. We identified gene modules associated with each intrinsic subset using biweight midcorrelation of the module eigengenes. Further results of this analysis and additional details are shown in Supplemental Materials (Figure S5).
We find that several modules significantly correlate to the intrinsic subsets and represent the previously defined distinct biological processes (Figure 4). Module 10 highly correlates to the inflammatory intrinsic subset, and the module eigenvalues are significantly higher for the inflammatory samples compared to the other subsets (p=6.433E-9, Wilcoxon). This module contains 739 genes, enriched in GO biological processes such as inflammatory response, leukocyte activation, and response to stress. Many of the genes in Module 10 have been shown to be important for inflammatory processes and in SSc, including COL4A1, TGFB3, HLA-DRA, COMP, and IL10RB (26–28). Module 6 is highly correlated to the fibroproliferative subset, and the module eigenvalues are significantly higher for samples labeled fibroproliferative compared to the other subsets (p=1.124E-5, Wilcoxon) and is enriched for biological processes including cell cycle, cellular process, and chromosome segregation. Among this module’s 1431 genes are CDC20, STAT3, CDK10, APOE, IRF3, USP4, MYST1, CYC1, and FBRS. Module 5 highly correlated to the normal-like subset, and the module eigenvalues are significantly higher for the normal-like samples compared to the other subsets (p=1.458E-8, Wilcoxon). This module is enriched for general cellular processes including organelle organization, RNA processing, and metabolic process. These results corroborate the findings of Mahoney et al., where the most consistent biological processes relevant to SSc were enriched in genes with increased expression in SSc that were significantly correlated with the inflammatory and fibroproliferative subsets (5).
Figure 4: Module analysis of Assassi, et al. (2015).

Expression values are shown for gene annotated to modules 10, 6, and 5 (A). The module eigengene of each module is shown in a stem-plot below each heatmap. The module eigenvalues were further compared for the inflammatory samples vs all others (B), fibroproliferative samples vs all others (C), and normal-like samples vs all others (D) using Wilcoxon Rank Sum Test (***p<0.0005).
Additionally, we performed unsupervised hierarchical clustering using the genes from the GLMnet classifier (Figure S6). The clustering shows distinct gene expression signatures associated with the intrinsic subset calls. Interestingly, there are some samples (particularly in the fibroproliferative subset) which appear to also have upregulated inflammatory signatures, which is consistent with the findings in Assassi et al. (25) and with our results from earlier publications.
Finally, we mapped the GLMnet genes to the gene modules identified in the WGCNA analysis (Figure S7). The GLMnet genes are fairly evenly dispersed throughout many modules. This indicates that the genes selected in the model provide a whole-genome summary of gene expression and include important genes with non-redundant information.
Overall, these analyses result in a GLMnet classification method that is reproducible across multiple DNA microarray platforms and experiments, by which we can assign intrinsic gene expression subsets to SSc patients in clinical trials or for diagnostic purposes. This tool will allow for the identification of the patients most likely to respond to a given therapy using molecular measures.
Discussion
To our knowledge, this is the first published classifier for the intrinsic molecular subsets in SSc. This study represents improvement over previous approaches which required paired samples from many individuals and rigorous computational analyses through unsupervised clustering algorithms to identify intrinsic gene expression subsets. Moreover, previous studies relied on the assumption that multiple intrinsic subsets were present in the cohort. Our classifier uses defined criteria based on gene expression signatures trained from a large compendium of curated data. It accurately classifies single samples and does not make the assumption that all intrinsic subsets are present in all datasets. This is particularly important for small, pilot clinical trials in SSc.
SSc Subsets, Clinical Outcomes, and Translation to the Clinic.
In the context of immunosuppressive therapy, molecular heterogeneity may explain improvement in select SSc patients (11). Representing distinct pathway signatures, the intrinsic subsets are a logical and meaningful way to interpret the overall picture of global gene expression in patients with SSc. Our validated model accurately classifies single samples which will ultimately improve the speed and reproducibility of computational analyses and guide interpretation of clinical response in the context of intrinsic subsets.
The ability to classify a single sample from individual patients, as needed, is key to implementing such methods in routine clinical care. Performing genomic assays and assigning subsets in a rigorously controlled CLIA-certified laboratory is also needed to carefully oversee all aspects of the process and ensure that accurate results are generated. We believe the classification model we have developed here may allow personalized medicine in SSc by using intrinsic subsets to help guide the treatment and management of SSc. Our classification model is already being applied in SSc clinical trials and results for each trial will be published separately as part of those consortia. The method has been designed to work on a wide range of genomic platforms so that it is possible to classify any SSc patient with genomic level mRNA expression data (see below). We have previously shown that SSc intrinsic subset can predict response in small investigator-initiated clinical trials (4, 12, 13), and we are further testing this prediction in large randomized, placebo-controlled clinical trials. If intrinsic subset is shown to predict therapeutic response for a particular therapy in a rigorously controlled clinical trial, then intrinsic subset assignment could be done early in a patient’s disease to determine the patients most likely to benefit from certain therapies. This could have the benefit of getting patients onto the most effective therapy early, ultimately leading to faster and improved patient outcomes. It may be most impactful for therapies that have significant adverse side effects or which are of very high cost (e.g. stem-cell transplant and biologics).
Cross-Platform Considerations.
Over time, updates in gene expression profiling technology have improved overall data detection and quality. Namely, RNA-sequencing (RNA-seq) enables detection of novel transcripts as well as better detection of highly and lowly expressed transcripts, which leads to increased sensitivity and specificity. However, due to differences in methods of transcript quantification, there are significant differences in data distributions, which violate statistical assumptions important for machine learning methods. Thus, several considerations should be made in applying our methods to data generated from a different platform. Data should be examined for the existence of platform-related batch effects. We recommend feature specific quantile normalization (FSQN) for eliminating platform-based bias and increasing the comparability between two platforms (29). FSQN is a powerful and robust method that allows for highly accurate intrinsic subtype classification even in small datasets, which is an important factor for SSc analyses. Additionally, the SVM reported in this study, which retains all genes in the model, may provide more accurate results when assigning samples to intrinsic subsets from RNA-seq data. See Supplemental Materials for additional details (Table S7).
Molecular Biomarker Identification.
Intrinsic molecular subsets are a reproducible feature of SSc skin gene expression (2–5), and this study further validates the previously defined subsets through analysis of an independent gene expression dataset generated on a different DNA microarray platform. Our study is the first to build a classification model for accurate intrinsic subset classification for single samples in SSc skin. Although most bioinformatics efforts have focused on profiling the gene expression in SSc skin, there is substantial evidence to suggest that the intrinsic subsets and the immune-fibrotic axis span multiple affected SSc organs (7, 8). Further efforts are needed to explore molecular heterogeneity and intrinsic subsets in other tissues and particularly in peripheral blood, given its accessibility. The results of our study are proof of principle that it is feasible to identify a common set of genes sufficient for SSc subset classification. As with many rare diseases, the amount of gene expression data is quite limited and identifying a smaller set of genes for a biomarker panel is very difficult. With many more features than samples, overfitting the training data is of great concern. Additional work, including the integration of more gene expression data, will be necessary to further refine a gene expression-based biomarker panel for SSc intrinsic subset classification. In conclusion, this body of work represents an important step toward diagnostic testing for precision medicine in SSc.
Supplementary Material
Acknowledgements:
The authors would like to thank Jaclyn Taroni and Diana Toledo for helpful discussions.
Funding: This work was supported by the Scleroderma Research Foundation, Burroughs-Wellcome Big Data in the Life Sciences Training Program, the National Institutes of Health Big Data to Knowledge (5T32LM012204-03), and the Dr. Ralph and Marian Falk Medical Research Trust Catalyst and Transformational Awards.
Dr. Whitfield has received consulting fees from Bristol-Myers Squibb, Boehringer Ingelheim, Corbus Pharmaceuticals, Third Rock Ventures (less than $10,000 each) and from Celdara Medical LLC and UCB Biopharma (more than $10,000 each).
Footnotes
Disclosures: Ms. Franks, Dr. Martyanov, Dr. Cai, Dr. Wang, Dr. Li, and Ms. Wood have nothing to disclose.
References
- 1.Varga J, Denton CP, Wigley FM, Alanore Y, Kuwana M. Scleroderma : from pathogenesis to comprehensive management Second edition. ed. Cham: Springer; 2017. xxxv, 743 pages p. [Google Scholar]
- 2.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One 2008;3(7):e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol 2012;132(5):1363–73. doi: 10.1038/jid.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 2013;133(8):1979–89. doi: 10.1038/jid.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol 2015;11(1):e1004005. doi: 10.1371/journal.pcbi.1004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A 2003;100(21):12319–24. doi: 10.1073/pnas.1635114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taroni JN, Martyanov V, Huang CC, Mahoney JM, Hirano I, Shetuni B, et al. Molecular characterization of systemic sclerosis esophageal pathology identifies inflammatory and proliferative signatures. Arthritis Res Ther 2015;17:194. doi: 10.1186/s13075-015-0695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, et al. A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med 2017;9(1):27. doi: 10.1186/s13073-017-0417-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson ME, Mahoney JM, Taroni J, Sargent JL, Marmarelis E, Wu MR, et al. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS One 2015;10(1):e0114017. doi: 10.1371/journal.pone.0114017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390(10103):1685–99. doi: 10.1016/S0140-6736(17)30933-9. [DOI] [PubMed] [Google Scholar]
- 11.Martyanov V, Whitfield ML. Molecular stratification and precision medicine in systemic sclerosis from genomic and proteomic data. Curr Opin Rheumatol 2016;28(1):83–8. doi: 10.1097/BOR.0000000000000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakravarty EF, Martyanov V, Fiorentino D, Wood TA, Haddon DJ, Jarrell JA, et al. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res Ther 2015;17:159. doi: 10.1186/s13075-015-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon JK, Martyanov V, Franks JM, Bernstein EJ, Szymonifka J, Magro C, et al. Belimumab for the Treatment of Early Diffuse Systemic Sclerosis: Results of a Randomized, Double-Blind, Placebo-Controlled, Pilot Trial. Arthritis Rheumatol 2018;70(2):308–16. doi: 10.1002/art.40358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 16.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet 2006;38(5):500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 17.Wand M KernSmooth: Functions for Kernel Smoothing Supporting Wand & Jones (1995) 2015.
- 18.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
- 19.Liaw A, Wiener M Classification and Regerssion by randomForest. R News 2002;2(3):18–22. [Google Scholar]
- 20.Kuhn M Building Predictive Models in R Using the caret Package. J Stat Softw 2008;28(5):1–26.27774042 [Google Scholar]
- 21.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 2016;44(W1):W83–9. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reese SE, Archer KJ, Therneau TM, Atkinson EJ, Vachon CM, de Andrade M, et al. A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics 2013;29(22):2877–83. doi: 10.1093/bioinformatics/btt480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon JK, Martyanov V, Magro C, Wildman HF, Wood TA, Huang WT, et al. Nilotinib (Tasigna) in the treatment of early diffuse systemic sclerosis: an open-label, pilot clinical trial. Arthritis Res Ther 2015;17:213. doi: 10.1186/s13075-015-0721-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 2015;67(11):3016–26. doi: 10.1002/art.39289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lafyatis R Transforming growth factor beta--at the centre of systemic sclerosis. Nat Rev Rheumatol 2014;10(12):706–19. doi: 10.1038/nrrheum.2014.137. [DOI] [PubMed] [Google Scholar]
- 27.Hesselstrand R, Kassner A, Heinegard D, Saxne T. COMP: a candidate molecule in the pathogenesis of systemic sclerosis with a potential as a disease marker. Ann Rheum Dis 2008;67(9):1242–8. doi: 10.1136/ard.2007.082099. [DOI] [PubMed] [Google Scholar]
- 28.Hikami K, Ehara Y, Hasegawa M, Fujimoto M, Matsushita M, Oka T, et al. Association of IL-10 receptor 2 (IL10RB) SNP with systemic sclerosis. Biochem Biophys Res Commun 2008;373(3):403–7. doi: 10.1016/j.bbrc.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 29.Franks JM, Cai G, Whitfield ML. Feature Specific Quantile Normalization Enables Cross-Platform Classification of Molecular Subtypes using Gene Expression Data. Bioinformatics 2018. doi: 10.1093/bioinformatics/bty026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.