

Abstract
As a model organism for studies of cell and environmental biology, the free-living and cosmopolitan ciliate Euplotes vannus shows intriguing features like dual genome architecture (i.e. separate germline and somatic nuclei in each cell/organism), “gene-sized” chromosomes, stop codon reassignment, programmed ribosomal frameshifting (PRF) and strong resistance to environmental stressors. However, the molecular mechanisms that account for these remarkable traits remain largely unknown. Here we report a combined analysis of de novo assembled high-quality macronuclear (MAC; i.e. somatic) and partial micronuclear (MIC; i.e. germline) genome sequences for E. vannus, and transcriptome profiling data under varying conditions. The results include: 1) the MAC genome contains more than 25,000 complete “gene-sized” nanochromosomes (~85 Mb haploid genome size) with the N50 ~2.7 kb; 2) though there is a high frequency of frameshifting at stop codons UAA and UAG, we did not observe impaired transcript abundance as a result of PRF in this species as has been reported for other euplotids; 3) the sequence motif 5’-TA-3’ is conserved at nearly all internally-eliminated sequence (IES) boundaries in the MIC genome, and chromosome breakage sites (CBSs) are duplicated and retained in the MAC genome; 4) by profiling the weighted correlation network of genes in the MAC under different environmental stressors, including nutrient scarcity, extreme temperature, salinity and the presence of ammonia, we identified gene clusters that respond to these external physical or chemical stimulations; 5) we observed a dramatic increase in HSP70 gene transcription under salinity and chemical stresses but surprisingly, not under temperature changes; we link this temperature-resistance to the evolved loss of temperature stress-sensitive elements in regulatory regions. Together with the genome resources generated in this study, which are available online at Euplotes vannus Genome Database (http://evan.ciliate.org), these data provide molecular evidence for understanding the unique biology of the highly adaptable microorganisms.
Keywords: Ciliated protist, genome rearrangement, environmental stress, frameshifting
1. INTRODUCTION
Single-celled microorganisms were the first forms of life that developed on Earth approximately ~3.2 billion years ago, firstly as prokaryotic forms and then evolved into eukaryotic cells between 1.4–2.0 billion years ago (Cavalier-Smith 2006; Schopf et al., 2018). As an important part of the bulk biodiversity on Earth, microbes play crucial roles in biogeochemical cycles and ecosystems, and can be found almost anywhere, even in extreme environments (Hu 2014). Therefore, microbes have been evolving and developed a wide variety of biological mechanisms to survive in the long history of the Earth. One of the most diverse and highly differentiated group among single-celled microorganisms are ciliates, which emerged approximately one billion years ago (Parfrey et al., 2011) and are abundant in diverse habitats across the globe, where they are among the most important components of food webs in aquatic ecosystems (Gao et al., 2017; Lynn 2009). Ciliate diversity, physiology and abundance have been linked to studies of environmental change (Gong et al., 2005; Xu et al., 2014), pollution monitoring (Gutiérrez et al., 2003; Jiang et al., 2011; Stoeck et al., 2018), biogeography (Foissner et al., 2008; Liu et al., 2017; Petz et al., 2007), adaptive evolution (Clark & Peck 2009; He et al., 2019), cell biology (Jiang et al., 2019; Wang et al., 2017a; Zheng et al., 2018) and epigenetics (Wang et al., 2017c; Xiong et al., 2016; Zhao et al., 2017).
As a monophyletic clade, ciliates show several intriguing features. First, ciliates possess both the compact germline micronucleus (MIC) and the transcriptionally active somatic macronucleus (MAC) within each cell (Katz 2001; Prescott 1994). The streamlined and efficient MAC genome is developed from a zygotic nucleus during sexual reproduction (conjugation) through a series of genome wide rearrangements, including chromosome fragmentation, micronuclear DNA elimination, and DNA amplification (Chalker & Yao 2011; Chen et al., 2014; Nowacki et al., 2011; Riley & Katz 2001). However, conflicting models suggest a variety of mechanisms for genome rearrangement within the investigated ciliates (Chen et al., 2014; Feng et al., 2017; Maurer-Alcalá et al., 2018), Second, the nuclear genetic code in ciliates is diversified and flexible as standard stop codons are often reassigned to amino acids; even stranger, in some ciliates all three standard stop codons can either code for amino acid or terminate translation in a context-dependent manner (Swart et al., 2016). More relevant for the current study, euplotid ciliates exhibit widespread programmed ribosomal frameshifting (PRF) at stop codons, 60-fold higher than other organisms, for instance, human, mouse, flies, C. elegans, yeast and E. coli (Wang et al., 2016). Stop codon is indicated not sufficient for translation termination in euplotids and frameshifting is sequence context-dependent (Lobanov et al., 2017). Third, ciliates have great ability to survive in a wide range of harsh conditions, and feature strong tolerance to an array of environmental stressors. Many of these conditions, such as heavy metal contamination, are believed to induce evolutionarily conserved molecular defense mechanisms (Kim et al., 2018). It is therefore important to elucidate the molecular mechanisms employed by the single cells in response to external stresses.
In the present study, we analyze the germline (MIC) and somatic (MAC) genomes of euplotid ciliates to reveal the deeper mechanisms of how single celled organisms survive in diverse environments. Euplotids, one of the most common families of free-living ciliates, play important roles as both predators of microalgae and preys of multicellular eukaryotes in global waters (Dhanker et al., 2013; Sheng et al., 2018; Zhao et al., 2018). They have been widely used for decades as model organisms in studies of predator/prey relationships (Kusch 1995; Wiąckowski & Szkarłat 1996), cell signaling (Hadjivasiliou et al., 2015; Jerka-Dziadosz et al., 1987), toxicology of marine pollutants (Trielli et al., 2007) and experimental ecology (Day et al., 2017; Walton et al., 1995; Xu et al., 2004). Euplotids are notable for their ability to survive in extreme environmental stresses. Euplotes vannus, the focus of this work, in particular is known to tolerate the high levels of ammonia surrounding its microzooplanktonic prey, potentially causing great damage in the ammonium-rich aquaculture systems necessary to the microalgal industry (Day et al., 2017; Xu et al., 2004).
Here we have produced and analyzed sequence data for the somatic genome and a partial germline genome of a new marine model organism Euplotes vannus, and performed transcriptomic profiling for this species under varying harsh conditions. Through bioinformatics analysis, we revealed its extensively fragmented somatic genome and high frequency of programmed ribosomal frameshifting (PRF). By genome comparison, we analyzed its chromosomal rearrangements patterns focusing on IES excision and chromosome breakage, and compared with that of other ciliates. We then performed differential gene expression analysis to reveal the molecular basis of its strong tolerance to extreme environmental stresses. These data reveal insights into its unique features of the dual genomes, and provide clues to the ability of E. vannus to tolerate the different environmental stresses it encounters.
2. METHODS
2.1. Cell culture and sample preparation
Euplotes vannus was collected from seawater along the Yellow Sea coast at Qingdao (36°06′ N, 120°32′ E), China. Four cells were picked and cultured in filtered marine water (pH 7.8) at 20°C and 30 psu (practical salinity unit), with a monoclonal population of Escherichia coli as a food source, until reaching 106 cells/L. Cells used for macronuclear and micronuclear genomic sequencing, as well as control cells for transcriptome sequencing, were harvested from this stock population.
Experimental cells were further cultured under identical conditions, except for those noted below. Each group including the control began with 106 cells had two biological replicates. To simulate the stress from nutrient scarcity, 106 cells of E. vannus were starved in 1L of filtered seawater for 48 hours before harvest. For the temperature and salinity stresses, we chose the most extreme conditions that E. vannus can survive after a series of gradient test (Figure S1). For stresses from low and high temperature, 106 cells of E. vannus were cultured at 4 °C and 35 °C, respectively, for 6 hours before harvest. For stresses from low and high salinity, 106 cells were cultured under salinity levels of 10 psu and 60 psu, respectively, for 6 hours before harvest. For stress from the presence of free ammonia, 106 cells were cultured in filtered marine water with 100 mg/L NH4Cl (pH 8.2, 20 °C and 30 psu), as described previously (Xu et al., 2004). To account for the effect of an alkaline environment when free ammonia exists, parallel sample cells were grown and harvested in pH 8.2 marine water without the presence of free ammonia.
2.2. High-throughput sequencing and data processing
For transcriptomic sequencing and genomic sequencing to acquire macronucleus (MAC) genome information, cells were harvested by centrifugation at 300 g for 3 min. The genomic DNA was extracted using the DNeasy kit (QIAGEN, #69504, Germany). Total RNA was extracted using the RNeasy kit (QIAGEN, #74104, Germany) and digested with DNase. The rRNA fraction was depleted using GeneRead rRNA Depletion Kit (QIAGEN, #180211, Germany).
For single-cell whole-genome amplification to acquire micronucleus (MIC) genome information, a single vegetative cell of E. vannus was picked and washed in PBS buffer (without Mg2+ or Ca2+). MIC genomic DNA was enriched and amplified as described in Maurer-Alcalá et al., (2018), by using the REPLI-g Single Cell Kit (QIAGEN, #150343, Germany), which was based on the whole-genome amplification (WGA) technology and tended to amplify longer DNA fragments.
Illumina libraries were prepared from MAC genomic DNA, mRNA and amplified single-cell MIC genomic DNA of E. vannus using Nextera DNA Flex Library Prep Kit (Illumina #20018704) and TruSeq RNA Library Prep Kit (Illumina #RS-122–2001) according to manufacturer’s instructions. Paired-end sequencing (150 bp read length) was performed using an Illumina HiSeq4000 sequencer. The sequencing adapter was trimmed and low-quality reads (reads containing more than 50% bases with Q value <= 5) were filtered out by FASTX-Toolkit (-q 5 -p 0.5) (Gordon & Hannon 2010).
2.3. Genome assembly and annotation
MAC and MIC genomes were assembled using SPAdes v3.7.1 (-k 21,33,55,77), respectively (Bankevich et al., 2012; Nurk et al., 2013). Mitochondrial genomic sequences of ciliates and genome sequences of bacteria were downloaded from GenBank as BLAST databases to remove contamination caused by mitochondria or bacteria (BLAST E-value cutoff = 1e-5) and 6.6% of MAC and 1.6% of MIC genome assemblies were removed as potential contamination. CD-HIT v4.6.1 (CD-HIT-EST, -c 0.98 -n 10 -r 1) was employed to eliminate the redundancy of contigs (sequence identity threshold = 98%) (Fu et al., 2012). Poorly supported contigs (coverage < 5 and length < 300 bp) in the MAC genome were discarded by a custom Perl script. RNA-seq data of E. vannus were mapped to the MAC genome assembly by HISAT2 v2.0.4 (Kim et al., 2015). Telomeres were detected using a custom Perl script that recognized the telomere repeat 8-mer 5’-(C4A4)n-3’ at the ends of contigs, as described in a previous study (Swart et al., 2013). Contigs without neither telomeres nor RNA-seq reads mapped were further removed by a custom Perl script to avoid potential germline genome DNA contamination, which were about 10.8% of MAC genome. Repeats in the somatic MAC genome assembly were annotated by combining de novo prediction and homology searches using RepeatMasker (-engine wublast -species ‘Euplotes vannus’ -s -no_is) (Tarailo-Graovac & Chen 2009). The tRNA and other ncRNA genes were detected in the MAC genome by tRNAscan-SE v1.3.1 and Rfam v11.0, respectively (Burge et al., 2013; Lowe & Eddy 1997).
2.4. Gene modeling and stop codon detection
The transcriptome and gene models were generated using StringTie v1.3.3b (Pertea et al., 2015). Genome and transcriptome assemblies of E. octocarinatus (accession numbers: PRJNA294366) were acquired from NCBI and previously published studies (Wang et al., 2018; Wang et al., 2016). Mapping results for RNA-Seq reads were visualized on GBrowse v2.0 (Stein 2013). Predicted protein products were annotated by alignment to domains in the Pfam-A database by InterProScan v5.23 and to ciliate protein sequences from NCBI GenBank by BLAST+ v2.3.0 (E-value cutoff = 1e-5) (Camacho et al., 2009; Jones et al., 2014). Two-gene chromosomes were detected using a custom Perl script that recognized chromosomes containing multiple genes. The frequency of stop codon usage was estimated by a custom Perl script that recognized the stop codon TAA or TAG in transcripts of euplotids.
2.5. Detection of frameshifting events
Frameshifting events were detected using a custom Perl script modified from the protocol in a previous study (Wang et al., 2016). Transcripts of euplotids were compared using BLASTX to conserved protein sequences of other ciliates (E-value cutoff = 1e-5), and frame changes between adjacent BLASTX hits were identified. To avoid false-positives created by introns, the results were limited to adjacent hits separated by a strict inner distance of <= 10 bp. Sequences from 30 bp upstream and downstream of each type of frameshifting site (+1, +2 or −1) were extracted, and sequence motifs in information content (bits) within these regions were identified and illustrated by WebLogo 3 (Crooks et al., 2004). We assessed statistical significance in comparison of transcript with or without frameshifting using a two-tailed Student’s t-test, considering a value of p < 0.05 as significant.
2.6. Genome rearrangement analysis
The MIC and MAC genome assemblies and annotations for macronuclear-destined segments (MDSs), internally-eliminated segments (IESs), and pointers of Oxytricha trifallax and Tetrahymena thermophila were acquired from the database <mds_ies_db> (Burns et al., 2015). The MIC and MAC genome assemblies of Paramecium tetraurelia strain 51 were acquired from ParameciumDB (Arnaiz & Sperling 2010). Scrambled and non-scrambled MDSs were identified by homologous search between MAC and MIC genomic sequences in each species by MIDAS (Jonoska & Saito 2015), which utilized BLAST+ v2.3.0 (Camacho et al., 2009). Non-scrambled MDSs were further identified by analyzing qualified BLASTN hits (E-value cutoff = 1e-5 and match length cutoff = 100 nt) between MAC and MIC genomes. IESs were assigned between MDSs from the same MAC chromosomes. Non-scrambled pointers were identified as the overlap regions of adjacent MDSs on MAC chromosomes. Chromosome breakage sites (CBSs) were identified as the regions between MDSs from different MAC chromosomes mapping to the adjacent regions of MIC genome. Motifs in information content (bits) of pointers, CBSs and their flanking regions were illustrated by WebLogo 3 (Crooks et al., 2004).
2.7. Differential gene expression analysis
Transcript abundances were estimated by using featureCounts (Liao et al., 2013). Differential gene expression analysis and principal component analysis (PCA) were performed by R package “DESeq2” (adjusted p-value by Benjamini-Hochberg procedure < 0.01) (Love et al., 2014). Starvation induced genes were defined by an average RPKM value of gene expression in starved samples > 1 and an average value of RPKM of gene expression from vegetative samples < 0.1. Weighted gene co-expression eigengene network analysis was performed by WGCNA (Langfelder & Horvath 2008). Gene Ontology (GO) term enrichment analysis was performed by using BiNGO v3.0.3 (adjusted p-value by Benjamini-Hochberg procedure < 0.05), which was integrated in Cytoscape v3.4.0, and the plot was generated by the R package, ggplot2 (Kohl et al., 2011; Maere et al., 2005; Wickham 2016).
2.8. Homolog detection of environmental stress-related genes
Homologous Hsp70 (gene id: MSTRG.11315) and its two relatively distant homologs, BiP (Binding immunoglobulin protein, gene id: MSTRG.32307) and mtHsp70 (mitochondrial Hsp70, gene id: MSTRG.32363), were identified in E. vannus using BLAST+ v2.3.0 (E-value cutoff = 1e-5), according to the Hsp70 protein sequences of E. focardii and E. nobilii from previous studies (GenBank accession number: AAP51165 and ABI23727, respectively) (La Terza et al., 2001; La Terza et al., 2007) and NCBI Non-redundant protein sequences (nr) database. The complete sequences of the E. focardii and E. nobilii HSP70 genes are available at NCBI with the accession numbers AY295877 and DQ866998 (La Terza et al., 2004; La Terza et al., 2007). The consensus amino acid sequence of Hsp70 (La Terza et al., 2004; La Terza et al., 2007) and essential amino acid positions of Hsp70 (Morshauser et al., 1999; Sriram et al., 1997) were reported in previous studies.
2.9. Phylogenetic analysis
The DNA and amino acid sequences of Euplotes pheromone homologs (Vallesi et al., 2014) were acquired from NCBI, and aligned by MUSCLE v3.8.31 and ClustalW v2.1, respectively (Chenna et al., 2003; Edgar 2004). A Maximum Likelihood tree based on amino acid sequences was reconstructed by MEGA v7.0.20, using the LG model of amino acid substitution, 500 bootstrap replicates (Kumar et al., 2016; Le & Gascuel 2008).
Phylogenomic analysis was performed using the supertree method (Chen et al., 2018). Predicted protein sequences from Euplotes vannus identified in this work, from 31 other ciliates collected from previous studies (Aeschlimann et al., 2014; Keeling et al., 2014; Slabodnick et al., 2017; Wang et al., 2018), and from transcriptome sequencing by the Marine Microbial Eukaryote Transcriptome Sequencing Project (data available on iMicrobe: http://imicrobe.us/, accession number and gene ID see Table S1), were used to generate a concatenated dataset (Chen et al., 2018; Gentekaki et al., 2017). A Maximum Likelihood tree based on the concatenated dataset covering 157 genes was reconstructed by using GPSit v1.0 (relaxed masking, E-value cutoff = 1e-10, sequence identity cutoff = 50%) (Chen et al., 2018) and RAXML-HPC2 v8.2.9 (on CIPRES Science Gateway, LG model of amino acid substitution + Γ distribution + F, four rate categories, 500 bootstrap replicates) (Stamatakis 2014). Trees were visualized by MEGA version 7.0.20 (Kumar et al., 2016).
2.10. Euplotes vannus Genome Database
The genome, annotated gene and protein sequence files produced in this work are available at Euplotes vannus Genome Database (http://evan.ciliate.org), based on the architecture of Tetrahymena Genome Database (Stover et al., 2012). Sequence data are available for search using NCBI BLAST (Altschul et al., 1997) and display in GBrowse2 (Stein et al., 2002). Functional annotations including Gene Ontology, domains and gene names can be accessed by keyword search and updated using a community annotation interface.
3. RESULTS
3.1. General description of genome sequencing and assembly of Euplotes vannus
We assembled the 85.1 Mb somatic MAC genome and 120.0 Mb germline MIC genome of Euplotes vannus (Table 1, 2, S2 and Figure S2). We compared E. vannus to the previously sequenced euplotid E. octocarinatus to assess somatic genome size, gene number, telomere length, number of 2-telomere contigs and N50 value (Table 1, Figure 1 and Figure S3a). Most chromosomes in E. vannus are “nanochromosomes” bearing a single gene, with an average size distribution around 1.5 kb, and telomeric repeats of C4A4 and T4G4 on both ends (37501/38245, 98.1%) (Figure 1c), similar to that of E. octocarinatus (Wang et al., 2016). The distance between the transcription start site (TSS) for each gene and the upstream telomere is generally less than 80 nt (Figure S3b). 32755 protein-coding genes were identified in the final somatic genome assembly, along with 109 tRNAs comprising 48 codon types for 20 amino acids (Table S3). Most E. vannus introns are around 25 bp in size, with a canonical sequence motif 5’-GTR (N)nYAG-3’ at the respective ends (Figure 1d and Figure S4). The annotation of gene functions and repeat regions are summarized in Table S4 and Table S5. A model for nanochromosome structure in the E. vannus MAC is summarized and illustrated in Figure 1e.
Table 1.
MAC genome assembly and transcriptome-informed gene annotation of Euplotes vannus in comparison with that of E. octocarinatus (Wang et al., 2018).
| E. vannus | E. octocarinatus | |
|---|---|---|
| Genome size (Mb) | 85.1 | 88.9 |
| %GC | 37.0 | 28.2 |
| Contig # | 38245 | 41980 |
| Contig N50 (bp) | 2685 | 2947 |
| 2-telomere contig # | 25519 | 29532 |
| 1-telomere contig # | 7835 | 4842 |
| 0-telomere contig # | 4890 | 7606 |
| 2-telomere contig percentage (%) | 66.7 | 70.3 |
| Genome size (with telomere) (Mb) | 75.9 | 83.1 |
| Scaffold (with telomere) # | 33354 | 34374 |
| %Scaffold (with telomere) | 87.2 | 81.9 |
| Scaffold N50 (bp) | 2714 | 2999 |
| Gene # | 32755 | 29076 |
| Exon # | 175735 | 96843 |
| Transcript # | 43040 | 29076 |
Notes: A contig/scaffold with telomeres refers to containing telomere on at least one of its two ends.
Table 2.
MIC genome assembly information of Euplotes vannus and recognition of MDS-containing contigs and those that contain multiple MDSs.
| MIC genome | Total | With MDS | Multi-MDS |
|---|---|---|---|
| Genome size (Mb) | 120.0 | 49.8 | 31.8 |
| %GC | 36.0 | 35.7 | 35.9 |
| Contig # | 104988 | 13140 | 5166 |
| Contig N50 (bp) | 1953 | 5597 | 7718 |
FIGURE 1.
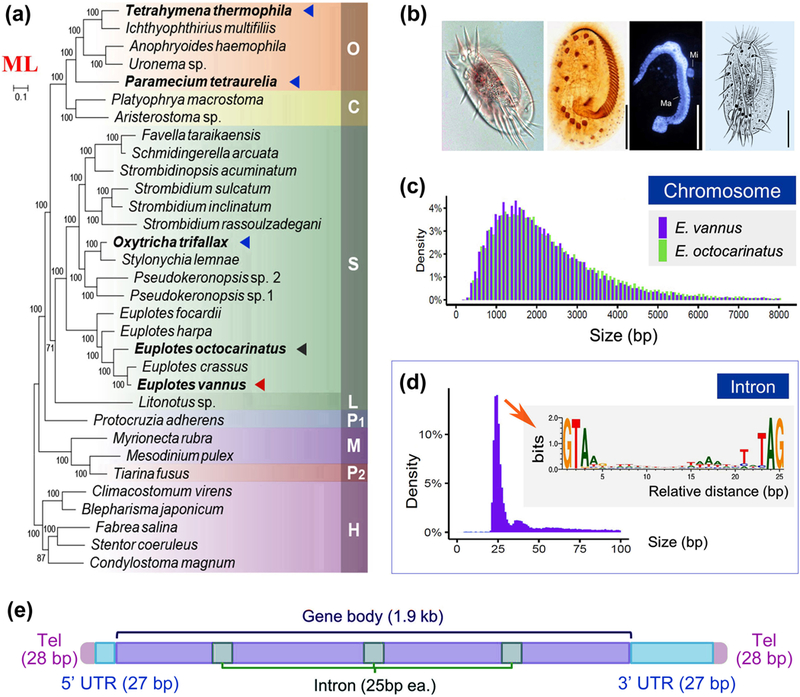
Macronucleus (MAC) genome assembly and features of chromosome and introns of Euplotes vannus as compared to other ciliates. (a) Maximum likelihood phylogenomic tree by supermatrix approach based on a 158-gene dataset. S: class Spirotrichea; L: class Litostomatea; O: class Oligohymenophorea; C: class Colpodea; P1: class Protocruzia; M: class Mesodiniea; P2: class Prostomatea; H: class Heterotrichea. The scale bar corresponds to 10 substitutions per 100 nucleotide positions. The red, black and blue triangles denote the positions of E. vannus, E. octocarinatus and three other model ciliates Tetrahymena thermophila, Paramecium tetraurelia and Oxytricha trifallax. (b) Photomicrographs in vivo, protargol-stained specimen, DAPI staining and morphology and infraciliature schema of E. vannus. Ma, macronucleus; Mi, micronucleus. Scale bars are 25 μm. (c) Size distribution of 2-telomere MAC scaffolds of E. vannus and E. octocarinatus (Wang et al., 2018). (d) Size distribution of introns within E. vannus MAC genes and sequence motif of tiny introns in most abundant size category (8792 introns of 25 bp in length). The weblogo is generated and normalized to neutral base frequencies in intron regions. (e) A schema illustrates the canonical structure of nanochromosomes in MAC of E. vannus. “Tel” denotes telomere and “gene body” denotes the gene transcription region. “UTR” denotes the untranscribed region. The mean sizes of different regions are shown in the parentheses.
A small proportion of chromosomes were found to contain more than one gene (Figure S3c). We divided these two-gene nanochromosomes into two groups according to the direction in which the genes they contained are transcribed, and compared these data to those of E. octocarinatus. Trans-nanochromosomes containing genes on different strands were similar in number between E. vannus (283; 0.74% of total chromosomes) and E. octocarinatus (373; 0.89%). However, our estimate of Cis-nanochromosomes where all genes on the chromosome are transcribed from the same DNA strand, were 2-fold more abundant in E. octocarinatus (1211; 2.89%) than E. vannus (461; 1.21%).
3.2. Frameshifting events in euplotids
Frameshifting events in E. vannus and E. octocarinatus, defined as recoding events that shifts the ribosome reading frame at a specific position during translation, and then continue translation in this frame, were detected by identifying adjacent BLASTX hits targeting the same protein sequence in different frames (illustrated by Figure 2a). An E-value cutoff (1e-5) ensured the accuracy of the prediction process and a small inner distance cutoff (10 nt) was applied to remove interference of introns, all of which are larger than 20 nt as described above (Figure 1d). 1,208 (2.8% of all transcripts) and 1,016 (3.5%) frameshifting events were detected in E. vannus and E. octocarinatus, respectively. Figure 2b and 2c showed that the high frequency of +1 programmed ribosomal frameshifting (PRF) at a canonical motif of 5’-AAA-TAR-3’ (R = A or G) is a conserved feature in euplotids. Intriguingly, more +2 and −1 PRF events were found in E. vannus (16.6% of all PRF events) than in E. octocarinatus (4.4%). In the cases of +2 and −1 PRF events, the novel motif 5’-WWW-TAR-3’ (W = A or T) rather than the +1 PRF signal was preferred (Figure 2bc). Frameshifting sites using a non-AAA upstream codon are associated less frequently with TAA codons, and are preferentially found upstream of TAG codons (Figure S5a). The impact of frameshifting on the abundance of transcripts with or without frameshifting was tested. No significant differences by T-test (p > 0.05) were observed among abundance of transcripts without frameshifts and those subject to +1, +2 or −1 frameshifting (Figure 2d).
FIGURE 2.
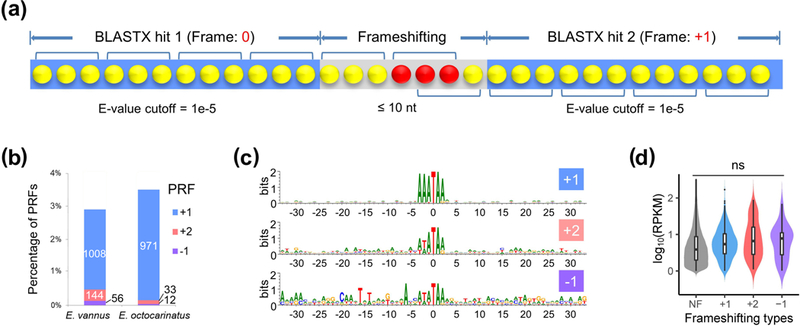
Programmed ribosomal frameshifting (PRF) does not impair the transcript abundance in Euplotes vannus. (a) A schema illustrates the criteria for detecting +1 frameshifting events. Blue boxes indicate the different BLASTX hits of a CDS region to a same target protein sequence (E-value cutoff = 1e-5). Grey boxes indicate the adjacent region between two BLASTX hits of a CDS region (inner distance cutoff = 10 nt). The brackets above denote the 0-frame codons and the brackets underneath denote the +1-frame codons. Yellow dots denote the nucleotides while the red ones denote the slippery site where frameshifting events occur. (b) Percentage of +1, +2 and −1 PRF frameshifting events detected among all transcripts in E. vannus (43040 transcripts) and E. octocarinatus (29076 transcripts). Numbers in the labels shows the frequencies of frameshifting events in these two species. (c) Conserved sequence motif associated with frameshift sites. Sizes of letters denote information content, or sequence conservation, at each position. The analysis is based on the alignment of 30 bp upstream and downstream the frameshifting motif from predicted frameshifting events that involves stop codon TAA or TAG. Note the canonical motif 5’-AAA-TAR-3’ (R = A or G) in +1 PRF and noncanonical motif 5’-WWW-TAR-3’ (W = A or T) in +2 and −1 PRF. (d) Abundance comparison of transcripts without frameshifting (NF) and with different types of frameshifting (+1 / +2 / −1) in E. vannus. “ns” denotes not significant.
Stop codon usage at translation termination sites in the non-slippery transcripts and at the slippery sites of PRF were compared between E. vannus and E. octocarinatus (Figure S5b). In these two euplotids, UAA was preferentially used at the termination signal (73.7% and 76.0%, respectively) and in the slippery signal (91.3% and 91.0%, respectively). The frequency of UAA codon usage in the slippery signal is significantly higher than that in the termination signal (p = 0.005024 < 0.01, by analysis of variance), indicating that UAA may be favorable for frameshifting in both E. vannus and E. octocarinatus.
3.3. A new model for genome rearrangement featuring conserved pointers and palindromic chromosome breakage sites
Genome rearrangement from MIC to MAC includes two important events: IES excision and chromosome breakage (Figure 3 and Figure 4). We analyzed MDSs in two ciliates in the class Spirotrichea – E. vannus and O. trifallax – and two in Oligohymenophorea – T. thermophila, and P. tetraurelia – through comparisons of the MAC and MIC genome sequences (Figure 3a). The results showed great variability among these ciliate lineages in both the number and size of MDSs (Figure 3b). E. vannus and P. tetraurelia have large MDSs (>1 kb) while O. trifallax and T. thermophila feature much smaller MDSs. The IES sequences that separate MDSs in the MIC also vary in number, but their lengths are comparable – most IESs are smaller than 200 nt (Figure 3c).
FIGURE 3.
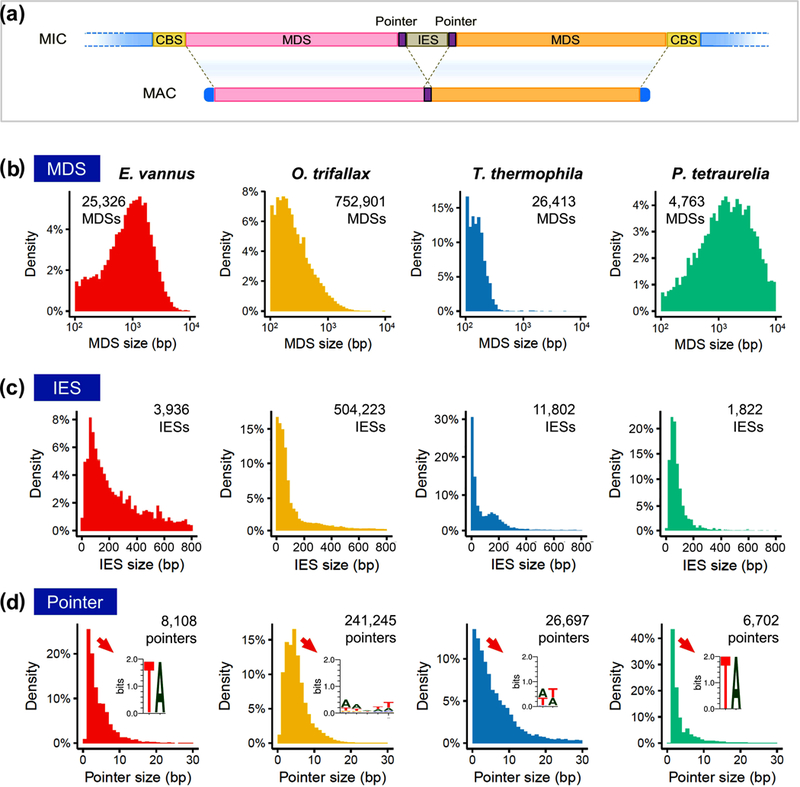
Comparison of micronuclear genome features among ciliates. The pointers joining adjacent macronuclear-destined sequences (MDSs) during the genome rearrangement are in a highly conserved “TA” motif in Euplotes vannus and Paramecium tetraurelia. (a) A cartoon illustrating the genome rearrangement model for MAC chromosome development from MIC. The MDS regions are joined by pointers that located at the boundaries of MDSs and internally-eliminated sequences (IESs) after IESs are removed. The blue boxes at the end of MAC chromosomes denote the telomeres. (b) The size distribution of MDSs of E. vannus, Oxytricha trifallax, Tetrahymena thermophila and P. tetraurelia. (c) The size distribution of IESs of E. vannus, O. trifallax, T. thermophila and P. tetraurelia. (d) The size distribution of pointers of E. vannus, O. trifallax, T. thermophila and P. tetraurelia. The weblogos show the sequence motif in information content (bits) of the pointers in most abundant size in each species.
FIGURE 4.
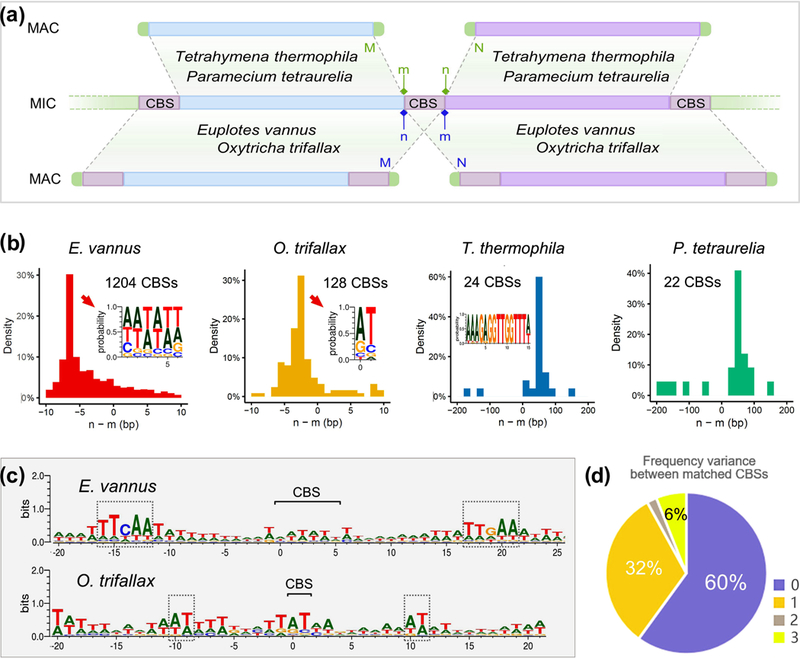
MIC chromosome breakage sites (CBSs) are retained in MAC genome of Euplotes vannus and Oxytricha trifallax, but not in Tetrahymena thermophila and Paramecium tetraurelia. (a) A cartoon illustrating two genome rearrangement models for chromosome breakage from MIC to MAC during ciliate sexual reproduction. “M” and “N” denote the end of two adjacent MAC chromosomes, corresponding to the breakage points “m” and “n” in the MIC genome. The blue boxes at the end of MAC chromosomes denote the telomeres. The CBS regions are identified by homologous search between MAC and MIC genomes in each species. (b) Distribution of the relative distance between the chromosome breakage points (“m” and “n”) in the MIC genome of E. vannus, O. trifallax, T. thermophila and P. tetraurelia. The positive value of relative distance between “n” and “m” indicates a CBS is not retained in the MAC genome while a negative value of relative distance means a CBS is overlapped in the MIC genome and retained in the MAC genome. The weblogos show the sequence motif in base possibilities of the CBSs in most abundant size in E. vannus and O. trifallax. (c) The sequence motif in information content (bits) of flanking regions (20 bp on each side) around the CBSs in most abundant size in E. vannus and O. trifallax. Grey boxes denote the consensus elements near CBSs. (d) The difference between frequencies of CBSs and their reverse complementary counterparts in the MAC genome of E. vannus. “Matched CBSs” refer to a pair of a CBS and its reverse complementary counterpart. 60% CBSs and their reverse complementary counterparts are present with equal frequency (difference = 0), 32% with the frequency difference of one (difference = 1), 2% with the frequency difference of two (difference = 2), 6% with the frequency difference of three (difference = 3).
Based on our analyses, 97.3% of genome rearrangement events are non-scrambled in E. vannus (Table S6). We identified the boundary repeats shared by adjacent MDS and IES, known as “pointers”, in non-scrambled genome rearrangement events (Figure 3d). The length of the pointer consensus sequence in these species is 2 bp, except for O. trifallax, where pointers average 5 bp. The 8,108 pointers identified in E. vannus have a highly conserved motif 5’-TA-3’. This 5’-TA-3’ motif is also seen in most P. tetraurelia pointers and in the 2 bp pointers flanking nonscrambled MDSs in O. trifallax, despite the vast evolutionary distances between these three species.
To investigate the chromosome breakage mechanism in E. vannus, we first identified complete MAC chromosomes containing telomeres at both ends, then mapped these to homologous regions in the MIC genome to determine CBS boundaries (denoted as “m” and “n” in Figure 4a). We identified the size of each CBS and calculated the distance between adjacent CBS boundaries using the value of “n - m”. Unlike the well-studied species T. thermophila and P. tetraurelia (both in class Oligohymenophorea), where most MAC chromosomes are separated by a positive number of base pairs that are excised and lost from the MAC genome, most of these are negative values in E. vannus (Figure 4b). O. trifallax, another spirotrich ciliate featuring nanochromosomes, also shows negative values in CBSs boundary distances. These negative values can be explained by overlapping loci in the MIC, resulting in duplicated sequences in the MAC genome.
To search the potential conserved sequence motifs, 20–25 bp upstream and downstream of the 1204 predicted CBS regions were extracted and analyzed using WebLogo. These loci in E. vannus and O. trifallax show the consensus CBS motifs and their flanking regions in an overall palindrome structure (Figure 4bc). Furthermore, we found that most (92%) CBSs and their reverse complementary counterparts are present at a similar frequency (difference ≤ 1) in the MAC genome (Figure 4d). The findings above suggest that ciliates with nanochromosomes follow a novel model of genome rearrangement, in which chromosome breakage sites are duplicated and retained in the somatic genome. This model bears similarity to IES deletion, whose removal depends on the direct repeats at their boundaries, lending support to the idea that these two processes may share a homologous mechanism in these species.
3.4. Molecular basis of strong tolerance to extreme environmental stresses
We conducted principal component analysis (PCA) based on the differential gene expression of E. vannus cells under different extreme environmental stresses (Figure S6 and Figure S7). The analysis revealed changes in the gene expression profile of cells under high temperature (35 °C), low temperature (4 °C), and high or low salinity (60 and 10 psu, respectively). The presence of ammonia also had a substantial impact on transcription patterns (Figure S7). Surprisingly, cells under high salinity and low salinity shared a similar gene expression profile (Figure S6a).
To further dissect the relationships between co-expressed genes associated with the regulation of cellular processes and pathways under substantial environmental changes, we constructed a weighted gene co-expression eigengene network (Figure 5a). The network clustered different eigengenes into six modules based on their co-expression profile (Figure S6b). A strongly co-expressed eigengene module was up-regulated in cells under both high and low salinity stresses (colored in steel blue in Figure 5a and Figure S6b). This module indicated an extensive activation of many pathways, mainly related to tRNA aminoacylation, tRNA and rRNA processing, nucleosome assembly and pseudouridine synthesis (adjusted p-value by Benjamini-Hochberg procedure < 0.05). In addition, two small eigengene modules were up-regulated in cells under high salinity stress (purple) and low salinity stress (dark green), respectively. Low salinity stress activated an extra pathway related to the glutamine metabolic process. Intriguingly, low temperature stress induced a large cluster of eigengenes (blue module in Figure 5 and Figure S6b) related to small GTPase mediated signal transduction. Cells exposed to high levels of ammonia were very similar to those under high temperature stress, and induced a small cluster of eigengenes related to lipid metabolic process (Figure 5 and Figure S6b). However, the normal expression of many genes also changes under high ammonia concentrations (Figure S6b and Figure S7), and the lipid metabolic pathway overall is severely impacted (Figure 5b).
FIGURE 5.
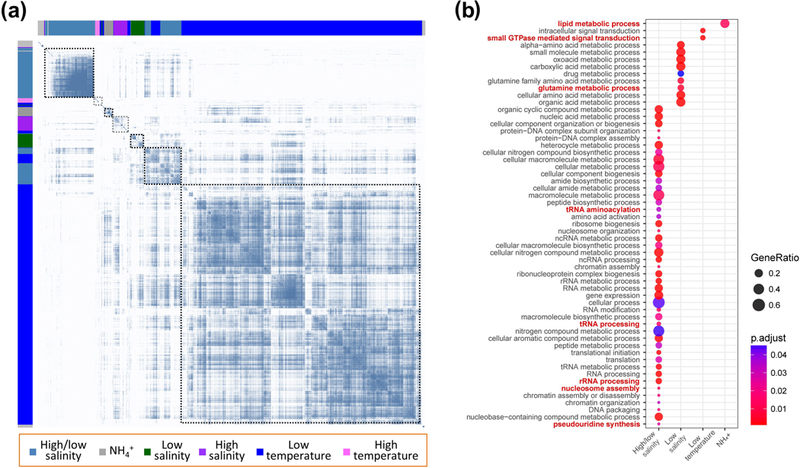
Differential gene expression analysis reveals several large cohorts of co-expressed genes under temperature, salinity and ammonia stresses. (a) Heatmap of weighted gene co-expression network of Euplotes vannus genes (both x-axis and y-axis), in accordance with different stress-response gene groups. Blue dots denote the co-expression relationship between different genes. (b) GO term enrichment analysis on different stress-response gene groups shows many pathways are activated when E. vannus faces environmental stresses (adjusted p-value by Benjamini-Hochberg procedure < 0.05). Dot size denotes the ratio of genes activated in each pathway. Dot color denotes the adjusted p value for activation of each pathway.
As many other organisms upregulate heat-shock protein 70 (HSP70) genes under heat stress (Clark & Peck 2009), we compared the sequence of the E. vannus HSP70 homolog to its counterparts in Euplotes nobilii and Euplotes focardii, as well as other two relatively distant homologs of Hsp70, BiP (Binding immunoglobulin protein) and mtHsp70 (mitochondrial Hsp70) (Figure S8). This comparison revealed that only the E. focardii HSP70 sequence, previously noted for its lack of response to temperature stress (La Terza et al., 2004), had numerous amino acid substitutions within its ATP-binding and substrate-binding domains (Figure 6a). While a few eigengenes co-expressed under high temperature (pink module in Figure 5a), transcription of either the highly conserved HSP70 or BiP gene in E. vannus did not respond to temperature stresses, whereas chemical stress significantly changed the expression of this gene in E. vannus (Figure 6b and Figure S7). However, the other HSP70 homologous gene mtHSP70 that located in mitochondria, responds actively when cell faces stress from low temperature.
FIGURE 6.
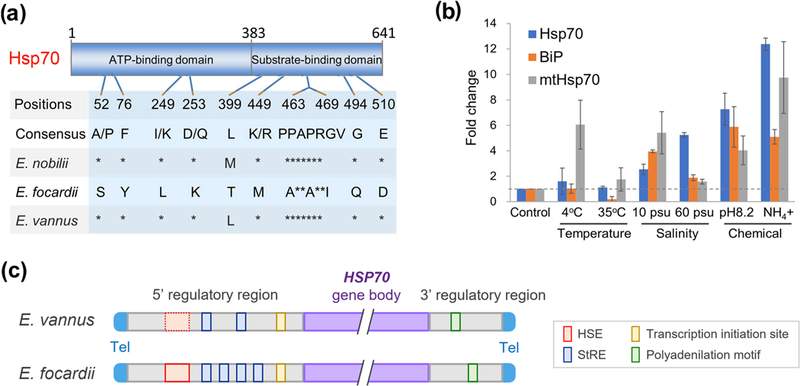
Sequence and gene expression analysis of HSP70 in Euplotes vannus shows changes that may explain its insensitivity to temperature change. (a) Amino acid substitutions that occur in Euplotes focardii at the level of its HSP70 ATP- and substrate-binding domains and are unique with respect to Euplotes nobilii and other organisms. Asterisks denote identities. Numbers indicate essential amino acid positions of Hsp70. (b) Fold change of gene expression level of E. vannus Hsp70 (gene id: MSTRG.11315) as well as its two relatively distant homologs, BiP (Binding immunoglobulin protein, gene id: MSTRG.32307) and mtHsp70 (mitochondrial Hsp70, gene id: MSTRG.32363), under different environmental stresses (4 °C, 35 °C, 10 psu, 60 psu, pH 8.2 or with the presence of free ammonia) with respect to the control (20 °C, 30 psu and pH 7.8). The dashed line in grey denotes the normal level of HSP70 and the other two homologous genes. (c) A schema illustrates the sequence alignment of 5’ and 3’ regulatory regions of Hsp70 genes in Euplotes vannus and E. focardii. Sequence motifs bearing agreement with HSE and StRE elements, putative sites for the transcription initiation and polyadenylation motifs are denoted by red, blue, yellow and green boxes, respectively; neither of E. vannus and E. focardii carries ARE elements in the 3’ regulatory region. The dashed red box shows the poorly conserved HSE element in E. vannus. “Tel” denotes telomere and “gene body” denotes the gene transcription region. The sequence alignment of 5’ and 3’ regulatory regions see Figure S9.
To investigate the molecular basis of HSP70 gene expression in euplotids, we analyzed the structure of non-coding regions flanking the gene in E. vannus and E. focardii (Figure 6c and Figure S9). We observed no substantial difference in the 5’ promoter region between the HSP70 gene between these two species. Both bear canonical regulatory cis-acting elements that bind transcriptional trans-activating factors, including heat-shock elements (HSE) and stress-response elements (StRE) (Figure 6c and Figure S9a). However, the sequences of HSEs in these two species are poorly conserved. Furthermore, neither E. vannus nor E. focardii contained the motif 5’-ATTTA-3’ in their 3’ promoter region, an mRNA destabilization adenine-uridine rich element (ARE) commonly found in other species (Figure 6c and Figure S9b).
4. DISCUSSION
4.1. Programmed ribosomal frameshifting does not affect transcript abundance
Comparative genome analysis in the present study reveals that E. vannus shares similar patterns of frameshifting and stop codon usage with E. octocarinatus (Figure 2). Our analyses are consistent with previous studies demonstrating that euplotids have a large number of genes requiring programmed ribosomal frameshifting (PRF), and this phenomenon is more prevalent in this clade of ciliates than in the viruses, prokaryotes and other eukaryotes where it has been studied (Karamysheva et al., 2003; Wang et al., 2016). Previous study reported a putative suppressor tRNA of UAA, which may play an important role in +1 frameshifting in E. octocarinatus (Wang et al., 2016). Unfortunately, we did not find strong evidence to show the presence of “suppressor” tRNA based on the present data. A more recent study suggested that the function of stop codons as frameshifting or termination is determined by their proximity to poly(A) tails (Lobanov et al., 2017). It was observed that ribosomal frameshifting was slower than the standard decoding of sense codons (Lobanov et al., 2017). Our results show no significant difference between the abundance of transcripts that incorporate a frameshifting event and those without frameshifting and thus implied that the decoding delay induced by ribosomal frameshifting would not be compensated by transcript abundance change.
Frameshifting will typically occur when the codon upstream of a UAA/UAG is AT-rich (usually a AAA codon) (Figure 2c). Our results here indicate that transcripts with +2 / −1 frameshifting (associated with non-AAA upstream codon) have a slightly higher abundance compared to transcripts with either a +1 (associated with AAA upstream codon) or no frameshifting (Figure 2d). Besides, frameshifting sites using a non-AAA upstream codon are preferentially found upstream of UAG codons (Figure S5a). The previous study demonstrated that the observed frameshifting efficiencies of loci with AAA and non-AAA upstream codons are similar, but the ribosome pausing signal at frameshifting sites was stronger for TAA codons than for TAG codons (Lobanov et al., 2017). One possible explanation for these observations is that the non-AAA upstream codon, when in combination with the following TAG stop codon, results in decreased ribosomal pausing and increased efficiency of frameshifting.
Euplotes MAC genome are extensively fragmented to gene-sized nanochromosomes, which facilitates the evolution of genetic code. Previous studies indicate that ciliates evolved diversified and flexible nuclear genetic code from their ancestors with ambiguous genetic codes (Swart et al., 2016). For most species, UGA remains as stop while UAA and UAG are reassigned to code glutamine, tyrosine or glutamic acid (Swart et al., 2016). It is opposite in Euplotes, whose UGA codon is reassigned to code cysteine while UAA and UAG are stops (Lozupone et al., 2001). However, euplotids evolves another important mechanism of programmed ribosomal frameshifting at the stop codons UAA and UAG, which can solve the same problem of canonical stop codons residing in the coding regions. It is proposed that translation (either through reassignment or frameshifting), rather than termination, is the default recognition mode for “stop”; codons while termination is due to the context-specific override provided by transcript ends (Lobanov et al., 2017; Swart et al., 2016). This would be much more robust when the MAC genome are extensively fragmented to nanochromosomes, where stop codons will potentially even be unnecessary.
4.2. Pointers and CBSs are duplicated after genome rearrangement to increase homology in MAC of euplotids
A novel insight from our study, based on more than 4,000 identified IESs, is that pointers with the motif 5’-TA-3’ are universal and highly conserved in E. vannus (Figure 3d). This is consistent with previous studies that IESs containing Tec transposable elements in euplotids were precisely excised at pointer sequences with a consensus motif 5’-TA-3’ (Jacobs & Klobutcher 1996; Karamysheva et al., 2003). This sequence is identical to that found in Paramecium, which also utilizes it as a universal dinucleotide pointer (Steele et al., 1994). Most Tetrahymena IESs are flanked instead by 5’-AT-3’, but a greater variety of pointer sequences are seen in this species, perhaps due to the existence of more IESs (Hamilton et al., 2016). An even more complicated set of pointers may be necessary in Oxytricha, which excises more than half a million IESs and reorders the MDS segments by inversion or permutation (Figure 3c). Together with a previous study (Chen et al., 2014), our result suggests that pointers evolve to larger sizes under selective pressure in genomes with higher rearrangement complexity.
Our study reveals that chromosome breakage sites (CBSs) are duplicated and retained in the Euplotes MAC genome. Previous studies identified a conserved CBS element with the sequence motif 5’-TTGAA-3’ at several breakage loci in euplotid MIC chromosomes (Baird & Klobutcher 1989; Klobutcher et al., 1998). However, the role of the conserved element in specifying chromosome fragmentation was still unclear, since a number of CBS regions lacked this sequence. Our study has determined 1,204 CBSs with high confidence and identified the sequence motif 5’-TTGAA-3’ within downstream (or 5’-TTCAA-3’ upstream) flanking regions, with an inner distance of 11 nt (Figure 4c). These observations, along with similar observations in Oxytricha, strongly support previous models of chromosome breakage that involve a short staggered cut in ciliates containing gene-sized nanochromosomes (Baird & Klobutcher 1989; Klobutcher et al., 1998).
We have also demonstrated that copies of the CBS region remain at the ends of both resulting MAC chromosomes after genome rearrangement, by showing the conservation of the frequencies of the CBS sequences and their reverse complement counterparts in the MAC genome (Figure 4d). Notably, the Euplotes nanochromosomes have extremely short 5’ untranscribed regions (the average size of 27 bp, Figure 1e), and the distance between the transcription start site (TSS) for each gene and the upstream CBS is generally less than 80 nt (Figure S3b). Furthermore, the first three periodic peaks of the distance between CBSs and TSSs is 10–11 bp (Figure S3b), i.e. the characteristic number of bases in one turn of DNA double helix. This might indicate regular spacing of transcription factors relative to the CBS/telomere addition site, indicating potential interactions or constraints between telomere-binding proteins and transcription complex. Based on these observations, we speculate that the AT-rich CBSs may serve as TATA boxes. These facts show that ciliates with nanochromosomes retain sizable regions of unique, repeated sequence in MIC-adjacent MAC chromosomes. These sequences may allow for the co-regulation of one or more aspects of MAC chromosome biology, such as telomere addition and gene co-expression.
Together with previous studies, the current result indicates that genome rearrangement patterns and mechanisms in ciliates are highly diversified. For example, the extent of chromosomal fragmentation varies from limitedly (e.g. Tetrahymena and Paramecium) to extensively (e.g. Oxytricha and Euplotes). Extensive fragmentation has been found to occur in members of three separate classes, Spirotrichea (including Oxytricha and Euplotes), Phyllopharyngea (e.g. Chilodonella), and Armophorea (e.g. Metopus), which demonstrates multiple origins within ciliates (Riley & Katz 2001). The types of DNA elimination are also quite diverse among different ciliates (Chalker & Yao 2011; Nowacki et al., 2011). Excision of IESs is generally not precise in Tetryhmena while must be precise in Paramecium, Chilodonella, Oxytricha and Euplotes. Moreover, gene scrambling is widespread in Oxytricha (Chen et al., 2014) and Chilodonella (Maurer-Alcalá et al., 2018; Zhang et al., 2018), but not frequently observed in Tetrahymena, Paramecium and Euplotes. As to the Euplotes in the present study, which is most closely related to Oxytricha based on morphology and phylogeny, its chromosomal fragmentation is similar to Oxytricha (Figure 4); however, its DNA elimination resembles Paramecium (Figure 3). The mechanisms underlie the genome rearrangement in Tetrahymena, Paramecium and Oxytricha are different, though that of Euplotes is still largely unknown (Wang et al., 2017b). This again demonstrates the plasticity of genome rearrangement among different species of ciliates, which likely contributes to the biodiversity and adaptability of ciliates.
4.3. Absence of trans-acting sequence elements prevents HSP70 response during temperature stress
Extensive gene networks are co-expressed in E. vannus cells under different extreme environmental stresses (Figure 5). However, the HSP70 gene of E. vannus can be activated when faced with chemical stresses, but is not upregulated in response to thermal changes (Figure 6b). This is similar to a previous study that E. focardii is able to tolerate temperature stresses without inducing a typical HSP70 response (La Terza et al., 2001). The previous study also found that HSP70 gene expression in E. nobilii differed from E. focardii in response to both gradual and abrupt temperature changes. When transferred from 4 to 20 °C, a strong transcriptional activity of HSP70 gene was induced in E. nobilii cells, whereas no measurable change was found in cells of E. focardii. In contrast, HSP70 expression in both species increased with oxidative and chemical stresses, such as tributyltin and sodium arsenite (La Terza et al., 2004). Furthermore, the HSP70 protein of E. focardii carries unique amino acid substitutions of potential significance for cold adaptation, which are absent in E. nobilii (La Terza et al., 2007). The current work indicates the protein product of E. vannus’ HSP70 gene is not evolved for cold adaptation and resembles that of E. nobilii (Figure 6a). However, the other HSP70 homologous gene mtHSP70 responds actively when cell faces stress from low temperature, which might be a survival strategy of E. vannus in low temperature.
Further, in our analyses neither HSP70 nor its two relatively distant homologs BiP and mtHsp70 respond to the stress of high temperature (Figure 6b). While it is possible that 35 °C is not high enough to induce heat shock, even though they cannot survive at higher temperatures in the lab. More likely, the divergence of trans-activating sequence elements controls HSP70 gene expression profiles. E. focardii harbors cis-acting elements like heat-shock elements (HSE) and stress-response elements (StRE) in the 5’ promoter region of its HSP70 gene. These elements bind trans-acting transcriptional activators and are associated with stress-inducible genes in a variety of organisms (Fernandes 1994; Kobayashi & McEntee 1993; Ruis & Schüller 1995). HSP70 gene transcription in response to temperature stress is believed to be modulated by HSE binding, whereas StRE binding mediates HSP70 transcription under a broad range of non-temperature stresses (La Terza et al., 2007). In E. vannus the HSP70 HSE element is poorly conserved, which may explain its insensitivity to temperature stress (Figure 6c and Figure S9). Furthermore, HSP70 mRNAs typically contain an adenine-uridine rich element (ARE) in the 3’ regulatory region to allow rapid degradation of the message; this signal is absent in both E. vannus (Figure 6c and Figure S9b) and E. focardii (La Terza et al., 2007). These observations together argue that divergence of trans-activating sequence elements underlies the lack of change in HSP70 gene expression in response to temperature stress in E. vannus.
5. CONCLUSION
In the current study, we de novo assembled a high-quality MAC genome of the single celled ciliate Euplotes vannus, which features “gene sized” nanochromosomes and is much more streamlined and efficient compared to the MIC genome. The MAC genome is produced by a genome rearrangement process including two important events: IES excision and chromosome breakage. Its MDS and IES boundaries are universally flanked by conserved 5’-TA-3’ pointer sequences and gene scrambling is not widespread. The chromosome breakage sites and their flanking regions display a consensus motif in an overall palindrome structure and CBS regions are duplicated on adjacent MAC chromosomes to increase homology in MAC genome. Programmed ribosomal frameshifting is widespread in this species, predominantly at 5’-AAATAA-3’ sites leading to a +1 frameshift, but with many examples of +2 and −1 shifts at 5’-ATATAA-3’ and 5’-ATATAG-3’ sites. PRF at these sequences was not shown to impair transcript abundance in this species. When facing dramatic environmental changes, especially low temperature and low/high salinity, extensive gene networks are co-expressed. Under osmotic and chemical stresses, E. vannus rapidly enhances transcription of the highly conserved HSP70 gene, but does not induce this gene due to temperature stresses. Although the putative E. vannus HSP70 protein does not contain cold-adapted amino acid substitutions proposed in E. focardii, it has lost the temperature stress-sensitive HSE and ARE elements used in other species to regulate HSP70 expression in response to temperature shifts. These results shed light on several of the most intriguing aspects of euplotid biology, and establish the genomic tools needed for future discoveries in this unique model organism.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank: Tengteng Zhang and Ruitao Gong (Ocean University of China, China), for the assistance of experimental verification; Chunyu Lian and Limin Jiang (Ocean University of China, China), for generously providing photographs of ciliate Euplotes vannus; Dr. Henglong Xu (Ocean University of China, China), for the advice on the experiment design of environmental stresses; Dr. Fengbiao Mao (University of Michigan, USA), for the advice on data visualization, Dr. Estienne Swart (Max Planck Institute, Germany) for the advice on the preparation of the manuscript, and three anonymous reviewers and editors for their insightful suggestions on the paper improvement. This work was supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2018SDKJ0406–1), National Natural Science Foundation of China (No. 31772428), Young Elite Scientists Sponsorship Program by CAST (2017QNRC001) and the Fundamental Research Funds for the Central Universities (201841013 and 201762017) to F.G. Research reported in this publication was also supported by the grants of the National Institutes of Health (award No. P40OD010964) and the National Science Foundation (grant No. 1158346) to N.A.S. and two grants to L.A.K. (NSF DEB-1541511 and NIH 1R15GM113177–01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Footnotes
DATA AVAILABILITY
All Illumina sequencing data are deposited in the NCBI Short Read Archive (SRA), under the BioProject PRJNA474770. Euplotes vannus MAC and MIC genome assemblies and gene annotation data including coding regions and predicted protein sequences are available at Euplotes vannus DB (EVDB, http://evan.ciliate.org). The custom scripts are available on GitHub (https://github.com/seanchen607/Evannus).
DATA CITATION
[dataset] Xiao Chen, Yaohan Jiang, Feng Gao, Weibo Zheng, Timothy J. Krock, Naomi A. Stover, Chao Lu, Laura A. Katz, Weibo Song; 2019; Euplotes vannus DB (EVDB); http://evan.ciliate.org.
REFERENCES
- Aeschlimann SH, Jonsson F, Postberg J, et al. (2014) The draft assembly of the radically organized Stylonychia lemnae macronuclear genome. Genome Biology and Evolution, 6, 1707–1723. 10.1093/gbe/evu139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Sperling L (2010) ParameciumDB in 2011: new tools and new data for functional and comparative genomics of the model ciliate Paramecium tetraurelia. Nucleic Acids Research, 39, D632–D636. 10.1093/nar/gkq918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird SE, Klobutcher LA (1989) Characterization of chromosome fragmentation in two protozoans and identification of a candidate fragmentation sequence in Euplotes crassus. Genes & Development, 3, 585–597. 10.1101/gad.3.5.585 [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology, 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge SW, Daub J, Eberhardt R, et al. (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Research, 41, 3 10.1093/nar/gks1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J, Kukushkin D, Lindblad K, et al. (2015) <mds_ies_db>: a database of ciliate genome rearrangements. Nucleic Acids Research, 44, D703–D709. 10.1093/nar/gki130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 1471–2105. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T (2006) Cell evolution and Earth history: stasis and revolution. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 361, 969–1006. 10.1098/rstb.2006.1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Yao M-C (2011) DNA elimination in ciliates: transposon domestication and genome surveillance. Annual Review of Genetics, 45, 227–246. 10.1146/annurev-genet-110410-132432 [DOI] [PubMed] [Google Scholar]
- Chen X, Bracht JR, Goldman AD, et al. (2014) The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell, 158, 1187–1198. 10.1016/j.cell.2014.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang Y, Sheng Y, Warren A, Gao S (2018) GPSit: An automated method for evolutionary analysis of nonculturable ciliated microeukaryotes. Molecular Ecology Resources, 4, 1–14. 10.1111/1755-0998.12750 [DOI] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, et al. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Research, 31, 3497–3500. 10.1093/nar/gkg500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MS, Peck LS (2009) HSP70 heat shock proteins and environmental stress in Antarctic marine organisms: a mini-review. Marine genomics, 2, 11–18. 10.1016/j.margen.2009.03.003 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Research, 14, 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JG, Gong Y, Hu Q (2017) Microzooplanktonic grazers - A potentially devastating threat to the commercial success of microalgal mass culture. Algal Research, 27, 356–365. 10.1016/j.algal.2017.08.024 [DOI] [Google Scholar]
- Dhanker R, Kumar R, Tseng L-C, Hwang J- S (2013) Ciliate (Euplotes sp.) predation by Pseudodiaptomus annandalei (Copepoda: Calanoida) and the effects of mono-algal and pluri-algal diets. Zoological Studies, 52, 34 10.1186/1810-522x-52-34 [DOI] [Google Scholar]
- Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang G, Hamilton EP, et al. (2017) A germline-limited piggyBac transposase gene is required for precise excision in Tetrahymena genome rearrangement. Nucleic Acids Research, 45, 9481–9502. 10.1093/nar/gkx652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes M (1994) Structure and regulation of heat shock gene promoters In: The Biology of Heat Shock Proteins and Molecular Chaperones, pp. 375–393. Cold Spring Harbor Laboratory Press. [Google Scholar]
- Foissner W, Chao A, Katz LA (2008) Diversity and geographic distribution of ciliates (Protista: Ciliophora). Biodiversity and Conservation, 17, 345–363. 10.1007/s10531-007-9254-7 [DOI] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Huang J, Zhao Y, et al. (2017) Systematic studies on ciliates (Alveolata, Ciliophora) in China: progress and achievements based on molecular information. European Journal of Protistology, 61, 409–423. 10.1016/j.ejop.2017.04.009 [DOI] [PubMed] [Google Scholar]
- Gentekaki E, Kolisko M, Gong Y, Lynn D (2017) Phylogenomics solves a long-standing evolutionary puzzle in the ciliate world: The subclass Peritrichia is monophyletic. Molecular Phylogenetics and Evolution, 106, 1–5. 10.1016/j.ympev.2016.09.016 [DOI] [PubMed] [Google Scholar]
- Gong J, Song W, Warren A (2005) Periphytic ciliate colonization: annual cycle and responses to environmental conditions. Aquatic Microbial Ecology, 39, 159–170. 10.3354/ame039159 [DOI] [Google Scholar]
- Gordon A, Hannon G (2010) FASTX-Toolkit: FASTQ/A short-reads preprocessing tools (unpublished). http://hannonlab.cshl.edu/fastx_toolkit.
- Gutiérrez JC, Martín-González A, Díaz S, Ortega R (2003) Ciliates as a potential source of cellular and molecular biomarkers/biosensors for heavy metal pollution. European Journal of Protistology, 39, 461–467. 10.1078/0932-4739-00021 [DOI] [Google Scholar]
- Hadjivasiliou Z, Iwasa Y, Pomiankowski A (2015) Cell-cell signalling in sexual chemotaxis: a basis for gametic differentiation, mating types and sexes. Journal of The Royal Society Interface, 12, 20150342. 10.1098/rsif.2015.0342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton EP, Kapusta A, Huvos PE, et al. (2016) Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife, 5, e19090. 10.7554/eLife.19090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Wang J, Fan X, et al. (2019) Genetic basis for the establishment of endosymbiosis in Paramecium. The ISME Journal, 1 10.1038/s41396-018-0341-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X (2014) Ciliates in extreme environments. Journal of Eukaryotic Microbiology, 61, 410–418. 10.1111/jeu.12120 [DOI] [PubMed] [Google Scholar]
- Jacobs ME, Klobutcher LA (1996) The long and the short of developmental DNA deletion in Euplotes crassus. Journal of Eukaryotic Microbiology, 43, 442–452. 10.1111/j.1550-7408.1996.tb04503.x [DOI] [PubMed] [Google Scholar]
- Jerka-Dziadosz M, Dosche C, Kuhlmann HW, Heckmann K (1987) Signal-induced reorganization of the microtubular cytoskeleton in the ciliated protozoon Euplotes octocarinatus. Journal of Cell Science, 87, 555–564. [Google Scholar]
- Jiang Y, Xu H, Hu X, et al. (2011) An approach to analyzing spatial patterns of planktonic ciliate communities for monitoring water quality in Jiaozhou Bay, northern China. Marine Pollution Bulletin, 62, 227–235. 10.1016/j.marpolbul.2010.11.008 [DOI] [PubMed] [Google Scholar]
- Jiang YH, Zhang T, Vallesi A, Yang X, Gao F (2019) Time-course analysis of nuclear events during conjugation in the marine ciliate Euplotes vannus and comparison with other ciliates (Protozoa, Ciliophora). Cell Cycle. 10.1080/15384101.2018.1558871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, et al. (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics, 30, 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonoska N, Saito M (2015) MDS/IES - DNA Annotation Software (MIDAS), http://knot.math.usf.edu/midas.
- Karamysheva Z, Wang L, Shrode T, et al. (2003) Developmentally programmed gene elimination in Euplotes crassus facilitates a switch in the telomerase catalytic subunit. Cell, 113, 565–576. 10.1016/S0092-8674(03)00363-5 [DOI] [PubMed] [Google Scholar]
- Katz LA (2001) Evolution of nuclear dualism in ciliates: a reanalysis in light of recent molecular data. International Journal of Systematic and Evolutionary Microbiology, 51, 1587–1592. 10.1099/00207713-51-4-1587 [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Burki F, Wilcox HM, et al. (2014) The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS biology, 12, e1001889. 10.1371/journal.pbio.1001889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B-M, Rhee J-S, Choi I-Y, Lee Y-M (2018) Transcriptional profiling of antioxidant defense system and heat shock protein (Hsp) families in the cadmium- and copper-exposed marine ciliate Euplotes crassus. Genes & Genomics, 40, 85–98. 10.1007/s13258-017-0611-y [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods, 12, 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klobutcher LA, Gygax SE, Podoloff JD, et al. (1998) Conserved DNA sequences adjacent to chromosome fragmentation and telomere addition sites in Euplotes crassus. Nucleic Acids Research, 26, 4230–4240. 10.1093/nar/26.18.4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, McEntee K (1993) Identification of cis and trans components of a novel heat shock stress regulatory pathway in Saccharomyces cerevisiae. Molecular and Cellular Biology, 13, 248–256. 10.1128/MCB.13.1.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl M, Wiese S, Warscheid B (2011) Cytoscape: software for visualization and analysis of biological networks. Methods in Molecular Biology, 696, 291–303. 10.1007/978-1-60761-987-1_18 [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch J (1995) Adaptation of inducible defense in Euplotes daidaleos (Ciliophora) to predation risks by various predators. Microbial Ecology, 30, 79–88. 10.1007/BF00184515 [DOI] [PubMed] [Google Scholar]
- La Terza A, Miceli C, Luporini P (2004) The gene for the heat-shock protein 70 of Euplotes focardii, an Antarctic psychrophilic ciliate. Antarctic Science, 16, 23–28. 10.1017/S0954102004001774 [DOI] [Google Scholar]
- La Terza A, Papa G, Miceli C, Luporini P (2001) Divergence between two Antarctic species of the ciliate Euplotes, E. focardii and E. nobilii, in the expression of heat-shock protein 70 genes. Molecular Ecology, 10, 1061–1067. 10.1046/j.1365-294X.2001.01242.x [DOI] [PubMed] [Google Scholar]
- La Terza A, Passini V, Barchetta S, Luporini P (2007) Adaptive evolution of the heat-shock response in the Antarctic psychrophilic ciliate, Euplotes focardii: hints from a comparative determination of the hsp70 gene structure. Antarctic Science, 19, 239–244. 10.1017/S0954102007000314 [DOI] [Google Scholar]
- Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Molecular Biology and Evolution, 25, 1307–1320. 10.1093/molbev/msn067 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Liu W, Jiang J, Xu Y, et al. (2017) Diversity of free-living marine ciliates (Alveolata, Ciliophora): faunal studies in coastal waters of China during the years 2011–2016. European Journal of Protistology, 61, 424–438. 10.1016/j.ejop.2017.04.007 [DOI] [PubMed] [Google Scholar]
- Lobanov AV, Heaphy SM, Turanov AA, et al. (2017) Position-dependent termination and widespread obligatory frameshifting in Euplotes translation. Nature Structural & Molecular Biology, 24, 61–68. 10.1038/nsmb.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15, 014–0550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight RD, Landweber LF (2001) The molecular basis of nuclear genetic code change in ciliates. Current Biology, 11, 65–74. 10.1016/S0960-9822(01)00028-8 [DOI] [PubMed] [Google Scholar]
- Lynn D (2009) Ciliates In: Encyclopedia of Microbiology (Third Edition) (ed. Schaechter M), pp. 578–592. Academic Press, Oxford. [Google Scholar]
- Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics, 21, 3448–3449. 10.1093/bioinformatics/bti551 [DOI] [PubMed] [Google Scholar]
- Maurer-Alcalá XX, Knight R, Katz LA (2018) Exploration of the germline genome of the ciliate Chilodonella uncinata through single-cell omics (transcriptomics and genomics). mBio, 9, e01836–01817. 10.1128/mBio.01836-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morshauser RC, Hu W, Wang H, et al. (1999) High-resolution solution structure of the 18 kDa substrate-binding domain of the mammalian chaperone protein Hsc701. Journal of Molecular Biology, 289, 1387–1403. 10.1006/jmbi.1999.2776 [DOI] [PubMed] [Google Scholar]
- Nowacki M, Shetty K, Landweber LF (2011) RNA-mediated epigenetic programming of genome rearrangements. Annual review of genomics and human genetics, 12, 367–389. 10.1146/annurev-genom-082410-101420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Bankevich A, Antipov D, et al. (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. Journal of Computational Biology, 20, 714–737. 10.1089/cmb.2013.0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJ, Knoll AH, Katz LA (2011) Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proceedings of the National Academy of Sciences, 108, 13624–13629. 10.1073/pnas.1110633108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, et al. (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology, 33, 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petz W, Valbonesi A, Schiftner U, Quesada A, Cynan Ellis-Evans J (2007) Ciliate biogeography in Antarctic and Arctic freshwater ecosystems: endemism or global distribution of species? Fems Microbiology Ecology, 59, 396–408. 10.1111/j.1574-6941.2006.00259.x [DOI] [PubMed] [Google Scholar]
- Prescott DM (1994) The DNA of ciliated protozoa. Microbiological reviews, 58, 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL, Katz LA (2001) Widespread distribution of extensive chromosomal fragmentation in ciliates. Molecular Biology and Evolution, 18, 1372–1377. 10.1093/oxfordjournals.molbev.a003921 [DOI] [PubMed] [Google Scholar]
- Ruis H, Schüller C (1995) Stress signaling in yeast. Bioessays, 17, 959–965. 10.1002/bies.950171109 [DOI] [PubMed] [Google Scholar]
- Schopf JW, Kitajima K, Spicuzza MJ, Kudryavtsev AB, Valley JW (2018) SIMS analyses of the oldest known assemblage of microfossils document their taxon-correlated carbon isotope compositions. Proceedings of the National Academy of Sciences, 115, 53–58. 10.1073/pnas.1718063115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y, He M, Zhao F, Shao C, Miao M (2018) Phylogenetic relationship analyses of complicated class Spirotrichea based on transcriptomes from three diverse microbial eukaryotes: Uroleptopsis citrina, Euplotes vannus and Protocruzia tuzeti. Molecular Phylogenetics and Evolution, 129, 338–345. 10.1016/j.ympev.2018.06.025 [DOI] [PubMed] [Google Scholar]
- Slabodnick MM, Ruby JG, Reiff SB, et al. (2017) The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Current Biology, 27, 569–575. 10.1016/j.cub.2016.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram M, Osipiuk J, Freeman B, Morimoto R, Joachimiak A (1997) Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure, 5, 403–414. 10.1016/S0969-2126(97)00197-4 [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele CJ, Barkocy-Gallagher GA, Preer LB, Preer JR (1994) Developmentally excised sequences in micronuclear DNA of Paramecium. Proceedings of the National Academy of Sciences, 91, 2255–2259. 10.1073/pnas.91.6.2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD (2013) Using GBrowse 2.0 to visualize and share next-generation sequence data. Briefings in Bioinformatics, 14, 162–171. 10.1093/bib/bbt001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, et al. (2002) The generic genome browser: a building block for a model organism system database. Genome Research, 12, 1599–1610. 10.1101/gr.403602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck T, Kochems R, Forster D, Lejzerowicz F, Pawlowski J (2018) Metabarcoding of benthic ciliate communities shows high potential for environmental monitoring in salmon aquaculture. Ecological Indicators, 85, 153–164. 10.1016/j.ecolind.2017.10.041 [DOI] [Google Scholar]
- Stover NA, Punia RS, Bowen MS, Dolins SB, Clark TG (2012) Tetrahymena Genome Database Wiki: a community-maintained model organism database. Database, 2012, bas007. 10.1093/database/bas007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Bracht JR, Magrini V, et al. (2013) The Oxytricha trifallax macronuclear genome: a complex eukaryotic genome with 16,000 tiny chromosomes. PLoS Biol, 11, 29 10.1371/journal.pbio.1001473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Serra V, Petroni G, Nowacki M (2016) Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell, 166, 691–702. 10.1016/j.cell.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 4, 4–10. 10.1002/0471250953.bi0410s25 [DOI] [PubMed] [Google Scholar]
- Trielli F, Amaroli A, Sifredi F, et al. (2007) Effects of xenobiotic compounds on the cell activities of Euplotes crassus, a single-cell eukaryotic test organism for the study of the pollution of marine sediments. Aquatic Toxicology, 83, 272–283. 10.1016/j.aquatox.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Vallesi A, Alimenti C, Federici S, et al. (2014) Evidence for gene duplication and allelic codominance (not hierarchical dominance) at the mating-type locus of the ciliate, Euplotes crassus. Journal of Eukaryotic Microbiology, 61, 620–629. 10.1111/jeu.12140 [DOI] [PubMed] [Google Scholar]
- Walton BM, Gates MA, Kloos A, Fisher J (1995) Intraspecific variability in the thermal dependence of locomotion, population growth, and mating in the ciliated protist Euplotes vannus. Physiological Zoology, 68, 98–113. 10.1086/physzool.68.1.30163920 [DOI] [Google Scholar]
- Wang C, Zhang T, Wang Y, et al. (2017a) Disentangling sources of variation in SSU rDNA sequences from single cell analyses of ciliates: impact of copy number variation and experimental error. Proceedings of the Royal Society B-Biological Science, 284, 20170425. 10.1098/rspb.2017.0425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Miao W, Wang W, Xiong J, Liang A (2018) EOGD: the Euplotes octocarinatus genome database. BMC Genomics, 19, 63 10.1186/s12864-018-4445-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Xiong J, Wang W, Miao W, Liang A (2016) High frequency of +1 programmed ribosomal frameshifting in Euplotes octocarinatus. Scientific Reports, 6, 21139 10.1038/srep21139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YR, Wang YY, Sheng Y, et al. (2017b) A comparative study of genome organization and epigenetic mechanisms in model ciliates, with an emphasis on Tetrahymena, Paramecium and Oxytricha. European Journal of Protistology, 61, 376–387. 10.1016/j.ejop.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Wang YY, Chen X, Sheng Y, Liu Y, Gao S (2017c) N6-adenine DNA methylation is associated with the linker DNA of H2A. Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Research, 45, 11594–11606. 10.1093/nar/gkx883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiąckowski K, Szkarłat M (1996) Effects of food availability on predator-induced morphological defence in the ciliate Euplotes octocarinatus (Protista). Hydrobiologia, 321, 47–52. 10.1007/Bf00018676 [DOI] [Google Scholar]
- Wickham H (2016) ggplot2: elegant graphics for data analysis Springer. [Google Scholar]
- Xiong J, Gao S, Dui W, et al. (2016) Dissecting relative contributions of cis- and trans-determinants to nucleosome distribution by comparing Tetrahymena macronuclear and micronuclear chromatin. Nucleic Acids Research, 44, 10091–10105. 10.1093/nar/gkw684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Song W, Warren A (2004) An investigation of the tolerance to ammonia of the marine ciliate Euplotes vannus (Protozoa, Ciliophora). Hydrobiologia, 519, 189–195. 10.1023/B:HYDR.0000026505.91684.ab [DOI] [Google Scholar]
- Xu H, Zhang W, Jiang Y, Yang EJ (2014) Use of biofilm-dwelling ciliate communities to determine environmental quality status of coastal waters. Science of The Total Environment, 470, 511–518. 10.1016/j.scitotenv.2013.10.025 [DOI] [PubMed] [Google Scholar]
- Zhao X, Wang Y, Wang Y, Liu Y, Gao S (2017) Histone methyltransferase TXR1 is required for both H3 and H3. 3 lysine 27 methylation in the well-known ciliated protist Tetrahymena thermophila. Science China Life Sciences, 60, 264–270. 10.1007/s11427-016-0183-1 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yi Z, Warren A, Song W. (2018) Species delimitation for the molecular taxonomy and ecology of a widely distributed microbial eukaryotes genus Euplotes (Alveolata, Ciliophora). Proceedings of the Royal Society B-Biological Science, 285, 20172159. 10.1098/rspb.2017.2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wang C, Katz LA, Gao F. (2018) A paradox: rapid evolution rates of germline-limited sequences are associated with conserved patterns of rearrangements in cryptic species of Chilodonella uncinata (Protist, Ciliophora). Science China-Life Science, 61, 1071–1078. 10.1007/s11427-018-9333-1 [DOI] [PubMed] [Google Scholar]
- Zheng W, Wang C, Yan Y, et al. (2018) Insights into an extensively fragmented eukaryotic genome: de novo genome sequencing of the multinuclear ciliate Uroleptopsis citrina. Genome Biology and Evolution, 10, 883–894. 10.1093/gbe/evy055 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.