

Abstract
Background:
Metastatic castration-resistant prostate cancer (mCRPC) is the lethal form of the disease. Several recent studies have identified genomic alterations in mCRPC, but the clinical implications of these genomic alterations have not been fully elucidated.
Objective:
To use whole-genome sequencing (WGS) to assess the association between key driver gene alterations and overall survival (OS), and to use whole-transcriptome RNA sequencing to identify genomic drivers of enzalutamide resistance.
Design, setting, and participants:
We performed survival analyses and gene set enrichment analysis (GSEA) on WGS and RNA sequencing results for a cohort of 101 mCRPC patients.
Outcome measurements and statistical analysis:
OS was the clinical endpoint for all univariate and multivariable survival analyses. Candidate drivers of enzalutamide resistance were identified in an unbiased manner, and mutations of the top candidate were further assessed for enrichment among enzalutamide-resistant patients using Fisher’s exact test.
Results and limitations:
Harboring two DNA alterations in RB1 was independently predictive of poor OS (median 14.1 vs 42.0 mo; p = 0.007) for men with mCRPC. GSEA identified the Wnt/β-catenin pathway as the top differentially modulated pathway among enzalutamide-resistant patients. Furthermore, β-catenin mutations were exclusive to enzalutamide-resistant patients (p = 0.01) and independently predictive of poor OS (median 13.6 vs 41.7 mo; p = 0.025).
Conclusions:
The presence of two RB1 DNA alterations identified in our WGS analysis was independently associated with poor OS among men with mCRPC. The Wnt/β-catenin pathway plays an important role in enzalutamide resistance, with differential pathway expression and enrichment of β-catenin mutations in enzalutamide-resistant patients. Moreover, β-catenin mutations were predictive of poor OS in our cohort.
Patient summary:
We observed a correlation between genomic findings for biopsy samples from metastases from men with metastatic castration-resistant prostate cancer (mCRPC) and clinical outcomes. This work sheds new light on clinically relevant genomic alterations in mCRPC and provides a roadmap for the development of new personalized treatment regimens in mCRPC.
Keywords: Advanced prostate cancer, Castration-resistant, Clinical outcomes, Enzalutamide, Genomics, Metastatic prostate cancer, Prognostic factors
1. Introduction
Metastatic castration-resistant prostate cancer (mCRPC) is the lethal form of PC and is predicted to lead to 29 000 PC-related deaths in the USA in 2019 [1]. As part of a broad effort to elucidate genomic drivers of mCRPC, large-scale next-generation sequencing studies have been conducted. Whole-exome sequencing (WES) studies have identified AR, ETS family (eg, ERG), PTEN, and TP53 as genes that are frequently affected by somatic alterations, with TP53 and AR alterations enriched in mCRPC compared to primary PC [2,3]. Recent whole-genome sequencing (WGS) studies revealed AR enhancer tandem duplications as an additional important genomic feature observed in a subset of mCRPCs [4,5]. These next-generation sequencing efforts identified recurrent variants in mCRPC and highlighted the genomic heterogeneity of the disease.
Despite recent advances in identifying genomic drivers of mCRPC, there is a paucity of studies with longitudinal outcomes examining the clinical implications of these genomic alterations. It has been shown that in primary prostate tumors, PTEN loss [6–9], focal deletions on chromosomes 13q (including RB1) and 18q [10], focal amplifications on chromosome 5p13 or 5p15 [10], and percentage genome alteration (PGA) [11] are associated with advanced disease and poor clinical outcomes. The prognostic value of other common variants, such as TMPRSS2-ERG fusions, remains equivocal [12–14]. It is likely that prognostic molecular determinants similarly exist in mCRPC, but they have not been fully elucidated to date.
Despite the overall efficacy of enzalutamide in treating mCRPC [15], individual patients have variable responses to treatment, and the eventual development of resistance is nearly universal. Recent studies have begun to investigate the variability in response to enzalutamide by focusing largely on genomic variants known to affect AR signaling or DNA damage repair [16,17]. However, an unbiased approach to identify dysregulated genes and pathways associated with the development of enzalutamide resistance has yet to be performed. To identify clinically relevant genomic markers in mCRPC, we undertook a subsequent detailed clinical analysis of 101 mCRPC patients for whom we had previously reported the results of whole-transcriptome RNA sequencing (RNA-seq) and WGS. This cohort included a group of patients who developed progressive disease following treatment with enzalutamide. In addition to evaluating the association between key driver gene alterations and overall survival (OS), we performed RNA expression–based gene set enrichment analysis (GSEA) to identify potential drivers of resistance to enzalutamide. We followed up this analysis with an investigation of DNA genomic events related to the top candidate pathway (Wnt/β-catenin), and examined the prognostic significance and enrichment of CTNNB1 mutations in enzalutamide-resistant patients.
2. Patients and methods
2.1. Clinical samples and data processing
Tissue biopsy samples from 101 mCRPC patients enrolled in the multi-institutional Stand Up 2 Cancer / Prostate Cancer Foundation-funded West Coast Prostate Cancer Dream Team underwent WGS as previously described (Supplementary material) [4]. Sequencing was performed using the Illumina HiSeq platform, and a mean tumor sequencing depth of 109× and a matched normal sample sequencing depth of 38× were achieved. The results of these sequencing efforts have previously been reported [4]. Characteristics for biopsied patients were compared between the WGS and no WGS groups using a χ2 independence test for categorical variables, a two-sample t test for age as a normally distributed continuous variable, and a Wilcoxon rank-sum test for continuous variables that were not normally distributed. The significance level was set to 0.05 for all comparisons.
2.2. Variable definitions
Genes of interest for survival analyses were selected a priori by choosing putative tumor suppressors and oncogenes in metastatic PC. We examined the prognostic significance of DNA alterations in TP53, PTEN, and RB1, the three most commonly aberrant and well-characterized tumor suppressors in mCRPC. In addition, we examined MYC amplification, TMPRSS2-ERG fusions, global copy number burden (PGA), and AR gain-of-function via activating mutation, copy number gain, or enhancer tandem duplication. Given that prior studies have reported that combinatorial loss of at least two of three genes TP53, PTEN, and RB1 was associated with more clinically aggressive disease [18–20], we examined two additional groups of interest: (1) two or more DNA alterations in each of at least two of the three tumor suppressor genes; and (2) no DNA alterations in any of these three genes. We defined PGA as in a previous report on localized prostate cancer [11] and stratified our cohort into PGA quartiles for survival analysis. We focused on these specific genomic alterations because they are prevalent and likely to be important in PC pathogenesis.
2.3. Mutation, copy number, and structural variant calls
Mutations, copy number alterations, and structural variants were identified and integrated into final estimates of the predicted number of altered alleles for each gene interrogated (as described in the Supplementary material). Predicted monoallelic and biallelic loss calls were made on the basis of the total number of DNA alterations due to inactivating mutations, shallow (1-copy) or deep (2-copy) deletions, and inactivating structural variants observed.
2.4. RNA-seq pathway analysis of enzalutamide resistance
To identify genomic features associated with enzalutamide resistance, we performed a cross-sectional GSEA on RNA-seq expression data using the standard GSEA tool (Supplementary material) [21]. Analyses were performed to identify gene sets that were enriched in enzalutamide-resistant patients relative to enzalutamide-naïve patients. Gene sets were considered significantly enriched if their false discovery rate (FDR) q value was < 0.25, as defined by the publishers of the GSEA tool. To further assess pathway-level expression differences between enzalutamide-resistant and enzalutamide-naïve patients, we computed a Wnt signaling pathway expression score for each patient based on expression of genes in the pathway (Supplementary material). A one-sided Mann-Whitney U test was performed to assess whether the median Wnt signaling pathway expression score was greater in the enzalutamide-resistant group than in the enzalutamide-naïve group.
2.5. Clinical endpoints and survival analysis
Enzalutamide resistance was defined as progression during treatment with enzalutamide according to the Prostate Cancer Clinical Trials Working Group-2 criteria [22]. OS from the time of mCRPC diagnosis was examined to evaluate the clinical implications of specific genomic alterations of interest. Survival analyses were conducted using the Kaplan-Meier method with log-rank testing for significance. When assessing the prognostic significance of DNA alterations in TP53, PTEN, and RB1, we decided a priori to perform two-group survival comparisons between patients with two or more DNA alterations and those with zero or one DNA alteration. When assessing the prognostic significance of low-frequency alterations, we used the exact log-rank test (ExaLT), as it has been shown that this has better properties than the log-rank test for comparing small or asymmetric groups [23]. To limit the number of statistical tests performed, we selected relatively few genomic features to investigate as potential prognostic factors. We also performed a more unbiased analysis examining the DNA alterations observed at different frequencies in the tertile of patients with the shortest OS relative to the tertile with the longest OS. Multivariable survival analysis with Cox proportional hazards models was performed to account for demographic, clinicopathologic, and treatment factors associated with survival. In addition to RB1 and CTNNB1 alteration status, the multivariable survival analysis included six of the eight clinical factors previously reported to be prognostic for OS [24], excluding opioid analgesic use and serum albumin (which were not available in our cohort). Unless otherwise stated, all independence and hypothesis tests were performed using a two-sided significance level of 0.05. R v.3.5.0 was used to perform all statistical analyses along with the survival (v.2.42–6), survminer (v.0.4.3), and ExaLT (v.1.0) R packages for survival analysis.
3. Results
3.1. Patient characteristics
A total of 256 patients with mCRPC were enrolled in a prospective biopsy cohort. Of these 256 patients, 78% (200) had biopsies positive for metastatic PC. Of these 200 biopsies, 101 with sufficient tumor volume were selected for WGS. The clinicopathologic and sequencing results for these patients have previously been reported [4]. Clinical outcomes for this cohort have since become available, and are the basis of this report. Overall, the clinical characteristics of the patients whose samples were selected for WGS were similar to those of the entire biopsy cohort with respect to all variables except site of metastasis at the time of biopsy (Table 1). Some 5.0% of patients had a transcriptomic profile consistent with treatment-emergent small-cell neuroendocrine prostate cancer (t-SCNC). Enzalutamide-naïve patients comprised 65% of the cohort, while 35% had received prior enzalutamide treatment. The mean length of clinical follow-up from the time of initial mCRPC diagnosis was 3.0 yr. Three WGS patients were excluded from survival analyses because of the date of their initial mCRPC diagnosis was unknown.
Table 1 –
Patient demographics and clinicopathologic featuresa
| Characteristic | No sequencing | Sequencing | p value |
|---|---|---|---|
| Patients (n) | 155 | 101 | |
| Mean age, yr (standard deviation) | 69 (7.9) | 71 (8.4) | 0.11 |
| Race, n (%) | 0.9 | ||
| Asian | 4 (2.9) | 4 (4.3) | |
| Black or African American | 9 (6.6) | 5 (5.3) | |
| Native American | 1 (0.7) | 0 (0) | |
| White | 122 (90) | 85 (90) | |
| Missing | 19 (12) | 7 (6.9) | |
| Gleason score at diagnosis, n (%) | 0.8 | ||
| ≥8 | 79 (54) | 52 (57) | |
| <8 | 66 (46) | 39 (43) | |
| Missing | 10 (6.5) | 10 (9.9) | |
| ECOG performance status, n (%) | 0.9 | ||
| 0 | 82 (54) | 57 (56) | |
| 1 | 65 (43) | 41 (41) | |
| ≥2 | 5 (3.3) | 3 (3) | |
| Missing | 3 (2.0) | 0 (0) | |
| Metastatic sites at time of biopsy, n (%) | 0.025 | ||
| Liver | 25 (16) | 15 (15) | |
| Visceral metastases (not liver) | 32 (21) | 11 (11) | |
| Bone ± lymph node | 93 (60) | 64 (63) | |
| Lymph node only | 5 (3.2) | 11 (11) | |
| Missing | 0 (0) | 0 (0) | |
| Enzalutamide treatment status, n (%) | 0.2 | ||
| Naїve | |||
| Prior abiraterone treatment | 58 (37) | 27 (27) | |
| No prior abiraterone treatment | 43 (28) | 38 (38) | |
| Resistant | |||
| Prior abiraterone treatment | 33 (21) | 20 (20) | |
| No prior abiraterone treatment | 21 (14) | 15 (15) | |
| Missing | 0 (0) | 1 (1) | |
| PSA response to AR targeted therapy (%) | 63 | 57 | 0.7 |
| Median PSA, ng/ml (IQR) | 41 (13–148) | 43 (15–148) | 0.7 |
| Median alkaline phosphatase, U/l (IQR) | 98 (65–176) | 91 (65–140) | 0.4 |
| Median lactate dehydrogenase, IU/l (IQR) | 190 (164–248) | 203 (166–291) | 0.13 |
| Median hemoglobin, g/dl (IQR) | 12 (11–13) | 13 (12–14) | 0.15 |
ECOG = Eastern Cooperative Oncology Group; IQR = interquartile range; PSA = prostate-specific antigen.
All clinicopathologic variables were measured at time of biopsy and p values are for comparison of the sequenced and not sequenced groups.
3.2. DNA alterations
As previously reported, two or more DNA alterations in TP53 were observed in 47/101 patients (47%), while two or more DNA alterations in PTEN or RB1 were observed in 36/101 (36%) and 12/101 patients (12%), respectively [4]. A total of 38/101 patients (38%) had MYC amplification, and 86/101 (86%) had AR gain of function. Closer examination of combinatorial alterations in individual patients revealed that 23/101 (23%) had two or more DNA alterations in at least two of the three genes (TP53, PTEN, RB1), 17 had two or more DNA alterations in each of TP53 and PTEN, and 7/101 (6.9%) had no DNA alterations in any of the three genes (Fig. 1).
Fig. 1 –

(A) Venn diagram showing the distribution of combinatorial biallelic loss of three key tumor suppressor genes. (B) Heat map showing the functional gene status of key oncogenes and tumor suppressors for each patient. Black denotes two or more DNA alterations, blue denotes one DNA alteration, grey denotes no DNA alterations, and red denotes the presence of a genomic feature or a gene gain of function.
3.3. Harboring two DNA alterations in RB1 is associated with shorter survival and a distinct transcriptomic profile
The median OS for patients with and without two DNA alterations in RB1 was 14.1 and 42.0 mo, respectively (ExaLT p = 0.007; Fig. 2). There was no significant difference in OS between groups when examining the prognostic significance of AR gain of function, two or more TP53 DNA alterations, two or more PTEN DNA alterations, MYC amplification, TMPRSS2-ERG fusion, or PGA (Fig. 3, Supplementary Fig. 2). Prior analyses have suggested that two DNA alterations in at least two of the RB1, PTEN, and TP53 genes (cluster B) may predict poor survival [20] or, inversely, that no DNA alterations in RB1, PTEN, and TP53 (cluster A) may predict better survival. However, neither the combinatorial gene alteration signatures (cluster A and cluster B) nor the large subcategory of cluster B with two or more alterations in each of TP53 and PTEN had prognostic significance with respect to OS (Fig. 3, Supplementary Fig. 3). These analyses suggest that DNA alterations in the RB1 gene alone may be associated with clinically aggressive mCRPC. Since prior studies found that RB1 haploinsufficiency may be an important phenomenon [25,26], we also assessed the prognostic significance of having one DNA alteration in RB1. We found that there was no difference in survival between patients with one DNA alteration in RB1 and those with no alterations (p = 0.7; Supplementary Fig. 4). Examination of DNA alteration frequency in the shortest-OS and longest-OS tertiles revealed that RB1 was among the top 20 genes with a higher prevalence of mutations in the shortest-OS tertile relative to the longest-OS tertile. Complete results for these OS-stratified tertile analyses are presented in Supplementary Table 1.
Fig. 2 –

Differences in overall survival between patients with two DNA alterations in RB1 and those with zero or one DNA alteration from the date of initial mCRPC diagnosis.
Fig. 3 –

Forest plot demonstrating the prognostic significance of genomic variants of interest with respect to overall survival. A hazard ratio >1 indicates poor prognosis in terms of overall survival.
Given our finding that the presence of two DNA alterations in RB1 was associated with poor clinical outcomes, we investigated the genomic characteristics of patients with two RB1 DNA alterations in greater detail. Assessment of transcriptomic changes previously shown to be associated with RB1 alterations revealed having two alterations in RB1 was associated with expanded E2F1 function and lower AR activity (Supplementary Fig. 5A,B). These findings are consistent with prior studies in RB1-deficient mCRPC [27,28]. We also assessed whether t-SCNC was enriched in patients with at least two DNA alterations in each of TP53 and RB1, as previously described [20]. While there were only four patients with dual TP53/RB1 alterations in our cohort, two of the four had gene expression profiles consistent with a previously published t-SCNC transcriptomic signature [29]. This was in contrast to three of 96 patients in the remainder of the cohort who had gene expression profiles consistent with t-SCNC (50% vs 3.1%; p = 0.01). Taken together, these analyses suggest that mCRPCs with two DNA alterations in RB1 had not only shorter OS but also a distinct transcriptomic profile that was consistent with the literature.
3.4. Alterations in the Wnt/β-catenin pathway may be associated with enzalutamide resistance
After assessing the prognostic impact of selected genomic alterations on OS, we sought to identify genomic features associated with enzalutamide resistance. To identify candidate genes that might confer resistance to enzalutamide, we performed RNA-seq expression-based GSEA [21] on our cohort. GSEA identified the Wnt/β-catenin pathway as the most highly enriched pathway among enzalutamide-resistant patients (normalized enrichment score 1.95, FDR = 0.15; Fig. 4A, Supplementary Table 2). Using the Wnt signaling pathway genes as a transcriptomic signature, we found that expression of these genes was significantly higher on average among enzalutamide-resistant patients (p = 0.035; Supplementary Fig. 6). To further investigate this finding, we examined genomic alterations in CTNNB1 (β-catenin), the main effector of the Wnt pathway. Four patients were identified with exonic missense mutations in CTNNB1. All four of the mutations were located in a similar genomic region (Fig. 4C), with three of these occurring at known pathogenic mutational hotspots [30]. Moreover, all four of these individuals were resistant to enzalutamide. This enrichment of CTNNB1 mutations among enzalutamide-resistant patients was significant on cross-sectional analysis (Fisher’s exact test: p = 0.01). Although some patients treated with enzalutamide had received prior therapy with abiraterone, none of the four patients with enzalutamide resistance and CTNNB1 mutations had been exposed to abiraterone. In addition to enzalutamide resistance, CTNNB1 alterations were associated with poor OS. The median OS for patients with and without CTNNB1 mutations was 13.6 and 41.7 mo, respectively (ExaLT p = 0.025; Fig. 4D).
Fig. 4 –
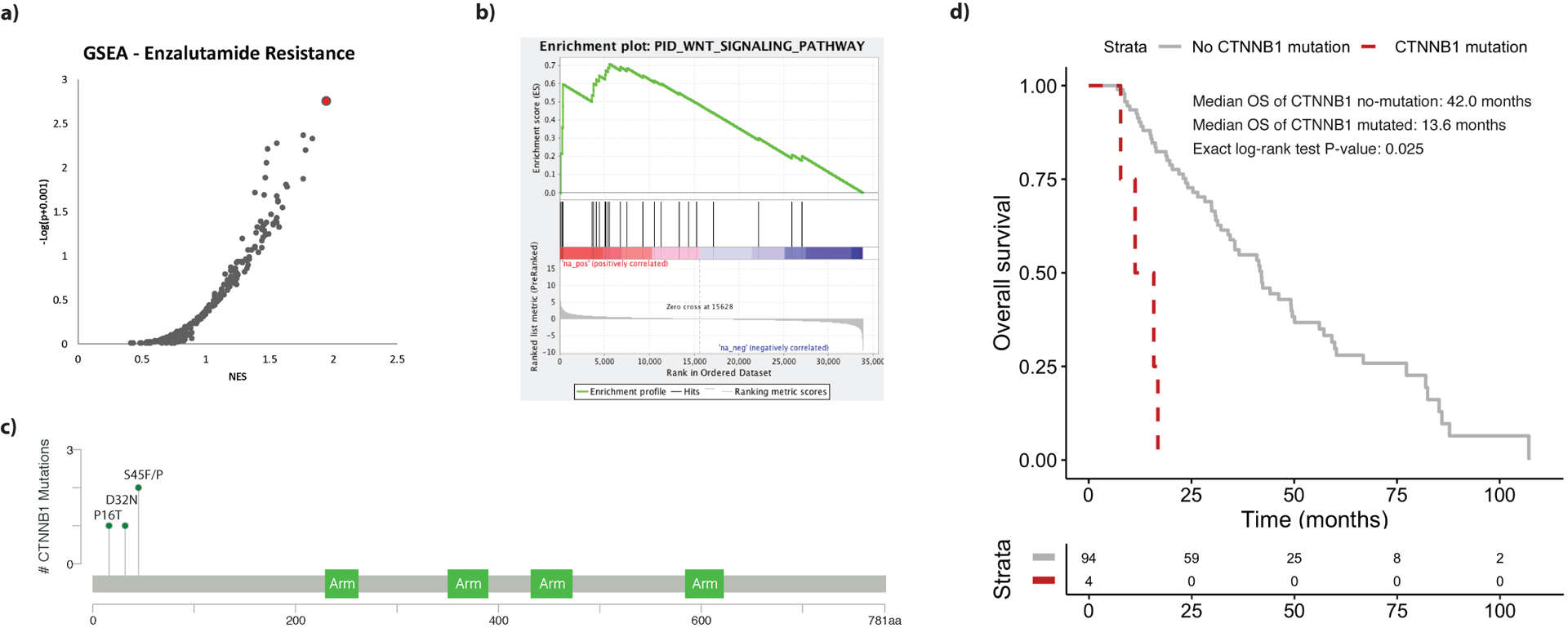
(A) Scatter plot of candidate gene sets ordered in increasing likelihood of enrichment among enzalutamide-resistant patients according to gene set enrichment analysis (GSEA). The top candidate pathway (Wnt/β-catenin) is highlighted in red. NES = normalized enrichment score. (B) GSEA enrichment plot demonstrating the degree of correlation between genes in the Wnt pathway gene set and enzalutamide resistance. (C) MutationMapper [40] lollipop plot highlighting genomic coordinates of the four CTNNB1 missense mutations in our cohort. (D) Overall survival differences between patients with mutated and wild-type CTNNB1.
3.5. Multivariable analyses demonstrate that RB1 and CTNNB1 alterations are prognostic after accounting for clinicopathologic variables
Important findings from univariate analysis were that two DNA alterations in RB1 (Fig. 2) and CTNNB1 activation (Fig. 4D) were associated with poor OS. To determine whether these genomic alterations were predictive of worse OS after accounting for demographic and other important clinical features known to be prognostic in mCRPC, we performed multivariable analysis using a multivariable Cox proportional hazards model with backward selection of variables. CTNNB1 mutations and the presence of two DNA alterations in RB1 were both independently prognostic when considered in the same multivariable model (two RB1 alterations, p = 0.003; CTNNB1 mutation, p < 0.001) after adjusting for clinicopathologic variables including serum alkaline phosphatase, serum hemoglobin, prostate-specific antigen (PSA), and the presence of liver metastases (Table 2).
Table 2 –
Multivariable analysisa
| p value | |
|---|---|
| Lactate dehydrogenase in IU/l | 0.2 |
| Prostate-specific antigen in ng/ml | 0.5 |
| Hemoglobin in g/dl | 0.9 |
| Alkaline phosphatase in U/l | 0.037 |
| Eastern Cooperative Oncology Group performance status >0 | 0.6 |
| Presence of visceral metastases | 0.009 |
| Two DNA alterations in RB1 | 0.003 |
| CTNNB1 mutation | <0.001 |
Laboratory values were all measured at the time of biopsy and were modeled as log values for multivariable analysis.
4. Discussion
We used WGS of biopsies to assess the association between key driver gene alterations and OS among patients with mCRPC, and subsequently used whole-transcriptome RNA-seq to identify genomic drivers of enzalutamide resistance.
We found that having two DNA alterations in RB1 (but not two alterations in PTEN, two alterations in TP53, MYC amplification, or AR gain-of-function) was associated with poor OS independently of other clinicopathologic factors. We observed two DNA alterations in RB1 in 12% of our cohort, as compared to a prevalence of 2–3% in recent primary PC cohorts [31–33]. This is consistent with studies that found that RB1 alterations were enriched in metastatic compared to primary PCs but present in still only a minority of mCRPCs [2,3,32]. It has been reported that RB1 loss predicts poor recurrence-free survival in localized PC [34], and a recent study of circulating tumor DNA (ctDNA) sequencing found that RB1 loss was a predictor of progression-free survival in post-enzalutamide mCRPCs [16]. A limitation of the previous analysis of RB1 in mCRPCs is that it only included patients for whom ctDNA was detectable, which potentially introduced a bias for patients with more advanced disease [35,36]. Our study potentially faced similar bias. By profiling only tumors with sufficient tumor volume for WGS, we may have inadvertently selected for patients with a higher total tumor volume and potentially more advanced disease. We addressed this by comparing clinicopathologic characteristics between patients whose tumors were sequenced and those whose tumors were not sequenced (Table 1), which revealed that the groups had comparable clinical characteristics and baseline parameters at the time of biopsy. The one exception to this observation was that, if anything, WGS patients had a lower prevalence of visceral involvement (26% vs 37%) and a higher prevalence of lymph node–only metastases (11% vs 3.2%) in comparison to the rest of the biopsy cohort (p = 0.025). There were no differences in baseline parameters known to be prognostic in mCRPC and that are generally felt to be associated with tumor volume, including PSA, alkaline phosphatase, lactate dehydrogenase, and hemoglobin. Finally, there was no difference in OS in our biopsy cohort between patients whose tumors were and were not subjected to WGS (p = 0.7). Taken together, these data suggest that the patients selected for sequencing were clinically representative of the entire biopsy cohort.
In our study we focused on DNA alterations directly affecting tumor suppressor genes because these are most likely to result in specific inactivation of the affected genes. However, we acknowledge that there may be other mechanisms responsible for inactivation of tumor suppressor genes. For example, it has been shown that RB1 may be inactivated via mechanisms such as CDK4/6 upregulation, cyclin D/E amplification, and CDKN2A copy number loss [37]. As RB1 is emerging as a clinically important gene in mCRPC, we analyzed the prevalence in our cohort of alterations in genes other than RB1 that have been shown to be associated with RB1 loss (Supplementary Table 3, Supplementary Fig. 7). Interestingly, cyclin D/E amplification and CDKN2A two-copy deletion tended to be observed in samples with fewer than two DNA alterations in RB1, which is consistent with literature suggesting that these different mechanisms of RB1 loss of function may tend to be mutually exclusive [37]. Additional studies are needed to assess the effects of these alterations on RB1 function in mCRPC.
Our analyses of enzalutamide resistance revealed that Wnt/β-catenin pathway expression and β-catenin (CTNNB1) mutations were associated with enzalutamide treatment resistance. Furthermore, CTNNB1 mutations were associated with worse OS, although this finding should be interpreted with caution given the low number of CTNNB1 mutations observed (n = 4). β-Catenin is a major downstream effector molecule of Wnt in the canonical Wnt pathway and has been implicated in many cancers [38]. It has been shown that CTNNB1 mutations are enriched in metastatic disease, with an estimated prevalence of 4% in mCRPC [2,4] compared to <1% in localized PCs [31,32]. In addition, a prior ctDNA study of enzalutamide resistance in mCRPC found CTNNB1 activating mutations in four of 13 (31%) enzalutamide-resistant PCs [16], suggesting that CTNNB1 mutations may be enriched in enzalutamide-resistant mCRPC. In our unbiased GSEA analysis of genomic pathways associated with enzalutamide resistance, we found that Wnt signaling was the top pathway that differentiated enzalutamide-resistant from enzalutamide-naïve patients. Moreover, since our cohort contained both enzalutamide-resistant and enzalutamide-naïve patients, we were able to perform a cross-sectional analysis to explicitly evaluate CTNNB1 mutation enrichment. Although CTNNB1 mutations were observed at a relatively low frequency in our cohort (n = 4), they were exclusive to enzalutamide-resistant patients. This enrichment in enzalutamide-resistant disease (11% compared to zero patients out of 65) was statistically significant. We report a lower absolute prevalence of activating CTNNB1 mutations among enzalutamide-resistant patients compared to the previous estimate of 31%. This difference might be attributable to the relatively low number of CTNNB1 mutational events observed in both cohorts. Nonetheless, the enrichment trend is consistent across cohorts and bolsters the theory that Wnt pathway activation involving β-catenin is a potential mechanism underlying enzalutamide resistance. Our findings are of particular interest in light of preclinical data demonstrating that Wnt/β-Catenin inhibition may overcome resistance to enzalutamide in CRPC [39]. Additional studies are needed to elucidate the emerging evidence that this pathway is potentially clinically targetable in PC.
A particular strength of our study design is the use of WGS data, which allowed us to incorporate high-confidence mutation, copy number, and structural variant calls into final assignments of functional copy status. We found that structural variants contributed to a substantial proportion of genomic events in our selected genes of interest. For example, six of the 12 patients with two DNA alterations in the RB1 tumor suppressor had an inactivating structural variant that contributed to the loss of one copy of the gene. These structural variants most likely disrupted gene function, as evidenced by associated low mRNA expression [4]. Similarly, we found that 21/36 patients with two PTEN DNA alterations and 14/47 patients with two TP53 DNA alterations had inactivating structural variants involving the respective genes. In addition to providing a more comprehensive view of alterations in specific driver genes, WGS allowed us to study the prognostic significance of genomic events that are otherwise difficult or even impossible to assess accurately with either whole-exome sequencing or targeted (mutational) panels. For example, we were able to examine the prognostic significance of tandem duplications in the enhancer upstream of the AR gene body, which is emerging as an important genomic event in advanced prostate cancer [4].
Whole-genome sequencing offers better resolution than other sequencing types for identifying structural variants, but as a tradeoff for coverage of the entire genome, tumors are often sequenced at a relatively lower depth. We achieved a mean sequencing depth of 109× in tumor samples, which is generally considered very high for WGS. Nonetheless, this coverage depth may be insufficient to identify certain subclonal mutations present in only a small fraction of cancer cells within a tumor. Future studies involving deep sequencing of genomic regions such as the RB1 and CTNNB1 loci are needed to assess the prevalence and clinical relevance of these subclonal mutations in a low fraction of cancer cells.
5. Conclusions
By using WGS, the gold standard for detection of structural variants, in conjunction with mRNA expression profiling in our study, we were able to link a comprehensive view of genomic alterations in men with mCRPC to clinical outcomes. The scope of the alterations we examined extends beyond what can be assessed with targeted panels or whole-exome sequencing and may be useful for designing targeted panels to conduct even larger clinical studies in the future. Our analysis suggests that emerging genomic variants of mCRPC can help to explain the clinical heterogeneity of the disease. We highlight RB1 and CTNNB1 as recurrently aberrant genes with clinical prognostic value, and we identified Wnt pathway activation as a potential mechanism underlying clinical enzalutamide resistance.
Supplementary Material
Acknowledgments
Funding/Support and role of the sponsor: This work was supported by Pacific Northwest Prostate Cancer SPORE/NCI (P50 CA097186 to J.J.A.), Department of Defense Synergistic Idea Awards (W81XWH-13–1–0420 and W81XWH-16–1–0597 to J.J.A.), a Prostate Cancer Foundation Challenge Award (to F.Y.F.), a Prostate Cancer Foundation Special Challenge Award (15CHAS03 to E.J.S.), and a Stand Up To Cancer-Prostate Cancer Foundation Prostate Cancer Dream Team Award (SU2C-AACR-DT0812 to E.J.S.). Stand Up To Cancer (SU2C) is a division of the Entertainment Industry Foundation. This research grant was administered by the American Association for Cancer Research, the scientific partner of SU2C. The sponsors played a role in the design and conduct of the study and in data collection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The presence of two DNA alterations in RB1 is associated with poor overall survival independently of other clinicopathologic factors in metastatic castration-resistant prostate cancer. In addition, Wnt/β-catenin pathway activation and β-catenin mutations are associated with enzalutamide resistance and poor overall survival.
Financial disclosures:
Eric J. Small certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- 2.Robinson D, Van Allen EM, Wu Y-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grasso CS, Wu Y-M, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239–43. 10.1038/nature11125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;174:758–69.e9. 10.1016/j.cell.2018.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018;174:433–47.e19. 10.1016/j.cell.2018.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boström PJ, Bjartell AS, Catto JWF, et al. Genomic predictors of outcome in prostate cancer. Eur Urol 2015;68:1033–44. 10.1016/j.eururo.2015.04.008 [DOI] [PubMed] [Google Scholar]
- 7.Leinonen KA, Saramaki OR, Furusato B, et al. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol Biomarkers Prev 2013;22:2333–44. 10.1158/1055-9965.EPI-13-0333-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCall P, Witton CJ, Grimsley S, Nielsen KV, Edwards J. Is PTEN loss associated with clinical outcome measures in human prostate cancer? Br J Cancer 2008;99:1296–301. 10.1038/sj.bjc.6604680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krohn A, Diedler T, Burkhardt L, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol 2012;181:401–12. 10.1016/j.ajpath.2012.04.026 [DOI] [PubMed] [Google Scholar]
- 10.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11–22. 10.1016/j.ccr.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lalonde E, Ishkanian AS, Sykes J, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol 2014;15:1521–32. 10.1016/S1470-2045(14)71021-6 [DOI] [PubMed] [Google Scholar]
- 12.Gopalan A, Leversha MA, Satagopan JM, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 2009;69:1400–6. 10.1158/0008-5472.CAN-08-2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomlins SA, Alshalalfa M, Davicioni E, et al. Characterization of 1577 primary prostate cancers reveals novel biological and clinicopathologic insights into molecular subtypes. Eur Urol 2015;68:555–67. 10.1016/j.eururo.2015.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nam RK, Sugar L, Yang W, et al. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer 2007;97:1690–5. 10.1038/sj.bjc.6604054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424–33. 10.1056/NEJMoa1405095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyatt AW, Azad AA, Volik SV, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol 2016;2:1598 10.1001/jamaoncol.2016.0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov 2018;8:444–57. 10.1158/2159-8290.CD-17-0937 [DOI] [PubMed] [Google Scholar]
- 18.Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355:84–8. 10.1126/science.aah4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83. 10.1126/science.aah4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aparicio AM, Shen L, Tapia ELN, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res 2016;22:1520–30. 10.1158/1078-0432.CCR-15-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59. 10.1200/JCO.2007.12.4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandin F, Papoutsaki A, Raphael BJ, Upfal E. Accurate computation of survival statistics in genome-wide studies. PLoS Comput Biol 2015;11:e1004071 10.1371/journal.pcbi.1004071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halabi S, Lin C-Y, Kelly WK, et al. Updated prognostic model for predicting overall survival in first-line chemotherapy for patients with metastatic castration-resistant prostate cancer. J Clin Oncol 2014;32:671–7. 10.1200/JCO.2013.52.3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Vasconcellos I, Anastasov N, Sanli-Bonazzi B, Klymenko O, Atkinson MJ, Rosemann M. Rb1 haploinsufficiency promotes telomere attrition and radiation-induced genomic instability. Cancer Res 2013;73:4247–55. 10.1158/0008-5472.CAN-12-3117 [DOI] [PubMed] [Google Scholar]
- 26.Coschi CH, Ishak CA, Gallo D, et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov 2014;4:840–53. 10.1158/2159-8290.CD-14-0215 [DOI] [PubMed] [Google Scholar]
- 27.McNair C, Xu K, Mandigo AC, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest 2017;128:341–58. 10.1172/JCI93566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis JN, Wojno KJ, Daignault S, et al. Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res 2006;66:11897–906. 10.1158/0008-5472.CAN-06-2497 [DOI] [PubMed] [Google Scholar]
- 29.Aggarwal R, Huang J, Alumkal JJ, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol 2018;36:2492–503. 10.1200/JCO.2017.77.6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155–63. 10.1038/nbt.3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Espiritu SMG, Liu LY, Rubanova Y, et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 2018;173:1003–13.e15. 10.1016/j.cell.2018.03.029 [DOI] [PubMed] [Google Scholar]
- 32.Wedge DC, Gundum G, Mitchell T, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet 2018;50:682–92. 10.1038/s41588-018-0086-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser M, Sabelnykova VY, Yamaguchi TN, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64. 10.1038/nature20788 [DOI] [PubMed] [Google Scholar]
- 34.Sharma A, Yeow W-S, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest 2010;120:4478–92. 10.1172/JCI44239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang Y, Tolani B, Nie X, Zhi X, Hu M, He B. Review of the clinical applications and technological advances of circulating tumor DNA in cancer monitoring. Ther Clin Risk Manage 2017;13:1363–74. 10.2147/TCRM.S141991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008;8:714–24. 10.1038/nrc2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17:9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Cheng L, Li J, et al. Inhibition of the Wnt/β-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res 2018;78:3147–62. 10.1158/0008-5472.CAN-17-3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.