

Abstract
Use of abundant feedstock pronucleophiles in catalytic carbonyl reductive coupling minimizes preactivation, enhancing efficiency in target-oriented synthesis. For reactions of this type, equally inexpensive reductants are desired or, ideally, corresponding hydrogen autotransfer processes may be enacted wherein alcohols serve dually as reductant and carbonyl proelectrophile.
Keywords: Petrochemical Feedstocks, Atom-Efficiency, Enantioselective Catalysis, Carbonyl Addition, Hydrogenation, Transfer Hydrogenation
Graphical Abstract
I. Introduction and Inspiration
The construction of complex small molecules often requires intervention of non-native structural elements to direct reactivity, regio- and stereoselectivity. For example, directing or protecting groups are frequently used to guide the site-selectivity of chemical processes, chiral auxiliaries are often required to direct the stereochemistry of covalent bond formations, and stoichiometric carbanion generation via halogenation-metalation-transmetallation sequences remains common. The additional manipulations required to prepare, install and remove such structural elements contribute to inefficiency.[1] More idealized views on chemical synthesis have been posited;[2] however, the relatively modest lexicon of synthetic methods available in the 20th century has necessitated a more arcane approach. For example, the commercial manufacturing route to eribulin, a skeletally and stereochemically complex chemotherapeutic agent used to treat breast cancer, demands a total of 65 steps, half of which are devoted to redox reactions and protecting group manipulations.[3] A vast number of “sacrificial reagents” are required. In contrast, dioctyl phthalate (an industrial plasticizer) is made through a 4-step sequence composed almost entirely of skeletal construction events with water as the sole byproduct.[4] This gap in efficiency, which may be quantified by the E-factor analysis,[5] should be viewed as a source of inspiration. While methods used in academic and discovery-related research are often poorly suited for large volume chemical manufacture, one may look to the largest volume applications of homogenous catalysis, hydroformylation[6] and methanol carbonylation,[7] and see that a principal characteristic of a scalable method resides in the ability to affect C-C bond formation between feedstock reactants in the absence of stoichiometric byproducts. Accordingly, one may narrow this gap in efficiency through the development of methods that enable complex molecule syntheses through routes composed exclusively of byproduct-free skeletal construction events (Scheme 1).
Scheme 1.

An inverse relationship between complexity and efficiency highlights the need for redox- and site-selective catalytic C-C bond formation.
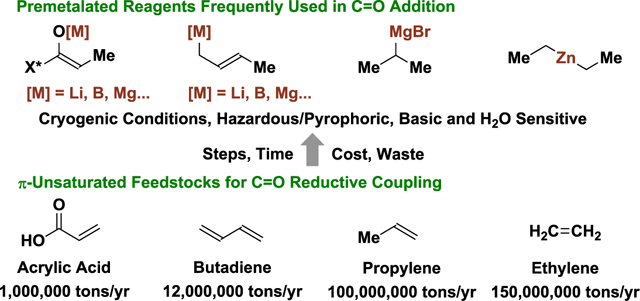
For both eribulin and dioctyl phthalate, the majority of the skeletal construction events exploit carbonyl addition, implicating the potential impact of developing catalytic methods for stereo- and site-selective carbonyl addition, especially processes that embody high levels of redox economy and exploit inexpensive feedstock reactants. In an effort to valorize carbonyl addition methodology for complex molecule synthesis, we have developed a broad, new class of metal-catalyzed carbonyl reductive C-C coupling reactions that occur through the addition or redistribution of hydrogen (Scheme 2).[8–10] These processes enable carbonyl addition from π-unsaturated pronucleophiles, bypassing the use of premetalated reagents and the attendant issues of safety, selectivity, and waste posed by their use. The terminal reductants utilized in these reactions are abundant, low molecular weight feedstocks (elemental hydrogen, 2-propanol and formic acid). This aspect is significant: metallic reductants (e.g. Mn, Zn) can be just as problematic as the organometallic reagents they are intended to replace,[8,9] and silane based reductants are orders of magnitude more expensive than the feedstocks they are used to activate.[11] Simply stated: don’t sacrifice something expensive to modify something cheap! For this reason, carbonyl reductive couplings wherein alcohols serve dually as reductant and carbonyl proelectrophile are uniquely efficient. Such hydrogen autotransfer reactions directly convert lower alcohols to higher alcohols without exogenous reductants or discrete alcohol-to-carbonyl redox reactions.
Scheme 2.

Catalytic reductive coupling of π-unsaturated feedstocks: beyond stoichiometric organometallic reagents. Production statistics shown here and elsewhere in this review are taken from the Kirk-Other Encyclopedia of Chemical Technologyor Ullmann’s Encyclopedia of Industrial Chemistry.
In this mini-review, efforts to exploit π-unsaturated feedstocks as pronucleophiles and reductants in target-oriented synthesis via metal-catalyzed carbonyl reductive coupling and related hydrogen autotransfer reactions are surveyed.[5] For detailed discussions of reaction mechanism, the reader is referred to the primary literature. This review is not exhaustive, but utilizes case studies to illustrate how this new carbonyl addition chemistry streamlines chemical synthesis. Indeed, as organic molecules are compounds of carbon and hydrogen, we feel catalytic C-C bond formation via H2-addition or H2-redistribution are natural endpoints in the evolution of synthetic methods for efficient chemical synthesis.
II. From Feedstocks to Building Blocks in Target-Oriented Synthesis
Polyketides and their derivatives are used more frequently in human medicine than any other natural product class.[9,12] However, with the exception of eribulin,[3] all commercial polyketides are manufactured via fermentation or semi-synthesis, suggesting the present lexicon of synthetic methods does not adequately address the challenges that arise by the construction of such complex structures. Asymmetric aldol additions[13] and carbonyl allylations[14] are among the most commonly utilized methods for polyketide construction. However, to guide relative and absolute stereochemistry, most methods for asymmetric aldol addition and carbonyl allylation require multi-step syntheses of discrete enol(ate) or allylmetal derivatives that involve numerous sacrificial reagents and, consequently, generate numerous stoichiometric byproducts. For example, the vast majority of methods for asymmetric carbonyl crotylation exploit stereospecific reactions of (E)- or (Z)-crotylmetal reagents to form anti-or syn-configured propionate substructures, respectively. As exemplified by Leighton’s reagent,[15] many of the requisite crotylmetal reagents derive from butadiene (12 × 106 tons/yr) through multi-step sequences (Scheme 3). Direct asymmetric butadiene-mediated carbonyl crotylation via hydrogen autotransfer would be significantly more efficient, as undesired degrees of separation between reagent and feedstock would be removed and the crotylation would be completely atom-efficient.
Scheme 3.

Removing degrees of separation between reagent and feedstock in asymmetric carbonyl crotylation.
In 2008, aldehyde crotylations via ruthenium-catalyzed butadiene-carbonyl reductive coupling mediated by 2-propanol were developed, along with mechanistically related hydrogen autotransfer reactions involving primary alcohols.[16a] In these processes, alcohol dehydrogenation generates a ruthenium(II) hydride, which promotes butadiene hydrometalation to form a transient crotylruthenium(II) nucleophile. While the initially reported reactions displayed high levels of regioselectivity, diastereo- and enantioselective variants required use of 2-trialkylsilyl-substituted dienes.[16b] It was later found that ruthenium catalysts modified by chiral phosphate counterions could affect highly enantioselective butadiene-mediated carbonyl crotylations with access to either the anti- or syn-diastereomers depending on the choice of chiral counter-ion (Scheme 4).[16c,d] This method was used in a concise total synthesis of 6-deoxyerythronolide B.[17,18] As will be shown (vide infra, Scheme 5), the triketide stereopolyad spanning C1–C6 is created using a related two-directional transfer hydrogenative carbonyl crotylation of 2-methyl-1,3-propane diol.
Scheme 4.

Direct butadiene-mediated carbonyl crotylation with chiral counterion-dependent stereoselectivity.
Scheme 5.

Hydrogen-mediated aldol reductive coupling for the total synthesis of swinholide A.
The total synthesis of the marine macrodiolide swinholide A enables juxtaposition of classical carbonyl addition chemistry and corresponding hydrogenative and transfer hydrogenative carbonyl additions.[19–22] Rather than utilizing a chiral auxiliary-based asymmetric aldol addition, as exemplified by Evans-type aldol reagents (Figure 1),[19] an asymmetric vinyl ketone-aldehyde reductive coupling catalyzed by a chiral phosphinite-modified rhodium complex is employed.[24,25] Methyl vinyl ketone (MVK) is manufactured from acetone (1.56 × 106 tons/yr) and formaldehyde (8.7 × 106 tons/yr), and is of commercial significance due to its frequent use as a comonomer in photodegradable plastics. The reductant is elemental hydrogen (60 × 106 tons/yr). To construct the swinholide A substructure spanning C2–C18, MVK is hydrogenated at ambient pressure in the presence of the indicated C15 aldehyde to deliver the aldol product with excellent control of relative and absolute stereochemistry. This method is completely atom-efficient, meaning no sacrificial reagents are required and no stoichiometric byproducts are generated. Furthermore, the hydrogen-mediated coupling is highly chemoselective and can be performed in the presence of olefins and diene functional groups without competing conventional hydrogenation (Scheme 5).
Figure 1.

Propionate synthons for polyketide construction.
To prepare the C26-C32 trisubstituted pyran of swinholide A, an iridium-catalyzed stereo- and site-selective carbinol C-H allylation of (S)-1,3-butane diol was performed (Scheme 6, bottom).[26] Unlike conventional carbonyl allylation methodology,[14] which typically combines preformed allylmetal and aldehyde reactants, the analogous reactive species are generated transiently from abundant, tractable precursors: (S)-1,3-butane diol is commercially available and allyl acetate is a feedstock (7 × 104 tons/yr) prepared via palladium-catalyzed aerobic allylic acetoxylation of propylene (1 × 108 tons/yr) using acetic acid (8 × 106 tons/yr). The same C-C bond construction has been affected using the Brown reagent, which requires multiple cryogenic steps and sacrificial reagents (Scheme 6, top).[27] Alcohol-mediated carbonyl allylation methodology[28] is also used to prepare the triketide stereopolyad spanning C19-C25 of swinholide A. Here, α-methyl allyl acetate serves as a crotylmetal surrogate[29] in a two-directional chain elongation of 2-methyl-1,3-propane diol – a process that forms predominantly one of 16 possible stereoisomers.[30] Syntheses of closely related stereopolyads have required 15 steps (Scheme 6, right).[31] This “double crotylation” has dramatically simplified the syntheses of other polyketide natural products, such as 6-deoxyerythronolide B[17] and zincophorin.[32]
Scheme 6.

Stereo- and site-selective alcohol-mediated carbonyl allylation for the total synthesis of swinholide A.
The catalytic enantioselective two-directional allylation of 1,3-propane diol (1.5 × 105 tons/yr) using allyl acetate (7 × 104 tons/yr) combines two abundant feedstocks to directly furnish a value-added C2-symmetric diol (Scheme 7).[33] The same C2-symmetric diol previously required a 7 step synthesis involving three protecting group manipulations, two separate alcohol oxidations and two separate carbonyl asymmetric allylborations.[34] As illustrated in concise total syntheses of (+)-roxaticin,[35,36] bryostatin 7,[37,38] cryptocaryol,[39,40] mandelalide (not shown),[41] and neopeltolide (not shown),[42,43] this C2-symmetric diol serves as a powerful building block for polyketide construction. Neopentyl glycol (6.8 ×105 tons/yr) is used extensively in the paints and coatings industry. The two-directional enantioselective allylation of neopentyl glycol delivers a C2-symmetric diol that served as a key intermediate in total syntheses of cyanolide A[44,45] and psymberin (irciniastatin A) (not shown).[46]
Scheme 7.

Polyketide construction via two-directional allylation of feedstock glycols using allyl acetate.
Acetylene (7 × 104 tons/yr) is a basic feedstock used in a variety of applications. Previously generated from calcium carbide (which is derived from coal), the majority of acetylene is now obtained as a side-product in the production of ethylene by hydrocarbon cracking. Hydrogenation of acetylene in the presence of aldehydes and imines using a chiral cationic rhodium catalyst results in 3-component reductive coupling to form enantiomerically enriched (Z)-butadienylated allylic alcohols and amines, respectively (Scheme 8).[47] Using this method, along with an alcohol-mediated crotylation to define the propionate-based stereotriad,[29] the triene-containing C17-benzene ansamycins, trienomycins A and F, were prepared in a short number of steps.[48,49] The hydrogen-mediated reductive coupling of acetylene to L-glyceraldehyde acetonide displays high levels of catalyst-directed diastereoselectivity, enabling preparation of all eight L-hexoses (not shown).[47c]
Scheme 8.

Hydrogen-mediated reductive coupling of acetylene with imines or aldehydes.
In an effort to extend the concepts of hydrogen autotransfer-mediated carbonyl addition to the synthesis of terpenoid natural products, catalytic couplings of alcohols with isoprene oxide were developed.[50] This process converts primary alcohols to products of carbonyl tert-(hydroxy)-prenylation, generating an acyclic quaternary carbon stereocenter with excellent control of diastereo- and enantioselectivity.[51] No stoichiometric byproducts are generated, as each atom in the reactants is integrated in the reaction product. Isoprene oxide is obtained upon regioselective metal-catalyzed epoxidation of the feedstock diene isoprene (8 × 105 tons/yr). Using this method, concise total syntheses of oridamycin A, triptoquinones B and C and isoiresin were completed from a common intermediate in significantly fewer steps than previously required (Scheme 9).[52,53]
Scheme 9.

Synthesis of terpenoid natural products using a byproduct-free hydrogen auto-transfer addition.
Beyond alkanes, which are so plentiful they are used as fuel, the most abundant petrochemical feedstocks are ethylene (1.5 × 108 tons/yr) and higher olefins such as propylene (1.0 × 108 tons/yr) and 1-octene (1 × 106 tons/yr). Use of these compounds as non-stabilized carbanion equivalents in metal-catalyzed reductive coupling to unactivated carbonyl partners and related hydrogen autotransfer additions remains an unmet challenge.[8h] Progress has been made; however, vicinal dicarbonyl proelectrophiles are required. For example, ruthenium(0) catalyzed couplings of 3-hydroxy-2-oxindole to ethylene, propylene and 1-octene occur in excellent yield with complete levels of branched regioselectivity (Scheme 10).[54] Styrene (2.5 × 107 tons/yr) is also a competent partner in these transformations, and in couplings with simple aliphatic alcohols (not shown).[55] Styrene also participates in reductive couplings to anhydrides mediated by elemental hydrogen.[56] These processes have yet to be deployed in target-oriented synthesis, but demonstrate important proof-of-concept vis-à-vis use of feedstock alkenes as non-stabilized carbanion equivalents.
Scheme 10.

Ethylene and higher feedstock olefins as non-stabilized carbanion equivalents.
III. Conclusion
The current dichotomy between transformations used routinely in academic and discovery-related research vs large volume chemical manufacture defines a technological gap and, most importantly, an opportunity to innovate.[57] To achieve more ideal chemical syntheses and fulfill the longstanding goal of enacting synthetic routes composed solely of skeletal construction events,[2,9b] it is necessary to minimize preactivation by eliminating the degree of separation between reactant/reagent and the parent feedstock.[58] Hence, the development of catalytic methods that employ feedstock pronucleophiles in combination with feedstock reductants in stereo- and site-selective (protecting group-free) C-C bond formations represents an important objective.[59] Even more efficient, however, are related hydrogen autotransfer processes wherein native structural elements serve dually as redox mediators and coupling partners, as in hydrogen autotransfer additions of alcohols and π-unsaturated reactants. As demonstrated by the catalytic processes highlighted in this account, many traditional transformations that exploit preformed carbanions already have catalytic counterparts that bypass stoichiometric organometallic reagents and the multi-step sequences required for their preparation. We believe human ingenuity and economic-aesthetic selective pressure will cause this field to grow.
Acknowledgments
The Welch Foundation (F-0038) and the NIH (R01 GM093905) are acknowledged for partial support of this research.
References
- [1].As any reaction may be optimized, we posit that the most fundamental means of assessing synthetic efficiency is to tally the total number of chemical transformations associated with the preparation of each stoichiometric reactant and each stoichiometric reagent from its parent chemical feedstock. As compounds closer to the parent feedstock are more abundant and less expensive, commercial availability and cost is not explicitly considered. It is our hope that this metric will further inform computer-aided route design:; Klucznik T, Mikulak-Klucznik B, McCormack MP, Lima H, Szymkuć S, Bhowmick M, Molga K, Zhou Y, Rickershauser L, Gajewska EP, Toutchkine A, Dittwald P, Startek MP, Kirkovits GJ, Roszak R, Adamski A, Sieredzińska B, Mrksich M, Trice SLJ, Grzybowski BA, Chem 2018, 4, 522–532. [Google Scholar]
- [2].“The ideal synthesis creates a complex skeleton… in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality.”; Hendrickson JB, J. Am. Chem. Soc 1975, 97, 5784–5800. [Google Scholar]
- [3].For a review on the synthesis of eribulin, see:; Yu MJ, Zheng W, Seletsky BM, Nat. Prod. Rep 2013, 30, 1158–1164. [DOI] [PubMed] [Google Scholar]
- [4].Slezak Z, Chemik 2003, 56, 153–157. [Google Scholar]
- [5].(a) Sheldon RA, Chem. Ind 1997, 12–15; [Google Scholar]; (b) Sheldon RA, Green Chem 2007, 9, 1273–1283; [Google Scholar]; (c) Sheldon RA, Chem. Soc. Rev 2012, 41, 1437–1451; [DOI] [PubMed] [Google Scholar]; Also, see:; (d) Trost BM, Science 1991, 254, 1471–1477; [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Angew. Chem 1995, 107, 285–307; [Google Scholar]; Angew. Chem. Int. Ed. Engl 1995, 34, 259–281. [Google Scholar]
- [6].For reviews on alkene hydroformylation, see:; (a) van Leeuwen PWNM, Claver C, Rhodium Catalyzed Hydroformylation, Kluwer Academic Publishers, Norwell, MA, 2000; [Google Scholar]; (b) Breit B, Seiche W, Synthesis 2001, 1–36. [Google Scholar]
- [7](a).For reviews on methanol carbonylation, see:; (a) Jones JH, Platinum Met. Rev 2000, 44, 94–105; [Google Scholar]; (b) Haynes A, Top. Organomet. Chem 2006, 18, 179–205. [Google Scholar]
- [8].For reviews on hydrogen-mediated carbonyl reductive coupling and related hydrogen autotransfer reactions:; (a) Bower JF, Krische MJ, Top. Organomet. Chem 2011, 34, 107–138; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hassan A, Krische MJ, Org. Proc. Res. Devel 2011, 15, 1236–1242; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moran J, Krische MJ, Pure Appl. Chem 2012, 84, 1729–1739; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ketcham JM, Shin I, Montgomery TP, Krische MJ, Angew. Chem 2014, 126, 9294–9302; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2014, 53, 9142–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sam B, Breit B, Krische MJ, Angew. Chem 2015, 127, 3317–3325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2015, 53, 3267–3274; [Google Scholar]; (f) Shin I, Krische MJ, Top. Curr. Chem 2016, 372, 85–101; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Perez F, Oda S, Geary LM, Krische MJ, Top. Curr. Chem 2016, 374, 365–387; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354 (6310), aah5133; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For reviews on the impact of catalytic reductive coupling and related hydrogen autotransfer reactions on target-oriented synthesis, see:; (a) Dechert-Schmitt A-MR, Schmitt DC, Gao X, Itoh T, Krische MJ, Nat. Prod. Rep 2014, 31, 504–513; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shin I, Montgomery TP, Krische MJ, Aldrichimica Acta 2015, 48, 15; [PMC free article] [PubMed] [Google Scholar]; (c) Feng J, Kasun ZA, Krische MJ, J. Am. Chem. Soc 2016, 138, 5467–5478; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schwartz LA, Krische MJ, Isr. J. Chem 2018, 58, 45–51. [Google Scholar]
- [10].For general reviews on metal-catalyzed carbonyl reductive coupling, see:; (a) Moragas T, Correa A, Martin R, Chem. - Eur. J 2014, 20, 8242–8258; [DOI] [PubMed] [Google Scholar]; (b) Holmes M, Schwartz LA, Krische MJ, Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Recently, a (MeO)2MeSiH-mediated allene-ketone reductive coupling was reported. Allene constitutes approximately 6% of the crude C3 fraction in petroleum cracking, but it is reduced and recycled (not isolated). More importantly, the requisite silane costs over $1/gram.; Liu RY, Zhou Y, Yang Y, Buchwald SL, J. Am. Chem. Soc 2019, 141, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For selected reviews on polyketide natural products, see:; (a) O’Hagan D, The Polyketide Metabolites, Ellis Horwood Ltd., Chichester, 1991; [Google Scholar]; (b) Rimando AM, Baerson SR, Polyketides: Biosynthesis, Biological Activity, and Genetic Engineering, ACS Symposium Series 955, 2007. [Google Scholar]
- [13].For selected reviews on asymmetric aldol addition, see:; (a) Knochel P, Molander GA, Comprehensive Organic Synthesis, Elsevier Ltd., Amsterdam, Vol. 2, 2014; [Google Scholar]; (b) Arya P, Qin H, Tetrahedron 2000, 56, 917–947; [Google Scholar]; (c) Romea P, Urpí F, Modern Methods in Stereoselective Aldol Reactions, (Ed.: Mahrwald R), Wiley-VCH, Weinheim, 2013, pp. 1–81; [Google Scholar]; (d) Yamashita Y, Yasukawa T, Yoo W-J, Kitanosono T, Kobayashi S, Chem. Soc. Rev 2018, 47, 4388–4480. [DOI] [PubMed] [Google Scholar]
- [14].For selected reviews on asymmetric carbonyl allylation, see:; (a) Ramachandran PV, Aldrichim. Acta 2002, 35, 23–35; [Google Scholar]; (b) Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794; [DOI] [PubMed] [Google Scholar]; (c) Yu C-M, Youn J, Jung H-K, Bull. Korean Chem. Soc 2006, 27, 463–472; [Google Scholar]; (d) Marek I, Sklute G, Chem. Commun 2007, 1683–1691; [DOI] [PubMed] [Google Scholar]; (e) Hall DG, Synlett 2007, 1644–1655; [Google Scholar]; (f) Hargaden GC, Guiry PJ, Adv. Synth. Catal 2007, 349, 2407–2424; [Google Scholar]; (g) Lachance H, Hall DG, Org. React 2008, 73; [Google Scholar]; (h) Han SB, Kim IS, Krische MJ, Chem. Commun 2009, 7278–7287; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; (j) Moran J, Krische MJ, Asymmetric Synthesis – The Essentials II (Eds.: Christmann M, Bräse S), Wiley-VCH, Weinheim, 2012, pp. 187; [Google Scholar]; (k) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2013, 113, 5595–5698; [DOI] [PubMed] [Google Scholar]; (l) Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320; [Google Scholar]; (m) Kumar P, Tripathi D, Sharma BM, Dwivedi N, Org. Biomol. Chem 2017, 15, 733–761; [DOI] [PubMed] [Google Scholar]; (n) Spielmann K, Niel G, de Figueiredo RM, Campagne J-M, Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- [15] (a).Hackman BM, Lombardi PJ, Leighton JL, Org. Lett 2004, 6, 4375–4377; [DOI] [PubMed] [Google Scholar]; (b) Kim H, Ho S, Leighton JL, J. Am. Chem. Soc 2011, 133, 6517–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] (a).Shibahara F, Bower JF, Krische MJ, J. Am. Chem. Soc 2008, 130, 6338–6339; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR, Moran J, Krische MJ, J. Am. Chem. Soc 2011, 133, 10582–10586; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ, Science 2012, 336, 324–327; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McInturff EL, Yamaguchi E, Krische MJ, J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].For the Krische total synthesis of 6-deoxyerythronolide B, see:; Gao X, Woo SK, Krische MJ, J. Am. Chem. Soc 2013, 135, 4223–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].For prior total and formal syntheses of 6-deoxyerythronolide B, see:; (a) Masamune S, Hirama M, Mori S, Ali SA, Garvey DS, J. Am. Chem. Soc 1981, 103, 1568–1571; [Google Scholar]; (b) Myles DC, Danishefsky SJ, J. Org. Chem 1990, 55, 1636–1648; [Google Scholar]; (c) Evans DA, Kim AS, Tetrahedron Lett 1997, 38, 53–56; [Google Scholar]; (d) Evans DA, Kim AS, Metternich R, Novack VJ, J. Am. Chem. Soc 1998, 120, 5921–5942; [Google Scholar]; (e) Stang EM, White MC, Nat. Chem 2009, 1, 547–551; [DOI] [PMC free article] [PubMed] [Google Scholar]; Formal Synthesis:; (f) Crimmins MT, Slade DJ, Org. Lett 2006, 8, 2191–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].For a review encompassing synthetic approaches to swinholide A, see:; Schwan J, Christmann M, Chem. Soc. Rev 2018, 47, 7985–7995. [DOI] [PubMed] [Google Scholar]
- [20].For the Krische total synthesis of swinholide A, see:; (a) Shin I, Krische MJ, Org. Lett 2015, 17, 4686–4689; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shin I, Hong S, Krische MJ, J. Am. Chem. Soc 2016, 138, 14246–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].For Paterson’s total synthesis of swinholide A, see:; (a) Paterson I, Yeung K-S, Ward RA, Cumming JG, Smith JD, J. Am. Chem. Soc 1994, 116, 9391–9392; [Google Scholar]; (b) Paterson I, Cumming JG, Ward RA, Lamboley S, Tetrahedron 1995, 51, 9393–9412; [Google Scholar]; (c) Paterson I, Smith JD, Ward RA, Tetrahedron 1995, 51, 9413–9436; [Google Scholar]; (d) Paterson I, Ward RA, Smith JD, Cumming JG, Yeung K-S, Tetrahedron 1995, 51, 9437–9466; [Google Scholar]; (e) Paterson I, Yeung K-S, Ward RA, Smith JD, Cumming JG, Lamboley S, Tetrahedron 1995, 51, 9467–9486. [Google Scholar]
- [22].For Nicolaou’s total synthesis of swinholide A, see:; (a) Nicolaou KC, Ajito K, Patron AP, Khatuya H, Richter PK, Bertinato P, J. Am. Chem. Soc 1996, 118, 3059–3060; [Google Scholar]; (b) Nicolaou KC, Patron AP, Ajito K, Richter PK, Khatuya H, Bertinato P, Miller RA, Tomaszewski MJ, Chem. - Eur. J 1996, 2, 847–868. [Google Scholar]
- [23].Evans DA, Bartroli J, Shih TL, J. Am. Chem. Soc 1981, 103, 2127–2129. [Google Scholar]
- [24].Bee C, Han SB, Hassan A, Iida H, Krische MJ, J. Am. Chem. Soc 2008, 130, 2746–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].For a review on metal-catalyzed reductive aldol addition, see:; Jang H-Y, Krische MJ in Comprehensive Chirality, Vol. 4 (Eds.: Yamamoto H, Carreira EM), Elsevier, Oxford, 2012, pp. 100–121. [Google Scholar]
- [26].Shin I, Wang G, Krische MJ, Chem. - Eur. J 2014, 20, 13382–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Judd TC, Bischoff A, Kishi Y, Adusumilli S, Small PLC, Org. Lett 2004, 6, 4901–4904. [DOI] [PubMed] [Google Scholar]
- [28].For catalytic asymmetric carbonyl allylation via alcohol-mediated hydrogen transfer using allyl acetate as the allyl donor, see:; (a) Kim IS, Ngai M-Y, Krische MJ, J. Am. Chem. Soc 2008, 130, 6340–6341; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS, Ngai M-Y, Krische MJ, J. Am. Chem. Soc 2008, 130, 14891–14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].For catalytic asymmetric carbonyl crotylation via alcohol-mediated hydrogen transfer using α-methyl allyl acetate as the allyl donor, see:; (a) Kim IS, Han SB, Krische MJ, J. Am. Chem. Soc 2009, 131, 2514–2520; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao X, Townsend IA, Krische MJ, J. Org. Chem 2011, 76, 2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gao X, Han H, Krische MJ, J. Am. Chem. Soc 2011, 133, 12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nakamura R, Tanino K, Miyashita M, Org. Lett 2003, 5, 3579–3582. [DOI] [PubMed] [Google Scholar]
- [32].Kasun ZA, Gao X, Lipinski RM, Krische MJ, J. Am. Chem. Soc 2015, 137, 8900–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ, Angew. Chem 2009, 121, 5118–5121; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2009, 48, 5018–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Smith AB III, Minbiole KP, Verhoest PR, Schelhaas M, J. Am. Chem. Soc 2001, 123, 10942–10953. [DOI] [PubMed] [Google Scholar]
- [35].For the Krische total synthesis of (+)-roxaticin, see:; Han SB, Hassan A, Kim I-S, Krische MJ, J. Am. Chem. Soc 2010, 132, 15559–15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].For prior total syntheses of (+)-roxaticin, see:; (a) Rychnovsky SD, Hoye RC, J. Am. Chem. Soc 1994, 116, 1753–1765; [Google Scholar]; (b) Mori Y, Asai M, Kawade J.-i., Okumura A, Furukawa H, Tetrahedron Lett 1994, 35, 6503–6506; [Google Scholar]; (c) Mori Y, Asai M, Okumura A, Furukawa H, Tetrahedron 1995, 51, 5299–5314; [Google Scholar]; (d) Mori Y, Asai M, Kawade J.-i., Furukawa H, Tetrahedron 1995, 51, 5315–5330; [Google Scholar]; (e) Evans DA, Connell BT, J. Am. Chem. Soc 2003, 125, 10899–10905. [DOI] [PubMed] [Google Scholar]
- [37].For the Krische total synthesis of bryostatin 7, see:; Lu Y, Woo SK, Krische MJ, J. Am. Chem. Soc 2011, 133, 13876–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].For prior total and formal syntheses of bryostatin family members, see:; bryostatin 1:; (a) Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA, J. Am. Chem. Soc 2011, 133, 744–747; [DOI] [PMC free article] [PubMed] [Google Scholar]; bryostatin 2:; (b) Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M, Angew. Chem 1998, 110, 2526–2530; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 1998, 37, 2354–2359; [DOI] [PubMed] [Google Scholar]; (c) Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M, J. Am. Chem. Soc 1999, 121, 7540–7552; [Google Scholar]; bryostatin 3:; (d) Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S, Angew. Chem 2000, 112, 2376–2379; [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2000, 39, 2290–2294; [PubMed] [Google Scholar]; (e) Ohmori K, Bull. Chem. Soc. Jpn 2004, 77, 875–885; [Google Scholar]; bryostatin 7:; (f) Kageyama M, Tamura T, Nantz M, Roberts JC, Somfai P, Whritenour DC, Masamune S, J. Am. Chem. Soc 1990, 112, 7407–7408; [Google Scholar]; (g) Manaviazar S, Frigerio M, Bhatia GS, Hummersone MG, Aliev AE, Hale KJ, Org. Lett 2006, 8, 4477–4480; [DOI] [PubMed] [Google Scholar]; (h) Kedei N, Lewin NE, Geczy T, Selezneva J, Braun DC, Chen J, Herrmann MA, Heldman MR, Lim L, Mannan P, Garfield SH, Poudel YB, Cummins TJ, Rudra A, Blumberg PM, Keck GE, ACS Chem. Biol 2013, 8, 767–777; [DOI] [PMC free article] [PubMed] [Google Scholar]; bryostatin 9:; (i) Wender PA, Schrier AJ, J. Am. Chem. Soc 2011, 133, 9228–9231; [DOI] [PMC free article] [PubMed] [Google Scholar]; bryostatin 16:; (j) Trost BM, Dong G, Nature 2008, 456, 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].For the Krische total synthesis of cryptocaryol A, see:; Perez F, Waldeck AR, Krische MJ, Angew. Chem 2016, 128, 5133–5136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2016, 55, 5049–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].For prior total and formal syntheses of cryptocaryol A, see:; (a) Reddy DS, Mohapatra DK, Chem. - Eur. J 2013, 1051–1057; [Google Scholar]; (b) Wang Y, O’Doherty GA, J. Am. Chem. Soc 2013, 135, 9334–9337; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brun E, Bellosta V, Cossy J, J. Org. Chem 2015, 80, 8668–8676; [DOI] [PubMed] [Google Scholar]; (d) Dias LC, Kuroishi PK, de Lucca EC Jr., Org. Biomol. Chem 2015, 13, 3575–3584. [DOI] [PubMed] [Google Scholar]
- [41].For the Fürstner total syntheses of the initially proposed and revised structures assigned to mandelalide, see:; (a) Willwacher J, Fürstner A, Angew. Chem 2014, 126, 4301–4305; [Google Scholar]; Angew. Chem. Int. Ed 2014, 53, 4217–4221; [DOI] [PubMed] [Google Scholar]; (b) Willwacher J, Heggen B, Wirtz C, Thiel W, Fürstner A, Chem. - Eur. J 2015, 21, 10416–10430. [DOI] [PubMed] [Google Scholar]
- [42].For the She total synthesis of neopeltolide, see:; Yang Z, Zhang B, Zhao G, Yang J, Xie X, She X, Org. Lett 2011, 13, 5916–5919. [DOI] [PubMed] [Google Scholar]
- [43].For a review detailing total and formal syntheses of neopeltolide, see:; Fuwa H, Mar. Drugs 2016, 14, 65–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].For the Krische total synthesis of cyanolide A, see:; Waldeck AR, Krische MJ, Angew. Chem 2013, 125, 4566–4569; [Google Scholar]; Angew. Chem. Int. Ed 2013, 52, 4470–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].For prior total and formal syntheses of cyanolide A, see:; (a) Kim H, Hong J, Org. Lett 2010, 12, 2880–2883; [DOI] [PubMed] [Google Scholar]; (b) Hajare AK, Ravikumar V, Khaleel S, Bhuniya D, Reddy DS, J. Org. Chem 2011, 76, 963–966; [DOI] [PubMed] [Google Scholar]; (c) Yang Z, Xie X, Jing P, Zhao G, Zheng J, Zhao C, She X, Org. Biomol. Chem 2011, 9, 984–986; [DOI] [PubMed] [Google Scholar]; (d) Pabbaraja S, Satyanarayana K, Ganganna B, Yadav JS, J. Org. Chem 2011, 76, 1922–1925; [DOI] [PubMed] [Google Scholar]; (e) Gesinski MR, Rychnovsky SD, J. Am. Chem. Soc 2011, 133, 9727–9729; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sharpe RJ, Jennings MP, J. Org. Chem 2011, 76, 8027–8032. [DOI] [PubMed] [Google Scholar]
- [46].For the De Brabander total synthesis of psymberin (irciniastatin A), see:; Feng Y, Jiang X, De Brabander JK, J. Am. Chem. Soc 2012, 134, 17083–17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47] (a).Kong J-R, Krische MJ, J. Am. Chem. Soc 2006, 128, 16040–16041; [DOI] [PubMed] [Google Scholar]; (b) Skucas E, Kong JR, Krische MJ, J. Am. Chem. Soc 2007, 129, 7242–7243; [DOI] [PubMed] [Google Scholar]; (c) Han SB, Kong J-R, Krische MJ, Org. Lett 2008, 10, 4133–4135; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Williams VM, Kong J-R, Ko B-J, Mantri Y, Brodbelt JS, Baik M-H, Krische MJ, J. Am. Chem. Soc 2009, 131, 16054–16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].For the Krische total synthesis of (+)-trienomycins A and F, see:; Del Valle DJ, Krische MJ, J. Am. Chem. Soc 2013, 135, 10986–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].For total and formal syntheses of triene-containing C17-benzene ansamycins trienomycins A and F and thiazinotrienomycin E, mycotrienol I and mycotrienin I and cytotrienin A, see:; (a) Smith AB III, Barbosa J, Wong W, Wood JL, J. Am. Chem. Soc 1995, 117, 10777–10778; [Google Scholar]; (b) Smith AB III, Barbosa J, Wong W, Wood JL, J. Am. Chem. Soc 1996, 118, 8316–8328; [Google Scholar]; (c) Smith AB III, Wan Z, Org. Lett 1999, 1, 1491–1494; [DOI] [PubMed] [Google Scholar]; (d) Smith AB III, Wan Z, J. Org. Chem 2000, 65, 3738–3753; [DOI] [PubMed] [Google Scholar]; (e) Del Valle DJ, Krische MJ, J. Am. Chem. Soc 2013, 135, 10986–10989; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Panek JS, Masse CE, J. Org. Chem 1997, 62, 8290–8291; [DOI] [PubMed] [Google Scholar]; (g) Masse CE, Yang M, Solomon J, Panek JS, J. Am. Chem. Soc 1998, 120, 4123–4134; [Google Scholar]; (h) Hayashi Y, Shoji M, Ishikawa H, Yamaguchi J, Tamura T, Imai H, Nishigaya Y, Takabe K, Kakeya H, Osada H, Angew. Chem 2008, 120, 6759–6762; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2008, 47, 6657–6660. [DOI] [PubMed] [Google Scholar]
- [50] (a).Feng J, Garza VJ, Krische MJ, J. Am. Chem. Soc 2014, 136, 8911–8914; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guo Y-A, Lee W, Krische MJ, Chem. - Eur. J 2017, 23, 2557–2559; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo Y-A, Liang T, Kim S-W, Xiao H, Krische MJ, J. Am. Chem. Soc 2017, 139, 6847–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].For a review on metal-catalyzed formation of acyclic quaternary carbon stereocenters, see:; Feng J, Holmes M, Krische MJ, Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].For the Krische total syntheses of oridamycin A, triptoquinone B and C and isoiresin, see:; Feng J, Noack F, Krische MJ, J. Am. Chem. Soc 2016, 138, 12364–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].For prior total and formal syntheses of oridamycin A, triptoquinone B and C and isoiresin, see:; oridamycin A and B, and xiamycin:; (a) Rosen BR, Werner EW, O’Brien AG, Baran PS, J. Am. Chem. Soc 2014, 136, 5571–5574; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meng Z, Yu H, Li L, Tao W, Chen H, Wan M, Yang P, Edmonds DJ, Zhong J, Li A, Nat. Commun 2015, 6, 6096–6103; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trotta AH, Org. Lett 2015, 17, 3358–3361; [DOI] [PMC free article] [PubMed] [Google Scholar]; triptoquinones B and C:; (d) Shishido K, Goto K, Tsuda A, Takaishi Y, Shibuya M, J. Chem. Soc., Chem. Commun 1993, 793–794; [Google Scholar]; (e) Yamamura I, Fujiwara Y, Yamato T, Irie O, Shishido K, Tetrahedron Lett 1997, 38, 4121–4124; [Google Scholar]; iresin and isoiresin:; (f) Pelletier SW, Prabhakar S, J. Am. Chem. Soc 1968, 90, 5318–5320; [Google Scholar]; (g) Wang B-L, Gao H-T, Li W-DZ, J. Org. Chem 2015, 80, 5296–5301. [DOI] [PubMed] [Google Scholar]
- [54].a) Yamaguchi E, Mowat J, Luong T, Krische MJ, Angew. Chem 2013, 125, 8586–8589; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2013, 52, 8428–8431; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park BY, Luong T, Sato H, Krische MJ, J. Org. Chem 2016, 81, 8585–8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xiao H, Wang G, Krische MJ, Angew. Chem 2016, 128, 16353–16356; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2016, 55, 16119–16122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hong Y-T, Barchuk A, Krische MJ, Angew. Chem 2006, 118, 7039–7042; [Google Scholar]; Angew. Chem. Int. Ed 2006, 128, 6885–6888. [Google Scholar]
- [57].Rossen K, J. Org. Chem 2019, 84, 4580–4582. [DOI] [PubMed] [Google Scholar]
- [58].Han SB, Kim IS, Krische MJ, Chem. Commun 2009, 7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].For selected reviews on site-selective catalysis and protecting group-free synthesis, see:; (a) Hoffmann RW, Synthesis 2006, 3531–3541; [Google Scholar]; (b) Young IS, Baran PS, Nature Chem 2009, 1, 193–205; [DOI] [PubMed] [Google Scholar]; (c) Roulland E, Angew. Chem 2011, 123, 1260–1262; [Google Scholar]; Angew. Chem. Int. Ed 2011, 50, 1226–1227; [DOI] [PubMed] [Google Scholar]; ‘Site-Selective Catalysis’; (d) in Topics in Current Chemistry, Vol. 372 (Ed.: Kawabata T), Springer, Cham, Switzerland, 2016; [Google Scholar]; (e) Hartwig JF, Acc. Chem. Res 2017, 50, 549–555; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Toste FD, Sigman MS, Miller SJ, Acc. Chem. Res 2017, 50, 609–615; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dimakos V, Taylor MS, Chem. Rev 2018, 118, 11457–11517. [DOI] [PubMed] [Google Scholar]