


Abstract
Real-time visualization of single-proteins or -complexes on nucleic acid substrates is an essential tool for characterizing nucleic acid binding proteins. Here, we present a novel surface-condition independent and high-throughput single-molecule optical imaging platform called ‘DNA skybridge’. The DNA skybridge is constructed in a 3D structure with 4 μm-high thin quartz barriers in a quartz slide. Each DNA end is attached to the top of the adjacent barrier, resulting in the extension and immobilization of DNA. In this 3D structure, the bottom surface is out-of-focus when the target molecules on the DNA are imaged. Moreover, the DNA skybridge itself creates a thin Gaussian light sheet beam parallel to the immobilized DNA. This dual property allows for imaging a single probe-tagged molecule moving on DNA while effectively suppressing interference with the surface and background signals from the surface.
INTRODUCTION
Real-time single particle tracking of probe-tagged proteins on DNA molecules is strongly advantageous for understanding their mechanistic behaviors in DNA transactions. To follow individual proteins going through their physical or chemical reactions on a DNA molecule, one or both ends of the DNA molecule extended by an external force are attached to the surface of an imaging chamber (1,2). This strategy allows DNA-bound target molecules to move within the penetration depth of the excitation beam in total internal reflection fluorescence (TIRF) microscopy, which provides a high signal-to-noise ratio (SNR) due to the thin excitation depth (<200 nm) of the incident beam (3).
However, the probe-tagged proteins bound to DNA may not be resolved from those bound nonspecifically to the surface since both are located in the same imaging plane. The probe-tagged proteins on the surface can introduce undesirable background signals that significantly reduce the resolution of the target molecules. Thus, DNA surface immobilization for single-molecule imaging using TIRF microscopy requires surface passivation to minimize the nonspecific binding of the probe-tagged proteins. Polyethylene glycol (PEG) has been widely used to passivate quartz slides or cover glasses for preventing nonspecific binding of proteins (4); a lipid bilayer is also a traditional alternative (5). In general, these methods efficiently block the nonspecific adhesion of biomolecules in a few tens of nanomolar range, which is much lower than the dissociation constant of typical biomolecular interactions (6,7). In addition, the thermal fluctuation of extended and immobilized DNA can result in nonspecific interactions between the DNA-bound proteins and the surface due to the proximity of the DNA to the surface. Although significant efforts have been dedicated to improve surface passivation for avoiding nonspecific interactions of biomolecules with the surface (8–10), the imperfection of the surface passivation commonly results in nonspecific sticking of proteins at a concentration close to the dissociation constant.
To minimize the potential interruption by nonspecifically surface-bound proteins, we developed a single-molecule fluorescence imaging platform that monitors single fluorescently-labeled proteins on DNA molecules that are located far away from the surface. We fabricated a 3D structure with thin quartz barriers of a few μm height on a quartz slide. DNA molecules were stretched and immobilized on two adjacent ridges by a laminar flow, which results in DNA molecules aligned along the ridges. We successfully imaged single fluorophore-labeled proteins on the DNA with a high SNR at the critical angle of the incident beam, while emitters bound nonspecifically to the bottom surface were out of focus. Surprisingly, a thin light sheet near the ridges was produced by the interference of excitation beams refracted from the barriers, which allowed us to visualize a single fluorophore. We named this surface-condition independent and high-throughput single-molecule fluorescence imaging platform as ‘DNA skybridge’. Using the DNA skybridge, we successfully demonstrated single molecule fluorescence imaging of Cy5-conjugated PCNA diffusing on DNA molecules.
MATERIALS AND METHODS
Fabrication of 3D structure
The 3D structure was formed by standard photolithography processes on a (100) quartz wafer. Before the processes, the quartz wafer was cleaned using the sulfuric acid peroxide mixture cleaning process. Then, a positive photoresist (TDMR AR87) was used for patterning; when coated at 4500 RPM, a thickness of 400 nm was obtained. After a soft bake at 88°C for 90 s, the patterns were exposed through a chrome mask by using a MA-6 aligner. For the MA-6 mask aligner, an i-line light source with a wavelength of 365 nm and 25 mW was used. We obtained a resolution of 2 μm using a soft contact mode. The exposed samples were developed by an AZ300 MIF developer, and a hard bake was performed at 113°C for 90 s. The patterns with 10 mm width used in the experiment were spaced from 3 mm. When a chemical etching process was performed through the thin gaps, the 3D structure with a concave edge and a high hill was formed through the undercut structure. The etching process was carried out using a diluted HF solution (deionized water:HF = 6:1) at room temperature without stirring, giving a resultant etching rate of 2000 Å/min. As one of the results, the 3D structure with a width of 0.5 μm of a barrier, a depth of 4.0 μm and a spacing of 13.1 μm was obtained through the 20 min etching process (Figure 1A). To make a strong adhesion between the quartz wafer and the patterned photoresist, an additional photoresist annealing was performed at 110°C for 90 s just before the wet etching process. After all etching processes, the quartz wafer was cut by 7.5 cm × 2.5 cm via a dicing process.
Figure 1.

DNA immobilization on a 3D structure. (A) Schematic representation of DNA skybridge and an image of a wet-etched substrate taken by a scanning electron microscope (FE-SEM; HITACHI S-4800). (B) The PDMS stamping with Alexa Fluor 488 (AF488)-conjugated streptavidin displays that the streptavidin molecules are only associated with biotin on the top of the barriers (Top). (C) After DNA infusion into the flow chamber, DNA molecules were stained with intercalators (Sytox Orange). Extended DNA molecules were aligned and organized on the barriers (Top). The top and bottom surfaces of the barriers were focused, respectively. (D) DNA molecules were imaged over a field of view at total 96× magnification using 60× objective with 1.6× magnification.
PDMS stamping
The 3D patterned quartz surface was passivated with a PEGylation method previously reported (4,11). The quartz slides were stringently washed by sequential sonication with 10% alconox for 20 min, deionized water (Milli-Q) for 5 min, acetone for 15 min, KOH for 20 min and methanol for 10 min, and then were burnt with a torch for 30 s. The clean quartz slides were incubated in a 2% (v/v) (3-aminopropyl)triethoxysilane (Sigma) for 20 min. Polyethylene glycol succinimidyl valerate (PEG-SVA, MW 5000, Laysan Bio) and Biotin-PEG-SVA (MW 5000) with a mass ratio of 40:1 were dissolved in 0.1 M sodium bicarbonate (pH 8.3). The PEG solution was dropped on the surface of the quartz slide and then incubated for 6 h. The functionalized quartz slides were washed with deionized water and then dried with nitrogen gas. To make a PDMS stamp, we mixed the PDMS base and the curing agent with a 9:1 ratio (Dow Corning, SYLGARD® 184 SILICONE ELASTOMER KIT), degassed the mixture by lowering the atmospheric pressure to 0.2 atm using an air pump, and baked it at 80°C for 1 h. The PDMS stamp surface was dipped and incubated with streptavidin (Sigma-Aldrich, S4762; 25 μg/ml) in 0.025× PBS solution for 30 min; the solution on the PDMS was removed from the surface with nitrogen gas. We gently put the PDMS on the 3D structured quartz by adding ∼200 g mass for 10 min to make a reliable contact between the top of the barriers and the PDMS stamp. The quartz was rinsed with deionized water and then dried with nitrogen gas.
Simulation for intensity profile
The algorithm of the OptiFDTD 32-bit software solves both electric and magnetic fields in the temporal and spatial domain using the full-vector differential form of Maxwell's equations for arbitrary geometries (Optiwave, photonic software), which was used for the simulation of the excitation intensity profile at the 3D structure. The simulation space was 100 × 40 μm, the grid size was 36.48 nm, the time step was 8.113e–17 s, and the total run time was 6468-time steps. The wavelength of the incident light was 532 nm, its FWHM was 80 μm, and the incident angle was as described in the text. The refractive indices of quartz and water were set to 1.4585 and 1.3310, respectively. Using MATLAB scripts (MathWorks), we calculated the intensity of an excitation beam with the electric field obtained from Opti-FDTD and drew the intensity profile in the space of a 3D patterned structure (Figure 2).
Figure 2.

Computer-aided simulation for the intensity profile of excitation in DNA skybridge. (A) A 3D structure for the simulation was built with angled slope blocks for 4.0 μm barriers in height and 13.1 μm spacing between two barriers. The angled boundary barrier resulted from the superposition of two equilateral trapezoids. One isosceles trapezoid has an upper base of 0.4 μm, a lower base of 5.0 μm and a height of 4.0 μm. The other trapezoid has an upper base of 0.4 μm, a lower base of 8.1 μm and a height of 2.0 μm. The center of the bottom line of both trapezoids is located at an identical position on the quartz surface for the simulation. The width of the bottom surface is 5.0 μm. (B) The intensity profile of the excitation light with a wavelength of 532 nm in a computer simulation. The incident angle and laser power are set to 75° and 1, respectively. The red line corresponds to the barrier boundary of the 3D structured quartz. The origin of the vertical distance is the top of the barriers (right). We took the average of the intensity of horizontal components, which is presented with the vertical distance (right).
SNR measurement
We determined SNR for DNA skybridge, conventional TIRF microscopy and epi-illumination by measuring an emission intensity (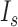 ) of Cy5 conjugated to PCNA diffusing on DNA and a background intensity
) of Cy5 conjugated to PCNA diffusing on DNA and a background intensity 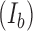 . To obtain emission signals by homogenous excitation, we selected only Cy5 signals in a circular area with a radius of 21.88 μm (128 pixels) around the center of a field of view because a Gaussian excitation beam results in an inhomogeneous excitation. Cy5 signals were at least 1.71 μm (10 pixels) far away from the ridges and lasted for 2 s. We applied the radial symmetric method to the intensity profiles of Cy5 signals to determine the position of the curve peak that corresponds to the center of the signal (12).
. To obtain emission signals by homogenous excitation, we selected only Cy5 signals in a circular area with a radius of 21.88 μm (128 pixels) around the center of a field of view because a Gaussian excitation beam results in an inhomogeneous excitation. Cy5 signals were at least 1.71 μm (10 pixels) far away from the ridges and lasted for 2 s. We applied the radial symmetric method to the intensity profiles of Cy5 signals to determine the position of the curve peak that corresponds to the center of the signal (12). 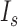 was determined by the intensity of the pixels in a circular area with a radius of 0.377 μm (2.2 pixels) around the center of the signal (Supplementary Figure S1). The signal area corresponds to two standard deviations (s.d.) of the Gaussian distribution.
was determined by the intensity of the pixels in a circular area with a radius of 0.377 μm (2.2 pixels) around the center of the signal (Supplementary Figure S1). The signal area corresponds to two standard deviations (s.d.) of the Gaussian distribution. 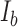 was obtained from the intensity of the outer pixels subtracting the Cy5 signal region from a circular area with a radius of 1.71 μm (Supplementary Figure S1). The resulting SNR is given by
was obtained from the intensity of the outer pixels subtracting the Cy5 signal region from a circular area with a radius of 1.71 μm (Supplementary Figure S1). The resulting SNR is given by 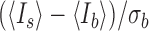 , where
, where 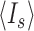 and
and 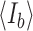 are the average intensities of the signal and the background, respectively, and
are the average intensities of the signal and the background, respectively, and 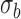 is one s.d. of the background intensity.
is one s.d. of the background intensity.
DNA substrate construction
We prepared λ-phage DNA molecules (48.5 kb; New England Biolabs) containing biotin at both ends of the DNA for all the experiments except the measurement in Figure 3A (Bi-biotin λ-phage DNA), and 44.5 kb long DNA molecules containing Cy3 and a 100 nt gap were used for the measurement of SNR in Figure 3A (Bi-biotin Cy3 gap DNA). Detailed information on the construction of the DNA substrates is described in Supplementary Data (see Supplementary Table S1 for information on oligos of the DNA substrates).
Figure 3.

Real-time visualization of a single fluorophore with DNA skybridge. (A) Cy3-labeled DNA molecules (44.5 kb) are extended and immobilized on top of the barriers. The SNR for DNA skybridge was measured at a given incident angle (θ0) that is greater than the critical incident angle (θc) of total internal reflection at a flat quartz slide. The SNR suddenly increases at a certain incident angle (θ0 + 1.7°) as the increase of the incident angle from θ0 and then gradually decreases as the incident angle increases. The error bars indicate s.d. (B) The top (Top) of the barriers and the bottom surface (Bottom) of the 3D structured quartz were focused. The blue arrows indicate Cy5-PCNA molecules on DNA. (C) The colocalization of Cy5-PCNA and DNA molecules immobilized on the top of the barriers. (D) Diffusion constant (D) of Cy5-PCNA at various concentrations of potassium glutamate (KGlu). D = 0.61 ± 0.05 μm2/s at 25 mM KGlu, 0.64 ± 0.05 μm2/s at 50 mM KGlu, 0.93 ± 0.09 μm2/s at 100 mM KGlu, 1.04 ± 0.06 μm2/s at 150 mM KGlu, 1.30 ± 0.07 μm2/s at 200 mM KGlu, 1.92 ± 0.12 μm2/s at 300 mM KGlu. These diffusion constants are similar to those measured in a previous report (16). Black box: s.d., Transparent white box: standard error, Red line: mean. n: the number of events.
Cy5 conjugation to PCNA
The PCNA ORF was cloned into the pET-Duet1 vector (Novagen) in MCS1 to encode PCNA with an N-terminal 6× His tag. The PCNA was purified over sequential chromatographic columns that included HisTrap HP, Q-Sepharose and finally Superdex 16/600–75 pg size exclusion using gel filtration. The buffer was 50 mM HEPES–KOH pH 7.5, 100 mM NaCl and 0.5 mM TCEP. To label PCNA, a 5-fold molar excess of Cy5 maleimide (GE Healthcare) was added to the PCNA and incubated for 6 h at 4°C. The labeling reaction was quenched by adding DTT to a final concentration of 10 mM. The excess free dye was separated from Cy5-labeled PCNA using a Superdex 75 10/300-GL gel filtration column. Fractions containing Cy5-PCNA were collected and dialyzed against storage buffer (25 mM Tris–HCl pH 7.5, 50% glycerol, 50 mM NaCl, 1 mM EDTA and 1 mM DTT). The purification of the PCNA loading protein, RFC, is described in the Supporting Information.
Single-molecule fluorescence imaging
DNA substrates (10 pM) in the 1× blocking buffer [20 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM EDTA and 0.0025% [v/v] Tween 20 (Sigma-Aldrich, P9416)] were infused into the flow chamber (30 mm in length, 4 mm in width and 100 μm in height) at a flow rate of 0.06 ml/min by a syringe pump (Harvard Apparatus). Then, 1 nM Cy5-PCNA and 1 nM RFC in 1× reaction buffer [25 mM Tris–HCl pH 7.5, 100 mM potassium glutamate, 5 mM MgCl2, 0.0025% [v/v] Tween 20, 0.1 mg/ml BSA (NEB, B9000S) and 1 mM DTT] with 1 mM ATP and 0.1 mM ADP were introduced and then incubated in the flow chamber for 1 min to load PCNA onto DNA molecules. After incubation, free PCNA and RFC molecules in the solution were washed out with 1× reaction buffer. We imaged Cy5-PCNA on DNA in the presence of the oxygen scavenging system [2 mM Trolox, 5 mM PCA (Sigma-Aldrich, P5630), 200 nM rPCO (Oriental Yeast, 46852004)] to minimize photobleaching and suppress photoblinking of the Cy5 fluorophore.
Emission signals of Cy5 excited with a 638 nm DPSS laser (Cobolt Samba, 100 mW) were imaged and enlarged by a 1.6× magnifier in a prism-type TIRF microscope (Olympus IX-71, water-immersion 60× objective, NA = 1.2) and recorded using EMCCD (ImagEM C9100-13, Hamamatsu) and MetaMorph 7.6 (Molecular Devices) imaging software at a 100 ms time resolution. DNA molecules stained with Sytox Orange (Thermo Fisher Scientific) were excited with a 532 nm DPSS laser (Cobolt Samba, 100 mW). The data were analyzed using DiaTrack 3.04 (13) and MATLAB 2013b (MathWorks). All experiments were performed at room temperature (22°C).
RESULTS
To place target proteins on DNA away from the surface, we constructed a 3D structure in a quartz slide with a 2D arrangement of ∼0.5 μm wide, ∼200 μm long and ∼4 μm high barriers, which were built by standard photolithography processes (Figure 1A; see ‘Fabrication of 3D structure’ in Materials and Methods). The space between adjacent barriers is ∼13 μm, which corresponds to ∼80% of the crystallographic length (0.34 nm/bp) of the 48.5 kb λ-phage DNA. To attach DNA molecules on the ridge, the surface of the quartz slide was coated with polyethylene glycol (PEG)-biotin (‘PDMS stamping’ in Materials and Methods), and streptavidin molecules were introduced on the top of the barrier to attach each biotinylated end of the DNA to two adjacent barriers via streptavidin-biotin interaction. To achieve this, streptavidin-coated polydimethylsiloxane (PDMS) was gently placed and incubated on the barrier-patterned surface, which induced the attachment of streptavidin to the PEG-biotin only on the top of the barriers (Supplementary Figure S2A; ‘PDMS stamping’ in Materials and Methods): we confirmed this attachment using AlexaFluor488 (AF488)-streptavidin (Figure 1B). The modified λ-phage DNA molecules containing biotin at both ends were then infused into the sample chamber, resulting in extended DNA molecules that were only arranged between two adjacent barriers (Figure 1C; Supplementary Figure S2B). We can visualize 247 ± 19 (mean ± s.d.; 10 experiments) of individual DNA molecules on eight barriers per field of view (8.3 × 103 μm2) at 96× magnification (Figure 1D). This indicates that a total of ∼900 000 individual DNA molecules can be immobilized on the ridges of the flow cell (10 mm × 3 mm). This yield is significantly higher when compared to the yield of double-tethered individual DNA molecules obtained by the high-throughput DNA curtains method; ∼100 DNA molecules were visualized at 60× magnification, indicating that only a few thousand DNA molecules can be immobilized in the DNA curtains flow cell (14). The bright signals at the ridges are likely to be caused by unstretched DNA molecules with both ends attached to the same ridge or only one end attached to the ridge. This 3D structural organization of the DNA on the top of the barriers is named DNA skybridge.
As shown in Figure 1C and D, intercalating dye-DNA complexes can be visualized by epi-fluorescence illumination due to the high density of fluorescent molecules inserted between DNA base pairs. However, in general, this epi-fluorescence microscopy does not allow high enough SNR to image a single fluorophore (15). Moreover, since fluorescently labeled target proteins on DNA in the DNA skybridge are located over the penetration depth of the evanescent wave in TIRF microscopy, an illumination method appropriate for single-molecule imaging in the DNA skybridge is required.
To obtain the excitation profile in our 3D structure, we carried out a computer-aided simulation using an OptiFDTD software (see ‘Simulation for intensity profile’ in Materials and Methods) to find numerical solutions of the time-dependent Maxwell's equations for the electric field of the incident beam at specific boundary conditions. For the computer simulation, we constructed a barrier-formed 3D structure that is similar to the fabricated one in Figure 1A (Figure 2A). The square of the electric field amplitude displays the intensity profile of an excitation beam in space (Figure 2B). Interestingly, an incident angle greater than the critical angle for total internal reflection at the bottom surface results in a thin layer of excitation with a full width half maximum (FWHM) of 1.1 μm (a light sheet with ∼1.1 μm thickness) at the top of the barriers (Figure 2B; Supplementary Figure S3). The center of the light sheet is located <500 nm from the top of the barriers and its intensity is 2.5 times brighter than that of the incident beam (Figure 2B, right). The simulation also shows that additional excitation beams can be formed near the bottom surface and ∼1.5 μm below the top barrier (Figure 2B). In fact, an evanescent wave is produced by the total internal refection at the bottom surface. However, this evanescent wave with a penetration depth of <200 nm was buried in the bright light sheet at the bottom surface (Supplementary Figure S4). In our 3D structure, the interference between incident and reflected light beams produced bright and dark fringes in the quartz region (Figure 2B). Therefore, the light sheet excitation beams result from the interference among incident light waves that are refracted at the interface of the barriers. Thus, the formation of a thin light sheet generated at the top of the barriers strongly depends on the incident angle and the boundary conditions of the 3D structure (Supplementary Figures S3 and S5).
The simulation predicts that at a certain incident angle, the probe-tagged proteins on the immobilized DNA can be imaged with high SNR. To illustrate the simulation result, we measured the SNR of a single Cy3 fluorophore attached to DNA at different incident angles using prism-based TIRF microscopy (Figure 3A), which is defined by the ratio of the change of mean intensities of the signal and the background to the s.d. of the background (see ‘SNR measurement’ in Materials and Methods). We first confirmed a rough critical incident angle (θc ∼ 68°) for total internal refection by visualizing a single Cy3 on the surface of a flat quartz slide since there is a poor accuracy to measure the θc in our TIRF microscopy system. To determine the SNR in the DNA skybridge as a function of the incident angle, we measured the SNR of a Cy3 fluorophore on the DNA extended between adjacent barriers at an arbitrary incident angle (θ0) that is certainly greater than θc and then at the incident angles increased by 0.7° ± 0.4° (mean ± s.d.). The result clearly shows that the greatest value of SNR occurs at a certain incident angle (θ0 + 1.7°) (Figure 3A), which is consistent with the simulation result that shows an optimal light sheet at the top of the barriers depending on the incident angle (Supplementary Figure S3).
We also expect that the background signals from the bottom surface will be dramatically reduced because emitters on the bottom surface appear to be out of focus, resulting in high SNR of the probe on the DNA. We confirmed this hypothesis by clearly visualizing a single Cy5 fluorophore conjugated to PCNA that is bound to DNA even though many Cy5-PCNA molecules are nonspecifically bound to the surface (Figure 3B; see ‘Single-molecule fluorescence imaging’ in Materials and Methods). Figure 3C displays individual Cy5-PCNA diffusing along DNA molecules that are aligned between two adjacent ridges, demonstrating a significant feasibility of a high-throughput method for studying protein-DNA interactions (Supplementary Video 1). The PCNA molecules move substantially faster along DNA as the salt concentration increases (Figure 3D), which is consistent with the previous measurement using the conventional surface-tethered TIRF microscopy (16). The diffusion constant (D) of PCNA on DNA was determined from the mean squared displacement (MSD) curve over time (t) using the diffusion equation (MSD = 2Dt). Taken together, these results are achieved by illuminating the fluorophores on the DNA with a thin light sheet perpendicular to the imaging axis, thereby only illuminating the focal plane of the imaging object. Unfocused emitters excited by other excitation beams see less light.
In the absence of free Cy5 fluorophores in a solution, the SNR for DNA skybridge is higher than that for conventional surface-tethered TIRF microscopy (Figure 4). This is surprising since the thickness of the light sheet (∼1.1 μm) in the DNA skybridge is greater than the penetration depth (∼100 nm) of an evanescent wave in our TIRF microscopy. These results suggest that autofluorescence on the quartz slide surface has a greater effect on the background noise than in the solution. However, since the excitation volume in the DNA skybridge is larger than in the surface-tethered TIRF microscopy, it is expected that SNR will be lower in the DNA skybridge when excess Cy5-PCNA is present in the solution. To evaluate this, we preloaded Cy5-PCNA onto modified λ-phage DNA and then added excess Cy5 fluorophores in the imaging chamber. As the concentration of Cy5 increases, the SNR for the DNA skybridge decreases faster than that for the TIRF microscopy (Figure 4). We found that the DNA skybridge can clearly resolve a single fluorophore in the presence of up to 3 nM of probe-labeled target proteins, which is 3-fold lower than in the TIRF microscopy (Figure 4). In epi-illumination, Cy5-PCNA on DNA was barely seen even in the presence of 0.02 nM Cy5 in the solution. Taken together, these results suggest that the DNA skybridge not only separates target proteins from false signals on the surface but it also has high SNR that allows for visualizing individual fluorophore-labeled proteins in the presence of low concentrations of protein. However, similar to conventional TIRF microscopy, DNA skybridge is not suitable for imaging transient interactions characterized by high dissociation constant.
Figure 4.
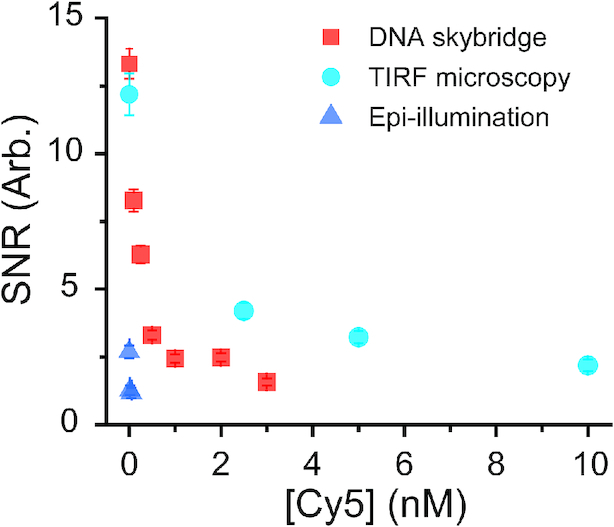
Cy5-PCNA diffuses on λ-phage DNA in the presence of various Cy5 concentrations in a solution. The SNRs for DNA skybridge were compared with those for conventional surface-tethered TIRF microscopy or epi-illumination. The error bars indicate s.d.
DISCUSSION
Here, we introduce a high-throughput single-molecule imaging platform called DNA skybridge for the interaction between proteins and nucleic acids. In the DNA skybridge, an optimal incident angle greater than the critical angle of the total internal reflection can generate a thin light sheet of the excitation beam (∼1.1 μm; Figure 2B) on the ridge of the skybridge that allows for imaging a single fluorophore with a high signal-to-noise ratio, such as a small organic dye, by reducing the background noise from fluorophores on the surface. This design could be extended for imaging molecular motors on protein filaments, such as actin filaments or microtubules extended on the top of barriers. We could achieve various shapes of barriers and spaces between the barriers (Supplementary Figure S5). They all produce a thin light sheet on the ridge at a critical incident angle, which can be used for single-molecule imaging for proteins dynamics on DNA molecules or actin/microtubule filaments of different lengths.
DNA substrates are extended and aligned between the barriers in this platform, which seems similar to the ‘DNA curtains’ method in which target proteins on DNA molecules are aligned on the flat surface of the imaging chamber and visualized by using TIRF microscopy (17,18). However, DNA curtains cannot avoid the interruption of targeted signals on the DNA by nonspecific binding of target proteins to the surface. In fact, there is an independent surface-condition method called ‘DNA tightrope assay’ (19). However, this assay is not suitable for use as a high-throughput platform. Moreover, since the target probe is excited by epi-fluorescence illumination, a very bright probe, such as a quantum dot, is required for single-molecule imaging. DNA skybridge combines the best features of light sheet imaging and high-throughput: SNR is enhanced by a thin light sheet when compared with a standard widefield illumination, false signals from fluorescently-labeled proteins bound nonspecifically to the surface appear to have no effect on imaging, and organized DNA substrates allow efficient data collection.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Ja Yil Lee (UNIST) for helpful discussion about DNA curtains.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CRG3 [URF1/2201-01-01 to S.H.]; National Research Foundation (NRF) of Korea [2018R1A2B2008995] and KIST Institutional Program (to C.J.); Global Research Lab Program through the NRF of Korea funded by the Ministry of Science and ICT [NRF-2017K1A1A2013241 to J.-B.L.]. Funding for open access charge: Global Research Lab Program through the NRF of Korea funded by the Ministry of Science; ICT [NRF-2017K1A1A2013241].
Conflict of interest statement. None declared.
REFERENCES
- 1. Blainey P.C., van Oijen A.M., Banerjee A., Verdine G.L., Xie X.S.. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad Sci. U.S.A. 2006; 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu J., Hanne J., Britton B.M., Bennett J., Kim D., Lee J.B., Fishel R.. Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature. 2016; 539:583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001; 2:764–774. [DOI] [PubMed] [Google Scholar]
- 4. Chandradoss S.D., Haagsma A.C., Lee Y.K., Hwang J.H., Nam J.M., Joo C.. Surface passivation for single-molecule protein studies. J. Vis. Exp. 2014; 86:e50549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glasmastar K., Larsson C., Hook F., Kasemo B.. Protein adsorption on supported phospholipid bilayers. J. Colloid Interface Sci. 2002; 246:40–47. [DOI] [PubMed] [Google Scholar]
- 6. van Oijen A.M. Single-molecule approaches to characterizing kinetics of biomolecular interactions. Curr. Opin. Biotechnol. 2011; 22:75–80. [DOI] [PubMed] [Google Scholar]
- 7. Holzmeister P., Acuna G.P., Grohmann D., Tinnefeld P.. Breaking the concentration limit of optical single-molecule detection. Chem. Soc. Rev. 2014; 43:1014–1028. [DOI] [PubMed] [Google Scholar]
- 8. Krieg A., Ruckstuhl T., Seeger S.. Towards single-molecule DNA sequencing: assays with low nonspecific adsorption. Anal. Biochem. 2006; 349:181–185. [DOI] [PubMed] [Google Scholar]
- 9. Hua B.Y., Han K.Y., Zhou R.B., Kim H.J., Shi X.H., Abeysirigunawardena S.C., Jain A., Singh D., Aggarwal V., Woodson S.A. et al.. An improved surface passivation method for single-molecule studies. Nat. Methods. 2014; 11:1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu L., Li C.M., Liu Y., Gao J., Wang W., Gan Y.. Flow-through functionalized PDMS microfluidic channels with dextran derivative for ELISAs. Lab Chip. 2009; 9:1243–1247. [DOI] [PubMed] [Google Scholar]
- 11. Jeong C., Cho W.K., Song K.M., Cook C., Yoon T.Y., Ban C., Fishel R., Lee J.B.. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat. Struct. Mol. Biol. 2011; 18:379–U174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parthasarathy R. Rapid, accurate particle tracking by calculation of radial symmetry centers. Nat. Methods. 2012; 9:724–726. [DOI] [PubMed] [Google Scholar]
- 13. Vallotton P., van Oijen A.M., Whitchurch C.B., Gelfand V., Yeo L., Tsiavaliaris G., Heinrich S., Dultz E., Weis K., Grunwald D.. Diatrack particle tracking software: review of applications and performance evaluation. Traffic. 2017; 18:840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gorman J., Fazio T., Wang F., Wind S., Greene E.C.. Nanofabricated racks of aligned and anchored DNA substrates for single-molecule imaging. Langmuir. 2010; 26:1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paige M.F., Bjerneld E.J., Moerner W.E.. A comparison of through-the-objective total internal reflection microscopy and epifluorescence microscopy for single-molecule fluorescence Imaging. Single Mol. 2001; 2:191–201. [Google Scholar]
- 16. Kochaniak A.B., Habuchi S., Loparo J.J., Chang D.J., Cimprich K.A., Walter J.C., van Oijen A.M.. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J. Biol. Chem. 2009; 284:17700–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gorman J., Chowdhury A., Surtees J.A., Shimada J., Reichman D.R., Alani E., Greene E.C.. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007; 28:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tutkus M., Rakickas T., Kopu Stas A., Ivanovaite S.N., Venckus O., Navikas V., Zaremba M., Manakova E., Valiokas R.N.. Fixed DNA molecule arrays for high-throughput single DNA-protein interaction studies. Langmuir. 2019; 35:5921–5930. [DOI] [PubMed] [Google Scholar]
- 19. Kad N.M., Wang H., Kennedy G.G., Warshaw D.M., Van Houten B.. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol. Cell. 2010; 37:702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.