

Abstract
Background
Endometrial adenocarcinoma (EAC) is one of the most commonly diagnosed gynaecological malignancies among female population of the developed countries. DUSP6 is a negative regulator of ERK signaling, which is a molecular switch involved in MAPK signaling during the progress of malignancies. DUSP6 was previously found to inhibit tumorigenesis and EMT-associated properties in several cancers, however, its exact role in EAC remains unclear
Methods
The level of DUSP6, (E-cad) and (N-cad) in EAC cancerous tissues and respective adjacent non-cancerous tissues were examined by western-blot or immunohistochemistry. The cell growth, invasion and migration abilities were measured in Ishikawa 3H12 endometrial cancer cell lines with overexpressed or knock down DUSP6. Protein levels of EMT-associated markers E-cadherin, N-cadherin and Vimentin were also determined. The impacts of DUSP6 on ERK signaling was assessed by detection of ERK and p-ERK.
Results
Down-regulation of DUSP6 was observed in EAC compared with the normal controls. The overexpression of DUSP6 significantly attenuated tumor cell growth, invasion, migration abilities and inhibited EMT-associated markers, while knock down of DUSP6 showed opposite trends. Overexpression of DUSP6 also down-regulated p-ERK and the knock down of DUSP6 inversely up-regulated p-ERK level.
Conclusions
DUSP6 inhibited cell growth, invasion and migration abilities in Ishikawa 3H12 cells as well as attenuating EMT-associated properties. This tumor suppressive effect of DUSP6 in EAC is achieved by inhibiting ERK signaling pathway.
Key words: DUSP6, ERK, EMT, endometrial adenocarcinoma
Introduction
Endometrial cancer (EC) is one of the most commonly diagnosed gynaecological malignancy among female population in the developed countries.1 Over 300.000 new cases of endometrial cancer were diagnosed in 2012 around the world according to the data of the World Cancer Research Fund (WCRF)2 and an increasing number of patients has been noted in recent years.3 Endometrial adenocarcinoma (EAC) frequently occurs in pre-and post-menopausal women, which initiate from the occurrence of endometrial hyperplasia. Current treatments for EAC include surgery, radiotherapy, chemotherapy and hormone therapy for early-stage diseases.5 However, therapeutic efficiency of above treatments was extremely limited, hence searching for potential therapeutic targets in EAC become urgent.
Tumor suppressive role of dual-specificity phosphatase 6 (DUSP6) in cancers is suggested to act through ERK/MAPK signaling pathway. Intracellular ERK/MAPK signaling pathway is involved in a variety of cancer transformation as well as their tumorigenesis such as breast cancer10, lung cancer11, ovarian cancer12, pancreatic cancer.13 MAP kinases phosphatases (MKPs) that belong to the DUSP family are involved in the ERK/MAPK signaling cascade. The DUSP family mediates the activity of MAPK signaling pathway through dephosphorylating both of tyrosine and threonine residues on their targets.14 Specifically, DUSP6 negatively regulates ERK1/2 by dephosphorylating the tyrosine and threonine residues.15, 16 Evidence demonstrates tumor suppressive roles of DUSP6 in different cancers. For example, the loss of DUSP6 was frequently observed in primary ovarian cancer and proved to induce chemoresistance in ovarian cancer.12 ERK1/2 has been previously shown to induce EMT process in esophageal squamous cell carcinoma and nasopharyngeal carcinoma.17 In addition, the tumorigenesis potential of ERK pathway has been reported in mounting studies regarding endometrial cancer recently.18, 19, 20 It is noteworthy that ERK pathway in human Ishikawa endometrial cancer was reported to induce cell migration and invasion via regulating EMT-associated factors such as MMP family. By the negative regulation of the ERK pathway, the underlying mechanisms of DUSP6 on EMT progress involved in EAC need further investigation.
Evidence suggests the role of DUSP6 in suppressing the cell invasion and EMT-associated properties in several carcinomas such as esophageal squamous cell carcinoma previously21, however, few studies reveal the impacts of DUSP6 on EMT process via regulating ERK pathway in endometrial adenocarcinoma. Therefore, the present study aims to investigate the potential functions of DUSP6 on EMT-associated properties in endometrial adenocarcinoma via mediating the ERK signaling with human Ishikawa endometrial cancer cell lines. Both overexpression and knockdown of DUSP6 were constructed to evaluate the functions on regulating the expressions of EMT- markers, cell growth, invasion and migration abilities. The protein levels of ERK and phosphorylated ERK were also analyzed to determine the regulatory role of DUSP6 on the ERK signaling. Our results suggest DUSP6 inhibits EMT process in endometrial adenocarcinoma in vitro via suppressing the ERK signaling pathway.
Materials and methods
Tissue samples, cell culture
Tumor and the corresponding adjacent tumor tissues (more than 5 cm from the tumor site without cancer cell infiltrations) were obtained from 35 endometrial adenocarcinoma patients from 2016 to 2018 in the Department of Obstetrics and Gynecology, Shandong Provincial Hospital. The study was authorized by the Ethics Committee of Shandong University, China. Written consents were obtained from all patients. Additionally, Ishikawa 3H12 (Purchased from ATCC, USA) high-differentiated endometrial adenocarcinoma cell line was used in in vitro study. The cells were cultured in DMEM supplemented with 5% charcoal-stripped fetal bovine serum (FBS), 100μ/ml of penicillin and 100 μ/ml of streptomycin at 37°C in a humidified environment with 5% CO2 in air.
Constructs of cell transfection
Short hairpin RNA (shRNA) plasmids which targets the coding region of DUSP6; or by transfecting cells with non-target shRNA control vector were constructed by GenePharma, Shanghai and cloned into pGPU/GFP/Neo. The constructs were transfected into Ishikawa cells by Lipofectamine 3000 according to the manufacturer’s protocol. cDNA of DUSP6 was obtained and synthetized by GenePharma, Shanghai and cloned into pcDNA3.1 plasmid. Ishikawa 3H12 cells were transiently transfected with either pcDNA3.1-Dusp6 plasmid or empty vector (EV) using Lipofectamine 3000 according to the manufacturer’s protocol. Whole cell lysates were collected at the indicated time points after transfection to verify the expression level of DUSP6.
MTT assay
Cell viability was evaluated with MTT (Qiagen, German) assays according to the manufacturer’s instructions. Transfected Ishikawa 3H12 cells were seeded at a density of 6.000 cells per well in 96-well plates in RPMI 1640 containing 10% FBS overnight and then maintained in 0.5% FBS media for 96h before testing. The absorbance was measured at a wavelength of 490 nm on a Synergy Multi-Mode Microplate Reader (Biotek, USA). Each assay was performed on five replicate wells.
Immunohistochemistry analysis
Fresh endometrial adenocarcinoma tumor tissue samples were washed using phosphate buffered saline (PBS) and fixed using formalin. After dehydration and paraffin-embedding, samples were cut and mounted onto glass slides. Formalin-fixed sections were deparaffinized and rehydrated and incubated with 0.6% hydrogen peroxide in methanol. Then antigen retrieval was performed and the sections were blocked and incubated overnight with rabbit anti-human DUSP6/E-cad/N-cad primary antibody (1:100, Abcam, USA) at 4°C. HRP-conjugated goat anti-rabbit IgG secondary antibody was deployed afterwards. DAB was used as chromogenic agent. Same sections incubated with non-immune serum instead of primary antibody were used as a negative control. The experiments were performed in duplicate. The intensity of the staining of DUSP6/E-cad/N-cad was scored by a clinical pathologist who was blinded with any knowledge of the pathological data.
Wound healing assay
To evaluate the migration of cells, Ishikawa 3H12 cells overexpressed or knockdown of DUSP6 were cultured until confluent beforehand. Then the cell monolayer was scratched with a pipette tip (Scar bar: 1mm) and incubated for 24 h, the relative wound areas were calculated through: migrated distance of DUSP6 expression or knockdown clones/ migrated distance of vector-alone stable clones x 100.
Transwell Matrigel assay
A micropore chamber assay was used to assess the invasion ability of Ishikawa 3H12 cells that with the overexpression/knockdown of DUSP6. Matrigel was diluted by pre-cold serum free DMEM medium at 1: 5 before use. A total number of 3x104 cells were seeded into the top chamber of a 24-well Matrigel-coated micropore polycarbonate membrane filter with 8μm pores (Becton Dickinson Labware, Franklin Lakes, NJ) for invasion assays. The DMEM containing 10% FBS was added to the bottom chamber as a chemoattractant. The cells were then incubated at 37°C for 22 h. The members were washed and fixed followed by stained by crystal violet. The cells migrated through the membranes were counted under the inverted light microscope at 10x magnification. All tests were performed in triplicate.
RNA extraction and RT-qPCR
To verify the expression of DUSP6 in transfected cell, total RNA was isolated with TRIzol reagent (Life Technologies, USA) and converted to first-strand cDNA using M-MLV Reverse Transcriptase (USB, Cleveland, OH). The mRNA level was analyzed with quantitative real-time PCR assay. Specific primers were synthesized as the following sequences show: DUSP (forward) 5’-GAACTGTGGTGTCTTGGTACATT-3′ and (reverse) 5′-GTTCATCGACAGATTGAGCTTCT-3’.22 The GAPDH gene was used as an internal control and was amplified from the same cDNA samples for 25 cycles. The cDNA of a normal endometrial cell line was used as a positive control.
Western blot analysis
The whole cell lysates were extracted using RIPA buffer with protein inhibitors cocktail (Complete Mini, Roche Diagnostics, Switzerland). The concentration of total cellular protein was measured using BCA assay kit (Pierce, IL, USA) according to manufacturer’s instructions. 30 μg of proteins from each sample were electrophoresed to separate on 10% sodium dodecysulfate-polyacryl-amide gels (SDS-PAGE) and then transferred to polyvinylidene difluoride membrane (PVDF, CA, USA). The PVDF membranes were blocked with 5% non-fat dry milk for 1 h at room temperature and incubated at 4°C overnight with the primary antibodies against: DUSP6, E-cadherin, N-cadherin, Vimentin, ERK, p-ERK, GAPDH (1:1000, Cell Signaling, USA). The membranes were then washed and incubated with the secondary antibody against rabbit at 37°C for 1 h and protein bands were detected by the chemiluminescence method.
Statistical analysis
Data are presented as means ±SD from three independent experiments. All statistical analyses were performed using SPSS 13.0 (SPSS Inc, USA). For comparisons between groups, one-way analysis of variance was performed. Western blot analysis was quantified by Image J. P values <0.05 were considered as statistically significant.
Results
Down-regulation of DUSP6 and E-cadherin and up-regulation of process-cadherin in endometrial adenocarcinoma
By determining the expressions of DUSP6, E-cadherin and N-cadherin in both endometrial adenocarcinoma cancerous and the corresponding adjacent non-tumor tissues using western blot and immunohistochemistry analysis, down-regulation of DUSP6 (Figure 1A) and E-cadherin (Figure 1B) were obeserved in cancerous specimens compared to corresponding normal counterparts. The results of immunohistochemical staining were in line with the results of western-blot in terms of the expressions of DUSP6 and E-cadherin. Contrarily, significant up-regulation of N-cadherin was observed in cancerous tissues than that in their non-cancerous counterparts (Figure 1C)
Figure 1.
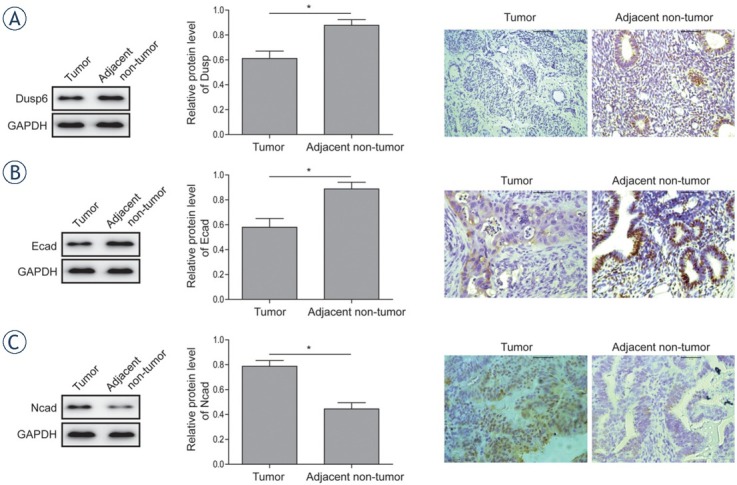
Expression analysis of Dusp6, E-cadherin and N-cadherin in EAC tissues and the adjacent non-tumor tissues. (A) The protein expression level of DUSP6 was analysed by western-blot and immunohistochemistry analysis in EAC tumor and adjacent non-tumor tissues. (B) The protein expression level of E-cadherin was analysed by western-blot and immunohistochemistry analysis in EAC tumor and adjacent non-tumor tissues. (C) The protein expression level of N-cadherin was analysed by western-blot and immunohistochemistry analysis in EAC tumor and adjacent non-tumor tissues.Data were presented as Mean ±SD. P < 0.05 was considered as significant.* P < 0.05. Each experiment was performed in triplicate.
Overexpression of DUSP6 attenuates tumor cell growth, invasion, migration and EMT process in endometrial adenocarcinoma cell line
To investigate the role of DUSP6 in the progress of endometrial adenocarcinoma, proliferation, invasion and migration of Ishikawa 3H12 cell line was assessed after overexpressing DUSP6 by transfecting pcDNA3.1-Dusp6 vector or empty vector control. The efficiency of transfection was validated by western-blot (Figure 2A) The MTT assay demonstrated that the cell viability was significantly decreased in DUSP6 overexpressed Ishikawa 3H12 cells than that of vector (Figure 2B) Further in vitro Transwell Matrigel assay (Figure 2C) and wound healing assay (Figure 2D) showed the attenuated invasion and migration ability in DUSP6 overexpressed tumor cell lines. DUSP6 overexpressed clones in Matrigel culture exhibited a non-cohesive and disorganized morphology compared to polarized and well-organized spheroids shown in vector-alone clones and controls. The width of wound formed in cultures of cells overexpressing DUSP6 significantly superior to that in cultures of vector-alone clones and controls together referred to differed EMT-associated properties.23 Increased protein level of E-cadherin and decreased Vimentin were observed in DUSP6 overexpressed cells (Figure 2E)
Figure 2.
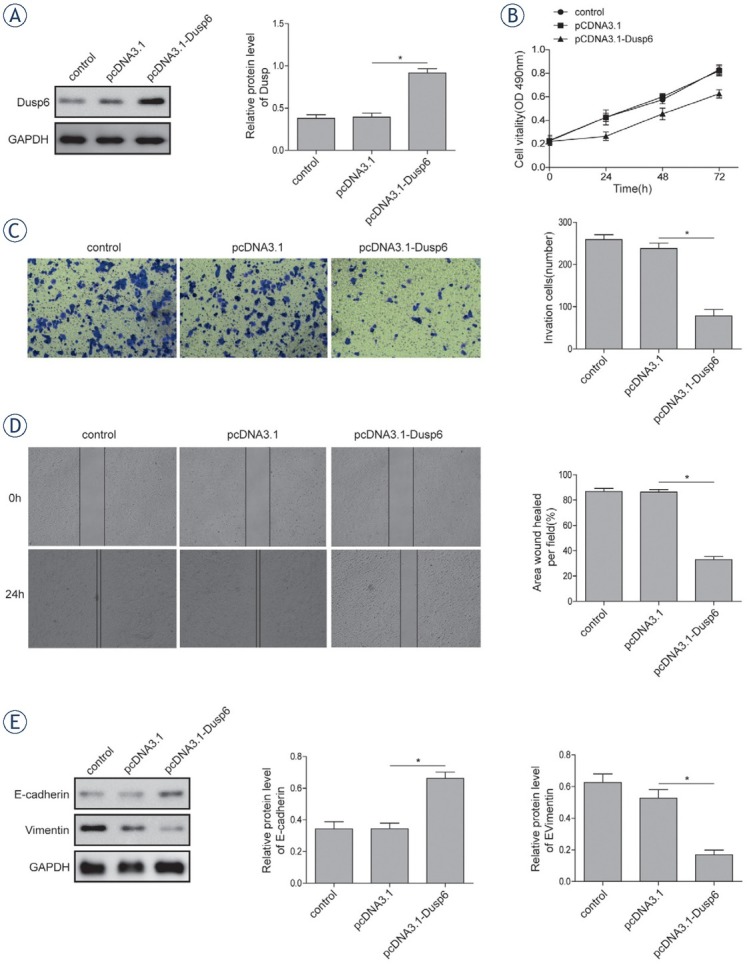
Effects of DUSP6 overexpression on cellular proliferation and EMT. (A) The protein expression level of DUSP6 was analysed using western-blot. (B) The cell viability of Ishikawa 3H12 cell lines measured using MTT assay. (C) Relative invasion ability of Ishikawa 3H12 cell lines measured using Transwell Matrigel assay. (D) Relative migration ability of Ishikawa 3H12 cell lines measured using wound healing assay. (E) The protein levels of E-cadherin and Vimentin in Ishikawa 3H12 cell lines measured using western-blot analysis. Groups: Negative control, pcDNA3.1 Vector-alone clones, pcDNA3.1-Dusp6 clones. Data were presented as Mean ±SD. P < 0.05 was considered as significant. *P < 0.05. Each experiment was performed in triplicate.
Knockdown of DUSP6 enhances tumor cell growth, invasion, migration and EMT process in endometrial adenocarcinoma cell line
Cell growth, migration and invasion were evaluated after silencing DUSP6 expression by shRNA in Ishikawa cell line. Cells were transfected with shRNA targeting DUSP6 or NC. The efficiency of knockdown was assessed by western blot (Figure 3A) Cell viability of DUSP6 knock down cell was increased significantly compared to that of NC (Figure 3B) The enhanced cell invasion and migration ability were also observed in tumor cells with the absence of DUSP6 expression based on transwell matrigel (Figure 3C) and wound healing assay (Figure 3D) respectively. EMT process was promoted in the DUSP6 knock down tumor cells indicated by decreased protein level of E-cadherin and increased level of Vimentin (Figure 3E)
Figure 3.
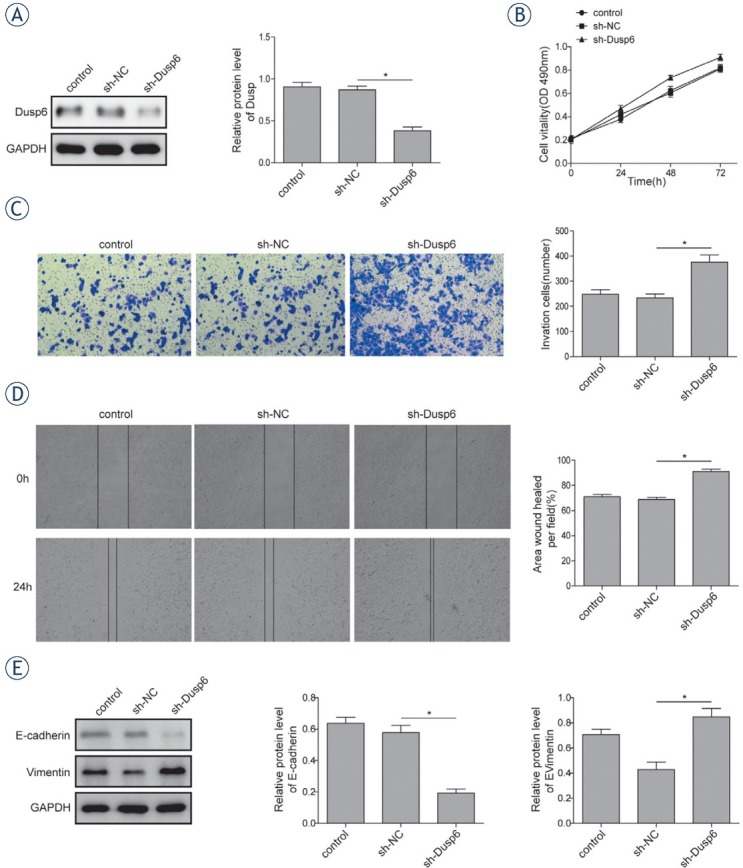
Effects of knocking down DUSP6 on cellular proliferation and EMT. The efficiency of transfection was validated by measuring protein expression level of DUSP6 using western blot. (B) The cell viability of Ishikawa 3H12 cell lines measured using MTT assay. (C) Relative invasion ability of Ishikawa 3H12 cell lines measured using Transwell Matrigel assay. (D) Relative migration ability of Ishikawa 3H12 cell lines measured using wound healing assay. (E) The protein levels of E-cadherin and Vimentin in Ishikawa 3H12 cell lines measured using western-blot analysis. Groups: Negative control, sh-NC clones, sh-Dusp6 clones. Data were presented as Mean ±SD. P < 0.05 was considered as significant. *P < 0.05. Each experiment was performed in triplicate.
DUSP6 inhibits activation of ERK pathway in endometrial adenocarcinoma
The activation of ERK pathway has been validated in tumorigenesis in varied human carcinoma and played an important role in endometrial adenocarcinoma. The present study confirmed that the dysregulation of DUSP6 induced aberrant activation of ERK signaling in Ishikawa 3H12 cell lines (Figure 4, Figure 5). According to western-blot results, the level of p-ERK/ERK was significantly down-regulated in overexpressed DUSP6 cells, indicating activation of ERK signaling was inhibited by DUSP6 (Figure 4). In contrary, the level of p-ERK/ERK was significantly up-regulated in DUSP6 knockdown cells compared to that in sh-NC (Figure 5).
Figure 4.
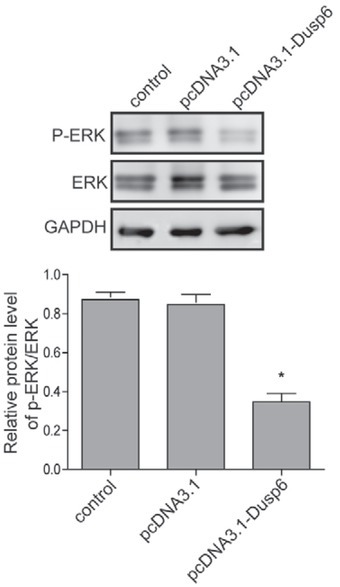
Assessment of effects of overexpressing DUSP6 on ERK signalling pathway. The protein level of phosphorylated ERK and ERK were measured using western blot analysis in Ishikawa 3H12 cell lines when DUSP6 was overexpressed. Β-actin served as an internal control. Groups: Negative control, pcD-NA3.1 Vector-alone clones, pcDNA3.1-Dusp6 clones. Data were presented as Mean ±SD. Each experiment was performed in triplicate.
Figure 5.
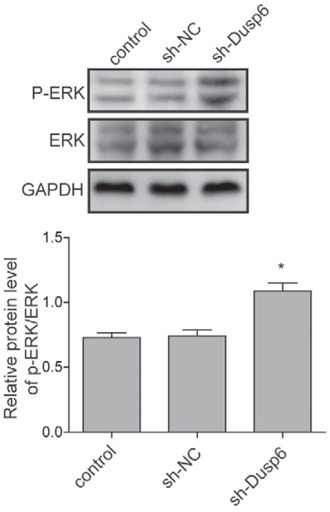
Assessment of effects of knocking down DUSP6 on ERK signalling pathway. The protein level of phosphorylated ERK and ERK were measured using western blot analysis in Ishikawa 3H12 cell lines when DUSP6 was knocked down. Β-actin served as an internal control. Groups: Negative control, sh-NC clones, sh-Dusp6 clones. Each experiment was performed in triplicate. Data were presented as Mean ±SD.
Discussion
Endometrial cancer usually initiates in the cells of the inner lining of the endometrium (uterus) and has developed into the most frequently occurring gynecological cancer in developed countries.24 Endometrial adenocarcinoma (EAC) is the most frequently occurring subtype of the endometrial cancer and accounts for approximately 75% of EC cases clinically. Dual-specificity phosphatase 6 (DUSP6) was reported to dephosphorylate phosphor-tyrosine and phosphor-threonine residues on ERK1/2 to inactivate the ERK kinase. Considering the facts that DUSP6 was reported to be involved in tumorigenesis of various cancers and the lower Dusp6 expression was observed in endometrial of post-menopausal age compared with that of reproductive age3, the potential role of Dusp6 during the development of EAC is hence worth investigating.
Accumulated evidence showed DUSP6 was down-regulated in various types of cancers, including ovarian cancer, lung cancer, esophageal squamous cell carcinoma and nasopharyngeal carcinoma.21 However, there is discrepancy on the expression of DUSP6 exists in other cancers including endometrial cancer. Previous studies indicated that the overexpression of DUSP6 is frequent in EAC, which is associated with enhanced pronecancer potential of estrogen in EAC.3 Also, the overexpression of DUSP6 was observed in EAC by the same research group and was found to promote 17β-estradiol stimulation in Ishikawa cell lines.25 It is interesting to find that DUSP6 was significantly down-regulated in Ishikawa 3H12 EAC cell lines based on the results of western blot, which was supported by results of immunochemistry analysis in EAC tumour biopsies as well in the present study. Previous evidence revealed the down-regulation of DUSP6 in cancers may be attributed to epigenetic silencing and specifically, the allelic loss of 12q21 where DUSP6 maps.21,26,27 It is possible that the down-regulation of DUSP6 in EAC is resulted from the similar mechanism whilst further investigation is needed with a larger sample size. Despite of this discrepancy, the potentially inhibitory functions of DUSP6 on growth and proliferation of Ishikawa 3H12 cell lines were verified in the present study, which suggested the therapeutic potential of DUSP6 in EAC.
EMT-associated properties were regulated by the overexpression and the knockdown of DUSP6 in EAC cancer cells. Since EMT causes the loss of apical-basal polarity and cell to cell adhesion28, the formation of polarized cell spheroid in DUSP6-overexpressed clones that indicates the reversion of EMT process, which may suggest the suppressive role of DUSP6 in EMT-associated properties in EAC. Moreover, the attenuation of cell invasion and migration ability in DUSP6 overexpressed clones suggested the inhibitory function of DUSP6 on cancer cell mobility. As important indicators of cell survival and prognosis, it was previously demonstrated that the loss of E-cadherin alongside upregulated levels of N-cadherin and Vimentin are involved in EMT process and morphological changes in pancreatic carcinoma9, also are associated with the better prognosis of tumor and lower death rate.29 Hence, higher level of N-cadherin detected in EAC cancerous specimens compared with the adjacent normal tissues alongside the loss of E-cadherin and DUSP6 indicates the inhibitory role of DUSP6 in EMT process and metastasis of EAC. Further study should be applied to confirm the in vivo suppressive effect of DUSP6.
DUSP6 is demonstrated as a negative regulator of ERK signalling pathway based on the present study, which is in accordance to the previous findings. It was reported that DUSP6 functionally and physically inhibited the activity of ERK2.30 Demonstrated promotive functions of ERK2 on cell invasiveness and EMT process in cancers17 further indicates the suppressive role of DUSP6 during the development of tumours. Here in this study the significant down-regulated phosphorylation of ERK in DUSP6 overexpressed clones alongside the up-regulated phosphorylated ERK in DUSP6 knocking down clones suggested DUSP6 impaired the activation of ERK pathway. This is also for the first time that DUSP6 was reported to regulate EMT process through affecting ERK signalling in endometrial adenocarcinoma.
In summary, the present study explored the role of Dusp6 in regulation of EMT process via ERK signaling pathway in endometrial adenocarcinoma. The decreased level of DUSP6 was verified in the EAC tumor samples compared to the endometrial normal tissues. Overexpression of Dusp6 was demonstrated to inhibit EAC cancer EMT, cell growth, invasion and migration abilities, while the knockdown of Dusp6 inversely enhanced the malignant phenotypes mentioned above. Additionally, the present study determined that DUSP6 impaired ERK signaling. The current findings provided theoretical basis of seeking for a novel therapeutic target for treating the endometrial adenocarcinoma.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No.81272858 and No.81671433).
Disclosure
No potential conflicts of interest were disclosed.
References
- 1.Jurcevic S, Klinga-Levan K, Olsson B, Ejeskär K. Verification of microRNA expression in human endometrial adenocarcinoma. BMC Cancer. 2016;16:261. doi: 10.1186/s12885-016-2296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. Cancer incidence and mortality worldwide: sources. methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. et al. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Yan L, Bai Y, Li C, Guo Q, Wang C. Dual-specificity phosphatase 6 predicts the sensitivity of progestin therapy for atypical endometrial hyperplasia. Gynecol Oncol. 2015;136:549–53. doi: 10.1016/j.ygyno.2014.11.008. et al. [DOI] [PubMed] [Google Scholar]
- 4.Emons G, Fleckenstein G, Hinney B, Huschmand A, Heyl W. Hormonal interactions in endometrial cancer. Endocr Relat Cancer. 2000;7:227–42. doi: 10.1677/erc.0.0070227. et al. [DOI] [PubMed] [Google Scholar]
- 5.Cavanagh D, Fiorica JV, Hoffman MS, Durfee J, Nicosia SV. Adenocarcinoma of the endometrium: an Institutional review. Cancer Control. 1999;6:354–60. doi: 10.1177/107327489900600405. [DOI] [PubMed] [Google Scholar]
- 6.Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005;366:491–505. doi: 10.1016/S0140-6736(05)67063-8. [DOI] [PubMed] [Google Scholar]
- 7.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 8.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. 2004;10:4125–33. doi: 10.1158/1078-0432.CCR-0578-03. et al. [DOI] [PubMed] [Google Scholar]
- 10.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–22. doi: 10.1038/sj.onc.1202367. et al. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–86. doi: 10.1093/carcin/bgq020. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan DW, Liu VW, Tsao GS, Yao KM, Furukawa T, Chan KK. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis. 2008;29:1742–50. doi: 10.1093/carcin/bgn167. et al. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807–15. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–89. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 15.Arkell RS, Dickinson RJ, Squires M, Hayat S, Keyse SM, Cook SJ. DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell Signal. 2008;20:836–43. doi: 10.1016/j.cellsig.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Jurek A, Amagasaki K, Gembarska A, Heldin CH, Lennartsson J. Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J Biol Chem. 2009;284:4626–34. doi: 10.1074/jbc.M808490200. [DOI] [PubMed] [Google Scholar]
- 17.Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010;38:114–27. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen HX, Xu XX, Tan BZ, Zhang Z, Zhou XD. MicroRNA-29b Inhibits angiogenesis by targeting VEGFA through the MAPK/ERK and PI3K/Akt signaling pathways in endometrial carcinoma. Cell Physiol Biochem. 2017;41:933–46. doi: 10.1159/000460510. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Wang D, Wang N, Long Z, Ren X. Long non-coding RNA BANCR promotes endometrial cancer cell proliferation and invasion by regulating MMP2 and MMP1 via ERK/MAPK signaling pathway. Cell Physiol Biochem. 2016;40:644–56. doi: 10.1159/000452577. [DOI] [PubMed] [Google Scholar]
- 20.Ma Z, Liu X, Li F, Wang Y, Xu Y, Zhang M. Perfluorooctanoic acid induces human Ishikawa endometrial cancer cell migration and invasion through activation of ERK/mTOR signaling. Oncotarget. 2016;7:66558–68. doi: 10.18632/oncotarget.11684. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong VC, Chen H, Ko JM, Chan KW, Chan YP, Law S. Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial-mesenchymal transition (EMT)-associated phenotype. Int J Cancer. 2012;130:83–95. doi: 10.1002/ijc.25970. et al. [DOI] [PubMed] [Google Scholar]
- 22.Wu QN, Liao YF, Lu YX, Wang Y, Lu JH, Zeng ZL. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 2018;412:243–55. doi: 10.1016/j.canlet.2017.10.007. et al. [DOI] [PubMed] [Google Scholar]
- 23.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–58. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 24.McGuire S. World Cancer Report. Vol. 7. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press; 2014. pp. 418–9. 2015. Adv Nutr 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Guo Q, Wang C, Yan L, Fu Y, Fan M. Dual-specificity phosphatase 6 (Dusp6), a negative regulator of FGF2/ERK1/2 signaling, enhances 17β-estrodial-induced cell growth in endometrial adenocarcinoma cell. Mol Cell Endocrinol. 2013;376:60–9. doi: 10.1016/j.mce.2013.02.007. et al. [DOI] [PubMed] [Google Scholar]
- 26.Mayama T, Fukushige S, Shineha R, Nishihira T, Satomi S, Horii A. Frequent loss of copy number on the long arm of chromosome 21 in human esophageal squamous cell carcinoma. Int J Oncol. 2000;17:245–52. et al. [PubMed] [Google Scholar]
- 27.Lo KW, Teo PM, Hui AB, To KF, Tsang YS, Chan SY. High resolution allelotype of microdissected primary nasopharyngeal carcinoma. Cancer Res. 2000;60:3348–53. doi: 10.3892/ijo.17.2.245. et al. [DOI] [PubMed] [Google Scholar]
- 28.Shaw KR, Wrobel CN, Brugge JS. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia. 2004;9:297–310. doi: 10.1007/s10911-004-1402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu LK, Jiang XY, Zhou XX, Wang DM, Song XL, Jiang HB. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: correlation with the clinicopathological features and patient outcome. Mod Pathol. 2010;23:213–24. doi: 10.1038/modpathol.2009. et al. [DOI] [PubMed] [Google Scholar]
- 30.Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–5. doi: 10.1126/sci-ence.280.5367.1262. et al. [DOI] [PubMed] [Google Scholar]