

ABSTRACT
The gap junction protein connexin 43 (Cx43) is a key player in wound healing, and inhibitors of Cx43, which speed epidermal wound healing, are currently in clinical trials. Here, we provide direct in vivo evidence that specific phosphorylation events on Cx43 change the physiological response during wound healing. Blocking phosphorylation, through mutation of serine residues in Cx43 at the protein kinase C (PKC) or casein kinase 1 (CK1) sites, significantly slowed the rate of wound closure in vivo and in vitro and resulted in a thicker epidermal layer after reepithelialization. Conversely, preventing Cx43 phosphorylation by mitogen-activated protein kinases (MAPKs) through mutation significantly increased the rate of wound closure in vivo. Defects in migration, but not proliferation, in all mutants were partially rescued in vitro by changing serine residues to aspartic or glutamic acid. These data prove that specific Cx43 phosphorylation events play an important role at different stages of wound healing. Thus, a clear physiological understanding of the spatiotemporal regulation of kinase activation and consequent effects on gap junctions could lead to a more targeted approach to modulating Cx43 expression during wound healing.
KEY WORDS: Connexin 43, Phosphorylation, Migration, Wound healing
Summary: Cx43 plays a key role in wound healing and Cx43 modulators are used in clinical trials. We tested the importance and roles of specific Cx43 phosphorylation sites during wound repair.
INTRODUCTION
Vertebrate gap junctions are composed of proteins from the connexin gene family, and they collectively form intercellular channels that permit passage of small molecules between cells (Goodenough et al., 1996). This communication is critical to many cellular processes, and connexins are expressed in every human tissue. Connexin 43 (Cx43, also known as GJA1) is the most abundant and widely expressed gap junction protein, and its essential role is highlighted by the lethality of Cx43 knockout in mice (Reaume et al., 1995). There are at least 14 phosphorylation sites in the C-terminal region of Cx43 and this post-translational modification has been shown to be an important regulator of gap junction intracellular communication (Solan and Lampe, 2009). The known Cx43 kinome involves at least protein kinase C (PKC), protein kinase A (PKA), casein kinase 1 (CK1), extracellular signal-related kinases 1/2 (ERK1/2), Src, cyclin-dependent kinase 2 (CDC2), Akt and Tyk2, though some may act indirectly; multiple additional unknown kinases that phosphorylate Cx43 remain to be discovered. Cx43 phosphorylation at S364 (TenBroek et al., 2001), S365 (Solan et al., 2007), S325, S328 and S330 (Cooper and Lampe, 2002; Lampe et al., 2006), and S373 (Dunn and Lampe, 2014) increases in response to stimuli that enhance gap junction assembly. Cx43 phosphorylation by mitogen-activated protein kinases (MAPKs) (at S255, S262, S279 and S282) (Cottrell et al., 2003; Warn-Cramer et al., 1998), PKC (at S368) (Lampe et al., 2000) and Src (at Y247 and Y265) (Lin et al., 2001) decrease gap junction communication and can, in some cases, increase gap junction turnover. Loss of the C-terminal region of Cx43 (where phosphorylation occurs) causes oculodentodigital dysplasia in humans (Vreeburg et al., 2007) and rigid skin in mice that readily peels off, killing almost all mice shortly after birth (Maass et al., 2004). Reflecting these dermatological phenotypes, Cx43 is abundantly expressed in skin and is known to play a key regulatory role in repair processes (Lampe et al., 1998; Becker et al., 2012; Montgomery et al., 2018).
Cx43 is extensively regulated during wound repair and there is ample evidence that regulating Cx43 expression and gap junctional communication can be beneficial to wound healing (Coutinho et al., 2003). In mice and humans, Cx43 expression decreases at the edge of a wound within 24 h in both epidermal and dermal layers and returns to homeostatic levels after wound closure. The wound healing process can also be disrupted by altering Cx43 expression. For example, mice with reduced levels of epidermal Cx43 show rapid healing (Nakano et al., 2008), while wound closure is delayed when Cx43 is overexpressed (Kretz et al., 2003). Transient reduction of Cx43 expression speeds wound healing in mice and humans (Qiu et al., 2003; Montgomery et al., 2018). Multiple clinical trials are investigating connexin modulators and, specifically, Cx43 mimetic peptides that target the intracellular loop and the C-terminal tail region appear promising (recently reviewed in Laird and Lampe, 2018).
During wound repair, Cx43 phosphorylation is also dynamically regulated through spatiotemporal interactions with a variety of kinases. Upon wounding, a sequence of phosphorylation events occur beginning with: (1) dephosphorylation at the CK1 sites S325, S328 and S330 and phosphorylation at the Akt site S373 within 5–30 min (Dunn et al., 2012; Dunn and Lampe, 2014), (2) phosphorylation at the MAPK sites S255, S262, S279 and S282 (Johnstone et al., 2012) at 15–60 min, and (3) phosphorylation at Src kinase site Y265 (Lin et al., 2001) at 30 min–24 h and at the PKC site S368 at 24 h (Richards et al., 2004). This sequence has demonstrated functional consequences including changes in (1) gap junction assembly driven by phosphorylation via CK1 on S325, S328 and S330 (Cooper and Lampe, 2002) and Akt on S373 (Dunn and Lampe, 2014); (2) channel selectivity by PKC-mediated phosphorylation on S368 (Axelsen et al., 2006; Lampe et al., 2000; Richards et al., 2004; Sáez et al., 1997); and (3) gap junction disassembly facilitated by MAPK-mediated phosphorylation on S279 and S282 (Warn-Cramer et al., 1996) and Src-mediated phosphorylation on Y247 and Y265 (Lin et al., 2001).
Since the impact of the aforementioned phosphorylation events during wound healing is unknown, we sought to definitively prove whether these coordinated responses are physiologically relevant. We performed wounding studies on knock-in mice expressing Cx43 mutants where the CK1, MAPK or PKC sites were mutated to eliminate serine phosphorylation. We found differential effects on wound healing in each mutant. We also measured differences in skin thickness and proliferation in wounded and unwounded tissue. Furthermore, in vitro modeling of these mutations showed differences in migration and proliferation that explained some of the in vivo effects. Analysis of proliferation and migration in both tissue and cell culture argue that Cx43 phosphorylation at distinct sites is critical during the wound healing process.
RESULTS AND DISCUSSION
Eliminating Cx43 serine phosphorylation at MAPK, PKC or CK1 sites alters wound healing in vivo
Phosphorylation at multiple sites in the C-terminal tail of Cx43 have been observed after wounding, but evidence for the necessity and functional relevance of these sites during wound repair has not been shown. We wanted to directly test the link of phosphorylation events on Cx43 to three specific aspects of epithelial biology during wounding: (1) migration, (2) proliferation, and (3) Cx43 expression. To do this we utilized three genetically engineered mouse models with Cx43 containing mutations at putative MAPK (S255, S262, S279 and S282, termed Cx43MAPK), PKC (S368, termed Cx43PKC) or CK1 (S325, S328 and S330, termed Cx43CK1) sites to block serine phosphorylation and examined whether these mutations affected wound healing compared to what was seen with wild-type Cx43 (Cx43WT). Mice received 3.5 mm punch biopsies on the back in between the shoulder blades, and wound closure was monitored over 13 days. Representative images of wounds at days 0, 5, 10 and 13 are shown in Fig. 1A and in Fig. S1. Wound closure in Cx43PKC mice was significantly delayed compared to Cx43WT mice at as soon as day 6 and continued through to the end of the experiment (day 13) (Fig. 1B). This is consistent with previous observations showing that Cx43 phosphorylation on S368 is dramatically upregulated in human keratinocytes over the first 24 h after wounding (Pollok et al., 2011; Richards et al., 2004) and the wound healing benefits of the Cx43 mimetic peptides Gap26 and Gap27 are at least partially dependent on phosphorylation at S368 (Pollok et al., 2011). Additionally, phosphorylation at S368 can be prevented by prior phosphorylation at S365 (Solan et al., 2007). Wound healing in Cx43CK1 mice was similar to that in Cx43WT during the first week after wounding but then experienced a significant delay in wound closure starting at day 8. While this phosphorylation event has not been well studied in skin wounding, it has been shown to be important in other types of cellular injury. For example, in the normal mouse heart, Cx43 is heavily phosphorylated at CK1 sites, but upon ischemia, these sites rapidly become dephosphorylated followed by gap junction disassembly (Axelsen et al., 2006; Lampe et al., 2006). Cx43CK1 mice develop deleterious cardiac phenotypes (Remo et al., 2011) and differ in their response to ischemia reperfusion injury (Solan et al., 2019) owing to alterations in cardiac gap junction formation and function, lending support to the importance of these phosphorylation sites during the injury response. In the latter case, increased phosphorylation of Cx43 at MAPK sites S262, S279 and S282 was observed in cardiac tissue in the Cx43CK1 mice (Solan et al., 2019). Interestingly, Cx43MAPK mice closed wounds ∼20% faster than Cx43WT. Recently, Cx43MAPK mice were shown to have reduced pathology after ischemic stroke (Freitas-Andrade et al., 2019), suggesting loss of phosphorylation at this site plays a protective role in injury responses. These results show for the first time that Cx43 phosphorylation events directly and differentially regulate epidermal wound healing in vivo.
Fig. 1.

Blocking Cx43 phosphorylation at MAPK, PKC or CK1 sites alters wound healing in vivo. (A) Representative images of wounds in Cx43WT, Cx43MAPK, Cx43PKC and Cx43CK1 mice 0, 5, 10 and 13 days after wounding. Scale bars: 5 mm. (B) Two 3.5 mm punch biopsies were performed on the back of mice as in A and the width of wounds were measured over the course of 13 days. n=5 (10) Cx43WT, 8 (16) Cx43MAPK, 5 (10) Cx43PKC, and 6 (12) Cx43CK1 mice (wounds) per group. Results are mean±s.e.m. *P<0.05 compared to WT (one-way ANOVA).
Cx43 phosphorylation plays a role in skin epithelial thickness
We examined the histopathology of the healed wounds. We first looked at the degree of immune cell infiltration into the healed wounds since downregulation of Cx43 expression has been shown to reduce leukocyte infiltration during wound healing (Mori et al., 2006). There were no significant differences between phospho-mutant mice (Fig. S2A-B), and a preliminary investigation demonstrated a high macrophage but low neutrophil or myofibroblast presence in all healed wounds (Fig. S2C–F). Further investigation into specific immune cell subtypes could illuminate differences. In agreement with wounding data from heterozygous Cx43-knockout mice (Cogliati et al., 2015), we did not observe any differences in the content of early or mature collagen (Fig. S3A). In healed wounds treated with Cx43 antisense DNA, collagen bundles show a more basket-weave appearance rather than the typical aligned parallel bundles seen in most scars (Becker et al., 2012); we did not observe any significant differences in the pattern or orientation of collagen in the healed wounds from different genotypes (Fig. S3B–D). We also did not observe any significant differences in the total area of the healed wounds (Fig. S3E). However, we did observe that the epidermal layer in the healed wound region was noticeably different between phospho-mutant mice (Fig. 2A,B). While the thickness of epidermal layer was comparable between Cx43WT and Cx43MAPK mice, the epidermal layer in Cx43PKC and Cx43CK1 mice was almost twice as thick (Fig. 2C). Treatment of human wounds with Cx43 antisense RNA not only decreases wound healing time, but also reduces scar appearance after healing (Qiu et al., 2003; Ghatnekar et al., 2009). Both Cx43CK1 and Cx43PKC mice had slower wound healing time, as well as significant increases in epidermal thickness, which can be associated with scar severity (Limandjaja et al., 2017). We next went back and compared homeostatic skin in these mouse models. Interestingly, Cx43CK1 mice had a small, but statistically significant increase in the thickness of the epidermal layer compared to all other genotypes, indicating a novel role for this site in regulating epidermal thickness (Fig. 2D,E).
Fig. 2.

Cx43PKC and Cx43CK1 have thicker skin epidermis after reepithelization of wounds. (A,B) Representative immunohistochemical images of Masson's Trichrome staining in the healed wounds of Cx43WT, Cx43MAPK, Cx43PKC and Cx43CK1 mice. Black boxes represent the origin of the magnified image shown in B. (C) Quantification of the epidermal thickness in healed wounds. n=4 (43) Cx43WT, 5 (96) Cx43MAPK, 3 (27) Cx43PKC, and 5 (54) Cx43CK1 mice (sites) per group. (D) Representative immunohistochemical images of Masson's Trichrome staining in the skin of Cx43WT, Cx43MAPK, Cx43PKC and Cx43CK1 mice. (E) Quantification of the epidermal thickness in normal skin. n=4 (55) Cx43WT, 5 (58) Cx43MAPK, 5 (62) Cx43PKC, and 6 (82) Cx43CK1 mice (sites) per group. Results are mean±s.e.m., *P<0.01, **P<0.005, ***P<0.0005 (one-way ANOVA). Scale bars: 500 µm (A,D), 100 µm (B).
Cx43MAPK mice have reduced Cx43 expression and Cx43PKC mice have lower proliferation rates in epidermal skin cells
Since there was a difference in the wound healing capabilities between mouse models, we next examined the homeostatic levels of Cx43 expression in the epidermis of these mice by immunohistochemistry. Cx43MAPK mice had a significantly lower level of Cx43 expression compared to Cx43WT, Cx43PKC or Cx43CK1 (Fig. 3A,B). Given a previous publication correlating reduced Cx43 expression with increased keratinocyte proliferation during wound healing (Kretz et al., 2003), we looked at homeostatic proliferation levels in the epidermis of the Cx43 phospho-mutant mice. Despite a reduction in Cx43 expression in Cx43MAPK mice, we did not observe a difference in the proliferation marker Ki67 between Cx43MAPK and Cx43WT mice (Fig. 3C,D). However, Cx43PKC and Cx43CK1 mice had a significant decrease in Ki67 staining in the skin epidermis compared to both Cx43MAPK and Cx43WT mice. Intriguingly, although we observed more cells in the epidermal layer in Cx43CK1 mice, this was not due to increased proliferation. It is possible that epithelial cells in Cx43CK1 mice may be protected from apoptosis in a similar manner to astrocytes and cardiomyocytes in ischemic injury models (Slavi et al., 2018). This suggests that regardless of total Cx43 expression there are inherent physiological consequences in the epidermis when Cx43 is mutated at specific phosphorylation sites in vivo.
Fig. 3.

Cx43MAPK mice have reduced Cx43 expression and Cx43PKC mice have decreased proliferation in the epidermis. (A) Representative immunohistochemical images of Cx43 expression in the skin of Cx43WT, Cx43MAPK, Cx43PKC and Cx43CK1 mice. (B) Quantification of Cx43 expression by immunohistochemistry in the epidermal layer of mouse skin. n=8 (66) Cx43WT, 9 (44) Cx43MAPK, 10 (49) Cx43PKC, and 10 (51) Cx43CK1 mice (sites) per group. (C) Representative immunohistochemical images of Ki67 expression in the skin of Cx43WT, Cx43MAPK, Cx43PKC and Cx43CK1 mice. Right and left quad-panels are identical except the right is pseudocolored (red, Ki67+; blue, Ki67−) for visual clarity. (D) Quantification of Ki67 expression by immunohistochemistry in the epidermal layer of mouse skin. n=8 (50) Cx43WT, 9 (44) Cx43MAPK, 10 (50) Cx43PKC, and 10 (50) Cx43CK1 mice (sites) per group. Results are mean±s.e.m. *P<0.05, **P<0.005, ***P<0.0005 (one-way ANOVA). Scale bars: 100 µm.
Delayed wound closing in MDCK cells with CK1 and PKC phospho-block mutants is rescued by phospho-mimetic mutations in vitro
Having established that blocking phosphorylation at CK1 and PKC sites slows wound healing in vivo, we wanted to determine: (1) whether we could model this phenotype in vitro, and (2) whether we could reverse this phenotype by mimicking constitutive phosphorylation at these sites. To this end we used a Madin–Darby canine kidney (MDCK) cell line which is null for Cx43 expression (Jordan et al., 1999), which upon stable transfection expresses functional Cx43 (Dunn et al., 2012). We stably transfected the parental MDCK cells with WT Cx43 or Cx43 containing mutations at the MAPK, PKC or CK1 sites to mimic (mutation to aspartic or glutamic acid residues, indicated with a -D or -E suffix) or block (mutation to alanine residues, indicated with an -A suffix) phosphorylation. Mutant cell lines were cloned and selected based on membrane Cx43 expression. Cells were uniformly scratch wounded and followed for 48 h to determine the rate of cell migration and proliferation (Fig. 4A). Compared to WT, all phospho-block mutations caused a significant delay in wound closure (i.e. a wider wound width) with MAPK sites causing the biggest and PKC sites the smallest delay (Fig. 4B). Delays in wound closure in MAPK-A, PKC-A and CK1-A cells were predominantly due to defects in cell migration, as proliferation levels remained comparable to those of WT MDCK cells (Fig. 4C). In agreement, we have previously shown that treating human keratinocytes with the PKC inhibitor bisindolylmaleimide at the time of wounding, and thus preventing Cx43 phosphorylation at the PKC-specific S368 site, inhibited migration (Richards et al., 2004). Changing MAPK, PKC or CK1 sites to phospho-mimetic mutations increased the rate of cell migration in all three groups, although only the PKC site mutant restored migration to WT levels. The increase in cell migration was predominantly due to increased motility, as MAPK-D cells showed no differences in proliferation compared to MAPK-A or WT and both PKC-D and CK1-E cells actually had a decrease in cell proliferation. Interestingly, the mutation mimicking constitutive phosphorylation at S368 (PKC-D) on Cx43 decreased proliferation compared to all other genotypes except CK1-E, indicating the importance of the site in the cell cycle. We have previously shown that cells in the G0 phase of the cell cycle contain very little Cx43 that is phosphorylated S368 and but have this phosphorylation increased ∼7-fold during the transition to the S and G2/M phases (Solan et al., 2003). It is possible that this site acts as a molecular switch during phases of proliferation and warrants further investigation.
Fig. 4.
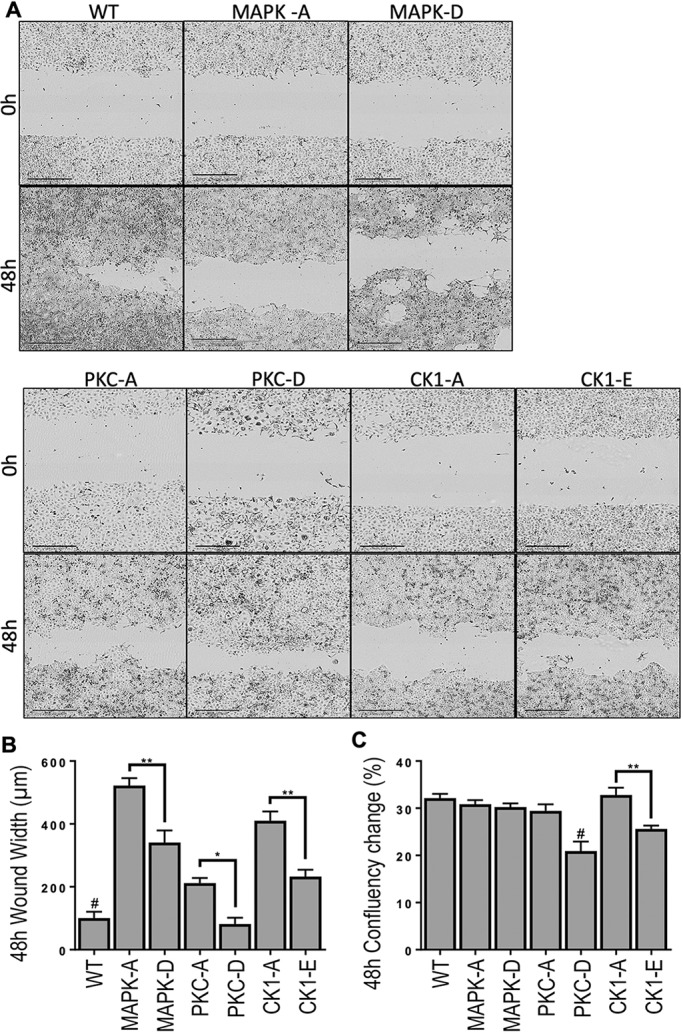
The wound closing delay in phospho-block mutants is rescued by phospho-mimetic mutations in vitro. MDCK cells transfected with WT Cx43 or mutant Cx43 [MAPK-A (S255A, S262A, S279A and S282A), MAPK-D (S255D, S262D, S279D and S282D), PKC-A (S368A), PKC-D (S368D), CK1-A (S325A, S328A and S330A) or CK1-E (S325E, S328E, S330E)]. (A) Representative images of scratched cells immediately and 48 h post scratch. Scale bars: 400 µm. (B) Cell migration into scratch was quantified after 48 h. n=62 (8) WT, 49 (4) MAPK-A, 51 (4) MAPK-D, 74 (4) PKC-A, 26 (4) PKC-D, 46 (3) CK1-A and 65 (4) CK1-E biological (experimental) replicates per cell line. Results are mean±s.e.m. *P<0.03, **P<0.005; #P<0.007 comparing WT to all groups except PKC-D (one-way ANOVA). (C) Overall cell proliferation was quantified 48 h after scratch wounding. n=180 (10) WT, 74 (6) MAPK-A, 100 (3) MAPK-D, 46 (4) PKC-A, 57 (5) PKC-D, 165 (10) CK1-A, and 197 (9) CK1-E biological (experimental) replicates per cell line. Results are mean ±s.e.m. **P<0.001; #P<0.01 comparing PKC-D to all groups except CK1-E (one-way ANOVA).
While we were able to phenotypically mimic the wound responses of Cx43PKC and Cx43CK1 mice in vitro, we observed differences in the effects of preventing phosphorylation on Cx43 at MAPK-specific sites in vivo versus in vitro. We can think of a few potential explanations for this discrepancy. We see a significant reduction in Cx43 expression in Cx43MAPK epidermal cell layers, whereas the MAPK-A and -D mutant Cx43 MDCK cells are selected for expression of Cx43, so the effects of reduced levels of Cx43 expression are lost in vitro. Thus, given the known benefit to healing rates for reduced levels of Cx43 (Montgomery et al., 2018), this could explain the difference. Another possibility is that the increased rate of wound healing might involve cell types (e.g. dermal or immune cells) other than keratinocytes since all of the Cx43 in the mice has mutations at these phosphorylation sites. For example, the Cx43MAPK mice wounds might close faster due to better wound contraction in the first several days due to mutant Cx43 in dermal cells.
Conclusions
Collectively, these findings indicate kinase-specific Cx43 phosphorylation events play discrete roles that influence the physiological process of wound healing. While a general role for Cx43 expression in wound repair is well established, mechanistic linkages between Cx43 phosphorylation and gap junction stability with cell behaviors, such as proliferation and motility, and downstream physiological consequences were previously unknown. Here, we began to connect these events through modulation of specific phosphorylation events on Cx43 in vitro and in vivo. We demonstrated that phosphorylation of Cx43 at PKC- and CK1-specific sites regulates epithelial migration, proliferation and epithelial thickness during wound healing. At least in vivo, preventing Cx43 phosphorylation at MAPK sites increased the rate of wound healing possibly due to the reduced expression of total Cx43 in the epidermis. These results support the critical role of phosphorylation in regulating Cx43 during wound repair and potentially could help create more targeted drugs to influence Cx43 behavior during wound healing.
MATERIALS AND METHODS
Mice
All mouse studies were conducted under Institutional Animal Care and Use Committee approval (FHCRC). All mice were on a C57BL/6J strain background, under 1 year of age at wounding and euthanasia, and male with the exception of one Cx43CK1 female mouse. Creation of the knock-in mouse lines has been previously described (Freitas-Andrade et al., 2019; Huang et al., 2011).
In vivo wounding
Mice were wounded as previously described (Sullivan et al., 2004). Personnel performing wounding procedures and measurements were blind to the genotype. Briefly, 2–8-month-old mice were anaesthetized, hairs were removed by use of depilatory cream and two 3.5 mm punch biopsies were performed on the back between the shoulder blades. At 14 days after wounding, sections of the skin containing the mostly healed wounds were fixed in formalin. Wounding studies were completed in two sets of experiments for a total of n=5 Cx43WT, n=8 Cx43MAPK, n=5 Cx43PKC and n=6 Cx43CK1 mice per group (two wounds per mouse).
Immunohistochemistry
Formalin-fixed tissues were embedded in paraffin and 5 µm sections were prepared. Standard methods for Masson's Trichrome staining were used, and hematoxylin and eosin (H&E) staining was completed via Tissue-Tek Prisma (Sakura, Torrance, CA) and sections with identifiable wounds were included in healed wound analyses. Using the automated Discovery ULTRA Staining Module (Roche Diagnostics, Indianapolis, IN), Cx43 was labeled with the rabbit anti-Cx43 C6219 antibody (Sigma-Aldrich, St Louis, MO; 1:500), and signal was detected using the Discovery anti-HQ, anti-HQ HRP and DAB system from Roche. Ki67 was labeled with Cell Signaling (Danvers, MA) antibody (12202, 1:1000) using the automated Leica Bond RX system and detected with anti-rabbit-IgG conjugated to HRP and DAB from Leica (Buffalo Grove, IL). Assessment of staining colocalization was performed using the OPAL seven-color fIHC kit (PerkinElmer, San Jose, CA) with the following antibodies and fluorophores: SMA (M0851, Dako, Santa Clara, CA; 1:600) and OPAL 520, F4/80 (70076S, Cell Signaling; 1:1000) and OPAL 520, Ly6G (127601, Biolegend, San Diego, CA; 1:2000) and OPAL 650, and Spectral DAPI FP1490 (PerkinElmer; 1:5000).
Inflammatory infiltrate in H&E-stained sections of healed wounds was graded in a blinded manner by a board-certified veterinary pathologist as 3 (low) when there were scant scattered inflammatory cells, 2 (moderate) when few clusters of cells are present and high (1) when there was extensive inflammation that extended up to deep subcutis in some cases (Cx43WT=4, Cx43MAPK=6, Cx43PKC=5, Cx43CK1=5 mice per group). Early (light blue) and mature (dark blue) collagen from Masson's Trichrome staining was digitally defined and quantified in whole-tissue sections for normal skin (Cx43WT=8, Cx43MAPK=9, Cx43PKC=10, Cx43CK1=10 mice per group) and defined as healed wound areas in wounded skin (Cx43WT=4, Cx43MAPK=6, Cx43PKC=5, Cx43CK1=5 mice per group). Orientation of collagen (vertical-1, mixed-2, or horizontal-3) in reference to the epidermis was determined based on Masson's Trichrome-stained sections in healed wounds. The pattern of collagen in healed wounds was determined as: (1) fine fibrils of collagen, (3) fascicle, when they organized to form bundles, or (2) mixed. Healed wound epidermis measurements were made with the ruler feature in Aperio ImageScope (Leica Biosystems) on Masson's Trichrome-stained sections (Cx43WT=4, Cx43MAPK=6, Cx43PKC=5, Cx43CK1=5 mice per group) approximately every 100 µm across the length of the healed wound. The same ruler feature was used to measure the epidermal layer of normal skin with Masson's Trichrome staining in ten sections per slide where the full length of hair follicles was present (Cx43WT=4, Cx43MAPK=5, Cx43PKC=5, Cx43CK1=6 mice per group). Cx43 and Ki67 (Cx43WT=8, Cx43MAPK=9, Cx43PKC=10, Cx43CK1=10 mice per group) expression in mouse skin epidermis was measured in five or more 500-µm-wide tissue sections per slide. Cx43 expression was quantified through DAB staining in skin epidermis with Halo imaging analysis software (Indica Labs, Albuquerque, NM). Ki67 staining was quantified per nucleus in the epidermal layer and the resulting positive or negative staining pseudocolored red or blue using HALO software. All quantification was completed by investigators blinded to mouse genotype.
Cell line maintenance and transfection
Serine-to-alanine or serine-to-aspartic acid or -glutamic acid mutations at S368 (PKC), S325, S328 and S330 (CK1) and S255, S262, S279 and S282 (MAPK) were made using the GeneTailor site-directed mutagenesis system (Invitrogen) on full-length Cx43 that had been cloned into the mammalian expression vector pIREShyg (Takara, Mountain View, CA). MDCK.2 cells obtained from the ATCC (CRL-2936) were tested for mycoplasma contamination and cultured in DME (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and antibiotics in a humidified 5% CO2 environment. Wild-type and mutant Cx43 were electroporated into the MDCK cell lines via a Nucleofector apparatus (Lonza Bioscience). Stably transfected clones were selected with hygromycin (200 μg/ml). Multiple individual clones were used for WT, MAPK-D, PKC-D, CK1-A and CK1-E cells experiments. Owing to long-term genetic instability, MAPK-A and PKC-A lines were established via bulk culture and all experiments were completed within 5 weeks after selection.
In vitro scratch wounding
MDKC cells were seeded at a density of 3200 cells in each well of a 96-well plate. After 24 h in culture, scratch wounds were made using the WoundMaker™ (Essen Biosciences) device and after washing, 100 µl of serum-free OPTI-MEM (Invitrogen) was added to each well. Cells were imaged in the IncuCyte™ live-cell imaging system for 48 h to monitor cell migration and proliferation. Only wounds with a starting width between 600 and 850 μM were included in the analysis with IncuCyte Cell Migration and Invasion software (WT=62, MAPK-A=49, MAPK-D=51, PKC-A=74, PKC-D=26, CK1-A=46, CK1-E=65). Cell proliferation was quantified using IncuCyte Confluency Processing software and only wells with a starting confluency of 20-50% were included in the analysis (WT=180, MAPK-A=74, MAPK-D=100, PKC-A=46, PKC-D=57, CK1-A=165, CK1-E=197).
Statistical analyses
All in vivo comparisons used one-way ANOVA. Student's two-tailed Welch's t-test was used to compare wound width between serine-to-alanine or serine-to-aspartic acid or -glutamic acid at the same sites in the MDCK cell line.
Supplementary Material
Acknowledgements
We thank Smitha P.S. Pillai, a board-certified comparative pathologist, for analyzing collagen orientation and pattern, the scoring of immune infiltration and thoughtful discussion.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.J.L., C.A.D., J.L.S., P.D.L.; Methodology: K.J.L., P.D.L.; Formal analysis: K.J.L., J.L.S., P.D.L.; Investigation: K.J.L., C.A.D., J.L.S.; Writing - original draft: K.J.L., P.D.L.; Writing - review & editing: K.J.L., P.D.L.; Supervision: P.D.L.; Funding acquisition: P.D.L.
Funding
This work was supported by grants from the National Institutes of Health (GM055632 to P.D.L. and a Cancer Center Support grant P30 CA015704). The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.234633.supplemental
References
- Axelsen L. N., Stahlhut M., Mohammed S., Larsen B. D., Nielsen M. S., Holstein-Rathlou N.-H., Andersen S., Jensen O. N., Hennan J. K. and Kjølbye A. L. (2006). Identification of ischemia-regulated phosphorylation sites in connexin43: a possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J. Mol. Cell. Cardiol. 40, 790-798. 10.1016/j.yjmcc.2006.03.005 [DOI] [PubMed] [Google Scholar]
- Becker D. L., Thrasivoulou C. and Phillips A. R. J. (2012). Connexins in wound healing; perspectives in diabetic patients. Biochim. Biophys. Acta 1818, 2068-2075. 10.1016/j.bbamem.2011.11.017 [DOI] [PubMed] [Google Scholar]
- Cogliati B., Vinken M., Silva T. C., Araújo C. M. M., Aloia T. P. A., Chaible L. M., Mori C. M. C. and Dagli M. L. Z. (2015). Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 79, 50-56. 10.1016/j.jdermsci.2015.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper C. D. and Lampe P. D. (2002). Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 277, 44962-44968. 10.1074/jbc.M209427200 [DOI] [PubMed] [Google Scholar]
- Cottrell G. T., Lin R., Warn-Cramer B. J., Lau A. F. and Burt J. M. (2003). Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 284, C511-C520. 10.1152/ajpcell.00214.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P., Qiu C., Frank S., Tamber K. and Becker D (2003). Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol. Int. 27, 525-541. 10.1016/S1065-6995(03)00077-5 [DOI] [PubMed] [Google Scholar]
- Dunn C. A. and Lampe P. D. (2014). Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 127, 455-464. 10.1242/jcs.142497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn C. A., Su V., Lau A. F. and Lampe P. D. (2012). Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 287, 2600-2607. 10.1074/jbc.M111.276261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas-Andrade M., Wang N., Bechberger J. F., De Bock M., Lampe P. D., Leybaert L. and Naus C. C. (2019). Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 216, 916-935. 10.1084/jem.20171452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatnekar G. S., O'Quinn M. P., Jourdan L. J., Gurjarpadhye A. A., Draughn R. L. and Gourdie R. G. (2009). Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 4, 205-223. 10.2217/17460751.4.2.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough D. A., Goliger J. A. and Paul D. L. (1996). Connexins, connexons, and intercellular communication. Annu. Rev. Biochem. 65, 475-502. 10.1146/annurev.bi.65.070196.002355 [DOI] [PubMed] [Google Scholar]
- Huang G.-Y., Xie L.-J., Linask K. L., Zhang C., Zhao X.-Q., Yang Y., Zhou G.-M., Wu Y.-J., Marquez-Rosado L., McElhinney D. B. et al. (2011). Evaluating the role of connexin43 in congenital heart disease: screening for mutations in patients with outflow tract anomalies and the analysis of knock-in mouse models. J. Cardiovasc. Dis. Res. 2, 206-212. 10.4103/0975-3583.89804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone S. R., Kroncke B. M., Straub A. C., Best A. K., Dunn C. A., Mitchell L. A., Peskova Y., Nakamoto R. K., Koval M., Lo C. W. et al. (2012). MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ. Res. 111, 201-211. 10.1161/CIRCRESAHA.112.272302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K., Solan J. L., Dominguez M., Sia M., Hand A., Lampe P. D. and Laird D. W. (1999). Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol. Biol. Cell 10, 2033-2050. 10.1091/mbc.10.6.2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz M., Euwens C., Hombach S., Eckardt D., Teubner B., Traub O., Willecke K. and Ott T (2003). Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J. Cell Sci. 116, 3443-3452. 10.1242/jcs.00638 [DOI] [PubMed] [Google Scholar]
- Laird D. W. and Lampe P. D. (2018). Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 17, 905-921. 10.1038/nrd.2018.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., Nguyen B. P., Gil S., Usui M., Olerud J., Takada Y. and Carter W. G. (1998). Cellular interaction of integrin α3β1 with laminin 5 promotes gap junctional communication. J. Cell Biol. 143, 1735-1747. 10.1083/jcb.143.6.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., TenBroek E. M., Burt J. M., Kurata W. E., Johnson R. G. and Lau A. F. (2000). Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149, 1503-1512. 10.1083/jcb.149.7.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., Cooper C. D., King T. J. and Burt J. M. (2006). Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 119, 3435-3442. 10.1242/jcs.03089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limandjaja G. C., van den Broek L. J., Waaijman T., van Veen H. A., Everts V., Monstrey S., Scheper R. J., Niessen F. B. and Gibbs S (2017). Increased epidermal thickness and abnormal epidermal differentiation in keloid scars. Br. J. Dermatol. 176, 116-126. 10.1111/bjd.14844 [DOI] [PubMed] [Google Scholar]
- Lin R., Warn-Cramer B. J., Kurata W. E. and Lau A. F. (2001). v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J. Cell Biol. 154, 815-827. 10.1083/jcb.200102027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K., Ghanem A., Kim J.-S., Saathoff M., Urschel S., Kirfel G., Grümmer R., Kretz M., Lewalter T., Tiemann K. et al. (2004). Defective epidermal barrier in neonatal mice lacking the C-terminal region of Connexin43. Mol. Biol. Cell 15, 4597-4608. 10.1091/mbc.e04-04-0324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery J., Ghatnekar G. S., Grek C. L., Moyer K. E. and Gourdie R. G. (2018). Connexin 43-based therapeutics for dermal wound healing. Int. J. Mol. Sci. 19, 1778 10.3390/ijms19061778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori R., Power K. T., Wang C. M., Martin P. and Becker D. L. (2006). Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 119, 5193-5203. 10.1242/jcs.03320 [DOI] [PubMed] [Google Scholar]
- Nakano Y., Oyamada M., Dai P., Nakagami T., Kinoshita S. and Takamatsu T (2008). Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Invest. Ophthalmol. Vis. Sci. 49, 93-104. 10.1167/iovs.07-0255 [DOI] [PubMed] [Google Scholar]
- Pollok S., Pfeiffer A.-C., Lobmann R., Wright C. S., Moll I., Martin P. E. M. and Brandner J. M. (2011). Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J. Cell. Mol. Med. 15, 861-873. 10.1111/j.1582-4934.2010.01057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C., Coutinho P., Frank S., Franke S., Law L., Martin P., Green C. R. and Becker D. L. (2003). Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 13, 1697-1703. 10.1016/j.cub.2003.09.007 [DOI] [PubMed] [Google Scholar]
- Reaume A. G., de Sousa P. A., Kulkarni S., Langille B. L., Zhu D., Davies T. C., Juneja S. C., Kidder G. M. and Rossant J (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831-1834. 10.1126/science.7892609 [DOI] [PubMed] [Google Scholar]
- Remo B. F., Qu J., Volpicelli F. M., Giovannone S., Shin D., Lader J., Liu F.-Y., Zhang J., Lent D. S., Morley G. E. et al. (2011). Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 108, 1459-1466. 10.1161/CIRCRESAHA.111.244046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards T. S., Dunn C. A., Carter W. G., Usui M. L., Olerud J. E. and Lampe P. D. (2004). Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 167, 555-562. 10.1083/jcb.200404142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáez J. C., Nairn A. C., Czernik A. J., Fishman G. I., Spray D. C. and Hertzberg E. L. (1997). Phosphorylation of Connexin43 and the regulation of neonatal rat cardiac myocyte gap junctions. J. Mol. Cell. Cardiol. 29, 2131-2145. 10.1006/jmcc.1997.0447 [DOI] [PubMed] [Google Scholar]
- Slavi N., Toychiev A. H., Kosmidis S., Ackert J., Bloomfield S. A., Wulff H., Viswanathan S., Lampe P. D. and Srinivas M (2018). Suppression of connexin 43 phosphorylation promotes astrocyte survival and vascular regeneration in proliferative retinopathy. Proc. Natl. Acad. Sci. USA 115, E5934-E5943. 10.1073/pnas.1803907115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2009). Connexin 43 phosphorylation–structural changes and biological effects. Biochem. J. 419, 261-272. 10.1042/BJ20082319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Fry M. D., TenBroek E. M. and Lampe P. D. (2003). Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 116, 2203-2211. 10.1242/jcs.00428 [DOI] [PubMed] [Google Scholar]
- Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R. and Lampe P. D. (2007). Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 179, 1301-1309. 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Márquez-Rosado L. and Lampe P. D. (2019). Cx43 phosphorylation mediated effects on ERK and Akt protect against ischemia reperfusion injury and alter stability of stress-inducible protein NDRG1. J. Biol. Chem. 294, 11762-11771. 10.1074/jbc.RA119.009162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan S. R., Underwood R. A., Gibran N. S., Sigle R. O., Usui M. L., Carter W. G. and Olerud J. E. (2004). Validation of a model for the study of multiple wounds in the diabetic mouse (db/db). Plast. Reconstr. Surg. 113, 953-960. 10.1097/01.PRS.0000105044.03230.F4 [DOI] [PubMed] [Google Scholar]
- TenBroek E. M., Lampe P. D., Solan J. L., Reynhout J. K. and Johnson R. G. (2001). Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 155, 1307-1318. 10.1083/jcb.200102017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreeburg M., de Zwart-Storm E. A., Schouten M. I., Nellen R. G. L., Marcus-Soekarman D., Devies M., van Geel M. and van Steensel M. A. M. (2007). Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am. J. Med. Genet. A. 143, 360-363. 10.1002/ajmg.a.31558 [DOI] [PubMed] [Google Scholar]
- Warn-Cramer B. J., Lampe P. D., Kurata W. E., Kanemitsu M. Y., Loo L. W. M., Eckhart W. and Lau A. F. (1996). Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J. Biol. Chem. 271, 3779-3786. 10.1074/jbc.271.7.3779 [DOI] [PubMed] [Google Scholar]
- Warn-Cramer B. J., Cottrell G. T., Burt J. M. and Lau A. F. (1998). Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 273, 9188-9196. 10.1074/jbc.273.15.9188 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.