

 This work is licensed under a
This work is licensed under a Summary
A 26-year-old woman presented with persistent headache and tiredness. Biological investigations disclosed a moderate inflammatory syndrome, low PTH-hypercalcemia and complete anterior hypopituitarism. A magnetic resonance imaging (MRI) of the pituitary gland was performed and revealed a symmetric enlargement with a heterogeneous signal. Ophthalmological examination showed an asymptomatic bilateral anterior and posterior uveitis, and a diagnosis of pituitary sarcoidosis was suspected. As the localization of lymphadenopathies on the fused whole-body FDG-PET/computerized tomography (CT) was not evoking a sarcoidosis in first instance, an excisional biopsy of a left supraclavicular adenopathy was performed showing classic nodular sclerosis Hodgkin’s lymphoma (HL). A diagnostic transsphenoidal biopsy of the pituitary gland was proposed for accurate staging of the HL and surprisingly revealed typical granulomatous inflammation secondary to sarcoidosis, leading to the diagnosis of a sarcoidosis–lymphoma syndrome. The co-existence of these diseases constitutes a diagnostic challenge and we emphasize the necessity of exact staging of disease in order to prescribe adequate treatment.
Learning points:
The possibility of a sarcoidosis–lymphoma syndrome, although rare, should be kept in mind during evaluation for lymphadenopathies.
In the case of such association, lymphoma usually occurs after sarcoidosis. However, sarcoidosis and lymphoma can be detected simultaneously and development of sarcoidosis in a patient with previous lymphoma has also been reported.
An accurate diagnosis of the disease and the respective organ involvements, including biopsy, is necessary in order to prescribe adequate treatment.
Patient Demographics: Adolescent/young adult, Female, White, Belgium
Clinical Overview: Pituitary, Pituitary, PTH, FSH, ACTH, Cortisol, IGF1, LH, Thyroxine (T4), TSH, Hypopituitarism, Hypercalcaemia, Lymphadenopathy
Diagnosis and Treatment: Headache, Fatigue, Hypercalcaemia, Hypopituitarism, Lymphadenitis, Sarcoidosis, CT scan, MRI, PET scan, PTH, C-reactive protein, 25-hydroxyvitamin-D3, X-ray, Cortisol (9am), ACTH, FT4, FSH, LH, IGF1, TSH, Histopathology, Haematoxylin and eosin staining, Immunohistochemistry, Chemotherapy, Hydrocortisone, Glucocorticoids, Levothyroxine, Dacarbazine, Methylprednisolone
Related Disciplines: General practice
Publication Details: New disease or syndrome: presentations/diagnosis/management, September, 2019
Background
Sarcoidosis is a systemic disease of unknown etiology characterized by non-caseified granulomatous reaction that can involve multiple organs. It is more frequent in young and middle-aged adults and typically presents with pulmonary infiltrates, bilateral hilar and mediastinal lymphadenopathy, uveitis, and more rarely affects other organs, including the hypothalamic–pituitary axis.
Malignancy rates in sarcoidosis patients have been reported as 1.2–2.5% (1, 2), chronic inflammation being a risk factor. The risk of hematologic malignancy is especially higher among these patients, so that the existence of a specific sarcoidosis-lymphoma syndrome has been suggested.
Our paper reports one such rare case of co-occurrence of pituitary sarcoidosis and Hodgkin’s lymphoma (HL).
Case presentation
We report the case of a 26-year-old Caucasian female patient with no significant medical history except obesity. Her only medication was dydrogesterone prescribed 1 year before for irregular cycles. The patient had been well until approximately 10 days before her first evaluation, when persistent headache and tiredness developed.
Investigation
The first laboratory analyses revealed a moderately increased C-reactive protein level (57.3 mg/L; normal range (NR) <5.0 mg/L) with no increased leukocytosis, mild hypercalcemia (2.60 mmol/L; NR: 2.15–2.50) with low PTH (19 pg/ml; NR 15–80), normal phosphate concentration (1.26 mmol/L; NR 0.81–1.45), low 25(OH)vitamin D3 levels (14 ng/mL; NR 30–100) and deficiency of the thyreotrope axis (Table 1). She was referred to our hospital for further investigation. On examination, the vital signs were normal. There were no compelling features to suggest systemic infection, no hepatosplenomegaly and no clinically palpable lymph node. She did not present any polyuria-polydipsia syndrome, nor had any biological sign of diabetes insipidus. The hormonal profile confirmed anterior hypopituitarism with deficiencies of the thyreotrope, corticotrope and gonadotrope axes but normal prolactin levels (Table 1). A hormone replacement therapy with hydrocortisone acetate (30 mg/day) and levothyroxine (50 µg/day) was introduced. A brain MRI was performed, revealing a symmetric enlargement of the pituitary gland with a heterogeneous signal (Fig. 1). Ophthalmological examination showed an asymptomatic bilateral anterior and posterior uveitis. An extensive workup allowed excluding infection with Mycobacterium tuberculosis, human immunodeficiency virus, toxoplasma and bartonella. Syphilitic serology was also negative. The angiotensin-converting enzyme was slightly elevated (45 UECA, NR: <42). A chest X ray showed mediastinal enlargement with left hilar lymphadenopathy. As sarcoidosis became our first diagnostic hypothesis, a 18-fluorodeoxyglucose positron emission tomography (FDG-PET) fused with a CT scan was performed and showed large hypermetabolic left unilateral hilar and mediastinal lymphadenopathies, left supraclavicular and axillar adenopathies and an enhanced signal in the pituitary gland (Fig. 2). As the non-symmetrical distribution of lymphadenopathies was not typical for sarcoidosis, an excisional biopsy of the left supraclavicular adenopathy was performed. Pathological examination of the operative specimen showed classic nodular sclerosis HL (Fig. 3). A bone marrow biopsy revealed no infiltration. A diagnostic transsphenoidal biopsy of the pituitary gland was proposed for staging of the HL. Surprisingly, pituitary biopsy specimens revealed inflammation with non-caseating granuloma formation which was pathognomonic of sarcoidosis (Fig. 4). A puncture of the anterior chamber was also performed for measurements of IL-10 concentration which was low (1.01 pg/mL) and IL-6 concentration which was high (199.5 pg/mL). These levels were consistent with uveitis of non-neoplastic etiology. According to these results, the HL was staged as IIAa and our final diagnostic was a sarcoidosis-lymphoma syndrome.
Table 1.
Main biological and hormonal data at the first evaluation of the patient and normal values.
| Parameters | Patient’s value | Normal range |
|---|---|---|
| CRP (mg/L) | 57.3 | <5.0 |
| Calcemia (mmol/L) | 2.60 | 2.15–2.60 |
| PTH (ng/L) | 19 | 15–80 |
| Morning cortisol (nmol/L) | 94.7 | 130–500 |
| Morning ACTH (ng/L) | 16.4 | 5.0–49 |
| TSH (mU/L) | 0.20 | 0.27–4.20 |
| FT4 (pmol/L) | 3.9 | 12.0–22.0 |
| FT3 (pmol/L) | 4.0 | 3.1–6.8 |
| Prolactin (μg/L) | 14.9 | 5.8–23.0 |
| IGF-1 (μg/L) | 127.7 | 131–320 |
| LH (IU/L) | 0.6 | 2.4–12.6 |
| FSH (IU/L) | 1.4 | 3.5–12.5 |
Figure 1.

(A, B and C). Coronal T1-weighted view of the brain MRI (panel A) showing a symmetric enlargement of the pituitary gland with a marked and heterogeneous signal enhancement after gadolinium injection (panel B). Coronal T2-weighted view of the pituitary lesion is shown on panel C.
Figure 2.

(A, B and C). Fused FDG-PET/CT scan showing enhanced metabolic signal at the level of the pituitary gland (arrow, panel A), as well as large metabolic unilateral hilar and mediastinal lymphadenopathies (panel B), as well as left supraclavicular and axillar adenopathies (arrows, panel C)
Figure 3.
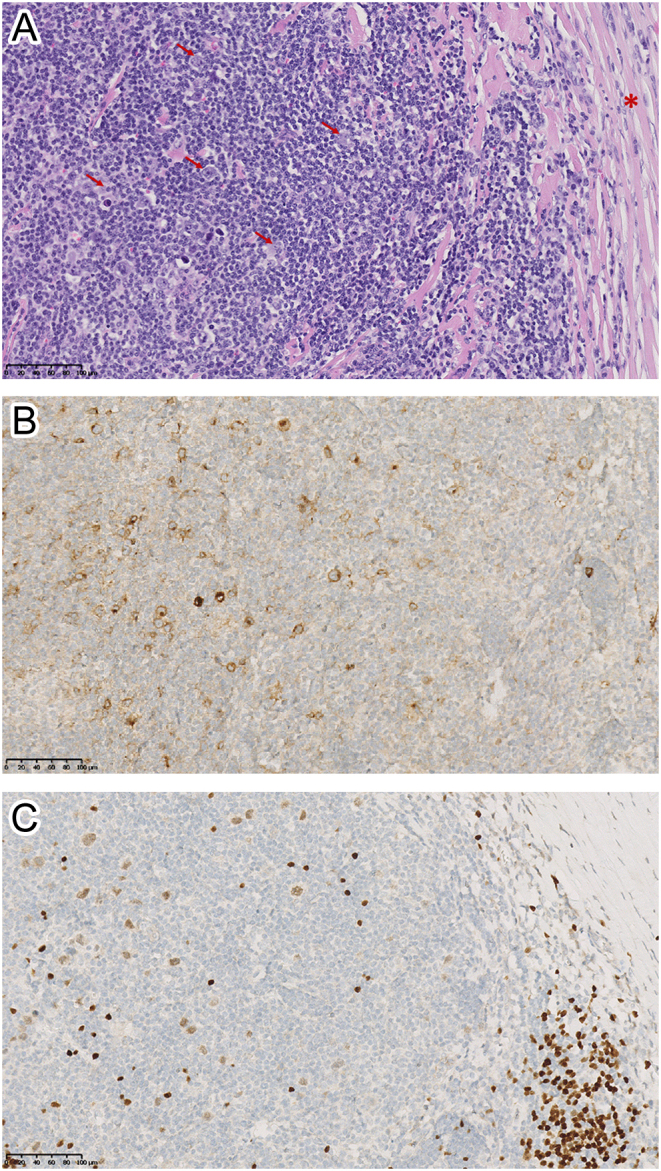
(A, B and C). Pathological examination of the supraclavicular lymph node biopsy specimen. Panel A (magnification ×20): hematoxylin-eosin staining showing a lymph node structure distorted by fibrotic bands (red asterisk) surrounding nodular reactive cell infiltrate composed of variable proportions of lymphocytes, histiocytes, eosinophils, and plasma cells and containing scattered large, malignant lymphoid cells. The abnormal large cells include bi-nucleated Reed-Sternberg cells (indicated by the red arrows), mononucleated Hodgkin cells and mummified cells. Panels B and C (magnification ×20): immunohistochemistry staining showing that the malignant lymphoid cells expressed CD30, a marker for HL (panel B), and PAX5, a transcription factor specific of B-cell lineage (panel C).
Figure 4.
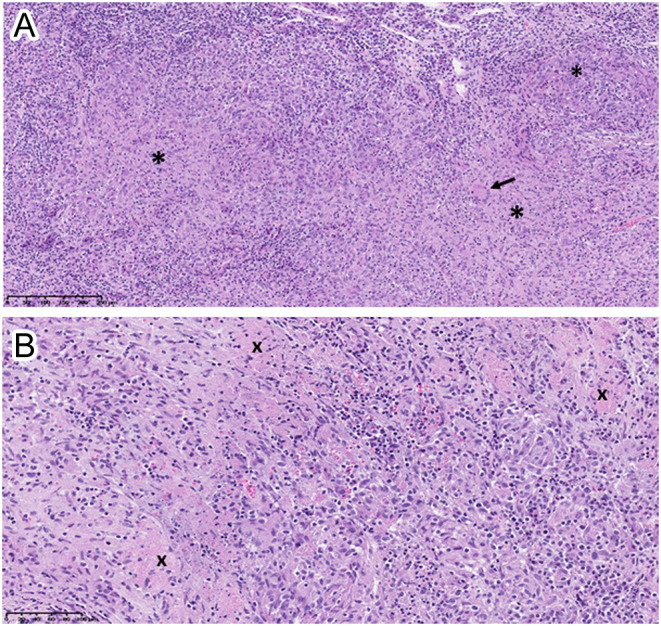
(A and B). Pathological examination of the pituitary biopsy specimen. Panel A (magnification ×10): hematoxylin and eosin staining showing diffuse lymphocytic inflammation with granuloma formation (indicated by the black asterisks). A multinucleated giant cell is also seen (arrow). Panel B (magnification x20): typical granulomatous inflammation with scattered areas of non-caseating necrosis (indicated by the black crosses). Normal pituitary tissue is not identified in the specimen and there are no histological signs of lymphoma.
Treatment
After ovarian tissue cryopreservation, the patient was treated by ABVD chemotherapy (doxorubicin, bleomycin, vinblastine and dacarbazine), followed by high doses of oral methylprednisolone.
Outcome and follow-up
No residual hypermetabolic lesion (Deauville score of 1) was observed on the intermediate PET/CT performed 2 months later, after two cycles of ABVD. Tolerance to treatment was satisfactory except for some digestive disorders in the immediate course of the treatment. Given this, four additional cycles of ABVD will be delivered.
Discussion
To our knowledge, this is the first reported case of co-occurrence of HL and proven pituitary sarcoidosis. On the basis of low PTH-hypercalcemia, bilateral uveitis, the pituitary MRI aspect (symmetric enlargement with heterogeneous contrast enhancement) and hilar lymphadenopathies detected on fused FDG-PET/CT, a diagnosis of systemic sarcoidosis was first considered. Sarcoidosis is a multisystem disease of unknown origin characterized by non-caseified granulomas and lymphoid hyperplasia resulting in the dysfunction of the immunoregulatory pathways. The disease usually presents as pulmonary infiltrates with bilateral and symmetric hilar and mediastinal lymphadenopathies, and may affect other organs including the eyes (uveitis), the central or peripheral nervous system (neurosarcoidosis) and, less commonly, the hypothalamic–pituitary (HP) axis (granulomatous inflammation). This last involvement may result in irreversible pituitary hormone deficiencies. In a recent review by Anthony et al. of 46 patients with HP sarcoidosis (3), central hypogonadism occurred most frequently (89%), followed by deficiency of the thyreotrope axis (67%) and diabetes insipidus (66%). Extraneurologic manifestations were also reported in the majority of these patients (80 %), with pulmonary, sinus and ocular locations being the most frequent. Our patient indeed suffered from complete anterior hypopituitarism, but had no clinical manifestations or biological signs of diabetes insipidus.
Furthermore, in our patient, the localization and asymmetry of lymphadenopathies was not typical of sarcoidosis. Excisional biopsy of a left supraclavicular adenopathy was therefore performed to exclude another diagnosis and indeed showed typical pathological features of nodular sclerosis HL.
HL is a hematolymphoid neoplasm, primarily of B cell lineage and typically affecting cervical, mediastinal and/or abdominal lymph nodes. Extranodal sites of the disease may be observed in about 10% of the cases and this presentation may be accompanied by general symptoms, including fever, night sweats, and weight loss in association with lymphoma called B symptoms. Central nervous system (CNS) involvement remains however very uncommon in HL being reported with low prevalence of 0.2–0.5% (4). In a recent review, prevalence of CNS-HL was reported to be even lower (0.02%) in patients with systemic HL (5). CNS-HL occurs more frequently in recurrent disease or at initial diagnosis in immunocompromised patients, but may also be seen at initial diagnosis in immunocompetent patients (6). The mechanisms of CNS involvement are either by skull contiguity, meningeal invasion or via hematogenous route. In a review of 36 patients with CNS-HL, the most frequent sites of involvement were brain parenchyma (64%) and dura or leptomeninges (19%), while the pituitary gland was affected in only one single case (7).
A baseline whole body FDG-PET is recommended for HL (and NHL) staging according to the 2014 Lugano classification (8). Given the physiologic avidity of the normal brain, for FDG, the CNS should be however evaluated by brain MRI and lumbar puncture. As involvement of the CNS is very uncommon in HL, any lesion in the brain of a patient known for HL should be investigated by biopsy for confirmation and to rule out a second disease process, as it was the case in our report (9). A pituitary biopsy was indeed performed and pathology confirmed our first diagnosis of pituitary sarcoidosis.
Of note, uveitis may occur both in association with systemic inflammatory diseases such as sarcoidosis and with neoplastic masquerade syndromes including lymphomas, although usually of the non-Hodgkin type (10). As the eye derives embryologically and anatomically from the brain, ocular lymphoma may be considered as another site of CNS-LH involvement (11). Ocular pathology assessment may require ocular biopsy. However, the most limiting factor is specimen acquisition and these paucicellular specimens with fragile population of lymphocytes can lead to misdiagnosis. Interleukin-10 (IL-10) is a growth factor and differentiation factor for B-lymphocytes, while IL-6 is a multifunctional cytokine playing a role in host inflammatory defense mechanisms. Ocular lymphomas are therefore often associated with elevated levels of IL-10 relative to levels of IL-6 in the vitreous (12). This ratio was very low in the anterior chamber liquid of our patient, thus consistent with uveitis of inflammatory and non-neoplastic origin.
An association of sarcoidosis and lymphoproliferative disease has previously been reported as the sarcoidosis-lymphoma syndrome. It was first described by Brinker in 1986 (13), who noticed that patients who have a chronic active form of sarcoidosis have a 5-fold higher risk of developing lymphoma. His findings were supported by a population-based case–control study which showed an increased risk of HL of 14.1% in patients with sarcoidosis. Sarcoidosis and lymphoma may be detected simultaneously, but usually lymphoma occurs after sarcoidosis with a median interval of 24 months (14). However, the development of sarcoidosis in patients with lymphoma has also been reported. In a series of 39 patients presenting with sarcoidosis following lymphoproliferative disease, the median delay between lymphoma and sarcoidosis was 18 months and patients frequently had a mild form of sarcoidosis (15). Therefore sarcoidosis reaction must be included in the differential diagnosis of lymphoma recurrence.
There are many immunological similarities between sarcoidosis and lymphoma. Both have cutaneous anergy, peripheral lymphadenopathies and infiltration of T helper cells in tissues. The pathogenesis of this co-occurrence may be explained by the inflammatory response in sarcoidosis increasing mitotic activity in lymphocytes and the greater risk of lymphocytes undergoing mutations. Another hypothesis is that some immunological abnormalities seen in sarcoidosis may predispose to lymphoproliferative disorders (16).
In our case, the co-existence of both diseases at first evaluation constituted a diagnostic challenge. It is not possible to determinate which entity was the first and drove the other one. The suspicion of sarcoidosis led to the diagnosis of HL and staging of the malignancy led to the diagnosis of pituitary sarcoidosis. Since clinical manifestations of both diseases can be quite similar and both rarely affect the pituitary gland (although more frequently for sarcoidosis), differentiating between these two conditions is mandatory for prognosis and treatment and should prompt a pituitary biopsy.
In conclusion, the pituitary sarcoidosis-lymphoma syndrome, although rare, should be kept in mind. We emphasize the pitfalls and necessity of accurate diagnosis of the disease and the respective organ involvement in order to prescribe adequate treatment.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Informed consent was obtained from the patient for publication of this article and images.
Author contribution statement
C Delcourt wrote the case report. R Furnica and D Maiter reviewed and corrected the manuscript. A Camboni prepared and commented on the pathological images. H Yildiz, E Van den Neste, V Roelants, A Kozyreff and J P Thissen contributed significantly to the diagnostic work-up of the patient.
References
- 1.Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clinics in Dermatology 2007. 326–333. ( 10.1016/j.clindermatol.2007.03.010) [DOI] [PubMed] [Google Scholar]
- 2.Ji J, Shu X, Li X, Sundquist K, Sundquist J, Hemminki K. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Annals of Oncology 2009. 1121–1126. ( 10.1093/annonc/mdn767) [DOI] [PubMed] [Google Scholar]
- 3.Anthony J, Esper GJ, Ioachimescu A. Hypothalamic pituitary sarcoidosis with vision loss and hypopituitarism: case series and literature review. Pituitary 2016. 19–29. ( 10.1007/s11102-015-0678-x) [DOI] [PubMed] [Google Scholar]
- 4.Himiz K, Foyle A, Wilke D, Burrell S, Brownstone R, Ago C, Pahil R, Couban S. Intracranial presentation of systemic Hodgkin’s disease. Leukemia and Lymphoma 2004. 1667–1671. ( 10.1080/10428190410001673409) [DOI] [PubMed] [Google Scholar]
- 5.Guermazi A, Brice P, de Kerviler E, Fermé C, Hennequin C, Meignin V, Frija J. Extranodal Hodgkin disease: spectrum of disease. RadioGraphics 2001. 161–179. ( 10.1148/radiographics.21.1.g01ja02161) [DOI] [PubMed] [Google Scholar]
- 6.Gerstner ER, Abrey LE, Schiff D, Ferreri AJM, Lister A, Montoto S, Tsanq R, Thiel E, Grasu F, Behringer D, et al. CNS Hodgkin lymphoma. Blood 2008. 1658–1661. ( 10.1182/blood-2008-04-151563) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Re D, Fuchs M, Schober T, Engert A, Diehl V. CNS involvment in Hodgkin’s Lymphoma. Journal of Clinical Oncology 2007. 3182 ( 10.1200/JCO.2007.12.5088) [DOI] [PubMed] [Google Scholar]
- 8.Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, Lister TA, Alliance, Australasian Leukemia and Lymphoma Group; Eastern Cooperative Oncology Group; European Mantle Cell Lymphoma Consortium, et al Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodkin lymphoma: the Lugano classification. Journal of Clinical Oncology 2014. 3059–3068. ( 10.1200/JCO.2013.54.8800) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anselmo AP, Proia A, Cartoni C, Baroni CD, Maurizi Enrici RM, Delfini R, Avvisati G. Meningeal localization in a patient with Hodgkin’s disease: description of a case and review of the literature. Annals of Oncology 1996. 1071–1075. ( 10.1093/oxfordjournals.annonc.a010501) [DOI] [PubMed] [Google Scholar]
- 10.Grange LK, Kouchouk A, Dalal MD, Vitale S, Nussenblatt RB, Chan CC, Sen HN. Neoplastic masquerade syndromes in patients with uveitis. American Journal of Ophthalmology 2014. 526–531. ( 10.1016/j.ajo.2013.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitcup SM, Stark-Vancs V, Wittes RE, Solomon D, Podgor MJ, Nussenblatt RB, Chan CC. Association of interleukin 10 in the vitreous and cerebrospinal fluid and primary central nervous system lymphoma. Archives of Ophtalmology 1997. 1157–1160. ( 10.1001/archopht.1997.01100160327010) [DOI] [PubMed] [Google Scholar]
- 12.Park S, Adad S, Tulliez M, Monnet D, Merlat A, Gyan E, Bouscary D, Dreyfus F, Grimaldi D, Dhote R, et al. Pseudouveitis, a clue to the diagnosis of primary central nervous system lymphoma in immunocompetent patients. Medicine 2004. 223–232. ( 10.1097/01.md.0000134850.35118.46) [DOI] [PubMed] [Google Scholar]
- 13.Brincker H. The sarcoidosis-lymphoma syndrome. British Journal of Cancer 1986. 467–473. ( 10.1038/bjc.1986.199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maayan H, Ashkenazy Y, Nagler A, Izbocki G. Sarcoidosis and lymphoma: case series and literature review. Sarcoidosis Vasculitis and Diffuse Lung Diseases: Official Journal of WASOG 2011. 146–152. [PubMed] [Google Scholar]
- 15.London J, Grados A, Fermé C, Charmillon A, Maurier F, Deau B, Crickx E, Brice P, Chapelon-Abric C, Haioun C, et al. Sarcoidosis occurring after lymphoma report of 14 patients and review of the literature. Medicine 2014. e121 ( 10.1097/MD.0000000000000121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landgren O, Engels EA, Pfeiffer RM, Gridley G, Mellemkjaer L, Olsen JH, Kerstann KF, Wheeler W, Hemminki K, Linet MS, et al. Autoimmunity and susceptibility to Hodgkin lymphoma : a population-based case – control study in Scandinavia. Journal of the National Cancer Institute 2006. 1321–1330. ( 10.1093/jnci/djj361) [DOI] [PubMed] [Google Scholar]