

Abstract
Multiplex measurement of protein expression with the single-cell resolution has been challenging. Although a few conventional approaches including flow cytometry and immunofluorescence-based methods have been developed to detect proteins in individual cells, they are either dependent on bulky instrument or not multiplexed and high-throughput enough. Here we present a portable single-cell analysis system that is operable in a resource-limited environment. A stand-sit microchip housed in a clamp enables simple and instrument-free operation of all necessary steps, and the detection based on immunogold enhancement exonerates the reliance on fluorescence optics and electronics. The quantified sensitivity was found comparable to the conventional fluorescence approaches. We used this system to analyze five immune effector proteins and found the system is equally effective to detect those proteins in hundreds of single cells. Significant increase of cytokine protein production by THP1 monocytes was observed upon stimulation by lipopolysaccharide. Further study showed that a low-end imaging setup with low resolution can also detect signals without much loss of sensitivity. Taken together, this portable multiplex single-cell system may find broad biomedical applications in a field setting.
Keywords: microchip, point of care diagnosis, cytometry, single-cell analysis, multiplex detection
Graphical Abstract
Inflammation is the body’s response to damage to its tissues by pathogen infection, chemical stimulation or physical injury. Although this immune system activation process is normally suppressed by itself after certain duration, dysregulation often occurs and results in chronic inflammation. The dysregulated inflammation is closely associated with a wide array of systematic diseases such as allergy, atherosclerosis, cancer, Alzheimer’s disease, arthritis, and autoimmune diseases.1–4 The immune cells and their secreted cytokines are the major players in inflammation. But the current diagnosis of inflammatory diseases typically only relies on white blood cell counting or cytokine quantification in blood serum, without further pathophysiological information of immune cells. Since the immune cells are extremely heterogeneous in the spectrum and time of producing cytokines,5–7 a cytokine cytometry based method, if available, would precisely reflect disease status and drug targets, a step closer to personalized, predictive, and precision medicine.
Measurement of cytokine production by immune cells poses a great challenge to the single-cell detection technologies that are currently available. Immune cells are highly heterogeneous in cytokine secretion and polyfunctional, and the phenotypically identical cells are profoundly different in response to environmental stimulation.7–8 Conventional enzyme-linked immunospot (ELISpot) is a simple, robust method, but it only measures 1–3 cytokines at a time with limited throughput and is time consuming (24–48 h).9 Flow cytometry not only enumerates immune cell subtypes but also characterizes their functions with the single-cell resolution, and thus is superior to the ELISpot method in single-cell analysis.10 However, flow cytometry usually limits multiplexing to 3 in practice, while the multiparameter flow cytometry is bulky and expensive. Although the recently developed mass cytometry (CyTOF) has significantly improved the multiplexity up to 100, cytometric analysis of secreted signaling proteins requires blocking of surface transporters, which will inevitably alter the signaling networks and cell functions.11 Thus, flow cytometry is not popular for assaying secreted proteins. Microfluidic chips are currently the primary tools for single-cell cytokine secretion analysis. They detect the proteins secreted from a single cell instead of those retained inside of cell membrane, and in the meantime possess multiple other advantages over the conventional counterparts.12,13–14 The microengraved single-cell chips are able to assay up to 3 cytokines for single cells based on fluorescence imaging. Cells are isolated in nano-volume wells, and their secreted proteins are captured by an antibody array on the glass slide. The single-cell barcode microchip has dramatically increased multiplexity at the secretome level by integrating a miniaturized microarray into a chip.15 Such a technology has been used to monitor patients’ responses to adoptive T cell therapies by measuring T cell secretome.5, 16 However, those microchips use fluorescence microscope or microarray scanner to detect signal and thus are not convenient to use in a resource-limited environment. There are a few microchips that permit dynamic analysis of cytokine secretion using antibody coated beads.17–19 They are usually singleplex, and therefore they are not able to comprehensively profile cell functions yet.
Gold nanoparticles are one of the most used approach in point-of-care diagnosis through visualization of detection results by naked eyes.20 The advantages include relatively low cost and minimal education required for users. However, their applications are hindered by the lack of sensitivity and quantifiability. The immunogold enhancement technique desirably fills the gap via increasing the sensitivity by 100 to 500 times.21–22 The visual detection limit is comparable with that of conventional fluorescence based immunoassay, but immunogold enhancement technique does not require sophisticated instruments, or even no instrument. Besides, immunogold enhanced signal is very stable and does not fade under light or in the air, whereas fluorescent molecules suffer from photobleaching or inhibition.
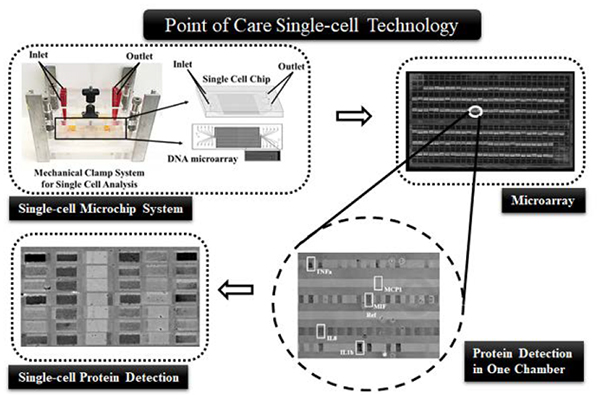
Here we introduce a portable single-cell measurement system combining the advantages of our single-cell stand-sit microchip and immunogold enhancement. The microchip design simplifies the operation procedure and makes the analysis robust and reliable. The result can be visualized by a common bright field microscope and can be recorded by a common camera. Thus, the whole system does not rely on complex facilities to assist single-cell assays and bulky instruments to read and record data. We demonstrate the systems on analyzing cytokine secretion of THP1 monocyte cells. The performance of the system is fully characterized, which was found to be similar to fluorescence method. We believe that this technology is an ideal platform to analyzed single-cell secreted protein and heterogeneity of cell populations in a field setting using only a low-end bright field microscope, or even a mobile microscope in the future.
EXPERIMENTAL DETAILS
Fabrication of splittable microchip.
The method for polydimethylsiloxane (PDMS; Ellsworth Adhesives) microchip fabrication was detailed elsewhere.23 Briefly, a multilayer mold was prepared by patterning SPR220 photoresists (Dow Chemical Company) and SU-8 (Microchem) on a 4″ wafer of silicon. The first layer acts as a base for next two layers, containing circular holes of 100 μm depth which changed to posts after casting of PDMS. The second layer (thickness of 3 μm) was constructed using SPR220. This layer fabricates the mold for the ducts that connect microchambers and microchannels. The third layer (thickness of 25 μm) containing microchambers was prepared from SU-8 2015. The final 50 μm thick SU-8 2025 layer shaped the mold for the microchannels at the gridlines’ intersections. The circular holes of the first layer aligned with the hollow center bowls to facilitate PDMS posts imprinted out of mold to attain 150 μm height. To confirm that the ducts were connected with the microchannels and microchambers, the features of second, third and fourth layer were made in a slightly overlapped manner. Then the mold features were transferred to PDMS (base: curing agent = 10:1), followed by aligning the PDMS replica with the antibody microarray to become a functional microchip. The fabricated microchip (75mm fixed height) consists of ~6,000 microchambers (1000 μm× 30 μm× 25 μm) for isolating single cells. An in-house designed clamp was used to apply pressure onto the microchip by adjusting the level of a screw (Fig. 1 A). Applied force can be adjusted to configure the collapsible PDMS posts between “sit” and “stand” states as well as ducts between “sit-closed” and “sit-open” states.24
Figure 1.

The components of the multiplex single-cell detection system. The operation requires a clamp that houses the single-cell microchip, a pipettor and biochemical reagents. Detection and recording of signal needs a low-end portable bright field microscope equipped with a digital camera. The top right image shows individual cells in microchambers, and the bottom right image shows sample single-cell protein data after gold enhancement. Scale bars in these two images are 200 μm and 100 μm, respectively.
Fabrication and design of antibody microarray.
Initially, DNA barcode microarray was patterned on a poly-L-lysine coated glass substrate and then converted to antibody microarray by hybridization with DNA-antibody conjugates. We have previously reported the procedure to prepare DNA-antibody conjugates through s-Hynic and SFB crosslinking.23 Soft lithography method was used to prepare PDMS mold for DNA patterning. The PDMS mold was mated to poly-L-lysine coated glass slide and baked at 80 °C for 1 h. Then six different types of amine-modified DNA (150 μM; Named as D, E, F, G, H and I) mixed with 1mM bis (sulfosuccinimidyl) suberate (BS3; Thermo Scientific) crosslinker were flowed into individual long and narrow microchannels for patterning six different DNA (DNA barcode). The DNA microarray was validated with Cyanine 3 (Cy3) conjugated complementary DNA (D’, E’, F’, G’, H’ and I’) to ensure no crosstalk and sufficient sensitivity. The DNA microarray was converted to antibody microarray by hybridizing with DNA-antibody conjugates cocktail solution [D’- interleukin 1 beta (IL1β), E’- interleukin 8, G’- macrophage migration inhibitory factor (MIF), H’- monocyte chemoattractant protein 1(MCP1) and I’- tumor necrosis factor alpha (TNFα); Table 1] at 37 °C for 1 h.23 After the conversion, the on-chip protein detection was performed immediately. The antibody microarray was validated and calibrated using known concentrations of recombinant protein (0.1 pg/ml to 2000 pg/ml).
Table 1.
List of antibodies and oligonucleotides for conjugation.
| Antibody | Oligo Name | Oligo Sequences |
|---|---|---|
| IL1β (eBioscience) | D’ | 5’-AAA AAA AAA AAA ATA CTC TGA CAT CTC GAC CAT-3’ |
| IL8 (R&D Systems) | E’ | 5’-AAA AAA AAA AAA ATA GAT ACT GCC ACT TCA CAT-3’ |
| MIF (Biolegend) | G’ | 5’-AAA AAA AAA AAA ATA CCG TGA ACC TTA CCT GAT-3’ |
| MCP1 (Biolegend) | H’ | 5’-AAA AAA AAA AAA AAA TGC TCG GGA AGG CTA CTC-3’ |
| TNFα (Biolegend) | I’ | 5’-AAA AAA AAA AAA ATG CCC TAT TGT TGC GTC GGA-3’ |
Cell culture and sample preparation.
THP-1 (ATCC® TIB-202™) human monocytes were cultured for a week before differentiation into macrophages in RPMI-1640 (Invitrogen) complete medium containing 10% (v/v) fetal bovine serum (Coring), 100 U/ml penicillin G and 100 U/ml streptomycin (Invitrogen), and 0.05 mM β-mercaptoethanol (Sigma) at 37 °C in an incubator supplied with 5% CO2. The THP1 cells were sub-cultured every 2 days to keep cell concentration lower than 5 million cells per ml. Cells were stained with Calcein AM (Invitrogen) at 0.2 μM for 10 min in serum free medium as per manufacturer’s instruction. Before microchip experiment, lipopolysaccharide (LPS, Sigma) at 1 μg/ml was used to stimulate the monocytes to differentiate into macrophages and produce cytokines. We have starved cells overnight without serum to synchronize the cell cycles.25
On-chip single-cell analysis and protein detection using gold enhancement.
The PDMS replica was sterilized by 70 % (v/v) ethanol under a laminar hood. The surface of the PDMS with features was oxidized using a plasma cleaner (Harrick Plasma) for 90 sec and then treated with 50 μg/ml of collagen (Corning) for 30 min in room temperature. The collagen coated PDMS surface was attached carefully with a DNA microarray glass slide to form a microchip with aligned barcode array and microchambers. The microchip at the “stand” state was blocked with 1% (w/v) bovine serum albumin (BSA, Fisher Scientific) in phosphate buffered saline (PBS) at pH 7.4. Then the assembled microchip was placed carefully inside the mechanical clamp. Both inlets and outlets were attached with 200 μl pipette tips to form reservoirs for culture medium or reagents. At the “stand” state, the bulk flow of buffer was driven by gravity. After blocking with BSA solution, a DNA-antibody cocktail was added to the microchip system and incubated at 37 °C for 1 h to hybridize the oligonucleotides of DNA-antibody conjugates with the complementary oligonucleotide DNA patterned on the glass slide, generating the antibody array. Unbound conjugates were washed off with the same BSA solution. The Calcein AM stained THP1 cells (2×105 cells/ml) in complete medium supplemented with 1 μg/ml of LPS were loaded into the microchip system through the inlets. The mechanical clamp was closed by adjusting the screw to convert the microchip to the “sit” state for isolating single cells in individual microchambers (Fig. 2). One cell per chamber was distinguished from zero cell per chamber by microscopic imaging (Fig. 1). Cell number and the microchamber address were recorded before incubation at 37 °C in 5% CO2 incubator for 6 h. The secreted proteins from the activated cells were captured by the antibody array.
Figure 2.

(A) Image of the single-cell microchip. Either side can be inlets or outlets. (B) Detailed scheme of gold enhanced sandwich immunoassay. Calcein AM stained THP1 cells were loaded into the single cell microchip system in the “stand” state. Microchamber was closed to isolate the single cell by adjusting the screws of mechanical clamp system. Gold enhanced sandwich immunoassay was performed to detect the signals.
After incubation, images were captured again to examine the viability of Calcein AM stained THP1 cells. Dead cells were excluded from the single-cell data. It’s found 90% cells were still alive. The chambers were stained with Alexa fluor 514 NHS ester (Invitrogen) to introduce grid pattern on the barcode glass slide which acts as a spatial address to the microchambers, when the ducts were closed at the “sit-close” state by exerting higher pressure. The mechanical pressure was released on the microchip by adjusting the screws, followed by injecting and washing three times with 1 % BSA in PBS. For protein detection, a cocktail of biotinylated secondary antibodies was introduced into the microchip and was incubated for 2 h in room temperature. After another thorough washing with 1 % (w/v) BSA in PBS, the glass slide was separated from the PDMS, and the biotin surface was blocked with 5% (w/v) non-fat milk and 0.1 % (v/v) tween 20 (Sigma) in PBS for 30 min at room temperature. Then 2 μg/ml Nanogold-streptavidin (Nanoprobes) in a blocking solution containing 1 % (w/v) non-fat milk and 0.05 % (v/v) tween 20 was added to the array slide and incubated at room temperature. The excess Nanogold-streptavidin was washed three times with PBS for 5 min and washed twice with 0.05 % (v/v) tween 20 in MiliQ water to reduce the background. GoldEnhance™ Blots (Nanoprobes) solution was used to enhance the signal as per manufacturer protocol. The slide was incubated with gold enhancement solution for 20 min at room temperature and washed with PBS to stop the reaction. The same procedure was performed for THP1 cells without LPS stimulation to generate the control data set.
Imaging, data acquisition and statistical analysis.
Images of cell loaded microchips and array slides were captured systematically using an inverted fluorescence microscope (Olympus IX73) with a digital camera (Zyla sCMOS, Andor). Fluorescence filter specification, objectives and other details were described before.24 We have also captured the images of cells and the barcode slides using a low-end bright field microscope (Micromaster, Fisher Scientific) and Moticam 2500 digital microscope portable camera (Motic) for comparative analysis. ImageJ (NIH) was used to process the images, and the mean grey scale intensity value of each array element was measured and documented in tabular format. On the other hand, the grey scale value of calibration barcode was also measured in similar way and the calibration curve was plotted by analyzing the corresponding recombinant protein concentration values. Grey scale value of control sample (background) was used to determine the detection limit of each cytokines.
GraphPad Prism 6 was used to process statistics of the one cell data vs. zero cell data. Statistical significance was determined by one-way ANOVA-test (non-parametric) and Bonferroni Post-hoc test between two groups (*p<0.05, **p<0.1 and ***p<0.001). Red bars on the scatter plots represent the medium value, whereas blue colored error bars denote the interquartile sample range. Cluster 3.0 (Stanford University) was used to hierarchically cluster single-cell data where the Euclidean Distance similarity metric method with centroid linkage clustering algorithm was used to execute the clustering. Clustered single-cell protein data were visualized by heatmaps using Java TreeView 3.0.
RESULTS AND DISCUSSION
Design of the single-cell measurement system.
The setup for the low-cost, portable single-cell diagnostic system is showed in Fig. 1. This system aims to be functionally comparable to conventional multiparameter flow cytometry, but it can work in a low-resource setting. Cytokine protein detection from immune cells is the focus of this platform, although the intracellular protein detection is also achievable by slight modification of the platform. All the instrument components including a clamp, a pipettor and a microscope are portable, and the operation does not need additional supporting facilities. Immunogold labeling and gold enhancement are employed here to avoid the dependence on fluorescence optics and electronics (which are normally bulky), so that a common bright field microscope is adequate to read the data. The detected protein signals are in grey to dark color, depending on the amount of detected antigens (Fig. 1). Through digitalization and calibration, we found the darkness of microarray elements is quantitatively correlated with the secreted proteins’ quantity.
The microchip design is the key to simplification of operation and instrument requisite. The chip is modified from our previous stand-sit design to accommodate the specific requirement in this work.23–24 The single cell microchip is comprised of a microarray glass slide and a PDMS replica. 6,720 microchambers of a microchip can be addressed by its unique spatial location. These spatial addresses of microchambers were used to match the cell numbers present in microchambers with the respective section profile of cytokines after detection. The small volume of each microchamber at 0.75 nL (1000 μm × 30 μm × 25 μm) contributes to high detection sensitivity, as many cytokines may not be produced by single cells in large quantity. A clamp is used to pressurize the microchip that contains inlet and outlet reservoirs (Fig. 1). The whole procedure of single-cell protein detection before imaging only needs to adjust the screw levels and use a pipettor to add and remove solutions in the reservoirs. Unlike other microchips, the buffer volume inside the microchip can be varied by applying mechanical force on the PDMS microchip. We have integrated every step of single-cell protein detection analysis such as cell culture, medium / buffer exchange, and multiple sandwich ELISA steps (including signal improvement using gold enhancement method) into one platform.
The antibody array was converted from a single-stranded DNA (ssDNA) array using DNA-antibody conjugates. The microchambers were carefully assembled with the stripe-shape DNA array to contain exactly 10 elements for each microchamber across the whole chip. This flow-patterned array is highly uniform, as the variation of detecting a homogeneous sample between any microchambers is <5%. Such a high uniformity ensures the detected heterogeneity is contributed only by biological difference but not measurement errors. The six oligo DNAs were selected to have no secondary structure and to be orthogonal to each other. We have used fluorophore-tagged complementary oligonucleotides to confirm no crosstalk between any pair of them. In the previous studies, we found that high loading of DNA-antibody is always beneficial to signal strength.24 Thus, ssDNAs were grafted onto PLL which was fixed on the glass slide surface. Ten times increase of signal was observed using this method. Fig. S1 shows no cross reactivity for measuring any individual proteins when recombinant proteins were used.
The stand-sit design takes advantage of PDMS elasticity to vary the chip’s inner volume and close or open the microchambers. The four layers of features stacking on the same mold have different thicknesses. Once the features are transferred to one-piece PDMS replica, they can collapse to certain degree depending on the strength of external mechanical force. Without pressure, the thin and tall posts, which are also the features on the one-piece PDMS, is strong enough to lift all the other PDMS features and leave an empty large space of ~100 μl within the microchip (Fig. 2). This “stand” state permits loading of cells or biochemical reagents as well as ELISA detection processes on the chip. The microchip adopts the “sit” state by applying pressure using mechanical clamp system. In this state cell chambers and microchannels are formed and single cells are entrapped inside the chambers. Material exchange between interconnected microchannels and cell chambers is possible if the mechanical force is not high enough, although the exchange is unnecessary for our current study since cells were cultured in the sealed microchambers for only 6 h. The solution replacement in the microchannels is governed by gravity-induced flow. Simply placing a solution in inlets will replace the preexisting solution in the entire microchannels within 10 min.
System characterization.
The general procedure of on-chip single-cell analysis is shown in Fig. 2. THP1 cells were selected in this study since they produce a variety of cytokines after LPS stimulation. These monocytes are in suspension before stimulation and become adherent on collagen coated surface immediately upon stimulation. Around 60–70% cells spread out and remain attached during the 6 h incubation time. Cells were sealed in the individual microchambers and incubated there at 37 °C in 5% CO2 incubator. The secreted cytokine proteins were captured by the integrated antibody array on the glass slide. A standard ELISA procedure was followed to detect the captured proteins by biotinylated detection antibodies. Gold nanoparticles conjugated with streptavidin (immunogold) were used to label the detection antibodies, and gold enhancement was employed to increase signal to visible level. During the enhancement process, gold ions first adhere to the surface of the catalytic gold nanoparticles anchored on the surface. Since the gold atoms deposit on the surface of gold nanoparticles possess the same catalytic capability, the gold enhancement process continues as long as there is supply of gold ions and reducing molecules in the vicinity of the gold grains, and thus expanding the size of the nanocrystals to >100 nm. The detection area would show grey to dark color, depending on the amount of antigens.
Five cytokines were examined on our platform including interleukin 1 beta (IL1β), interleukin 8 (IL8), macrophage migration inhibitory factor (MIF), monocyte chemoattractant protein 1(MCP1) and tumor necrosis factor alpha (TNFα). After activation of toll-like receptor (TLR) through LPS stimulation, those proinflammatory cytokines are usually secreted to the environment by macrophages.23, 26–27 The cytokines can induce the differentiation of monocytes via both antigen presentation and phagocytosis which is an important part of hosts’ immune system (innate immune response)28–31. The expression of those cytokines can dramatically increase after LPS stimulation as measured in the supernatant (Fig. S2). Note that the fold change of intensity is nonlinear with the presence of cytokines in the samples.
We have evaluated the performance of gold enhancement method using recombinant proteins. Five recombinant proteins at a gradient of concentrations were added to the microchip and sealed in microchambers, to simulate single-cell analysis process. The fluorescence method as the standard was compared with gold enhancement method in terms of sensitivity, dynamic range and specificity. All the antibodies and DNAs were validated to have no cross-reactivity and no non-specific binding in the assay performance (Fig. S1). The detection limits using gold enhancement method for IL1β, IL8, MIF, MCP1 and TNFα are 50, 25, 50, 25 and 100 pg/ml respectively (Fig. 3 A–B). They are slightly higher than the detection limits by fluorescence method (Fig. 3 C–D), mainly because the background of brightfield images is higher than that of fluorescence images. In both methods (gold enhancement vs. fluorescence), the detection limit of TNFα is lower than other cytokines, which is attributed to high quality of antibodies. The dynamic range of gold enhancement method at ~25 pg/ml to 500 pg/ml is also slightly smaller than that of fluorescence method. No obvious differences are observed for specificity/crosstalk between two methods, probably because they are based on the same ELISA detection strategy. During gold enhancement process, the gold nanoparticles are supposedly grow continuously. Thus, we tested the influence of incubation time and reagent amount on detected signal. The gold deposition actually occurs quickly within 5 min. The signal was increased slightly (~1.1–1.3 times) from 5 min to 20 min, and no change of signal was observed after 20 min (Fig. 3 E–F). Thus, for the above sensitivity test and all the single-cell experiments, we stick to 5 min for gold enhancement step.
Figure 3.

Immunoassay calibration curve and microarray readouts were prepared using different concentrations of recombinant IL1β, IL8, MIF, MCP1 and TNFα. (A&B) Microarray gold enhanced readouts and gold enhancement calibration curves using a series of concentrations of recombinant proteins and blank as a reference. (C&D) Microarray fluorescence readouts and fluorescence calibration curve using the same proteins, and Cy3 labeled DNA is used as a reference. (E&F) Microarray gold enhanced readouts and corresponding calibration curve by varying the incubation time of gold enhancement solution using 100 pg/ml of recombinant proteins cocktail. Error bar signifies the mean and standard deviation value of three repeats. Coefficient of Variation (CV) for IL1β, IL8, MIF, MCP1 and TNFα at various protein concentrations are 0.9–8.8%, 1–7.9%, 0.9–4%, 0.4–11.4% and 0.1–5.4%, respectively.
Single-cell analysis results.
The gold enhancement method was used to detect cytokine production by single THP1 cells on the microchip platform with point-of-detection setup. The procedure in Fig. 2 was followed, and a common bright field microscope with a low-end camera was applied to record signal. We select 6 h incubation time since major inflammatory cytokines could be secreted to a detectable amount according to literature.23, 32–33 The diffusion time within such a microchamber was measured to be around 15 min, which is negligible compared with 6 h incubation time. Thus, the cell location as the source of cytokine release should have no significant influence on cytokine assay as we studied before.24 Protein signals in microchambers with one cell and without cell were digitalized and generalized into scatter plots and heatmaps (Fig. 4A). Each of the one-cell datasets contains about 700 multiplexed single-cell protein measurements, and each of the zero-cell datasets have about 1,500 protein measurements. The control cells without stimulation do not show significant secretion of cytokines except MIF. This result is consistent with a report based on western blot using the same cell line.34 Unlike most cytokines that are tightly regulated, MIF can be substantially secreted in several cell types without stimulation through non-classical pathways.35–37 The level of MIF production is further increased after LPS stimulation. By contrast, the five cytokines in our panel are highly produced after stimulation, with some reaching saturation levels. They are also differentially expressed upon stimulation. We also quantified the percentage of single cells for cytokine expression and those with ≥ 1 ng/ml and 100 pg/ml secretion (Table 2). The cytokine signals from zero cell microchambers (mean± 2×standard deviation) was used as threshold to determine the percentage of single cell expressing cytokines. The single cells with protein levels above the threshold are considered as cytokine secreting cells. We also used the calibration curves in Fig. 3 to convert the grey scales to absolute secretion quantity, in order to show the percentage of the highly secreting cells. The single-cell data show that only a portion of cells produces cytokines, with only <1% secreting ≥ 1 ng/ml. In consideration of ≥ 1 ng/ml of TNFα and IL1β in bulk supernatant,38–39 most cytokines are produced by minor subpopulations. Thus the single monocyte cells are extremely heterogeneous. Fig. 4B shows the clustering and heterogeneity of single-cell secretion profiles. The intensity of color is correlated with relative expression level of proteins compared with blank (0-cell data). The control data of cells without stimulation appear to contain mainly two subpopulations, MIF secreting cells and silent cells. After activation, the cellular heterogeneity increases significantly, and no distinctive subpopulations can be identified. Most cells secrete at least one type of cytokines and may simultaneously produce other cytokines.
Figure 4.

Analysis of single cell data sets. (A) Scatter dot plots showing the gold enhanced data for LPS stimulated and control single THP1 cell secreted IL1β, IL8, MIF, MCP1 and TNFα from 1-cell on-chip experiments in comparison with background 0-cell experiments. P values are 0.05 (*), 0.01 (**), and 0.001 (***), with 0.05 considered statistically significant. Each dot represents a multiplexed protein measurement of a single cell. (B) Heatmaps showing relative secretion level of five proteins from LPS stimulated and control single THP1 cell isolated in chambers of microchip. The one-cell data were normalized and then median-centered by by 0-cell data. The color scale indicates the relative abundance of proteins. Euclidean distance similarity metric method with centroid linkage clustering algorithm was used to examine the hierarchical clustering.
Table 2:
Population percentage representation of cytokines expression profile of single cell
| LPS stimulated single cell cytokines expression | IL1β | IL8 | MIF | MCP1 | TNFα |
|---|---|---|---|---|---|
| % single cell expressing cytokines | 63% | 85% | 99% | 12% | 55% |
| % single cell expressing ≥ 1 ng/ml cytokines | 0.5% | 0.2% | 0.5% | 0.1% | 0.2% |
| % single cell expressing ≥ 100 pg/ml cytokines | 13% | 20% | 25% | 1% | 18% |
| Single cell cytokines expression (No LPS, Control) | |||||
| % single cell expressing cytokines | 14% | 17% | 90% | 2% | 11% |
| % single cell expressing ≥ 1 ng/ml cytokines | <0.1% | <0.1% | <0.1% | <0.1% | <0.1% |
| % single cell expressing ≥ 100 pg/ml cytokines | <0.1% | 1% | 7% | <0.1% | 1% |
Comparison of imaging by high-end microscope and low-end portable microscope setups.
We compared the performance of different bright field imaging setups, to demonstrate the feasibility of point-of-detection single-cell analysis in a field setting. Images taken by a high-end Olympus IX73 microscope (Fig. 5A) with Zyla 4.2 camera were compared with those by a low-end bright filed microscope with portable USB camera. Except for less resolution and more granularity, the image qualities are very similar, and all five cytokines are clearly detected. To estimate the signaling to noise ratios, the reverse grey scale intensities of all visible detected signals (E.g., the framed blocks) were averaged, and this mean value was used to divide the average reverse grey scale intensity of reference stripe (labeled as Ref in Fig. 5) to estimate the signal to noise ratios. For high end microscopic images, the estimated signal to noise ratio is 1.7±0.08 whereas it is 1.5±0.04 for low end microscopic images.
Figure 5.

Gold enhanced cytokines detection in microchambers of single cell microarray from LPS stimulated and control single THP1 cells using (A) high-end and (B) low-end bright field microscopes and cameras.
CONCLUSION
Our portable multiplex single-cell analysis technology not only allows for full set of complex on-chip manipulation of single cells and multiplex detection of several cytokines secreted from single cells with simple procedure, but also permits data reading of single-cell microarray result by simple bright field microscope, making it ideal for limited resource environment. The whole system overcomes the demanding facility requirement of present multiparameter flow cytometers and other formats of cytometers. We have designed this technology in such a way that it can be used for point-of-care diagnosis purposes at the single-cell level. Compared with the existing fluorescence-based technologies, the grey scale method is intrinsically less sensitive and may lose dynamic range to certain degree, although the gold enhancement has recovered majority of the lost sensitivity. Besides, the light source and background for imaging can also influence the data quality, while fluorescence images are usually cleaner. But, the gold enhanced slides can be kept without any noticeable degradation or change of signal for months, whereas the fluorescence-labeled slides could lose most of signal within few days and suffer from moisture. That makes our method valuable since the gold enhancement method is much more resistant to environmental influence, and thus is more suitable for out-door measurement. The future work should be directed towards standardization of the integrated single-cell measurement system and improving the dynamic range, while keeping the current merits of low cost and low instrument requirement.
We found that single cells differentially secrete cytokines upon stimulation by LPS, which is consistent with our prior observations using other cell lines.23–24 The single-cell analysis of cytokines has been emerging as one of the most important approaches to gauge the immune responses. The cytokines may bind to the same cell type through paracrine or autocrine signaling. Thus, the single-cell analysis would be expected to be different from bulk test where all cells are freely communicating with each other.40 In our technology, individual cells are confined in nano-size microchambers without communication with any of others, and thus paracrine signaling is not available. This method gives a clean evaluation of single-cell activity with no interference of other neighboring cells in the bulk assays. Zhou, J. reported that T cells can increase cytokine secretion by >10 times in the bulk assay over single-cell cytokine measurement.41 More biological studies should be conducted to find the mechanisms of cell signaling through communication, which is beyond the scope of our current work. In all, our single-cell analysis is robust and sensitive, and the assay results are comparable to conventional approaches. We believe that this multiplexed portable single-cell analysis system may find broad diagnostic applications in future as no clinical single-cell analysis technology hitherto exists.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the startup fund from SUNY Albany, National Institute of Health (R01GM12898401), NYSTEM (C32574GG) to J.W.
Footnotes
Supporting Information
Supporting Information Available: The following files are available free of charge.
SupportingInformation.pdf. Cross-reactivity of the cytokines assayed in the article, and cytokine secretion level of LPS stimulated THP-1 cells compared with control.
The authors declare no competing financial interest.
REFERENCES:
- (1).Elenkov IJ; Iezzoni DG; Daly A; Harris AG; Chrousos GP Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 2005, 12, 255–269, DOI: 10.1159/000087104. [DOI] [PubMed] [Google Scholar]
- (2).Stark AK; Sriskantharajah S; Hessel EM; Okkenhaug K PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol 2015, 23, 82–91, DOI: 10.1016/j.coph.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shaw AC; Goldstein DR; Montgomery RR Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol 2013, 13, 875–887, DOI: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fullerton JN; Gilroy DW Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov 2016, 15, 551–567, DOI: 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]
- (5).Ma C; Fan R; Ahmad H; Shi QH; Comin-Anduix B; Chodon T; Koya RC; Liu CC; Kwong GA; Radu CG; Ribas A; Heath JR A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Med 2011, 17, 738–744, DOI: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vivanco I; Robins HI; Rohle D; Campos C; Grommes C; Nghiemphu PL; Kubek S; Oldrini B; Chheda MG; Yannuzzi N; Tao H; Zhu SJ; Iwanami A; Kuga D; Dang JL; Pedraza A; Brennan CW; Heguy A; Liau LM; Lieberman F; Yung WKA; Gilbert MR; Reardon DA; Drappatz J; Wen PY; Lamborn KR; Chang SM; Prados MD; Fine HA; Horvath S; Wu N; Lassman AB; DeAngelis LM; Yong WH; Kuhn JG; Mischel PS; Mehta MP; Cloughesy TF; Mellinghoff IK Differential Sensitivity of Glioma-versus Lung Cancer-Specific EGFR Mutations to EGFR Kinase Inhibitors. Cancer. Discov 2012, 2, 458–471, DOI: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhao JL; Ma C; O’Connell RM; Mehta A; DiLoreto R; Heath JR; Baltimore D Conversion of Danger Signals into Cytokine Signals by Hematopoietic Stem and Progenitor Cells for Regulation of Stress-Induced Hematopoiesis. Cell Stem Cell 2014, 14, 445–459, DOI: 10.1016/j.stem.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Shin YS; Remacle F; Fan R; Hwang K; Wei W; Ahmad H; Levine RD; Heath JR Protein Signaling Networks from Single Cell Fluctuations and Information Theory Profiling. Biophys. J 2011, 100, 2378–2386, DOI: 10.1016/j.bpj.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Czerkinsky CC; Nilsson LA; Nygren H; Ouchterlony O; Tarkowski A A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 1983, 65, 109–121. [DOI] [PubMed] [Google Scholar]
- (10).Majumder B; North J; Mavroudis C; Rakhit R; Lowdell MW Toward new insights on the white blood cell differential by flow cytometry: A proof of concept study on the sepsis model. Cytometry B Clin. Cytom 2012, 82, 353–359, DOI: 10.1002/cyto.b.21027. [DOI] [PubMed] [Google Scholar]
- (11).Yee C; Greenberg P Modulating T-cell immunity to tumours: New strategies for monitoring T-cell responses. Nat. Rev. Cancer 2002, 2, 409–419, DOI: Doi 10.1038/Nrc820. [DOI] [PubMed] [Google Scholar]
- (12).Hong S; Pan Q; Lee LP Single-cell level co-culture platform for intercellular communication. Integr. Biol 2012, 4, 374–380, DOI: 10.1039/c2ib00166g. [DOI] [PubMed] [Google Scholar]
- (13).Wong IY; Javaid S; Wong EA; Perk S; Haber DA; Toner M; Irimia D Collective and individual migration following the epithelial-mesenchymal transition. Nat. Mater 2014, 13, 1063–1071, DOI: 10.1038/NMAT4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sun C; Cao Z; Wu M; Lu C Intracellular tracking of single native molecules with electroporation-delivered quantum dots. Anal. Chem 2014, 86, 11403–11409, DOI: 10.1021/ac503363m. [DOI] [PubMed] [Google Scholar]
- (15).Lu Y; Xue Q; Eisele MR; Sulistijo ES; Brower K; Han L; Amir ED; Pe’er D; Miller-Jensen K; Fan R Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc. Natl. Acad. Sci. USA 2015, 112, 607–615, DOI: 10.1073/pnas.1416756112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lin L; Ma C; Wei B; Aziz N; Rajalingam R; Yusung S; Erlich HA; Trachtenberg EA; Targan SR; McGovern DPB; Heath JR; Braun J Human NK Cells Licensed by Killer Ig Receptor Genes Have an Altered Cytokine Program That Modifies CD4(+) T Cell Function. J. Immunol 2014, 193, 940–949, DOI: 10.4049/jimmunol.1400093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Son KJ; Rahimian A; Shin DS; Siltanen C; Patel T; Revzin A Microfluidic compartments with sensing microbeads for dynamic monitoring of cytokine and exosome release from single cells. Analyst 2016, 141, 679–688, DOI: 10.1039/c5an01648g. [DOI] [PubMed] [Google Scholar]
- (18).Junkin M; Kaestli AJ; Cheng Z; Jordi C; Albayrak C; Hoffmann A; Tay S High-Content Quantification of Single-Cell Immune Dynamics. Cell Rep 2016, 15, 411–22, DOI: 10.1016/j.celrep.2016.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).An XY; Sendra VG; Liadi I; Ramesh B; Romain G; Haymaker C; Martinez-Paniagua M; Lu YB; Radvanyi LG; Roysam B; Varadarajan N Single-cell profiling of dynamic cytokine secretion and the phenotype of immune cells. Plos One 2017, 12, DOI: ARTN e0181904 10.1371/journal.pone.0181904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Linares EM; Kubota LT; Michaelis J; Thalhammer S Enhancement of the detection limit for lateral flow immunoassays: evaluation and comparison of bioconjugates. J. Immunol. Methods 2012, 375, 264–270, DOI: 10.1016/j.jim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- (21).Yang W; Li XB; Liu GW; Zhang BB; Zhang Y; Kong T; Tang JJ; Li DN; Wang Z A colloidal gold probe-based silver enhancement immunochromatographic assay for the rapid detection of abrin-a. Biosens. Bioelectron 2011, 26, 3710–3713, DOI: 10.1016/j.bios.2011.02.016. [DOI] [PubMed] [Google Scholar]
- (22).Wada A; Sakoda Y; Oyamada T; Kida H Development of a highly sensitive immunochromatographic detection kit for H5 influenza virus hemagglutinin using silver amplification. J. Virol. Methods 2011, 178, 82–86, DOI: 10.1016/j.jviromet.2011.08.017. [DOI] [PubMed] [Google Scholar]
- (23).George J; Wang J Assay of Genome-Wide Transcriptome and Secreted Proteins on the Same Single Immune Cells by Microfluidics and RNA Sequencing. Anal. Chem 2016, 88, 10309–10315, DOI: 10.1021/acs.analchem.6b03214. [DOI] [PubMed] [Google Scholar]
- (24).Ramirez L; Herschkowitz JI; Wang J Stand-sit microchip for high-throughput, multiplexed analysis of single cancer cells. Sci. Rep 2016, 6, DOI: 10.1038/srep32505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen M; Huang J; Yang X; Liu B; Zhang W; Huang L; Deng F; Ma J; Bai Y; Lu R Serum starvation induced cell cycle synchronization facilitates human somatic cells reprogramming. Plos One 2012, 7, e28203, DOI: 10.1371/journal.pone.0028203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Grishman EK; White PC; Savani RC Toll-like receptors, the NLRP3 inflammasome, and interleukin-1[beta] in the development and progression of type 1 diabetes. Pediatr. Res 2012, 71, 626–632, DOI: 10.1038/pr.2012.24. [DOI] [PubMed] [Google Scholar]
- (27).Yoshimura T; Takahashi M IFN-γ-Mediated Survival Enables Human Neutrophils to Produce MCP-1/CCL2 in Response to Activation by TLR Ligands. J. Immunol 2007, 179, 1942–1949, DOI: 10.4049/jimmunol.179.3.1942. [DOI] [PubMed] [Google Scholar]
- (28).Schenk M; Fabri M; Krutzik SR; Lee DJ; Vu DM; Sieling PA; Montoya D; Liu PT; Modlin RL Interleukin-1β triggers the differentiation of macrophages with enhanced capacity to present mycobacterial antigen to T cells. Immunol 2014, 141, 174–180, DOI: 10.1111/imm.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Varney ML; Olsen KJ; Mosley RL; Bucana CD; Talmadge JE; Singh RK Monocyte/macrophage recruitment, activation and differentiation modulate interleukin-8 production: a paracrine role of tumor-associated macrophages in tumor angiogenesis. In vivo (Athens, Greece) 2002, 16, 471–477. [PubMed] [Google Scholar]
- (30).Spoettl T; Hausmann M; Herlyn M; Gunckel M; Dirmeier A; Falk W; Herfarth H; Schoelmerich J; Rogler G Monocyte chemoattractant protein-1 (MCP-1) inhibits the intestinal-like differentiation of monocytes. Clin. Exp. Immunol 2006, 145, 190–199, DOI: 10.1111/j.1365-2249.2006.03113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wen F; Zheng J; Yu J; Gao M; Gao S; Zhou Y; Liu J; Yang Z Macrophage migration inhibitory factor in the regulation of myoblast proliferation and differentiation. Biosci. Biotechnol. Biochem 2016, 80 (7), 1313–1320, DOI: 10.1080/09168451.2016.1153951. [DOI] [PubMed] [Google Scholar]
- (32).Han Q; Bagheri N; Bradshaw EM; Hafler DA; Lauffenburger DA; Love JC Polyfunctional responses by human T cells result from sequential release of cytokines. Proc. Nat. Acad. Sci 2012, 109, 1607–1612, DOI: 10.1073/pnas.1117194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Björkbacka H; Fitzgerald KA; Huet F; Li X; Gregory JA; Lee MA; Ordija CM; Dowley NE; Golenbock DT; Freeman MW The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol. Genomics 2004, 19, 319–330, DOI: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- (34).Flieger O; Engling A; Bucala R; Lue HQ; Nickel W; Bernhagen J Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. Febs. Lett 2003, 551, 78–86, DOI: 10.1016/S0014-5793(03)00900-1. [DOI] [PubMed] [Google Scholar]
- (35).Bernhagen J; Calandra T; Mitchell RA; Martin SB; Tracey KJ; Voelter W; Manogue KR; Cerami A; Bucala R Mif Is a Pituitary-Derived Cytokine That Potentiates Lethal Endotoxemia. Nature 1993, 365, 756–759, DOI: Doi 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- (36).Merk M; Baugh J; Zierow S; Leng L; Pal U; Lee SJ; Ebert AD; Mizue Y; Trent JO; Mitchell R; Nickel W; Kavathas PB; Bernhagen J; Bucala R The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J. Immunol 2009, 182, 6896–906, DOI: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Skurk T; Herder C; Kraft I; Muller-Scholze S; Hauner H; Kolb H Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinol 2005, 146, 1006–1011, DOI: 10.1210/en.2004-0924. [DOI] [PubMed] [Google Scholar]
- (38).Sousa-Vasconcelos PD; Seguins WD; Luz ED; de Pinho RT Pattern of cytokine and chemokine production by THP-1 derived macrophages in response to live or heat-killed Mycobacterium bovis bacillus Calmette-Guerin Moreau strain. Mem. Inst. Oswaldo Cruz 2015, 110, 809–813, DOI: 10.1590/0074-02760140420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Segura M; Vadeboncoeur N; Gottschalk M CD14-dependent and -independent cytokine and chemokine production by human THP-1 monocytes stimulated by Streptococcus suis capsular type 2. Clin. Exp. Immunol 2002, 127, 243–254, DOI: DOI 10.1046/j.1365-2249.2002.01768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang J; Tham D; Wei W; Shin YS; Ma C; Ahmad H; Shi Q; Yu J; Levine RD; Heath JR Quantitating cell–cell interaction functions with applications to glioblastoma multiforme cancer cells. Nano Lett 2012, 12, 6101–6106, DOI: 10.1021/nl302748q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou J; Bethune MT; Malkova N; Sutherland AM; Comin-Anduix B; Su Y; Baltimore D; Ribas A; Heath JR A kinetic investigation of interacting, stimulated T cells identifies conditions for rapid functional enhancement, minimal phenotype differentiation, and improved adoptive cell transfer tumor eradication. Plos One 2018, 13, e0191634, DOI: 10.1371/journal.pone.0191634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.