

Marfan syndrome (MIM 154700) is an autosomal dominant connective tissue disorder caused by pathogenic variants in FBN1 (MIM 134797), encoding the fibrillin 1 (FBN1) protein. A rare early-onset form of MFS (early-onset MFS), which presents in the neonatal period, has been identified1 and is most often associated with de novo missense or exon-skipping pathogenic variants in FBN1 clustered in exons 24-40, particularly exons 24-32. 2 It is thought that pathogenic variants in this “neonatal region” (exons 24-32) may make FBN1 more susceptible to cleavage by physiologic proteases, thus resulting in a more severe presentation than classic MFS.3 Arachnodactyly, joint laxity, joint contractures, and ectopia lentis have commonly been seen in early-onset MFS patients1, and the cardiac complications, including tricuspid and/or mitral insufficiency and aortic root dilation, are associated with high mortality1.
A diagnosis of MFS is based on the revised Ghent nosology, which considers symptomology, family history, and molecular analysis of FBN14. Features that are commonly seen in the infantile presentation have previously been summarized1. Confirming a molecular diagnosis of either MFS or early-onset MFS typically begins with sequencing of FBN1, which uncovers the causative variant in approximately 91% of all MFS patients including early-onset MFS5. If FBN1 sequencing is unrevealing, deletion/duplication testing is typically performed to evaluate for disease-causing copy number variants (CNVs). Additionally, further testing for connective tissue conditions with clinical manifestations that overlap with MFS can also be considered. Here we describe a patient with a clinical presentation typical of early-onset MFS who underwent an extensive yet inconclusive genetic workup, including exome sequencing. Six years postmortem, genome sequencing ultimately revealed a pathogenic variant within the “neonatal region” of FBN1 that had been missed on prior testing.
Case Presentation
The child’s parents were initially seen at the Advanced Fetal Care Center at our institution due to a fetal anatomic survey at 20 weeks’ gestation with findings concerning for craniosynostosis and renal pelviectasis. Fetal magnetic resonance imaging (MRI) performed at 26 weeks’ gestation revealed an abnormal bulging appearance at the coronal sutures bilaterally and an increased binocular distance related to large-appearing globes. No cardiac or other structural abnormalities were noted with the exception of mild to moderate bilateral hydronephrosis. Amniocentesis revealed a normal karyotype (46,XY) and a normal (negative) craniosynostosis panel, including sequencing of FGFR2 exons 8 and 10, FGFR3 exon 7, and TWIST.
The infant was born via spontaneous vaginal delivery at 40+6/7 weeks’ gestation without complications to healthy, non-consanguineous parents of Northern European ancestry. Family history was not significant. On day of life 1, physical examination revealed distinctive facial features including hypertelorism, deeply set eyes, large ears with prominent lobes, and retrognathia (Figure 1, A and B). He was also noted to have severe arachnodactyly of the fingers and toes, joint contractures in the knees, elbows, and fingers, and pectus excavatum. A heart murmur was noted and echocardiogram revealed marked aortic root dilatation at the level of sinuses of Valsalva (1.92 cm, z-score: +7.11). He was admitted to the neonatal intensive care unit and started on oral propranolol and losartan, ultimately being discharged home at 10 days of age. At the time, his working clinical diagnosis was early-onset MFS.
Figure 1.
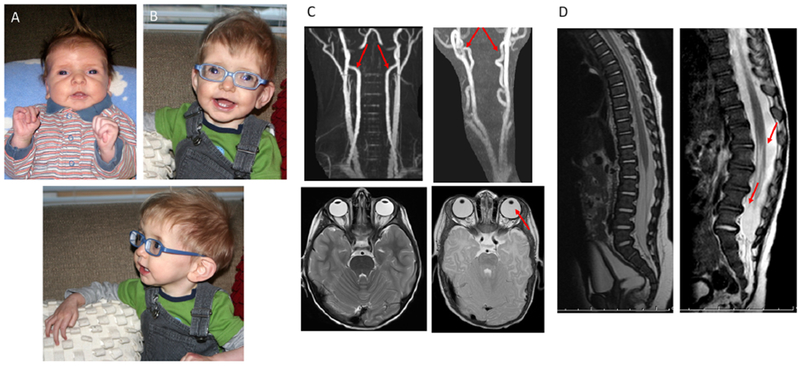
Patient photographs as (A) a neonate and (B) at 9 months of age revealed a phenotype consistent with early-onset Marfan syndrome, including marked arachnodactyly, hypertelorism, large ears with prominent lobes, and retrognathia. (C) MRI of healthy vertebral arteries and eyes (left) in comparison to patient’s tortuous vertebral arteries and absent zonule fibers (right). (D) MRI of healthy spine (left) in comparison to patient’s spine (right) demonstrating dural ectasia at 6 months of age.
At 5 weeks of age, the infant was evaluated in the Genetics clinic at our institution, where his length was 62.7 cm (z-score + 3.5), weight was 5.21 kg (73rd percentile), and head circumference was 40.5 cm (z-score + 2.3). Physical examination was notable for a positive thumb (Steinberg) sign bilaterally and a finger-to-total hand ratio of 49.3% (>97th percentile), confirming his severe arachnodactyly. Additional findings noted at this time included diastasis recti and severe thoracolumbar kyphoscoliosis. At 6 months of age, MRI of the brain and spine and magnetic resonance angiography of the head and neck were obtained and notable for marked tortuosity of the vertebral arteries in the neck bilaterally as well as in the internal carotid arteries at the region of the carotid siphon6. Dural ectasia was also noted in the thoracic spinal canal (Figure 1, C and D). At 2 years of age, he underwent spinal fusion for his scoliosis as well as duraplasty for repair of dural ectasia. Ophthalmologic evaluation at 3 months of age revealed astigmatism with significant myopia (OD: −13.00D sph, OS: −12.00D sph by cycloplegic refraction) and bilateral iridodonesis. Anteriorly subluxing lenses were noted by 9 months of age for which he underwent bilateral lensectomy at 1.5 years of age. Unfortunately, the aortic root dilation continued to progress rapidly despite appropriate medical therapy, enlarging to 3.10 cm (z-score +13.5) by 3 months, at which time a valve-sparing aortic root replacement surgery was performed without complications. At 12 months of age, he underwent mitral and tricuspid annuloplasty for worsening regurgitation. During this procedure, bilateral pulmonary blebs were noted and resected. He developed progressive and irreversible emphysematous changes throughout both lungs. In the child’s last few months of life, he required constant oxygen via high flow nasal cannula during the daytime and bilevel positive airway pressure (BiPAP) at night. He had difficulty ambulating and required support for most activities. Ultimately, the decision was made to redirect his management toward comfort care, and he died at home at 3 years of age.
In addition to early-onset MFS (highest on the differential as he exhibited nearly all of the features presented by Morse et al1), diagnoses considered by his clinical genetics team included Beals syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Postnatal genetic testing included sequencing of coding exons of FBN1 (for Marfan syndrome), FBN2 (Beals syndrome), COLA3A1 (vascular Ehlers-Danlos syndrome), TGFBR1 and TGFBR2 (Loeys-Dietz syndrome), FBLN4 and THSD4 (both genes having putative roles in connective tissue function and integrity), and multiplex ligation-dependent probe amplification (MLPA) of FBN1 for deletion/duplication analysis. No pathogenic variants or variants of uncertain significance were found. Chromosomal microarray via oligonucleotide-based targeted array comparative genomic hybridization and G-banded karyotype were also performed and were unrevealing.
Methods
Due to his negative molecular diagnostic evaluation in a clinical diagnostic laboratory, the patient and his parents were enrolled in the Manton Center for Orphan Disease Research, an IRB-approved gene discovery program at Boston Children’s Hospital, and informed consent was obtained. Peripheral blood was collected and trio ES and data processing was performed by the Genomics Platform at the Broad Institute of MIT and Harvard with an Illumina exome capture (38 Mb target) and sequenced (150 base pair paired reads) to cover more than 90% of targets at 20x and a mean target coverage of greater than 100x. ES data were processed through a pipeline using Picard for processing, and quality control of the aligned reads and mapping was done using the Burrows-Wheeler Aligner (BWA version 0.5.9-tpx) to the human genome build 37 (hg19). Variants were called using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.4. CNVs were ascertained using the Germline Copy Number Variant caller (gCNV Spark), a coverage-based CNV detection method that normalizes coverage across the exome by adjusting for systematic bias and uses a probabilistic framework to infer copy number from the normalized coverage.
Subsequently, GS and data processing for the proband only was performed by the Genomics Platform at the Broad Institute of MIT and Harvard. PCR-free preparation of sample DNA was accomplished using Illumina HiSeq X Ten v2 chemistry. Libraries were sequenced to a mean target coverage of greater than 30x. GS data were processed through a pipeline using Picard for processing and quality control of the aligned reads, using base quality score recalibration and local realignment at known indels. The BWA aligner (version 0.7.15-r1140) was used for mapping reads to the human genome build 38 (GRCh38). Single Nucleotide Variants (SNVs) and insertions / deletions (indels) were jointly called across all samples using GATK HaplotypeCaller package version 3.4. Default filters were applied to SNV and indel calls using the GATK Variant Quality Score Recalibration approach. Annotation was performed using Variant Effect Predictor (Ensembl Release 85). Structural variants were called using Manta v1.3.2 with the germline analysis default parameters7 and Alamut Visual (Interactive Biosoftware, Rouen, France) was used for splice effect prediction.
For confirmation of the variant described below, the region of FBN1 known to be affected by the child’s deletion was amplified using a set of primers designed using NCBI Primer-BLAST (forward primer 5’-GTGCATCATTACTCCATAGTTGAC-3’ and reverse primer 5’-TGCAGTCCTTGATAAGCAACC-3’) to produce an amplicon of 548 base pairs (bps). PCR was performed by standard methods. PCR products were resolved by gel electrophoresis on a 1% agarose gel to confirm amplification. The bands were excised from the gel and extracted using QIAQuick Gel Extraction kit and protocol (Qiagen, Hilden, Germany). Extracted DNA was then sent to GENEWIZ (GENEWIZ Boston Lab, Cambridge, MA) for Sanger sequencing.
Results
Analysis of ES data did not reveal any candidate disease-causing variants. By analysis of the GS data, a heterozygous 385 bp deletion was found in FBN1 (chr15:48474600-48474985 [GRCh38]), overlapping exon 33 and extending into the intron, that was confirmed through visualization via the Integrative Genomics Viewer (IGV) (Figure 2, A)8, 9. Follow-up Sanger sequencing of gel-extracted bands confirmed the deletion in addition to an 8 bp insertion (this 8 bp portion of the deleted region was likely flipped and re-inserted into the patient’s variant allele) on chromosome 15 (c.3965--335_4015delinsTCGATCAT). Manual curation of the variant with confirmation using Alamut Visual determined that it is predicted to result in skipping of exon 32 due to loss of a splice site. It is not present in the parents, so it was de novo in the proband, further supporting its pathogenicity (Figure 2, B). We then manually reviewed CNV results from the child’s ES data, which demonstrated evidence of this CNV (Figure 2, C) that was not found during the initial analysis; the deletion was identified but was filtered out by the quality filter used in our bioinformatics pipeline at the time (the quality score was 101.7000 and scores <300 are filtered out). Manual inspection of the exome data (Figure 2, D) in IGV demonstrated variable coverage of exon 33 between samples.
Figure 2.

A, The patient’s deletion as visualized in the Integrated Genomics Viewer using his genome sequencing data. Aligned reads are shown as gray horizontal bars and an area of relative depletion is seen where the deletion occurs (black arrow), overlapping exon 33, with the split reads highlighting the breakpoints (red arrows). B, Primers were designed to yield a 548-bp amplicon surrounding the region of the patient’s (P) deletion within FBN1 at chr15:48474600-48474985. This deletion of 385 bp would yield a 163 bp amplicon in the variant allele. Both the patient’s mother (M) and father (F) data revealed homozygosity for wild-type FBN1 without this deletion, indicating that the patient has a de novo variant. C, Germline Copy Number Variant caller results from the patient’s exome data demonstrating relative depletion in the area of the deletion (arrow). The y-axis denotes the copy number as inferred by Germline Copy Number Variant caller and the x-axis shows the position along the gene in kilobases. The blue line represents the sample of interest and the grey lines represent the other samples in the same batch of CNV calling. Each blue dot on the plot represents a probe for exon capture, which roughly represents exons. The green dots in this plot represent 2 probes that span exon 33. (The copy number estimation is expected to hover around 2.0 for autosomes.) D, Exome data for the trio as seen in Integrated Genomics Viewer revealed lower coverage for the affected child (proband) surrounding exon 33. The location of the deletion is demonstrated by the yellow bar.
Discussion
We present a child whose phenotype at birth was typical for early-onset MFS, a diagnosis confirmed by GS identifying a novel CNV: a de novo 385 bp deletion that encompassed part of exon 33 and the adjacent intron of FBN1. Prior to the result found on GS, a thorough clinical molecular genetic evaluation had been unrevealing, including sequencing of FBN1 and an assay (MLPA) ordered specifically to detect CNVs in FBN1, illustrating the importance of understanding both the limitations of molecular genetic testing techniques and how clinical diagnostic approaches change over time. The traditional genetic diagnostic odyssey often takes a tiered approach, first utilizing techniques for chromosomal analysis such as karyotype and chromosomal microarray, followed by sequencing of single genes, then panels of genes, and finally ES or GS if prior rounds of testing have been unrevealing. In recent years, rapid GS as a first-tier technique has been successfully employed for diagnosis in critically-ill neonates10 and pediatric patients11, and a meta-analysis has shown the diagnostic yield of ES and GS to be higher than that of a chromosomal microarray, though the yield of ES versus GS was not significantly different12. Although certain types of variants – such as structural variants, CNVs, and intronic variants – are more easily found using GS as opposed to ES, the incremental benefit of GS (which is more costly) compared with ES has been found to be relatively low. In a recent study, it was found that performing GS for a cohort of patients with non-diagnostic ES led to an additional 10 patients out of 105 (7%) being diagnosed by GS, and only three involved variants that were considered not detectable by ES13. Interestingly, 2 of these cases involved CNVs (the third was a deep intronic variant), as was found in our patient, illustrating one of the unique utilities of GS for diagnosis.
There are multiple ways in which a CNV may be detected. Sanger sequencing, one of the original methods used for determining the nucleotide sequence of a gene, is able to detect small insertions or deletions (indels)14 though may miss heterozygous deletions because one normal copy of the gene still remains. As primers used for Sanger sequencing are typically designed to align to intronic sequences just outside the exon, in this case, one of the primers was presumably aligned within the 385 bp deletion, so the allele with the deletion was not amplified for exon 33 and only the wild-type exon 33 was successfully sequenced, thus returning a “normal” result. For MLPA analyses, there is a similar challenge. In this assay, probes are designed to hybridize to certain segments of the DNA; probes are designed in pairs that must be adjacent to each other on the DNA sequence in order to be amplified by polymerase chain reaction (PCR). Therefore, in most cases, if a portion of the DNA sequence is missing, the probe for that region will not be able to bind and will not be amplified. By measuring the relative signals of amplified probes, deletions or duplications can be detected15. This was the technique for CNV detection most commonly available at the time of our patient’s initial diagnostic evaluation and likely failed to identify the molecular diagnosis due to the position of the MLPA probes, which are often placed within the exon, while this deletion encompassed only part of the exon and extended into the intron. Array comparative genomic hybridization (aCGH) can also be used to detect genomic deletions and gains. This method relies on labeling both patient and control DNA with a fluorescent dye and putting both on a chip containing multiple probes across the genome; the patient and control DNA will then hybridize to the chip and relative amounts of each color can be used to assess for relative depletion (i.e., deletion) or over-representation (i.e., gain) at a certain region. Finally, massively-parallel sequencing technologies, often referred to as “next-generation sequencing,” and used for ES and GS, can employ various methods such as read-depth comparisons or assessment of reads that map to the genome up to a consistent position and then abruptly to a different position (termed split reads) to determine whether or not a segment has been deleted or duplicated and can typically detect indels, larger deletions encompassing multiple exons, and inversions. As massively-parallel sequencing is performed by breaking up the coding sequence of the DNA being tested into multiple pieces that are then aligned to a reference sequence and re-processed to create the patient’s sequence, deletions can be seen when fragments are observed to be depleted within a specific region after alignment. A benefit to this method is that the sequencing and deletion/duplication analysis can be performed using the same assay.
ES, in which the coding regions of the genome are sequenced, was also unrevealing in this case owing to the location of a substantial portion of this deletion being outside of the FBN1 coding region and variable coverage of the affected exon. Although the CNV was identified when gCNV was used to analyze the exome data (Figure 1, C), it did not pass quality filters and was therefore excluded. This demonstrates the drawbacks of a “bioinformatics”-based approach to analysis. It also reveals the limitation of detecting CNVs in exome data using a read-depth approach; as coverage can vary across the exome data, differentiating between artificial changes in coverage and bona fide copy number events may be challenging. Based on this case, we have now changed our practices to visually evaluate strong candidate genes for a presenting phenotype. GS, which includes not only exonic but also the intronic sequences of the patient’s DNA, and has more consistent coverage in the sequencing reads across the genome, allows for a more comprehensive analysis for CNVs, and it was thus by this method that our patient’s deletion was identified. This case therefore highlights not only the limitations of current technology for CNV detection, but also the possible limitations of ES, which is becoming more widely used for genetic diagnosis.
The molecular confirmation of this child’s diagnosis occurred several years after his death. It is important to emphasize that in this case, the clinical diagnosis was sufficient to guide his management, and that finding the pathogenic variant during his lifetime was unlikely to have affected his clinical outcome. However, continuing to pursue the molecular diagnosis provided many benefits. First, confirming the etiology of the patient’s disease as a de novo genetic variant rather than an inherited variant allowed for appropriate reproductive counseling to the child’s parents regarding recurrence risks. Additionally, confirming the diagnosis might help to provide the family, who had continued to seek out answers even after his death, with a sense of closure. Finally, the diagnosis in this case also highlighted the unique value of GS as a diagnostic tool and served as an important reminder of the limitations of commonly-used molecular diagnostic techniques. Importantly, a “negative” genetic evaluation does not necessarily mean that the patient’s condition is not due to an underlying genetic condition, and the success in securing a diagnosis in this case is a testament to the dedication of the child’s family and providers who never gave up on this diagnostic odyssey.
Acknowledgments:
We thank Eleina England (supported by NIH R01 NEI EY027421) and Ellie Seaby for their assistance in variant interpretation and Dr. Sanjay Prabhu for providing the radiographic images. We also thank the patient’s family for their willingness to share this story.
Supported by the National Institutes of Health (NIH T32 GM007748 [to M.W.], NIH/NICHD (K12 HD052896 [to A.L.]), NIH/NIAMS (1R01AR068429-01 [to P.A.]), and NICHD/NHGRI/NIH (U19HD077671 [to P.A.]). Additional support provided by Boston Children’s Hospital/Harvard Medical School and the Brody School of Medicine at East Carolina University (to K.T.). Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute (UM1 HG008900 to Daniel MacArthur and Heidi Rehm). The authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Portions of this study were presented as a poster at the American College of Medical Genetics meeting, March 2010, Albuquerque, NM.
We describe an infant with a phenotype typical of early-onset Marfan syndrome whose genetic evaluation, including Sanger sequencing and deletion/duplication testing of FBN1 and exome sequencing, was negative. Ultimately, genome sequencing revealed a deletion missed on prior testing, demonstrating the unique utility of genome sequencing for molecular genetic diagnosis.
References
- [1].Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, Bieber FR, Lin A, et al. Diagnosis and management of infantile marfan syndrome. Pediatrics. 1990;86:888–95. [PubMed] [Google Scholar]
- [2].Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, et al. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24-40. Eur J Hum Genet. 2001;9:13–21. [DOI] [PubMed] [Google Scholar]
- [3].Kirschner R, Hubmacher D, Iyengar G, Kaur J, Fagotto-Kaufmann C, Bromme D, et al. Classical and neonatal Marfan syndrome mutations in fibrillin-1 cause differential protease susceptibilities and protein function. J Biol Chem. 2011;286:32810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–85. [DOI] [PubMed] [Google Scholar]
- [5].Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24:140–6. [DOI] [PubMed] [Google Scholar]
- [6].Morris SA, Orbach DB, Geva T, Singh MN, Gauvreau K, Lacro RV. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation. 2011;124:388–96. [DOI] [PubMed] [Google Scholar]
- [7].Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2. [DOI] [PubMed] [Google Scholar]
- [8].Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20:435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alfares A, Aloraini T, Subaie LA, Alissa A, Qudsi AA, Alahmad A, et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med. 2018;20:1328–33. [DOI] [PubMed] [Google Scholar]
- [14].Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin Genet. 2018;93:508–19. [DOI] [PubMed] [Google Scholar]
- [15].Eijk-Van Os PG, Schouten JP. Multiplex Ligation-dependent Probe Amplification (MLPA®) for the detection of copy number variation in genomic sequences. Methods Mol Biol. 2011;688:97–126. [DOI] [PubMed] [Google Scholar]