

Abstract
Raltegravir readily crosses the placenta to the fetus with maternal use during pregnancy. After birth, neonatal raltegravir elimination is highly variable and often extremely prolonged, with some neonates demonstrating rising profiles after birth despite removal from the source of extrinsic raltegravir. To establish an appropriate dosing regimen, an integrated maternal–neonatal pharmacokinetics model was built to predict raltegravir plasma concentrations in neonates with in utero raltegravir exposure. Postnatal age and body weight were used as structural covariates. The model predicted rising or decreasing neonatal elimination profiles based on the time of maternal drug administration relative to time of birth and degree of in utero drug disposition into the central and peripheral compartments. Based on this model, it is recommended to delay the first oral dose of raltegravir until 1–2 days of age in those neonates born to mothers who received raltegravir during pregnancy, labor, and delivery.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑Pharmacokinetics (PK) of raltegravir in pregnant women are altered. Raltegravir is metabolized by uridine 5′‐diphospho‐glucuronyltransferase 1A1 isoform, known to have very low activity at birth, which increases dramatically during the first few weeks of life.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ How to account for drug disposition in neonates if mothers received raltegravir prior to delivery? How does this knowledge affect the treatment of neonates with raltegravir?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This is the first population PK model of raltegravir that predicts the PK of both mother and fetus/neonate before and after delivery. This study examines the impact of last dose administration to mothers prior to delivery on raltegravir exposure in neonates after birth and the risk of overexposure if the first dose in the neonate is administered immediately after birth.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This model can be generally applied to describe the PK of drugs in neonates whose mothers received medications during pregnancy, labor, and delivery, which may impact the timing for the initial neonatal dose administration if the same medication is indicated or for potentially interacting medications.
Although progress has been made in identifying strategies to treat pregnant women living with HIV and to prevent the transmission of HIV to their infants, there were an estimated 160,000 infants who acquired HIV infections in 2016.1 In HIV endemic settings, up to 10% of pregnant women become infected during pregnancy, placing their infants at even higher risk for acquiring HIV infection.2 It is critically important to develop more effective postnatal prophylaxis regimens to prevent HIV‐1 infection of infants born to mothers living with HIV. In addition, many experts now advocate for early treatment with suppressive antiretroviral (ARV) combination therapy for neonates at high risk for or with documented HIV infection.3 The number of ARVs with formulations suitable for use in neonates and with neonatal safety and pharmacokinetic (PK) data is small. Integrase strand transfer inhibitors, which block the integration of viral DNA into the host cell and rapidly reduce HIV plasma viral load in infected individuals, hold promise as part of neonatal prophylaxis and treatment regimens.
Raltegravir (Isentress, Merck & Co Inc. Kenilworth, NJ, USA ) is an integrase strand transfer inhibitor indicated, in combination with other ARVs, for the treatment of HIV‐1 infection previously approved in infants and children 4 weeks of age and older at a dose of 6 mg/kg twice daily (b.i.d.). In November 2017, the US Food and Drug Administration provided marketing authorization for the use of raltegravir in neonates based on PK and safety results from the International Maternal, Pediatric, Adolescent AIDS Clinical Trials (IMPAACT, Icon Development Solutions, Ellicott City, MD, USA) Network P1110 protocol.4 The approved dose regimen in neonates is body weight (BW)‐based and age‐based and approximates to 1.5 mg/kg once daily (q.d.) for the first week of life and 3 mg/kg b.i.d. in infants 1–4 weeks of age, followed by 6 mg/kg b.i.d. after 4 weeks.5 Dose‐selection modeling was used in P1110, which enrolled full‐term infants. Based on PK modeling and simulations presented here, the US and Europe product labeling recommends to delay the first oral dose of raltegravir until 1–2 days of age in those neonates born to mothers who received raltegravir during pregnancy, labor, and delivery.
Raltegravir clearance (CL) is mainly nonrenal. The major elimination pathway is direct conversion of raltegravir into its glucuronide. The major enzyme involved in this metabolic reaction is the uridine 5′‐diphospho‐glucuronyltransferase 1A1 isoform (UGT1A1), whose activity is very low at birth and increases exponentially during the first weeks and months of life.6, 7, 8
Pregnant women receiving raltegravir just before labor and delivery probably have reduced bioavailability (F) and slower absorption than in the third trimester of pregnancy and postpartum.9, 10 The washout elimination of raltegravir in neonates exposed in utero from maternal pregnancy dosing demonstrates strikingly different rising or decreasing PK profiles after birth, which may be explained by postnatal enterohepatic recirculation or another unknown mechanism.11
The starting point for the neonate population PK model described here was a model developed for infants and children aged 4 weeks to 12 years.12 The data set was enriched by PK data from an adult population who received adult 400 mg doses. Raltegravir PK was adequately described by a two‐compartment linear disposition and first‐order absorption, with interindividual variability (IIV) on CL, intercompartmental clearance (Q), peripheral volume (V) of distribution, absorption rate constant (KA), and F. In addition, CL, Q and central V terms were scaled to BW using fixed allometric exponents of 0.75, (CL and Q) and 1.0, respectively.13, 14
Whether assessed from gene expression, protein measurement, or enzyme activity, liver UGT1A1 is not detectable preterm, becomes hardly detectable at birth, and achieves 90% of its maximum value by approximately 4 months of age.14, 15, 16 Therefore, a PK model for neonates and infants would require accounting for UGT1A1 maturation kinetics in addition to the allometric corrections for physical growth.
It is important to avoid excessive plasma concentrations of raltegravir in neonates, as raltegravir competes with unconjugated bilirubin for albumin‐binding sites.17 Displacement of unconjugated bilirubin from albumin by excessive raltegravir concentrations could place the neonate at risk for bilirubin‐induced neurologic dysfunction or for kernicterus, as seen with sulfisoxazole in neonates.18
The objective of this work was to use an integrated population PK analysis that incorporated data from pregnant women who received raltegravir as part of an ARV regimen, raltegravir‐unexposed neonates (infants born to mothers who did not receive raltegravir), and raltegravir‐exposed neonates (infants born to mothers who received raltegravir during pregnancy, labor, and delivery) to select a time after birth for the administration of initial raltegravir doses in raltegravir‐exposed neonates.
Methods
Local institutional review boards approved the protocol at all participating sites. Written informed consent was obtained from parents or guardians of all participants prior to enrollment.
A population PK model was developed empirically by using first‐order equations and maturation functions for CL and KA to design a raltegravir dosing regimen of raltegravir‐unexposed neonates.19 A proposed 6‐week dosing regimen (1.5 mg/kg q.d. in the first week of life, 3 mg/kg b.i.d. at weeks 2–4, and 6 mg/kg at weeks 5 and 6) was validated,20 and subsequently the model was further developed to include raltegravir‐exposed neonates.21
To help guide dose selection, PK target exposures were prespecified based on prior raltegravir experience in adults with HIV‐1 infection. Specifically, the PK exposure targets considered for efficacy and safety were a minimum geometric mean trough concentration of 75 nM (33.3 ng/mL using 0.4444 unit conversion factor)7 and a maximum area under the plasma concentration‐time curve from dose to 12 hours (AUC0–12)of 45 μM·hour (20 μg·hour/mL) and from dose to 24 hours (AUC0‐24) of 90 μM/hour (40 μg·hour/mL).22 Another safety target was to maintain peak concentrations below 19.63 μM (8.72 μg/mL). SAS version 9.4 (SAS Institute, Cary, NC) was applied for data set construction. Nonlinear Mixed Effects Modelling (ICON Inc., USA) was used for the population analyses and simulations. R version 3.1.3 (R Foundation for Statistical Computing, Vienna, Austria) was used for postprocessing and the generation of graphs.
Data
Study data
The data set included raltegravir plasma concentration data in raltegravir‐unexposed and raltegravir‐exposed term neonates up to 6 weeks of age from IMPAACT P1110,23 enriched by additional data from infants and toddlers from IMPAACT P106624 and 19 pairs of mother and raltegravir‐exposed neonates from IMPAACT P1097.11, 25 The design of these three studies and the definition of relevant cohorts is summarized in Supplementary Material S1 .
In all three trials, the plasma concentrations of raltegravir were determined by a validated liquid chromatography assay method with tandem mass spectrometry detection as described in Supplementary Material S2 .
Handling of missing data
The data set contained no missing information related to dosing or sampling. All concentrations below the lower limit of quantitation (LLOQ) were imputed to half the LLOQ (11.25 nM), and postdose concentrations below the LLOQ were included in the analysis (40 in total). One subject had consecutive LLOQ measurements, which had a negligible impact on model development.
Development of the maternal–neonatal population PK model
The paucity of maternal PK data warranted the development of an integrated population PK model that combined fetus/neonate and maternal data. The model was composed of two parts: one for raltegravir PK in the fetus/neonate and one for raltegravir PK in mothers (Figure 1). Raltegravir PK in mothers prior to and just after delivery was assumed to be comparable to the PK of pregnant women during the third trimester.9, 10 Because only 19 maternal PK samples were available, a fit‐for‐purpose maternal PK model was implemented, which was described by a two‐compartment linear disposition model with first‐order absorption rate, where disposition parameters were fixed to previous model parameter estimates.12 The purpose of the maternal PK component of the model was to use the maternal PK observations to inform about the raltegravir distribution in the raltegravir‐exposed neonate at birth. Maternal absorption parameters were estimated in an attempt to reflect the known decrease in raltegravir concentration and bioavailability during labor and delivery.9, 10, 26 The model did not account for potential changes in maternal raltegravir PK during labor and delivery. The unobserved raltegravir PK in the fetus was represented by a two‐compartment linear disposition based on the previously developed neonate PK model.20 The lack of raltegravir concentration data from fetuses imposed the simplifying assumption of an instantaneous equilibrium between placental and umbilical cord blood. In model terms, the intercompartmental CL linking the maternal and fetal central compartments was set to 1,000 L/hour. The fetus was assumed to have a BW as recorded at birth (2.2–3.4 kg).
Figure 1.

Compartmental structure of the final mother–neonate model. CL, apparent clearance; KA, absorption rate constant; Q, apparent intercompartmental clearance; V2, apparent central volume of distribution; V3, apparent peripheral volume of distribution.
The raltegravir exchange between the central compartments of the neonate and mother decoupled at birth, and mother and neonate continued as two independent entities. Hence, the depot compartment of the neonate (Figure 1) became available for oral absorption of additional doses. The PK in neonates was modeled as previously described and included BW‐based allometric exponents applied to CL, Q, and V and maturation functions for both the CL and KA.16, 20 Initial conditions depended on prior in utero exposure to raltegravir.
The IIV was modeled by a log‐normal distribution on CL and KA. Residual variability was modeled as combined additive and proportional errors. The pivotal development steps to the maternal–neonate population PK model are summarized in Table S1 .
Covariate analysis
The effects of postnatal age (PNA) on CL and KA, and the effects of neonate BW on CL, Q, and V were included in the base model as structural covariates. Additional covariates were tested based on the following hypotheses: (i) exposure to raltegravir in utero may have an impact on fetal development of liver function, potentially affecting raltegravir CL at birth or the rate constant of CL maturation after birth; (ii) feeding pattern may impact KA, the development of KA, as well as bioavailability; (iii) the UGT1A1 isoform may mature at different rates in boys and girls; (iv) the baseline body length at the time of first measurement may influence raltegravir CL; (v) assuming the presence of a relationship between body surface area (BSA) and intestine surface area, the baseline BSA may impact raltegravir KA. Although mother–infant pairs were enrolled at IMPAACT sites in the United States, Brazil, South Africa, and Thailand, race was not explored as a covariate because of the small sample size and data imbalance (the large majority of the infants included in the analysis were African American or black).
A univariate model–based covariate analysis was conducted, where the decision to retain a covariate effect in the model was based on a statistically significant (P < 0.05) decrease in the minimum objective function value as well as considerations about biological plausibility, plausibility of extrapolation beyond the range of the observed covariates, and clinical relevance. The effects of continuous covariates were coded as a power function (exponent θ covpar), where the covariate of interest was normalized to the median value in the population. The effects of categorical covariates were coded as a fractional change from the typical parameter value in the population.
Model evaluation
The evaluation of the population PK model was based on standard statistical criteria of goodness of fit (GOF) such as the log‐likelihood difference between alternative models (e.g., a decrease in the minimum objective function value), accuracy of parameter estimation (i.e., 95% confidence interval excluding 0), successful model convergence, and standard diagnostic plots. Confidence intervals of parameter estimates were confirmed by nonparametric bootstrap resampling analysis using 1,000 replicates, stratified by study, cohort, and status of in utero exposure.
Model qualification also included prediction‐corrected visual predictive checks27 based on 1,000 replicates, where raltegravir concentrations were grouped into meaningful time intervals. For each time interval, the observed median of raltegravir concentrations and 25% and 75% percentiles were calculated.
PK simulations
Simulations were performed to predict the typical plasma concentration‐time profiles of raltegravir in the mother and fetus in the 48 hours preceding birth and in the raltegravir‐exposed neonate during the first week after birth. The time span between the last dose administration to mother and birth was allowed to vary between 2 and 24 hours with a 2‐hour granularity. The time of first administration to neonate was assumed either 12 or 36 hours postpartum. The growth of the typical neonate postpartum was obtained by fitting the BW with age (PNA) for the dosing records of the infants using the following equation, empirically derived from the observations:
Results
PK analysis set
The PK analysis set included 759 PK data points from 19 mothers, 61 neonates up to 2 days of age at enrollment (36 raltegravir unexposed and 25 raltegravir exposed), and 24 infants between 4 weeks and 2 years of age at enrollment. The majority (60%) of neonates and infants were boys. All available PK samples were included in the analysis except one outlier maternal PK concentration attributed to a missed dose. The data set composition stratified by population, study, and cohort is summarized in Table 1. Each mother contributed to one PK data point only.
Table 1.
Number of subjects and observations included in the integrated mother–neonate PK analysis set and demographic variables, stratified by population, study, and cohort
| Population | Raltegravir‐unexposed neonates | Infants | Raltegravir‐exposed neonates | Mothers | |||
|---|---|---|---|---|---|---|---|
| Study | P1110 | P1110 | P1066 | P1066 | P1110 | P1097 | P1097 |
| Total number of subjects | 10 | 26 | 13 | 11 | 6 | 19 | 19 |
| Raltegravir administration | 2 × single dose | Multiple dose 0–6 weeks | Multiple dose b.i.d. | Multiple dose b.i.d. | In utero + 2 × single dose | In utero | 400 mg b.i.d. |
| Number of PK samples | 79 | 288 | 121 | 123 | 54 | 75 | 19 |
| Age range at enrollment | 0–2 days | 0–2 days | 6 months to <2 years | 4 weeks to <6 months | 0–2 days | 0–1 day | Unknown |
| Age range for PK sampling | 0–11 days | 0–6 weeks | 6 months to <2.4 years | 5 weeks to <1 year | 0–11 days | 0–2 days | Samples taken <1 hour postpartuma |
| Weight range, kg | 2.3–4.2 | 2.2–5.3 | 5.5–14 | 3.7–10.4 | 2.2–3.4 | 2.2–4.1 | Unknown |
| Sex, M/F | 4/6 | 14/12 | 8/5 | 7/4 | 4/2 | 14/5 | 0/19 |
PK, pharmacokinetic; b.i.d., twice daily.
Maternal sample of one mother was obtained 2.8 hours postpartum.
Covariate analysis
Other than BW and PNA, which were included as structural covariates, no additional covariate effects were retained in the model because they did not statistically significantly improve the model fit or, in the case of feeding pattern, led to contradictory results. Parameter estimates of the final population PK model are summarized in Table 2.
Table 2.
Parameter estimates of the integrated mother–neonate population PK model of raltegravir
| Parameter | Unit | Estimate | CI95 | Bootstrap result | |||
|---|---|---|---|---|---|---|---|
| Low | High | Median | P2.5 | P97.5 | |||
| Neonate (raltegravir unexposed and raltegravir exposed) | |||||||
| V2 | L | 7.04 | 5.07 | 9.75 | 7.16 | 4.85 | 9.91 |
| V3 | L | 10.3 | 7.97 | 13.4 | 10.3 | 7.37 | 13.4 |
| CLmax | L/hour | 9.44 | 7.44 | 11.4 | 9.34 | 7.44 | 11.8 |
| Q | L/hour | 0.786 | 0.559 | 1.11 | 0.8 | 0.538 | 1.2 |
| KAmax | 1/hour | 0.43 | 0.306 | 0.555 | 0.452 | 0.315 | 0.875 |
| F4 (fixed) | – | 1 | – | – | 11.3 | 7.38 | 15.9 |
| CLbase | L/hour | 0 | – | – | 0.0876 | 0.0216 | 0.247 |
| CLtau | 1/year | 11.3 | 7.56 | 15.1 | 60.8 | 6.7 | 135 |
| Kabase | 1/hour | 0.0915 | 0.0343 | 0.245 | 0.314 | 0.132 | 0.483 |
| Katau | 1/year | 63.2 | 1.4 | 125 | 0.178 | 0.0802 | 0.291 |
| IIV on CL | – | 0.33 | 0.108 | 0.552 | 7.16 | 4.85 | 9.91 |
| IIV on KA | – | 0.196 | 0.103 | 0.289 | 10.3 | 7.37 | 13.4 |
| Mothera | |||||||
| V2 (fixed) | L | 3.52 | – | – | |||
| V3 (fixed) | L | 27 | – | – | |||
| CL (fixed) | L/hour | 9.73 | – | – | |||
| Q (fixed) | L/hour | 0.866 | – | – | |||
| KA | 1/hour | 0.175 | 0.0888 | 0.261 | 0.178 | 0.0576 | 0.42 |
| F | – | 0.517 | 0.404 | 0.631 | 0.527 | 0.381 | 0.734 |
| IIV on F | – | 0.311 | 0.0834 | 0.538 | 0.283 | 0.101 | 1.01 |
| Residual error | |||||||
| RUV‐prop | – | 0.54 | 0.498 | 0.582 | 0.536 | 0.489 | 0.577 |
| RUV‐add | nM | 11.9 | 9.11 | 14.7 | 11.6 | 9.95 | 40.9 |
| Shrinkage | |||||||
| IIV CL (neonate) | 9.3% | ||||||
| IIV KA (neonate) | 24.0% | ||||||
| IIV F (mother) | 51.4% | ||||||
| ε | 5.6% | ||||||
Typical values of clearances and volumes refer to a subject weighing 25 kg.
CI95 low, lower limit of the 95% confidence interval; CI95 high, upper limit of the 95% confidence interval; CLbase, typical value of apparent clearance at birth; CLmax, maximum increase in apparent clearance from CLbase; CLtau, first‐order rate constant for the age‐related changes in apparent clearance; F, oral bioavailability mother relative to granules for suspension formulation25; F4, oral bioavailability neonate (after birth); IIV, interindividual variability; KAbase, typical value of absorption rate constant at birth; KAmax, maximum increase in absorption rate constant from KAbase; KAtau, first‐order rate constant for the age‐related changes in absorption rate constant; P2.5, 2.5% percentile; P97.5, 97.5% percentile; PK, pharmacokinetic; Q, typical value of apparent intercompartmental clearance; RUV‐add, additive term of the residual error; RUV‐prop, proportional term of the residual error; V2, typical value of apparent central volume of distribution; V3, typical value of apparent peripheral volume of distribution.
Mother PK component of the integrated model was based on limited information and was used only to inform about the initial raltegravir concentrations in the neonate of each mother at birth.
In the neonate, the CL increased from nil at birth (i.e., no elimination capacity) to an estimated 9.44 L/hour at full maturation, with a maturation rate constant of 11.3 year−1, implying 90% of CL maturation by approximately 11 weeks of age. At birth, the estimated KA was 0.0915 hour−1, which corresponded to an absorption half‐life of approximately 7.6 hours. The KA increased with PNA and reached 90% of its maximum by 12 days of age. The estimates of central V and peripheral V of distribution were 7.03 and 10.4 L, respectively. The IIV on neonate CL and KA was 33% and 20%, respectively. The proportional residual error accounted for approximately 54%.
Evaluation of the final model
The bootstrap analysis (N = 1,000) results were reported after omitting 237 runs with minimization terminated and three runs with estimates near a boundary. With more than 75% of runs that finished successfully, the bootstrap analysis provided a useful qualification of model robustness. The results are summarized in Table 2. Overall, the medians of bootstrapped parameter estimates deviated by less than ± 10% from population parameter estimates for both the fixed and random effects.
The GOF plots of the population PK model for raltegravir are presented in Figure S1 . Model diagnostics plots did not reveal any trend toward systematic deviations. The GOF plots further suggested that data from raltegravir‐unexposed and raltegravir‐exposed neonates were equally well captured by the model.
The maternal–neonate population PK model was also qualified by prediction‐corrected visual predictive checks (Figure S2 ). The between‐subject variability was well captured despite the sparse data. The median simulated data largely overlapped the median of the grouped observations and remained within the 25% and 75% confidence intervals of the observations. The confidence intervals of the simulated data showed a tendency of slightly higher concentrations for the simulated time periods (days 1 and 14 for raltegravir‐unexposed and days 1–2 for the raltegravir‐exposed neonates) but captured the median of the binned observed data. Representative individual model‐predicted and observed concentration‐time profiles are shown in Figure 2.
Figure 2.
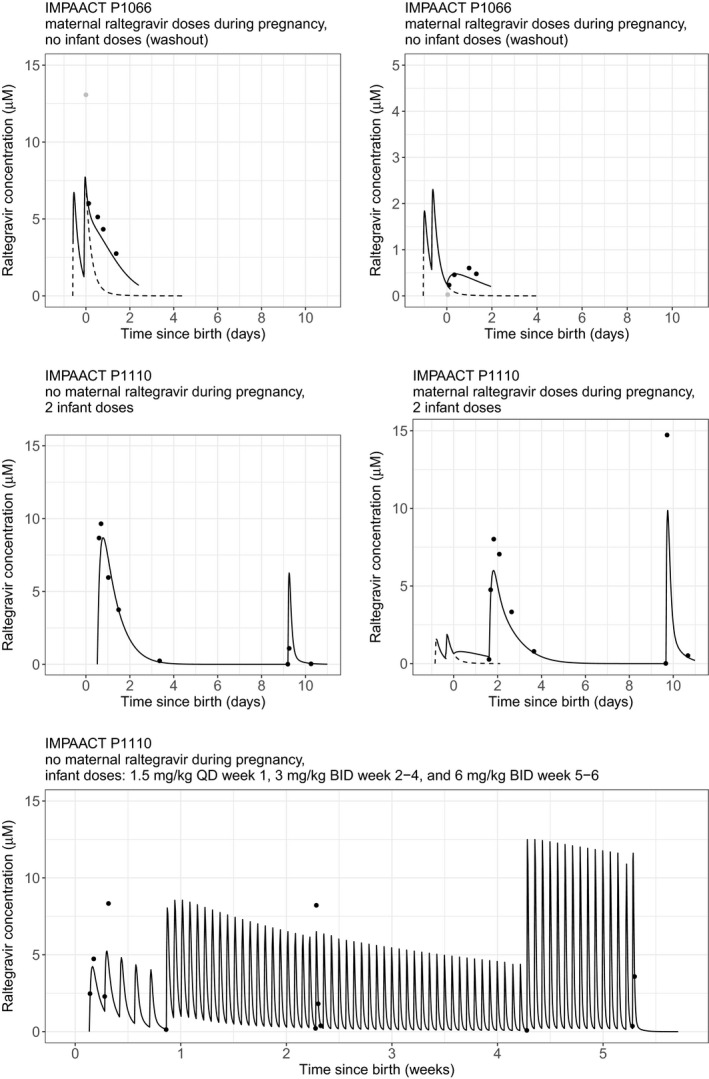
Representative individual model‐predicted concentration‐time profiles for neonates and mothers for IMPAACT P1110 and IMPAACT P1097. Observations are shown by dots. Solid line is for neonates; dotted line is for mothers. BID, twice‐daily dosing; IMPAACT, International Maternal, Pediatric, Adolescent AIDS Clinical Trials; QD, once‐daily dosing.
Simulations to support the time of first administration in neonates
Simulations investigating the influence of the time span between the last dose administration to the mother and the birth of the neonate on raltegravir concentration‐time profiles in the mother and the neonate are shown in Figure 3.
Figure 3.
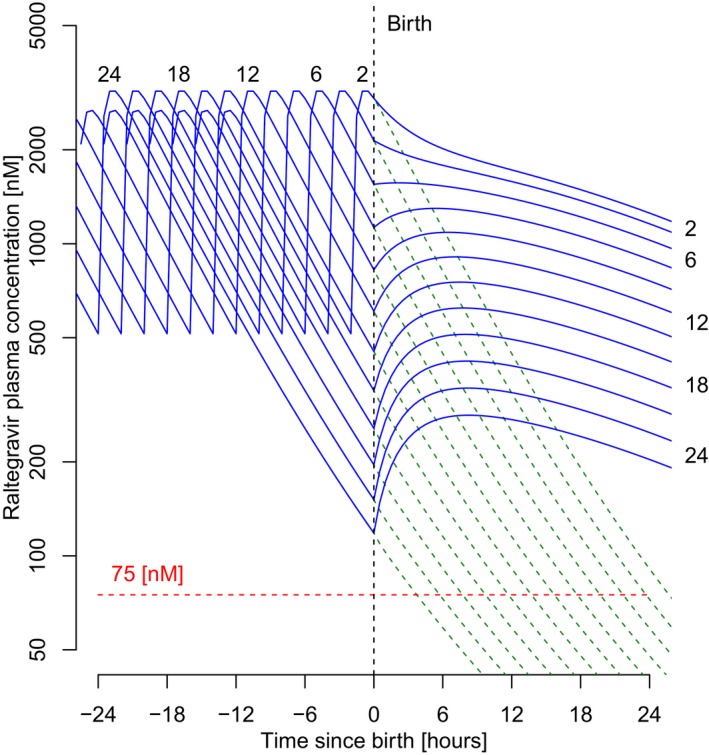
Predicted superimposed concentration‐time profiles of raltegravir (semilog scale) in mothers and neonates, with last dose administration 2–24 hours before giving birth. Dotted green line is for mothers; solid blue line is for neonates. Before birth and by model structure, predicted mother and neonate pharmacokinetic profiles superimpose. Numbers in the graph represent the time interval between the simulated last dose administered to the mother and delivery (hours).
During pregnancy, raltegravir plasma concentrations in the mother and fetus were assumed to be in equilibrium and therefore were indistinguishable from Figure 3. At birth and from a model perspective, the central compartments of the mother and neonate were separated, which mimicked the severance of linked physiological compartments and became two separate individuals with differing PK yet with a shared prepartum exposure history. Subsequently, the maternal plasma concentrations of raltegravir were predicted to decrease. In the neonate, however, the shape of the concentration‐time profile was predicted to vary. If the time span between the last dose administered to the mother and birth was more than 6 hours, the profile was predicted to initially rise as a result of a predominant flow from the neonate peripheral compartment back into the neonate central compartment. Conversely, if the time span of last dose administration to the mother was less than 6 hours, the neonate PK time course profile was predicted to decline. Because of this contrasted predicted PK in the neonate, additional simulations were performed to investigate the impact on the achievement of target PK endpoints. The predicted time‐course of trough concentrations and AUC0‐24 during the first week after birth are illustrated in Figures 4 and 5, respectively, for various assumptions of time span between the last dose administration to the mother and birth, ranging from 2–24 hours.
Figure 4.
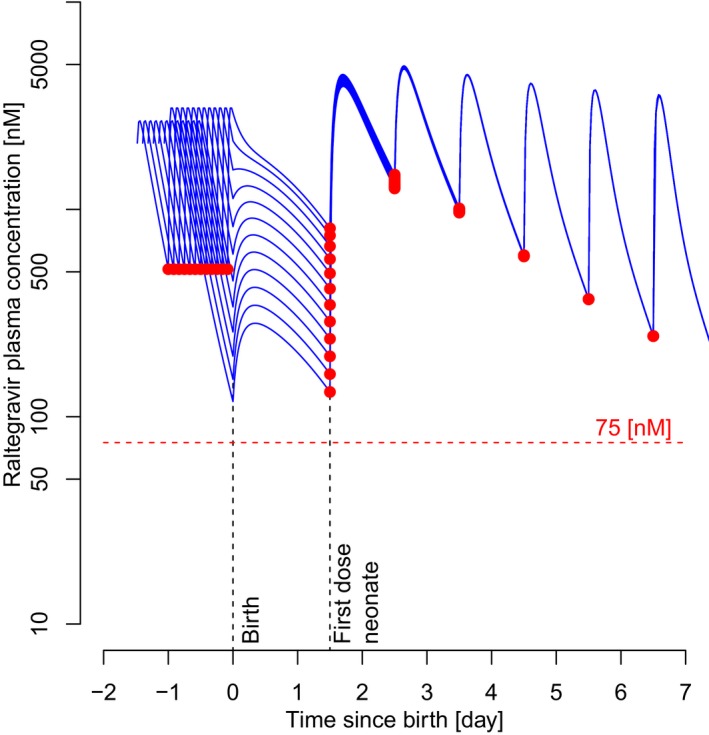
Influence of the time interval between the last dose administration to mothers and birth on raltegravir trough concentrations in neonates. Red dots represent simulated trough concentrations. Blue curves represent time‐course pharmacokinetic profiles of the neonate before and after birth, which depend on the time interval between the simulated last dose administered to the mother and delivery (2–24 hours, see Figure 1). First dose in the neonate is 36 hours postpartum.
Figure 5.
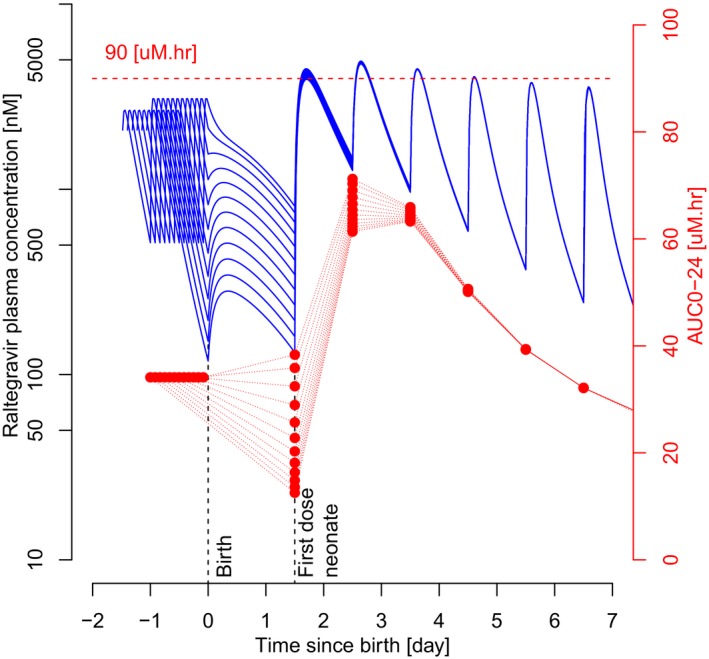
Influence of the time interval between the last dose administration to the mother and the birth of the neonate on raltegravir area under the plasma concentration‐time curve from dose to 24 hours (AUC0‐24) in the neonate. Red dots represent simulated AUC0‐24, connected by red dotted lines for each neonate. Blue curves represent time‐course pharmacokinetic profiles of the neonate before and after birth, which depend on the time interval between the simulated last dose administered to the mother and delivery (2–24 hours, see Figure 1). First dose in neonate is 36 hours postpartum. Horizontal red‐dotted line is safety criterion set at 90 μM·hour.
Depending on the time interval between the last dose administration to the mother and birth and the scenario that the first dose in raltegravir‐exposed neonate was given 36 hours postpartum, the trough concentration of raltegravir in the neonate was predicted to range between 140 nM (timespan 24 hours) and 1,500 nM (timespan 2 hours only). In all cases, these trough concentrations exceeded the PK target trough of 75 nM.
For the same first dose scenario, the AUC0‐24 of raltegravir in the neonate was predicted to stay within the upper limit of 90 μM·hour, including after the second dose, where AUC0‐24 would reach a maximum (Figure 5).
If the first dose administration in the neonate was changed to 12 hours postpartum, then the AUC0‐24 of raltegravir in the neonate was predicted to exceed 90 μM·hour by a maximum of 20% for a maximum of 2 days (Figure S3 ).
Discussion
IMPAACT P1097 demonstrated that raltegravir readily crosses the placenta and that the elimination of transplacentally‐acquired raltegravir in infants whose mothers received raltegravir during pregnancy was highly variable and prolonged.11 To provide dosing recommendations for infants born to mothers who received raltegravir during pregnancy, labor, and delivery, an integrated maternal–neonatal population PK model was built to predict raltegravir plasma concentrations in neonates with in utero raltegravir exposure. The model predicted rising or decreasing neonatal elimination profiles based on the time of maternal drug administration relative to time of birth and degree of in utero drug disposition into the central and peripheral compartments. Based on this model, it is recommended to delay the first oral dose of raltegravir until 1–2 days of age in those neonates born to mothers who received raltegravir during pregnancy, labor, and delivery.
The actual maternal information was limited to one PK observation per mother; therefore, the maternal component of the PK model is unlikely to appropriately describe the general PK of women who received raltegravir during pregnancy, labor, and delivery. This, for example, resulted in a high shrinkage on maternal bioavailability (51.4%; Table 2). However, the maternal observations do carry information about prebirth raltegravir exchange between the mother and fetus. The purpose of the maternal model was to capture this information to better estimate the initial raltegravir concentrations in the central and peripheral compartments of raltegravir‐exposed neonates. It was shown that these initial concentrations are determined by the timing of raltegravir dosing to the mother.
Consistent with infant UGT1A1 maturation kinetics,15, 16 the population PK model set raltegravir CL to be nil at birth and to gradually increase with age to reach 90% of its maximum value in approximately 11 weeks. The KA was slower in neonates during the first days of life when compared with older infants, children, and adults.28 This was accounted for by the implementation of a hyperbolic function to describe an age‐related increase in raltegravir KA, with KA reaching a maximum within 2 weeks of age. The covariate analysis did not identify any significant and clinically relevant effect of previous exposure to in utero raltegravir, feeding pattern, sex, or BSA on PK parameters. Only age and BW were retained as structural covariates. The final model passed a range of diagnostic tests. However, model qualification was achieved at the expense of multiple assumptions because of the sparsity of data inherent in PK trials in neonates.
The lack of capacity to eliminate raltegravir at birth had consequences on the shape of the concentration‐time profiles in the raltegravir‐exposed neonates. It was captured by the integrated mother–neonate PK model that concentrations may have decreased, initially remained steady, or even increased after birth, depending on whether birth occurred shortly after, approximately 6 hours after, or late after the last raltegravir dose administered to the mother, respectively. The rise of plasma concentrations after birth was attributed to concentrations in the peripheral compartment being greater than in the central compartment, resulting in a net flow back into the central compartment, while CL was approximately nil at birth. Plasma concentration was projected to continue to rise until the concentrations in the peripheral and central compartment were approximately equal (ignoring CL)and plasma concentrations began to decrease as a result of CL by glucuronidation.
Despite this variety of PK profiles, the simulations supported that dosing in raltegravir‐exposed neonates within 24–48 hours after birth resulted in trough concentrations consistently above the target trough of 75 nM. One limitation of the model was that raltegravir disposition was assumed to be the same during labor and delivery as during the third trimester of pregnancy. The rigors of labor may have an impact on drug disposition.29 Delayed gastric emptying and decreased gut blood flow during labor could delay drug absorption. Alterations in liver blood flow during labor could have an impact on drug metabolism. These effects may be different in women having a vaginal delivery when compared with those delivering by cesarean section without labor or following labor. There also may be differences in maternal drug disposition during labor and delivery because of medications administered for anesthesia and pain management. Because there are no data on the effects of labor and delivery on raltegravir disposition, it was assumed that raltegravir disposition during labor and delivery was unchanged when compared with the third trimester of pregnancy.
The analysis of observed and predicted trough concentrations demonstrated the necessity to increase total daily dose administration during the first 6 weeks in life to maintain the raltegravir trough concentrations in plasma above 75 nM. The recommended approximate dose of raltegravir granules for oral suspension in raltegravir‐unexposed infants is birth to day 7 of life, 1.5 mg/kg q.d.; 1–4 weeks of age, 3 mg/kg b.i.d.; 4 weeks and older, 6 mg/kg b.i.d., which represents an eightfold increase in dose during the first 4 weeks of life. Based on the modeling described here, the initial raltegravir dose should be delayed until 24–48 hours after birth in raltegravir‐exposed neonates to avoid an accumulation of excessive raltegravir plasma concentrations as a result of raltegravir acquired across the placenta during pregnancy. Both the simulations and clinical results from IMPAACT P1110 support the US and Europe product labeling, which recommends to delay the first oral dose of raltegravir until 1–2 days of age in those neonates born to mothers who received raltegravir during pregnancy, labor, and delivery.
Funding
Overall support for the International Maternal, Pediatric, Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases with cofunding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health, all components of the National Institutes of Health (NIH), under Awards UM1AI068632 (IMPAACT Leadership and Operations Center), UM1AI068616 (IMPAACT Statistical and Data Management Center), and UM1AI106716 (IMPAACT Laboratory Center), and by NICHD contract number HHSN275201800001I. Financial support for the studies was also provided by Merck & Co. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
A.C., L.W., and H.T. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (license holder Isentress). J.L., H.M., H.W., and T.K. are paid consultants for Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. M.M. received research support from Merck & Co., ViiV Healthcare, and Gilead Sciences. D.C. received research support from Merck &Co. and ViiV Healthcare and is the spouse of M.M. All other authors declared no competing interests for this work.
Author Contributions
All authors wrote the manuscript. D.C., H.T., M.M., E.P.A., S.N., and A.C. designed the research. T.K., J.L., H.M., and H.W. performed the research. J.L. and H.W. analyzed the data.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Table S1.
Supplementary Material S1. Description of the studies used for analysis.
Supplementary Material S2. Raltegravir assay description.
Supplemental References.
Acknowledgments
The authors acknowledge the International Maternal Pediatric Adolescent AIDS Clinical Trials Network and Flowers Lovern, Medical Writing Innovations, LLC (writing support).
References
- 1. United Nations Programme on HIV and AIDS. UNAIDS fact sheet – 2017 global HIV statistics <http://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf> (2018). Accessed November 8, 2018.
- 2. Mofenson, L.M. Prevention of mother‐to‐child HIV‐1 transmission – why we still need a preventive HIV immunization strategy. J. Acquir. Immune Defic. Syndr. 58, 359–362 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Panel on Opportunistic Infections in HIV‐Exposed and HIV‐Infected Children. Guidelines for the prevention and treatment of opportunistic infections in HIV‐exposed and HIV‐infected children <http://aidsinfo.nih.gov/contentfiles/lvguidelines/oi_guidelines_pediatrics.pdf> (2018). Accessed November 8, 2018.
- 4. Clarke, D.F. et al Raltegravir pharmacokinetics and safety in HIV‐1 exposed neonates: dose‐finding study. Conference on Retroviruses and Opportunistic Infections, Seattle, WA, February 13‐16, 2017.
- 5. Isentress [prescribing information] . <https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/022145s038,205786s007,0203045s015lbl.pdf> (2017). Accessed November 8, 2018.
- 6. Kassahun, K. et al Metabolism and disposition in humans of raltegravir (MK‐0518), an anti‐AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 35, 1657–1663 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Podany, A.T. , Scarsi, K.K. & Fletcher, C.V. Comparative clinical pharmacokinetics and pharmacodynamics of HIV‐1 integrase strand transfer inhibitors. Clin. Pharmacokinet. 56, 25–40 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawade, N. & Onishi, S. The prenatal and postnatal development of UDP‐glucuronyltransferase activity towards bilirubin and the effect of premature birth on this activity in the human liver. Biochem. J. 196, 257–260 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blonk, M.I. et al Raltegravir in HIV‐1‐infected pregnant women: pharmacokinetics, safety, and efficacy. Clin. Infect. Dis. 61, 809–816 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Watts, D.H. et al Raltegravir pharmacokinetics during pregnancy. J. Acquir. Immune Defic. Syndr. 67, 375–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clarke, D.F. et al Raltegravir pharmacokinetics in neonates following maternal dosing. J. Acquir. Immune Defic. Syndr. 67, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rizk, M.L. et al Population pharmacokinetic analysis of raltegravir pediatric formulations in HIV‐infected children 4 weeks to 18 years of age. J. Clin. Pharmacol. 55, 748–756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holford, N.H. A size standard for pharmacokinetics. Clin. Pharmacokinet. 30, 329–332 (1996). [DOI] [PubMed] [Google Scholar]
- 14. Krekels, E.H. , Danhof, M. , Tibboel, D. & Knibbe, C.A. Ontogeny of hepatic glucuronidation; methods and results. Curr. Drug Metab. 13, 728–743 (2012). [DOI] [PubMed] [Google Scholar]
- 15. McCarver, D.G. & Hines, R.N. The ontogeny of human drug‐metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J. Pharmacol. Exp. Ther. 300, 361–366 (2002). [DOI] [PubMed] [Google Scholar]
- 16. Miyagi, S.J. & Collier, A.C. The development of UDP‐glucuronosyltransferases 1A1 and 1A6 in the pediatric liver. Drug Metab. Dispos. 39, 912–919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clarke, D.F. , Wong, R.J. , Wenning, L. , Stevenson, D.K. & Mirochnick, M. Raltegravir in vitro effect on bilirubin binding. Pediatr. Infect. Dis J. 32, 978–980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andersen, D.H. , Blanc, W.A. , Crozier, D.N. & Silverman, W.A. A difference in mortality rate and incidence of kernicterus among premature infants allotted to two prophylactic antibacterial regimens. Pediatrics 18, 614–625 (1956). [PubMed] [Google Scholar]
- 19. Lommerse, J. et al Raltegravir dosing in neonates (IMPAACT P1110)—use of allometry and maturation in PK modeling to develop a daily dosing regimen for investigation during the first weeks of life. 24th Annual Conference of the Population Approach Group Europe, Hersonissos, Greece, June 2–5, 2015.
- 20. Lommerse, J. et al Raltegravir PK in neonates—an adaptive trial design to define an appropriate regimen for neonates from birth to 6 weeks of age. 7th Annual American Conference on Pharmacokinetics, Bellevue, WA, October 23–26, 2016.
- 21. Lommerse, J. et al Raltegravir PK in neonates—modeling rising and declining PK profiles of newborns exposed to raltegravir in‐utero. 7th Annual American Conference on Pharmacokinetics, Bellevue, WA, October 23–26, 2016.
- 22. Nachman, S. et al Pharmacokinetics and 48‐week safety and efficacy of raltegravir for oral suspension in human immunodeficiency virus type‐1‐infected children 4 weeks to 2 years of age. J. Pediatric. Infect. Dis Soc. 4, 76–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. A phase 1 trial to evaluate the safety and pharmacokinetics of raltegravir in HIV‐1‐exposed neonates at high risk of acquiring HIV‐1 infection. An international and domestic trial of the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT). Clinical study P1110. <https://clinicaltrials.gov/ct2/show/NCT01780831?term=P1110&rank=1> (2013). Accessed November 8, 2018.
- 24. Safety and pharmacokinetics (PK) of raltegravir in HIV (Human Immunodeficiency Virus)‐infected children and adolescents. Clinical study P1066. <https://clinicaltrials.gov/ct2/show/NCT00485264?term=p1066&rank=1> (2013). Accessed November 8, 2018.
- 25. Raltegravir pharmacokinetics and safety in neonates. A multicenter, US domestic and international trial of the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT). Clinical study P1097. <https://clinicaltrials.gov/ct2/show/NCT01828073?term=P1097&rank=1> (2013). Accessed November 8, 2018.
- 26. Miochnick, M. Antiretroviral pharmacology in pregnant women and their newborns. Ann. N. Y. Acad. Sci. 918, 289–297 (2000). [DOI] [PubMed] [Google Scholar]
- 27. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Batchelor, H.K. & Marriott, J.F. Paediatric pharmacokinetics: key considerations. Br. J. Clin. Pharmacol. 79, 395–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ansari, J. , Carvalho, B. , Shafer, S. & Flood, P. Pharmacokinetics and Pharmacodynamics of Drugs Commonly Used in Pregnancy and Parturition. Anesth. Analg. 122, 786–804 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Table S1.
Supplementary Material S1. Description of the studies used for analysis.
Supplementary Material S2. Raltegravir assay description.
Supplemental References.