

Abstract
Moxifloxacin is a widely used fluoroquinolone for the treatment of complicated intra‐abdominal infections. We applied physiologically‐based pharmacokinetic (PBPK) and population pharmacokinetic (popPK) modeling to support dose selection in pediatric patients. We scaled an existing adult PBPK model to children based on prior physiological knowledge. The resulting model proposed an age‐dependent dosing regimen that was tested in a phase I study. Refined doses were then tested in a phase III study. A popPK analysis of all clinical pediatric data confirmed the PBPK predictions, including the proposed dosing schedule in children, and supported pharmacokinetics‐related safety/efficacy questions. The pediatric PBPK model adequately predicted the doses necessary to achieve antimicrobial efficacy while maintaining safety in the phase I and III pediatric studies. Altogether, this study retroactively demonstrated the robustness and utility of modeling to support dose finding and confirmation in pediatric drug development for moxifloxacin.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The antibiotic moxifloxacin is a good candidate for pediatric populations because of its well‐established pharmacokinetic (PK) properties and well‐documented antimicrobial efficacy in adults. Physiologically‐based PK (PBPK) modeling is a useful tool for predicting safe doses for first‐in‐children clinical studies.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can a PBPK model predict safe and efficacious doses of moxifloxacin in a pediatric population?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Age‐adjusted and body weight–adjusted dosing regimens of moxifloxacin for two pediatric clinical studies were successfully predicted by scaling a PBPK model from adults to children. Data from two clinical studies in children were used by means of a pediatric population PK model to retrospectively validate the PBPK model in a learn‐and‐confirm approach.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The use of modeling and simulation techniques could lead to fewer subjects, increased efficiency, and less time and money required to complete early‐phase pediatric drug development programs.
Moxifloxacin is an eight‐methoxy‐fluoroquinolone antimicrobial with a broad spectrum of activity against most pathogens in complicated intra‐abdominal infections (cIAIs).1, 2 The benefits of moxifloxacin in adults with cIAIs have been demonstrated in a number of prospective, randomized, controlled clinical studies,3, 4, 5, 6 and the substance is recommended by the Surgical Infection Society and the Infectious Diseases Society of America for the treatment of adult patients with mild to moderate community‐acquired cIAIs.7
Current regulations from the US Food and Drug Administration (FDA) and the European Medicines Agency call for development strategies for pediatric dosing recommendations for drugs that have been approved in adults with the aim to maintain the efficacy and safety of adults in children younger than 18 years old.8, 9, 10, 11 One of the main obstacles to successful pediatric drug development is the planning of studies that can arrive at safe and effective doses for various age groups. To recommend these doses, quantitative tools are required. Physiologically‐based pharmacokinetic (PBPK) modeling is useful for predicting pharmacokinetic (PK) parameters for pediatric clinical trials as it can account for developmental changes that affect the absorption, distribution, metabolism, and excretion of drugs in children. Workflows and procedures for scaling adult PBPK models to pediatric populations have been successfully developed and tested.12, 13, 14, 15
To best benefit from incorporating PBPK modeling into the pediatric drug development process, the models should (i) be able to optimize the design of the pediatric PK study, (ii) be appropriate for a specific age group, and (iii) determine the best dose through a learn‐and‐confirm paradigm.16, 17 Successful PBPK models could help to minimize the number of pediatric patients and clinical trials necessary for pediatric drug development.
Here, we discuss how two complementary model‐based approaches, PBPK modeling and population PK (popPK) modeling, synergistically supported dose finding and dose confirmation for moxifloxacin in pediatric patients.
Methods
All PBPK models were built using PK‐Sim (version 4.2) and MoBi (version 2.3; both from Bayer AG, Leverkusen, Germany, and now available as part of the Open Systems Pharmacology Suite, www.open-systems-pharmacology.org). All optimizations and batch mode simulations were done using MATLAB (R2009a; MathWorks, Natick, MA) and the MoBi Toolbox for MATLAB (version 2.2; Bayer AG, Leverkusen, Germany).
Data sources
Moxifloxacin PK data were obtained from the following two clinical studies covering an age range from birth to 18 years: a phase I study (N = 31) in pediatric patients18 and a phase III study (N = 451; 301 patients were exposed to moxifloxacin and 150 to a comparator) in pediatric and adolescent patients with cIAIs.19 Briefly, in the phase I study, subjects received a single dose IV treatment of moxifloxacin, ranging from 5 to 10 mg/kg. In the phase III study, 301 subjects received multiple i.v. followed by oral (p.o.) doses of moxifloxacin, and of those, 155 contributed samples for PK data. A switch to oral treatment was done in 28 subjects. The doses were selected to target exposure equivalent to that observed after the recommended 400 mg dose for adults.19 The studies were conducted in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. Study protocols and amendments were approved by independent ethics committees in all participating countries of the global study. All participants provided written informed consent prior to study enrollment.
Pediatric PBPK model
On the basis of the adult PBPK model, the pediatric PBPK model for children aged 1 week to 18 years was established solely by scaling anthropometric and physiological parameters (i.e., body weight and height and the resulting organ volumes and blood flow rates), clearance (CL) processes, and age‐dependent protein binding in children. This information was incorporated from the default databases of the software that was used for the simulations in children (PK‐Sim).13, 20 No changes to any of the other input parameters of the adult model (e.g., physicochemical parameters) were made. To scale the intrinsic CLs, the activity of the enzyme in children was compared with the activity of the enzyme in adults per surface area (for gastrointestinal organs) or per organ weight (for all other organs). The calculation of intrinsic CL from plasma CL was done assuming well‐stirred conditions21, 22 and incorporated physiological information about the age‐dependent changes in body weight, liver/kidney weight, and hepatic/kidney blood flow.
Hepatic sulfate‐conjugation via Sulfotransferase 2A1 (SULT2A1) and glucoronidation via UDP‐glucuronosyltransferase 1A1 (UGT1A1) was scaled with age as previously described.20 It was assumed that both SULT2A1 and UGT1A1 activity in the gastrointestinal mucosa are fully developed at birth, i.e., the same activity per gram tissue weight as in adults.
The biliary CL of moxifloxacin and its metabolites is mediated via an unknown enzyme and/or transporter and is cytochrome P450 independent. As no ontogeny information about enzyme activity was available, this process was scaled to children only physiologically (i.e., same activity per gram tissue weight as in adults). The method of Hayton,23 as modified by Edginton et al.,20 was used to scale glomerular filtration.
Simulations of metabolite M1 and metabolite M2 are not shown here; PK data are presented elsewhere.18
The pediatric PBPK model provides a relationship between dose and the expected exposure of moxifloxacin in a virtual pediatric target population. In combination with PK target criteria, for example, an exposure target range based on efficacy and safety considerations (see later), a dosing regimen per age or weight group can be derived that fulfills these criteria.
Virtual target populations
The following two virtual pediatric target populations were generated using the population module of PK‐Sim:24
Target population A: age range from 7 days to 14 years split into 22 age groups (7 and 14 days; 1, 2, 3, 6, and 9 months; 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, and 14 years), body weight less than 45 kg.
Target population B: age range from 12–18 years split into 7 age groups (12, 13, 14, 15, 16, 17, and 18 years), body weight more than 45 kg. Each age group consisted of 500 male and 500 female virtual individuals summing up to a total of 29,000 simulated virtual children.
Various different dosing schemes (absolute and normalized to body weight) have been explored by simulation including, e.g., administration as i.v. infusion for 60 minutes for a hypothetical dose of 5 mg/kg for target population A and 400 mg p.o. for target population B.
The population module creates a number of virtual individuals within a given age, body weight, and body height or body mass index range on a stochastic approach using age‐dependent distributions of demographic and physiological parameters.24
Target exposure ranges for moxifloxacin
Target exposure ranges to interpret the modeling results were based on the FDA Pediatric Study Decision Tree.25 For diseases with comparable progression, response to intervention, and PK/pharmacodynamic (PD) relationship, a clinical dose can be defined by equivalence in exposures between adults and pediatric patients in combination with phase III safety studies.26
The main clinical surrogate parameter for fluoroquino‐lones is the 24‐hour area under the concentration‐time curve (AUC(0–24 h))/minimum inhibitory concentration, which correlates with antimicrobial efficacy. AUC(0–24 h)/minimum inhibitory concentration ratios exceeding a clinical breakpoint indicate clinical efficacy, whereas values below this threshold indicate an increased risk of therapeutic failure and development of resistant bacteria. Conversely, the maximum plasma concentration (Cmax) is predominantly used to evaluate clinical safety (e.g., cutoff to avoid concentration‐related side effects); however, there is often no established PK/PD relationship available to enable concrete dosing decisions. Although the predictions based on AUC(0–24 h) may suggest clinical dosing schedules, Cmax‐based predictions are more likely to be safety indicators for concentration‐related side effects (e.g., central nervous system). The latter must therefore be confirmed in phase III clinical trials.
In this modeling study, AUC estimates were used as primary parameters; Cmax data were considered secondary. Target exposure ranges were defined based on the observed distributions of AUC: AUC(0–24 h) after study drug administration at steady state and Cmax in the pooled adult phase I population following i.v. (as 60‐minute infusion) or p.o. administration of 400 mg moxifloxacin:18, 27 The minimum and maximum that were observed in adults were 11.8 and 71.7 mg·hour/L for AUC(0–24 h) and 1.5 and 6.9 mg/L for Cmax, with more than 95% between 20–60 mg · hour/L and 2–6.5 mg/L, respectively.28
The primary target achievement criterion for predicting a clinically effective dose was a narrower AUC range of 30–60 mg · hour/L (representing more than 90% of the observed study values28). In relation to the PK/PD relationship, this is a more conservative approach to avoid dose predictions that may increase the risk of emerging resistance in clinical trials as a result of exposure and decrease the likelihood of exceeding the maximum exposure observed in adults. For Cmax, an unchanged range of 2–6.5 mg/L was defined, as model predictions for this parameter are rather descriptive because of the reasons outlined previously.
Dose predictions in children
For target population A, the plasma concentration‐time courses of moxifloxacin were predicted following a single i.v. infusion for 60 minutes for a hypothetical dose of 5 mg/kg. For target population B, the plasma PK were predicted following a single p.o. dose of 400 mg moxifloxacin. From these predictions, the distributions for AUC from time 0 to infinity (AUCinf) and Cmax, CL (CL = DOSE/AUCinf, following i.v. administration), and the volume of distribution at steady state (V ss = CL•mean residence time, following i.v. administration) of moxifloxacin were calculated.
The PBPK‐predicted AUCinf and Cmax distributions for children were compared with the target exposure ranges for moxifloxacin from adult data to provide appropriate starting doses for the phase I clinical trial.18, 28 During and after conclusion of the phase I trial followed by an assessment of the PK and safety data observed for the starting doses, the dosing regimens for a subsequent phase III clinical trial were defined as described in detail in ref.19 In addition to the PBPK predictions that related pediatric doses to the expected exposure of moxifloxacin in children, a plethora of other aspects relevant for safety and efficacy were also taken into consideration.19
Pediatric popPK model
Data from the pediatric phase I and III trials were pooled for a popPK analysis to characterize the variability of moxifloxacin PK in pediatric patients and to quantify the influence of potential covariates on variability. Model development was guided by previously developed popPK models for moxifloxacin.29, 30, 31, 32 Model selection was done on the basis of (i) successful minimization and covariance step, (ii) number of significant digits, (iii) standard error of estimates, (iv) acceptable gradients at the last iteration, (v) correlation between model parameters, (vi) objective function value, and (vii) visual inspection of diagnostic plots.33 Structurally, linear two‐compartmental and three‐compartmental models with first‐order elimination and intercompartmental CL were tested. As suggested by an exploratory analysis (for details, see the Supporting Information), all CL and volume parameters were a priori scaled by body weight using an allometric model with commonly accepted scaling coefficients of 0.75 (for CL) and 1.0 (for volumes). In the covariate analysis, the influence of age, serum creatinine, estimated glomerular filtration rate, study (phases I or III), and sex on the PK of moxifloxacin were investigated by the stepwise forward inclusion/backward elimination procedure at a P value of < 0.01 and < 0.001, respectively.34 During model development, the assessment of outlier concentrations was done according to Byon et al.33 PopPK analysis was carried out using nonlinear mixed effects modeling (NONMEM) (version 7.2; ICON Development Solutions, Dublin, Ireland) with the Navigator workbench (version 9.1.5146; Mango Solutions, London, UK) on a Red Hat Enterprise (Raleigh, NC) Linux 6.3 environment. The final popPK model was qualified using prediction‐corrected visual predictive checks by comparing the 90% prediction interval of simulated concentrations with the observed moxifloxacin concentrations valid for the final model estimation.35 Goodness‐of‐fit plots were created on the basis of all data and stratified for study and route of administration (phase I/i.v. administration only, phase III/i.v. administration only, and phase III/p.o. administration only).
Retrospective evaluation of the PBPK model
For the retrospective evaluation of the precision of the PBPK model, the individual PK parameter estimates for CL and V ss derived from the final popPK model were plotted against age and body weight together with the CL and V ss predictions previously derived from the PBPK model.
Confirmation of dose predictions
Individual AUC(0–24 h) and Cmax at steady state were estimated from the final popPK model and plotted along with the adult antimicrobial and safety target ranges for AUC(0–24 h) and Cmax, respectively. The proposed age‐dependent and weight‐dependent dosing scheme in the phase III study was assessed by the percentage of pediatric patients that fall within or outside of the target ranges for AUC(0–24 h) and Cmax as established based on the pooled adult phase I data.
Results
Demographic analysis
In the phase I study, two subjects were ≥12 years and 29 were <12 years of age. In the phase III study, 98 subjects were ≥12 years and 57 were <12 years of age. In total, only 11 subjects younger than 3 years of age contributed PK information (10 from phase I and 1 from phase III).
Prediction of the phase I starting doses
The PBPK predictions for the virtual target population A show that the moxifloxacin AUCinf for an exemplary 5 mg/kg i.v. dose was at the lower end of the efficacy target range, but still above the observed minimum of the adult phase I population for AUC(0–24 h) at steady state for almost all children (Figure S2 ). Only some 2‐year‐old and 3‐year‐old female and male individuals were predicted to be exposed to less than 11.8 mg · hour/L, and some 7‐day‐old male individuals were predicted to be exposed to more than 71.7 mg · hour/L. If the hypothetical dose is doubled (i.e., 10 mg/kg as 1‐hour infusion), the population median for AUCinf would still be within the target exposure range for efficacy in children down to 2 months of age. Higher doses were associated with an increased risk for Cmax observations outside of the safety exposure range unless the infusion time was prolonged and/or the daily dose was split into two or more doses.
On the basis of these PBPK simulations, the following age‐adjusted and body weight–adjusted moxifloxacin doses were recommended for the pediatric phase I trial: 5–6 mg/kg for school children (≥6 to ≤14 years), 7–8 mg/kg for preschool children (≥2 to <6 years), and 9–10 mg/kg for infants and toddlers (>3 months to <2 years).18 As starting doses, the lower end of the recommended dose range per age cohort (i.e., 5 mg/kg for school children, 7 mg/kg for preschool children, and 9 mg/kg for infants and toddlers) were tested.
The corresponding predictions for virtual target population B show that the centers of the distributions for moxifloxacin AUCinf and Cmax were within the target ranges for efficacy and safety for all of these age groups (Figure S3 ). Only some female and male 12‐year‐olds and 13‐year‐olds were predicted to be exposed to a slightly higher AUCinf than the observed adult phase I maximum of 71.7 mg · hour/L. For Cmax, only some 12‐year‐old females were predicted to be exposed to a marginally higher Cmax than 6.9 mg/L.
PopPK model in children
A total of 186 pediatric subjects with 1,562 moxifloxacin plasma concentrations were available for popPK model development. During model development, 33 concentrations were identified as influential outliers and thus excluded from the final model. Furthermore, 14 concentrations from 12 subjects were maintained as noninfluential outliers.
The final popPK model of moxifloxacin in the pediatric population aged 3 months to ˂18 years consisted of a linear three‐compartment model with elimination from the central compartment. Interindividual variability was identified for CL and the central volume of distribution (Table S2 ). Residual variability was described by a proportional error model, with separate estimates for the different studies and routes of administration. The covariate analysis did not yield any significant effect on top of the relation between body weight and the disposition parameters, which was a priori included in the model. Goodness‐of‐fit plots and prediction‐corrected visual predictive checks show that the pediatric popPK model adequately described the data observed in the pediatric clinical phase I and III trials (Figures S4–S6 ).
Retrospective evaluation of the PBPK model
The comparison of PBPK predictions with the individual popPK post hoc estimates shows that the individual popPK parameter estimates of V ss over age and body weight were consistent with the PBPK predictions (Figure 1 a,b). Similarly, the individual popPK parameter estimates of V ss normalized to body weight over age were consistent with the PBPK predictions (Figure 1 c). With regard to CL, the individual popPK parameter estimates vs. age and body weight were also in accordance with the PBPK predictions (Figure 1 d,e). Similarly, the individual popPK parameter estimates of CL normalized to body weight over age were consistent with the PBPK predictions (Figure 1 f).
Figure 1.
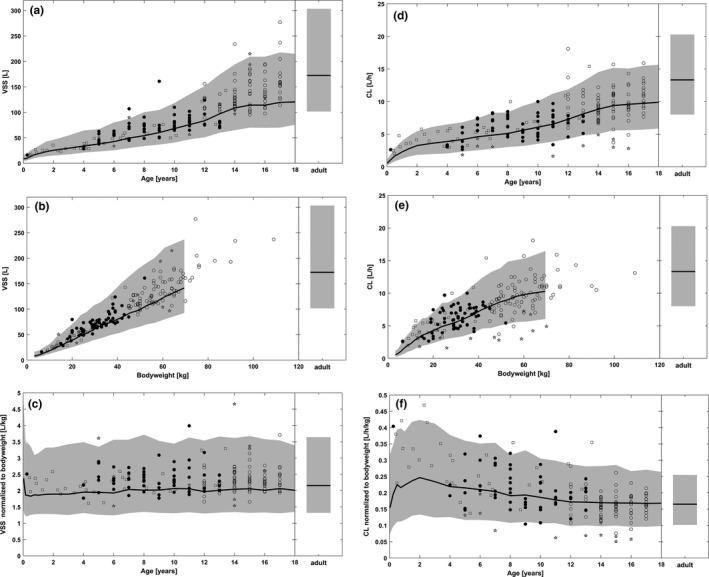
Comparison of PBPK–predicted
V
ss
to pop
PK
model‐derived
V
ss
vs. (a) age or (b) body weight and (c)
V
ss
normalized to body weight vs. age. Comparison of
PBPK
‐predicted
CL
to pop
PK
model‐derived
CL
vs. (d) age or (e) body weight and
(f) CL
normalized to body weight vs. age. Bold solid line indicates median of
PBPK
prediction; gray shaded area, percentiles 5–95 of the population
PBPK
prediction; symbols, pop
PK
model‐derived individual
PK
parameters for the subjects of the phase I study with single‐dose
intravenous (i.v.)
administration of 5–10 mg/kg (open squares), the subjects of the phase
III
study with multiple‐dose
i.v.
/
oral (p.o.
) administration of 400 mg once daily (open circles), the subjects of the phase
III
study with multiple‐doses
i.v.
/
p.o.
administration of 4–6 mg/kg twice daily (filled circles), and the subjects of the phase
III
study with any treatment and with at least one noninfluential outlier concentration included in the analysis (filled circles). CL, clearance; PBPK, physiologically-based pharmacokinetic; popPK, population pharmacokinetic;
V
ss
, volume of distribution at steady state.
 , median of simulated population;
, median of simulated population;  , 5th – 95th percentile of simulated population;
, 5th – 95th percentile of simulated population;  , values_11643_0;
, values_11643_0;  , values_11643_0_trea_2;
, values_11643_0_trea_2;  , values_11643_1;
, values_11643_1;  , values_11826
, values_11826
Confirmation of dose predictions
Plotted PK parameters at steady state derived from the popPK model showed that for AUC(0‐24 h), the median of all i.v. treatment groups combined was 41.7 mg · hour/L, with a deviation of −7.3% from the median of the target range of 45 mg · hour/L (Figure 2 a,b). Of the 143 subjects, 117 subjects (81.8%) were within the target range for AUC(0–24 h), 10 subjects (7.0%) were below the lower end, and 16 subjects (11.2%) were above the upper end of the target range for AUC(0–24 h), indicating a nearly even distribution (Table 1). For Cmax, the median value of all i.v. treatment groups combined was 4.83 mg/L. Of the 143 subjects, 103 subjects (72.0%) were within the target range for Cmax of 2–6 mg/L, and none were below the lower end. Of the 40 subjects (28.0%) that were above the upper end of the target range for Cmax, 39 were aged between 12 and 18 years, more than 45 kg body weight, and received 400 mg once daily as an i.v. treatment (Table 1). If peak concentrations outside the target range pose a safety risk, switching from once daily to twice daily dosing could be reasonable to avoid peak concentrations above the upper limit of the target range while maintaining the AUC(0–24 h). To test this concept, the effect of a 200 mg twice daily regimen on Cmax concentrations was simulated for the subjects in the phase III study who had been actually administered with 400 mg once daily. As expected, a 200 mg twice daily regimen improved the attainment with respect to the Cmax target range (Figure 3).
Figure 2.
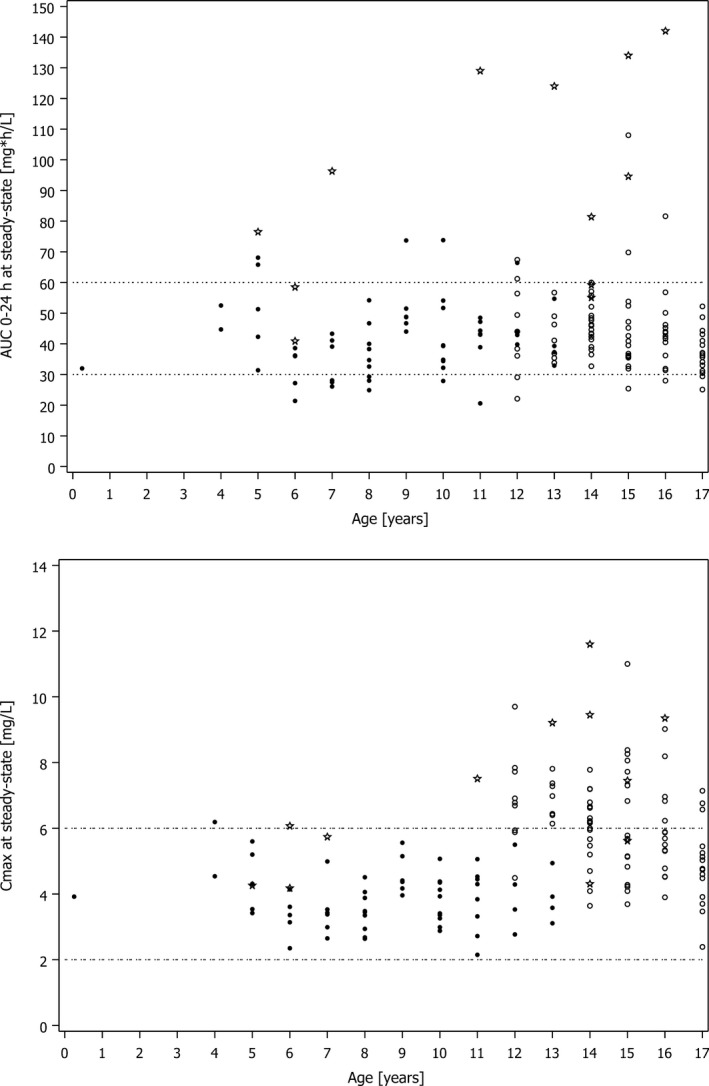
Pharmacokinetic (PK) parameters at steady state following intravenous (i.v.) administration vs. age in the phase III clinical study based on the population PK model for AUC (0–24 h) and C max . Open circles indicate subjects with multiple‐dose i.v. administration of 400 mg once daily; filled circles, subjects with multiple‐dose i.v. administration of 4–6 mg/kg twice daily; asterisks, subjects with any treatment and with at least one noninfluential outlier; dashed lines, limits of the C max target range. AUC (0–24 h) , 24‐hour area under the concentration‐time curve; C max , maximum plasma concentration.
Table 1.
Attainment rates with respect to the target ranges for AUC(0–24 h) and Cmax following i.v. and p.o. treatments, excluding subjects with noninfluential outlier concentrations
| Target range for AUC(0–24 h) (30–60 mg · hour/L) | i.v. treatment | p.o. treatment | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Within | Below | Above | Within | Below | Above | |||||||
| N | % | N | % | N | % | N | % | N | % | N | % | |
|
12 to <18 years (≥45 kg) Dose: 400 mg o.d. |
72 | 86.7 | 6 | 7.2 | 5 | 6.0 | 20 | 95.2 | 0 | 0.0 | 1 | 4.8 |
|
12 to <18 years (<45 kg) 6 to <12 years (all BWs) Dose: 4 mg/kg b.i.d. |
39 | 75.0 | 10 | 19.2 | 3 | 5.8 | 3 | 50.0 | 2 | 33.3 | 1 | 16.7 |
| 2 to <6 years (all BWs) Dose: 5 mg/kg b.i.d. | 5 | 71.4 | 0 | 0.0 | 2 | 28.6 | 0 | 0.0 | 0 | 0.0 | 1 | 100.0 |
|
3 months to <2 years (all BWs) Dose: 6 mg/kg b.i.d. |
1 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
|
All treatments N = 143 (i.v.);N = 28 (p.o.) |
117 | 81.8 | 16 | 11.2 | 10 | 7.0 | 23 | 82.1 | 2 | 7.1 | 3 | 10.7 |
| Target range for Cmax (2–6 mg/L) | i.v. treatment | p.o. treatment | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Within | Below | Above | Within | Below | Above | |||||||
| N | % | N | % | N | % | N | % | N | % | N | % | |
|
12 to <18 years (≥45 kg) Dose: 400 mg o.d. |
44 | 53.0 | 0 | 0.0 | 39 | 47.0 | 21 | 100 | 0 | 0.0 | 0 | 0.0 |
|
12 to <18 years (<45 kg) 6 to <12 years (all BWs) Dose: 4 mg/kg b.i.d. |
52 | 100.0 | 0 | 0.0 | 0 | 0.0 | 1 | 16.7 | 5 | 83.3 | 0 | 0.0 |
|
2 to <6 years (all BWs) Dose: 5 mg/kg b.i.d. |
6 | 85.7 | 0 | 0.0 | 1 | 14.3 | 1 | 100.0 | 0 | 0.0 | 0 | 0.0 |
|
3 months to <2 years (all BWs) Dose: 6 mg/kg b.i.d. |
1 | 100.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
|
All treatments N = 143 (i.v.); N = 28 (p.o.) |
103 | 72.0 | 0 | 0.0 | 40 | 28.0 | 23 | 82.1 | 5 | 17.9 | 0 | 0.0 |
AUC(0–24 h), 24‐hour area under the concentration‐time curve; b.i.d., twice daily; BWs, body weights; Cmax, maximum plasma concentration; i.v., intravenous; o.d., once daily; p.o., oral.
Figure 3.
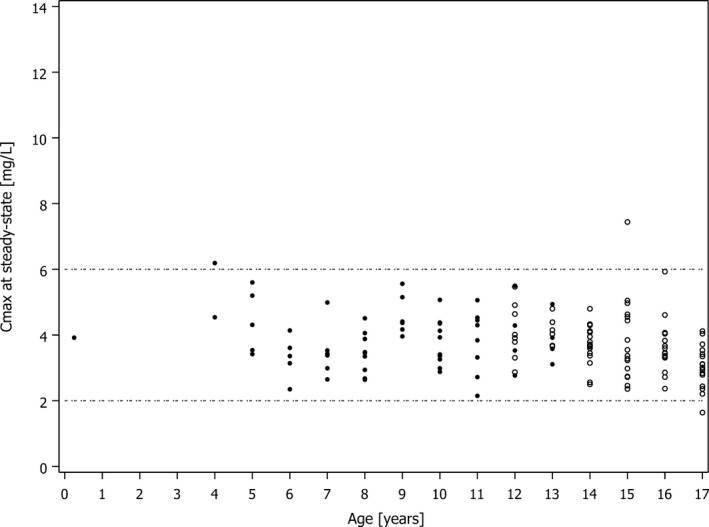
Simulated C max at steady state after intravenous (i.v.) administration of 200 mg moxifloxacin twice daily for subjects in the phase III study who actually had been administered with 400 mg once daily vs. C max at steady state after actual twice daily i.v. administration. Open circles indicate C max at steady state after i.v. administration of 200 mg twice daily for subjects in the phase III study who actually had been administered with 400 mg once daily; filled circles, C max at steady state after actual twice daily i.v. administration of 4 mg/kg to 6 mg/kg in the phase III study; dashed lines, limits of the C max target range. C max , maximum plasma concentration.
Discussion
Pediatric drug development remains a challenge for the pharmaceutical industry because of the various ethical, technical, regulatory, and legal requirements.36, 37, 38 Clinical trials in pediatric patients should be designed to maximize the amount of generated information while minimizing the risks for the patient. However, because of the difficulties of performing well‐conducted PK and PD studies in children, many drugs are used “off‐label,”39, 40 and dosing is often only based on body weight or body surface area. This allometric scaling approach ignores age‐related developmental changes in children such as enzyme ontogeny, CL, and protein binding. PBPK modeling takes these changes into account and allows for a more accurate prediction of PK parameters in children.41 The “learn and confirm” approach of PBPK modeling is suitable for future applications and allows optimization of dosing and sampling times in clinical studies. Recommended by US and European regulatory bodies,8, 9, 10, 11 pediatric PBPK modeling could lead to fewer subjects, increased efficiency, and less time and money required to complete drug development programs.
Here, we present how PBPK modeling and popPK modeling supported dose finding and dose confirmation in pediatric patients for the fluoroquinolone moxifloxacin. The mechanism of action of moxifloxacin and PK/PD parameters that correlate with efficacy for the treatment of cIAIs in adults should theoretically be similar for children. If we assume an identical exposure–response relationship in children and adults, dose optimization is reduced to the prediction of an age‐related and/or weight‐related dose–exposure relationship. This aims to obtain pediatric doses that result in the same exposure as observed in adults at therapeutically effective doses.42 To achieve antimicrobial efficacy while maintaining safety, pediatric dosing should lead to PK parameters (i.e., AUC, Cmax) within the 90% confidence intervals of the adult reference model. A dose of 400 mg moxifloxacin once daily is effective in treating bacterial infections including cIAIs in adults3, 4, 5, 6 and was used as the basis for establishing the target ranges for AUC and Cmax in children.
The pursued strategy is shown in Figure 4. In short, the starting point was a previously developed adult PBPK model for moxifloxacin43 that had been further refined and qualified using available adult PK data. This adult PBPK model was scaled to children using knowledge about age‐related changes of anatomical, physiological, and biochemical processes relevant for the absorption, distribution, metabolism, and excretion of moxifloxacin. The output of the resulting pediatric PBPK model was thus a solely physiologically informed estimation of the dose–exposure relationship of both i.v. and p.o. administered moxifloxacin in children of different ages. Based on the known pharmacological profile of moxifloxacin and a therapeutic target range established in adults, an age‐dependent dosing scheme for moxifloxacin could be proposed (learning step) and tested in the first‐in‐children trial.18, 44 The analysis of the uncertainties associated with model‐based predictions using phase I results as a reference resulted in refined doses that were confirmed in the larger phase III study investigating moxifloxacin treatment for cIAIs in children.19 After the completion of both trials, a pediatric popPK model for moxifloxacin was developed, and the resulting PK parameters were compared with the initial PBPK predictions to (i) retrospectively evaluate the PBPK model predictability, (ii) confirm the proposed dosing schedule of moxifloxacin in children, and (iii) support PK‐related clinical safety/efficacy questions (confirmation step).
Figure 4.
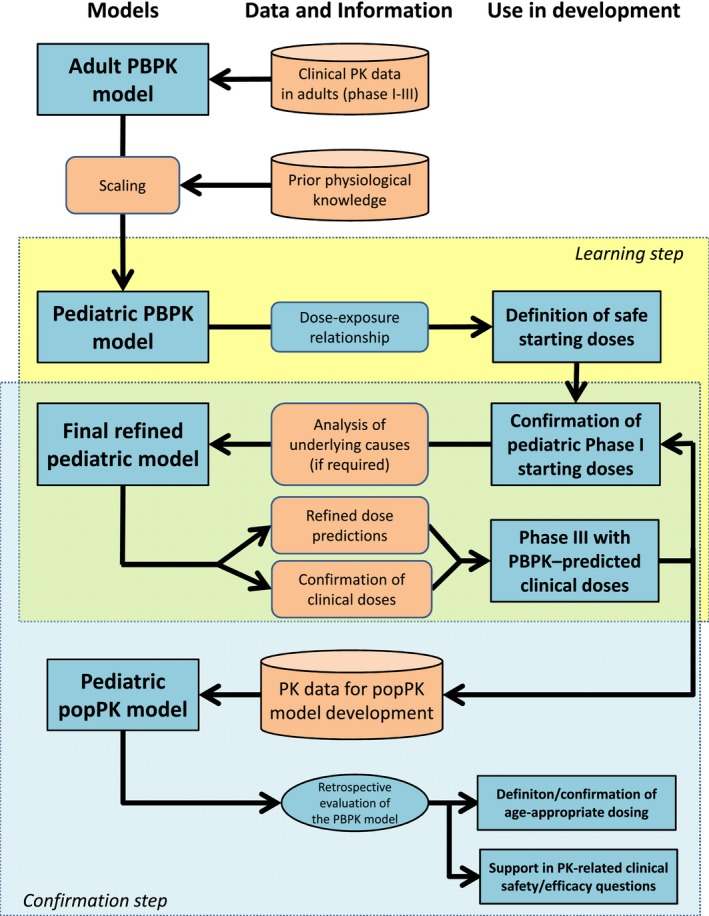
Schematic diagram of the modeling and simulation steps during pediatric development of moxifloxacin. PBPK, physiologically‐based pharmacokinetics; PK, pharmacokinetics; popPK, population pharmacokinetics.
The pediatric PBPK model for moxifloxacin presented here well predicted the doses necessary to achieve antimicrobial efficacy while maintaining safety in the phase I and phase III pediatric studies. In general, the predictions for the adult population provide a larger volume of distribution when compared with adolescents, reflecting a higher body weight for adults. In this study, however, the volume of distribution and CL at steady state from the retrospective popPK model were in good agreement with the PBPK predictions. The PBPK predictions did not cover the full body weight range, as some pediatric patients in the clinical studies were heavier than the heaviest predicted virtual patients. On average, CL was slightly underpredicted in children younger than 3 years of age; however, there was a very low number of subjects in this age group, which hampers the conclusions that can be made. No parameter optimization was performed for the pediatric PBPK model after the PK data in children became available.
As is often the case in pediatric trials, young children were underrepresented. In this investigation, there were only 10 individuals younger than 3 years of age included in the phase I trial, and only 1 subject in this age group contributed PK information in the phase III trial. Thus, the age group younger than 3 years only represented 5.9% of the total study population.
Comparison of the clinical study results to the antimicrobial and safety target ranges for AUC(0–24 h) and Cmax established for adults45 showed that the vast majority of pediatric patients were within these ranges. The upper limit of the peak plasma concentration target range was exceeded more often in the age group 12 ≤ 18 years (more than 45 kg) who received moxifloxacin once daily when compared with the other age groups who received a twice‐daily administration. There is a risk of underdosing or overdosing at the edges of the pediatric age range if only scaling to body weight is performed, and an altered safety profile is likely to be seen when only body weight is taken into account. The dose‐optimization strategy is discussed in detail in refs. 19,44. PK simulations based on the final popPK model show that an alternative dosing scheme (200 mg twice‐daily i.v. administration) can be developed for subjects aged 12 ≤ 18 years instead of 400 mg once‐daily i.v. administration to achieve lower peak plasma concentrations while keeping exposure constant. This may be used to improve safety margins for concentration‐related adverse effects if clinically indicated.
Thus, the PBPK model provided initial dosing proposals for the phase I study. During conduction of the phase I and III studies, these dose recommendations were subject to clinical validation and assessment through independent safety boards, allowing for the adjustment of the recommended doses if deemed necessary. The details of the clinical dose adjustment steps are presented in the clinical papers describing the phase I and III trials of moxifloxacin in children.19, 44
Summary and Conclusions
Modeling and simulation have been extensively applied during different phases of the pediatric development of moxifloxacin. Following the “learn and confirm” paradigm, PBPK modeling (learning step) and popPK modeling (confirmation step) have been successfully applied to the development of moxifloxacin in children and supported the dose selection for pediatric patients. This moxifloxacin case example demonstrates the usefulness of modeling and simulation approaches to support dose finding and dose confirmation in pediatric drug development.
Funding
The study was sponsored by Bayer AG, Leverkusen, Germany.
Conflict of Interest
S.W., M.F., G.S., K.C., T.W., T.E., J.L., and H.S. are employees and potential stockholders of Bayer AG.
Author Contributions
S.W., M.F., G.S., and H.S. wrote the manuscript. S.W., K.C., T.W., T.E., and J.L. designed the research. S.W., M.F., G.S., K.C., T.W., T.E., and H.S. performed the research. S.W., M.F., G.S., K.C., T.W., T.E., and J.L. analyzed the data.
Supporting information
Supplementary Material S1. Supplementary Methods, Supplementary Tables and Figures, Supplementary References.
Model Code.
Acknowledgments
The authors thank Maximilian Becker, PhD, (Bayer AG) for providing medical writing, data preparation, and editorial support.
References
- 1. Blot, S. , De Waele, J.J. & Vogelaers, D. Essentials for selecting antimicrobial therapy for intra‐abdominal infections. Drugs 72, e17–e32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rothrock, S.G. & Pagane, J. Acute appendicitis in children: emergency department diagnosis and management. Ann. Emerg. Med. 36, 39–51 (2000). [DOI] [PubMed] [Google Scholar]
- 3. De Waele, J.J. et al Randomised clinical trial of moxifloxacin versus ertapenem in complicated intra‐abdominal infections: results of the PROMISE study. Int. J. Antimicrob. Agents 41, 57–64 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Malangoni, M.A. , Song, J. , Herrington, J. , Choudhri, S. & Pertel, P. Randomized controlled trial of moxifloxacin compared with piperacillin‐tazobactam and amoxicillin‐clavulanate for the treatment of complicated intra‐abdominal infections. Ann. Surg. 244, 204–211 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Solomkin, J. , Zhao, Y.P. , Ma, E.L. , Chen, M.J. & Hampel, B. Moxifloxacin is non‐inferior to combination therapy with ceftriaxone plus metronidazole in patients with community‐origin complicated intra‐abdominal infections. Int. J. Antimicrob. Agents 34, 439–445 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Weiss, G. , Reimnitz, P. , Hampel, B. , Muehlhofer, E. & Lippert, H. Moxifloxacin for the treatment of patients with complicated intra‐abdominal infections (the AIDA Study). J. Chemotherapy 21, 170–180 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Solomkin, J.S. et al Diagnosis and management of complicated intra‐abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin. Infect. Dis. 50, 133–164 (2010). [DOI] [PubMed] [Google Scholar]
- 8. Committee for Medicinal Products for Human Use (CHMP) . European Medicines Agency Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population (CHMP/EWP/147013/04). https://www.ema.europa.eu/en/role-pharmacokinetics-development-medicinal-products-paediatric-population (2006). Accessed February 11, 2019.
- 9. US Food and Drug Administration . Summary minutes of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. https://wayback.archiveit.org/7993/20170404154933/https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM306989.pdf. (2012) Accessed February 11, 2019.
- 10. Wagner, C. et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA Public Workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao, P. et al Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 89, 259–267 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Edginton, A.N. Knowledge‐driven approaches for the guidance of first‐in‐children dosing. Paediatr Anaesth. 21, 206–213 (2011). [DOI] [PubMed] [Google Scholar]
- 13. Edginton, A.N. , Schmitt, W. & Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharmacokinet. 45, 1013–1034 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Johnson, T.N. , Rostami‐Hodjegan, A. & Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 45, 931–956 (2006). [DOI] [PubMed] [Google Scholar]
- 15. Willmann, S. et al Development of a paediatric population‐based model of the pharmacokinetics of rivaroxaban. Clin. Pharmacokinet. 53, 89–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chien, J.Y. , Friedrich, S. , Heathman, M.A. , de Alwis, D.P. & Sinha, V. Pharmacokinetics/pharmacodynamics and the stages of drug development: role of modeling and simulation. AAPS J 7, E544–E559 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 61, 275–291 (1997). [DOI] [PubMed] [Google Scholar]
- 18. Lettieri, J. et al . Safety, tolerability and pharmacokinetics (PK) of single dose intravenous moxifloxacin in pediatric patients. Clin. Pharmacol. Therapeut. 95(1 suppl.), S97–S98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wirth, S. et al Moxifloxacin in pediatric patients with complicated intra‐abdominal infections: results of the MOXIPEDIA randomized controlled study. Pediatr. Infect. Dis. J. 37, e207–e213 (2018). [DOI] [PubMed] [Google Scholar]
- 20. Edginton, A.N. , Schmitt, W. , Voith, B. & Willmann, S. A mechanistic approach for the scaling of clearance in children. Clin. Pharmacokinet. 45, 683–704 (2006). [DOI] [PubMed] [Google Scholar]
- 21. Ito, K. & Houston, J.B. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm. Res. 21, 785–792 (2004). [DOI] [PubMed] [Google Scholar]
- 22. Pang, K.S. & Rowland, M. Hepatic clearance of drugs. I. Theoretical considerations of a “well‐stirred” model and a “parallel tube” model. Influence of hepatic blood flow, plasma and blood cell binding, and the hepatocellular enzymatic activity on hepatic drug clearance. J. Pharmacokinet. Biopharm. 5, 625–653 (1977). [DOI] [PubMed] [Google Scholar]
- 23. Hayton, W.L. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS pharmSci. 2, 1–7 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willmann, S. , Lippert, J. & Schmitt, W. From physicochemistry to absorption and distribution: predictive mechanistic modelling and computational tools. Expert Opin. Drug Metab. Toxicol. 1, 159–168 (2005). [DOI] [PubMed] [Google Scholar]
- 25. US Food and Drug Administration . Exposure‐response relationships—study design, data analysis, and regulatory applications, guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/exposure-response-relationships-study-design-data-analysis-and-regulatory-applications (2003). Accessed February 11, 2019.
- 26. US Food and Drug Administration . General pharmacology considerations for drugs and biological products. Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-clinical-pharmacology-considerations-pediatric-studies-drugs-and-biological-products (2014). Accessed February 11, 2019.
- 27. Stass, H. , Kubitza, D. , Moller, J.G. & Delesen, H. Influence of activated charcoal on the pharmacokinetics of moxifloxacin following intravenous and oral administration of a 400 mg single dose to healthy males. Br. J. Clin. Pharmacol. 59, 536–541 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stass, H. et al Pharmacokinetics, safety, and tolerability of single‐dose intravenous moxifloxacin in pediatric patients: dose optimization in a phase 1 study. J. Clin. Pharmacol. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Florian, J.A. , Tornoe, C.W. , Brundage, R. , Parekh, A. & Garnett, C.E. Population pharmacokinetic and concentration–QTc models for moxifloxacin: pooled analysis of 20 thorough QT studies. J. Clin. Pharmacol. 51, 1152–1162 (2011). [DOI] [PubMed] [Google Scholar]
- 30. Kees, M.G. , Schaeftlein, A. , Haeberle, H.A. , Kees, F. , Kloft, C. & Heininger, A. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J. Antimicrob. Chemother. 68, 1331–1337 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Peloquin, C.A. et al Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob. Agents Chemother. 52, 852–857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu, L. , Wang, N. , Yang, W. , Zhang, Y. , Zhao, X. & Ji, S. Population pharmacokinetics of intravenous moxifloxacin 400 mg once‐daily dosage in infected patients. J. Infect. Chemother. 20, 621–626 (2014). [DOI] [PubMed] [Google Scholar]
- 33. Byon, W. et al Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT: Pharmacometr. Syst. Pharmacol. 2, 51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jonsson, E.N. & Karlsson, M.O. Automated covariate model building within NONMEM. Pharm. Res. 15, 1463–1468 (1998). [DOI] [PubMed] [Google Scholar]
- 35. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Institute of Medicine Roundtable on Research and Development of Drugs, Biologics, and Medical Devices . Raising awareness of regulatory, legal, and ethical issues In Rational Therapeutics for Infants and Children: Workshop Summary 5 (ed. Yaffe S.) 71–91 (National Academies Press, Washington, DC, 2000). [PubMed] [Google Scholar]
- 37. Leong, R. et al Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 91, 926–931 (2012). [DOI] [PubMed] [Google Scholar]
- 38. Turner, M.A. , Catapano, M. , Hirschfeld, S. & Giaquinto, C. Paediatric drug development: the impact of evolving regulations. Adv. Drug Deliv. Rev. 73, 2–13 (2014). [DOI] [PubMed] [Google Scholar]
- 39. Conroy, S. et al Survey of unlicensed and off label drug use in paediatric wards in European countries. BMJ 320, 79–82 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McIntyre, J. , Conroy, S. , Avery, A. , Corns, H. & Choonara, I. Unlicensed and off label prescribing of drugs in general practice. Arch. Dis. Child. 83, 498–501 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Edginton, A.N. & Willmann, S. Physiology‐based versus allometric scaling of clearance in children: an eliminating process based comparison. Paediatr. Perinat. Drug Ther. 7, 146 (2006). [Google Scholar]
- 42. Dunne, J. et al Extrapolation of adult data and other data in pediatric drug‐development programs. Pediatrics 128, e1242–e1249 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Edginton, A.N. , Ahr, G. , Willmann, S. & Stass, H. Defining the role of macrophages in local moxifloxacin tissue concentrations using biopsy data and whole‐body physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 48, 181–187 (2009). [DOI] [PubMed] [Google Scholar]
- 44. Stass, H. et al Pharmacokinetics, safety, and tolerability of single‐dose intravenous moxifloxacin in pediatric patients: dose optimization in a phase 1 study. J. Clin. Pharmacol. 59, 654–667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stass, H. & Dalhoff, A. The integrated use of pharmacokinetic and pharmacodynamic models for the definition of breakpoints. Infection 33(suppl. 2), 29–35 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1. Supplementary Methods, Supplementary Tables and Figures, Supplementary References.
Model Code.