

Abstract
Oligonucleotide-based agents have the potential to treat or cure almost any disease, and are one of the key therapeutic drug classes of the future. Bioconjugated oligonucleotides, a subset of this class, are emerging from basic research and being successfully translated to the clinic. In this Review, we first briefly describe two approaches for inhibiting specific genes using oligonucleotides—antisense DNA (ASO) and RNA interference (RNAi)—followed by a discussion on delivery to cells. We then summarize and analyze recent developments in bioconjugated oligonucleotides including those possessing GalNAc, cell penetrating peptides, α-tocopherol, aptamers, antibodies, cholesterol, squalene, fatty acids, or nucleolipids. These novel conjugates provide a means to enhance tissue targeting, cell internalization, endosomal escape, target binding specificity, resistance to nucleases, and more. We next describe those bioconjugated oligonucleotides approved for patient use or in clinical trials. Finally, we summarize the state of the field, describe current limitations, and discuss future prospects. Bioconjugation chemistry is at the centerpiece of this therapeutic oligonucleotide revolution, and significant opportunities exist for development of new modification chemistries, for mechanistic studies at the chemical–biology interface, and for translating such agents to the clinic.
Graphical abstract
INTRODUCTION
The molecular biology central dogma describes the transfer of biological information stored in genes to proteins using biomacromolecules.1 The biomacromolecule deoxyribonucleic acid (DNA) is found in all nucleated eukaryotic cells and it stores information via a specific sequence present in DNA. The first step in this unidirectional process of genes to proteins is transcription whereby ribonucleic acid (RNA) is generated from DNA (i.e., transcription). RNA is then spliced into the nucleus to retain only the coding part of the RNA, namely, the exons. The resulting messenger RNA (mRNA) is transferred to the cytoplasm, where the ribosome reads the information and produces a protein (i.e., translation). These biomacromolecules play key roles in all aspects of life from life cycle, pathogenesis, to healing and, thus, opportunities exist to control a biological outcome by intervening in transcription or translation.
In 1967, synthetic nucleoside derivatives were reported with the intent for specific base pairing with target sequences.2 In 1978, Summerton developed methods for inactivating sequences through cross-linking,3 and Zamecnik and Stephenson4,5 reported that short synthetic DNA fragments (called oligodeoxynucleotides, ODN) from the Rous sarcoma virus, possessing sequence binding complementary to RNA molecules, inhibited viral replication. This seminal discovery documented replication prevention of a viral RNA strain using a specific ODN—today known as antisense treatment.6 Several studies have also elucidated many of the molecular mechanisms underlying different human and animal pathologies. Thus, altering the expression of pathological genes, whether genetic or modified by mutation, is of keen interest and technologies are being developed for such purposes.
Antisense DNA (ASO) and RNA interference (RNAi) are two very promising technologies for inhibiting specific genes. The former utilizes a DNA fragment complementary to the target mRNA sequence. The latter approach, first described by R. Jorgensen7 and extensively studied by Andrew Z. Fire and Craig Mello,8 uses small interfering RNAs (siRNAs) to target the mRNA. Antisense oligonucleotides induce different inhibition mechanisms, which can occur in the cytoplasm and in the nucleus (Figure 1). After transcription of the DNA into pre-mRNA, several post-transcriptional modifications are performed, including splicing, capping at the 5′ end, or the polyadenylation of the 3′ end. The mRNA is then translocated into the cytoplasm by exportins, where it is then translated into proteins by ribosomes. A synthetic oligonucleotide elicits its activity in two cellular compartments, the cytoplasm or the nucleus. After hybridization of the oligonucleotide on mRNA, two inhibitory mechanisms are possible: (i) mRNA degradation or (ii) translational repression. mRNA is degraded upon hybridization with an oligonucleotide following RNase H recognition. Antisense DNA can also inhibit the translation step via a steric hindrance mechanism at the ribosome binding site9 or by prohibiting the ribosomal motion along the mRNA.10,11 Several mechanisms of inhibition are also possible in the nucleus, including the inhibition of post-transcriptional modifications, such as capping at the 5′ end, or by binding to polyadenylation sites at the 3′ UTR ends.12,13 Additionally, antisense may also inhibit or promote the inclusion of exons to alter the splicing of pre-mRNA (Figure 1).
Figure 1.

Mechanisms of antisense oligonucleotides. Normal expression of the gene and protein in the absence of ASO (1). After internalization, ASO hybridize to mRNA via complementary sequence pairing. The ASO/mRNA heteroduplex induces activation of RNase H and degradation of the mRNA (2) or ribosome blockage by steric hindrance (3). Both mechanisms result in inhibition of target protein production. Alternatively, penetration of ASO into the nucleus may lead to regulation of mRNA via inhibition of capping formation at the 5′ end (4), inhibition of splicing (5), or the activation of RNase H (6). Reproduced with permission from Chan, J. H. P., Lim, S., and Wong, W. S. F. (2006) Antisense Oligonucleotides: From Design to Therapeutic Application. Clin. Exp. Pharmacol. Physiol. 33, 533–540.12 Copyright 2006, Blackwell Publishing Asia Pty Lt.
In principle, protein regulation is accomplished by the use of either agonist14,15 or antagonist16–18 strategies, with the caveat that the target RNA sequence is known. Because they are exogenous molecules, synthetic oligonucleotides are substrates for endo- and exonucleases, and thus exhibit short half-lives both in vitro and in vivo. For example, after intravenous (IV) injection in monkeys, phosphodiester oligonucleotides are quickly degraded with a half-life of only 5 min.19 In order to overcome this low stability, oligonucleotides are chemically modified. Sites of chemical modification include the base, ribose, and the phosphate linkage. The types of modifications vary from classical to nonclassical bioisosteres.20 Additionally, one chemical modifications does not always address the same half-life limitation(s), and often two or more modifications are combined to increase oligonucleotide stability. During the last few decades, many different modification chemistries have been developed (Figure 2). One of the first chemical modifications reported is the replacement of the phosphodiester by a phosphorothioate linkage (PTO) to minimize oligonucleotide degradation. Interestingly, this modification does not interfere with the recruitment of RNase H and target RNA cleavage. Structures of the different chemical modifications and the knockdown mechanisms are shown in Figure 2.21–26 To improve the binding affinity and nuclease resistance, chemical modifications including the 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-MOE), and/or 2′-fluoro (2′-F) modifications of RNA are inserted into the structure. To enhance the binding affinity, locked nucleic acid (LNA) modifications are used where the conformational freedom of the ribose is restricted. More recently, Tricyclo-DNA (tcDNA) are reported as another constrained nucleotide featuring a three-ring scaffold. Importantly, these types of chemical modifications efficiently address the degradation issues. However, they do not improve the cellular uptake or targeting of the oligonucleotide to a specific site at the cellular, tissue, or organ level. Indeed, these hydrophilic polyanions do not easily cross a physiologic barrier (e.g., skin) or a cell membrane.
Figure 2.

Chemical modifications of synthetic oligonucleotides. Chemical modifications are incorporated at the phosphate, ribose, or at the 5′ or 3′ ends of the oligonucleotides. The type and the position of the modification induce three main mechanisms (steric blockers in green, activation of RNase H in blue, and RNA interference (RNAi) in purple). In the case of 2′ modification (2′-OMe, 2′-O-MOE, 2′NH2, 2′-F-RNA, and 2′-F-ANA), RNase H and RNAi mechanism are possible with a gapmer design. PTO: Phosphorothioate; CNA: Dioxaphosphorinane-Constrained Nucleic Acid; PNA: Peptide Nucleic Acid; PMO: Phosphorodiamidate Morpholino Oligonucleotide; E-VP: (E)-VinylPhosphonate; ddC: dideoxyCytosine; 2′-OMe: 2′ O-Methyl; 2′-O-MOE: 2′-O-MethOxy Ethyl; 2′-F RNA; 2′-F ANA: ArabinoNucleic Acid; LNA: Locked Nucleic Acid; ENA: 2′-O,4′-C-Ethylene-bridged Nucleic Acid; cEt: S-constrained Ethyl; tcDNA: tricycloDNA; FHNA: Fluoro Hexitol Nucleic Acid; UNA: Unlocked Nucleic Acid. Adapted with permission from Khvorova, A., and Watts, J. K. (2017) The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 35, 238–248.21 Copyright 2017, Nature Publishing.
Therapies must adhere to a wide range of stringent specifications in order to advance into clinical trials and succeed. An ideal oligonucleotide therapy is safe to administer, affordable to produce, exhibits a 2 year shelf life, possesses an extended half-life in blood and serum (>12 h), and inhibits intended targets effectively with low nonspecific or off-target interactions.27 That said, differing disease indications will require fine-tuning of the design requirements for optimal performance. As discussed below, a wide range of modifications to oligonucleotides improve one or many of these parameters, and the use of bioconjugation to oligonucleotides is primarily focused on improving the delivery of oligonucleotides to the cytosol or nucleus. Bioconjugates address a number of different oligonucleotide delivery challenges, such as improving biodistribution to a specific region or cell type (e.g., antibodies), promoting endosomal escape (e.g., CPP), increasing receptor-mediated transport (e.g., GalNAc), and/or increasing lipophilicity (e.g., cholesterol).
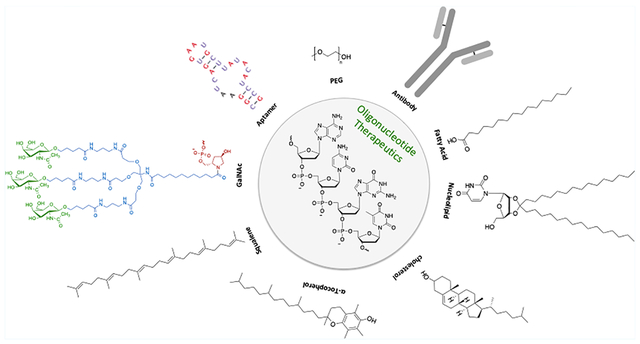
In this review, we discuss recent advances in the delivery of oligonucleotides. We briefly describe self-assembly approaches based on electrostatic, hydrophobic, or H-bonding interactions to give, for example, lipoplexes, and refer the reader to several comprehensive reviews on this topic.28,29 We focus on modified bioconjugated oligonucleotides for delivery, including GalNAc, cell penetrating peptides, α-tocopherol, aptamers, antibodies, cholesterol, squalene, fatty acids, nucleolipids, and bis-conjugates. We also describe relevant linking chemistries between the oligonucleotide and the bioconjugate with respect to molecular design. Additionally, the therapeutic applications of such bioconjugated oligonucleotides and outcomes from clinical trials are highlighted. Finally, we summarize the state of the field and discuss future prospects. Today, significant opportunities exist for development of new modification chemistries, for mechanistic studies at the chemical-biology interface, and for engineering systems for efficient delivery to the target site.
OLIGONUCLEOTIDE DELIVERY
Cellular uptake is one of the key steps in order for oligonucleotides to elicit their biological activity, as the target mRNAs and the cellular machinery are located in the cytoplasm and in the nucleus. Several transfection agents have been developed and commercialized for the intracellular delivery of oligonucleotides such as Oligofectamine 2000 (Invitrogen), JetPEI (Polyplus transfection), or K2 (Biontex). Many of these transfecting products are composed of cationic lipids, polyethylenimine, or DEAE-dextran, which self-organize and self-assemble in aqueous medium via electrostatic interactions with the negatively charged oligonucleotides. This strategy compacts the oligonucleotide into aggregates for subsequent endocytosis by the cell. Once inside the cell, the oligonucleotides are released into the cytoplasm. However, toxicity remains a disadvantage of these agents, as most transfecting agents are cationic, but alternatives are being investigated.30,31 Additionally, many of these complexes are not stable in the presence of serum, limiting or preventing their in vivo use.32,33 Following intravenous administration, the resultant oligonucleotide/transfecting agent complexes accumulate primarily in the liver, due to their size.34 Consequently, significant research efforts are dedicated to the delivery challenges in order to improve the transfection of nucleic acids.35–37 For example, poly(2-dimethylaminoethyl) methacrylate, when mixed with oligonucleotides,38 forms micelles of controlled sizes based on the N/P ratio. Upon adding an albumin coating to the surface, the cytotoxic effect of the polymer is minimized and cancerous cells are preferentially transfected relative to healthy cells.39 Another strategy employs modified viruses, which internalize nucleic acids in cells.40–42 However, this strategy can afford off-target toxicity since the oligonucleotides are not delivered in their synthetic form, but rather integrated into the viral genome. Lipid nanoparticles (LNP) are also being investigated as the nonviral transfecting cargo. However, the transfection efficiency is generally lower than that observed for viral transfection systems.43
Recently, LNPs loaded with siRNA targeting Polo-Like Kinase 1 (PLK1) protein, present in the triple negative breast cancer cell line (MDA-MB-231), have been modified with antibodies to target tumors. Biodistribution studies of labeled siRNA-LNPs demonstrated that antibody modified LNP (antibody against heparin-binding EGF-like growth factor, αHB-EGF) effectively delivered siRNA to tumor tissue in mice. Interestingly, the PLK1 protein expression was inhibited after intravenous injection of the LNPs and tumor growth was decreased. These results indicate that antibody modified LNPs loaded with siRNA are a promising therapeutic approach for breast cancer.44 Another recent article investigated the bioconjugation of antibodies to LNP for targeting endothelial cells lining the vascular lumen.45,46 These new particles were nontoxic both in vitro and in vivo as well as preferentially accumulated in the lungs. In contrast, the same nanoparticles without conjugated antibodies quickly accumulated into the liver, indicating that targeting moieties (antibodies/LNP bioconjugate) can avoid the hepatic uptake with endogenous serum protein like Apo-E.47
Alternatively, bioinspired molecules such as nucleolipids (NL) are being used to construct LNPs. NLs self-assemble to form unique supramolecular structures,48–52 and the NLs based LNPs loaded with nucleic acids successfully transfect plasmid DNA, siRNA, and antisense oligonucleotides to a number of different cell lines: human breast adenocarcinoma MCF-7 cells, human liver (HepG2), mouse fibroblast (NIH 3T3), Chinese hamster ovarian (CHO) cells, and human prostate cancer (PC-3) cells.51,53–55 Furthermore, these LNPs can be further modified to be stimuli-responsive, such as responding to changes in pH to enhance the delivery of nucleic acids.56 The above examples are representative and by no means comprehensive, as there are many formulations described for nucleic acid vectorization (Figure 3), and the reader is referred to several comprehensive reviews on the subject.57,58
Figure 3.

Strategies for the delivery of ASO and/or siRNA.57 A: Covalent conjugation between the agent and the oligonucleotide. B: Examples of stable and cleavable linkages. C: Formation of a complex between a biomolecule or a polymer and the oligonucleotide via electrostatic interactions and/or the hydrophobic effect. D: Formation of liposomal supramolecular structure with targeting or transfecting agent (PEG, peptide, CPP, protein, lipid glycoconjugate, etc.).
The retargeting of nucleic acids using viral vectors was investigated by Reynolds et al. in the 2000s.59,60 Viral vectors are attractive candidates for in vivo gene delivery because the infection efficacy is higher compared to other nonviral approaches. For example, an adenovirus vector was prepared containing both a Fab fragment of an anti-Ad5 knob antibody and the anti-ACE monoclonal antibody mAb 9B9. This bispecific conjugate exhibited enhanced pulmonary distribution by a synergic effect (transductional and transcriptional).59,60 The major drawback of using this vector is sequestration by Kupffer cells into liver tissue.
BIOCONJUGATED OLIGONUCLEOTIDE DELIVERY
The conjugation of specific molecules to oligonucleotides is a promising therapeutic approach for nucleic acid based drugs. Consequently, bioconjugates are of increasing presence in the pharmaceutical development pipeline. The major advantages of working with a bioconjugate include: (1) a new chemical entity; (2) of defined composition; and (3) synthesized using chemical methods as opposed to bioprocesses. These attributes, unlike formulations with polymers or other transfecting reagents61 that give heterogeneous mixtures requiring extensive multipronged characterization analyses, will facilitate translation to the clinic. Additionally, these covalently conjugated molecules play one or more roles in recognition, targeting of cells or tissues, cellular internalization, and pharmacokinetics.
The review of covalently conjugated oligonucleotides necessitates discussion on appropriate and effective linkers and linking chemistries, as interference strategies can be stymied by functionalization(s).62 Both cleavable and stable linkers are successfully used. Bio-orthogonal “click” chemistry approaches such as akyne-azide and thio-maleimide are two common approaches to form stable linkages with highly specific reactions. Cleavable bonds such as reducible disulfide linkages, esters (cleavable through hydrolysis or esterases), phosphodiesters (cleavable through nucleases), and peptides (cleavable through proteases) are also being utilized, and examples are discussed below. Additionally, alternatives such as end functionalization (5′ or 3′) and other cleavable strategies will be discussed in the context of different conjugated moieties.
PEG Modification.
Poly(ethylene glycol) (PEG) was first conjugated to oligonucleotides more than two decades ago by different academic groups, including Bonora63 and Burcovich,64 in order to improve stability, avoid rapid degradation, and enhance cellular uptake.65 In 2003, Ji et al. described PEG attached to an antisense oligonucleotide sequence (c-myb) to afford a surrounding corona, which would prohibit interactions with plasma proteins and increase aqueous solubility.66 Intracellular uptake is achieved by the formation of a micellar system resulting from the combination of PEG conjugates with fusogenic cationic peptides. These formulations exhibit higher antiproliferative activity against smooth muscle cells compared to the unconjugated c-myb antisense.
GalNAc Modification.
The GalNAc (N-acetylgalactosamine) modification, introduced by Nair et al., lead to hydrophilic glycoconjugates67 (Figure 4), increasing the cellular internalization in the liver due to binding to the ASialoGlycoProtein Receptor (ASGPR). These lectin type C receptors are highly expressed on the plasma membrane of hepatocytes and both galactose and GalNAc are the preferred ligands for these receptors. After binding to ASGPR, the complex is quickly internalized to form early endosomes via a clathrin dependent mechanism.68 As a result, ASGPR targeting conjugates are readily delivered to the liver through intraveneous injection.
Figure 4.

Molecular structures of GalNAc based conjugates.72 Diagram showing an oligonucleotide coupled to GalNAc (left) and molecular structure (right). The triantennular GalNAc is represented in the upper part; the GalNAc incorporated in sequence in the lower part.
Once inside the cell, the oligonucleotides must escape from the endosome intact to elicit their bioactivity. At neutral pH, the dissociation constant between these modified oligonucleotides and their receptor is very low (Kd in the nanomolar range), indicating a strong association with ASGPR receptors. Once inside the endosome, the acidic pH allows for dissociation of the ligand–receptor complex, resulting in the return of ASGPR to the membrane (Figure 5) and subsequent release of the ligand. The GalNAc modification facilitates cell internalization. Finally, the GalNAc modified oligonucleotides escape the endosome.69
Figure 5.

Cellular internalization of GalNAc oligonucleotide conjugates.69 The GalNAc moiety is a ligand of the ASGPR receptors. The formation of the complex induces a clathrin-dependent endocytosis mechanism. Acidification dissociates the siRNA-releasing substrate/receptor complex (3) into the cytoplasm (4). RNAi-mediated degradation of the targeted mRNA mechanism (5). Inhibition of protein translation (6). ASGPR receptors are recycled in the plasma membrane (7).
To prepare these bioconjugates, the GalNAc modification is introduced into the oligonucleotide either during DNA synthesis as a non-nucleoside monomer or at the end of the oligonucleotide synthesis as a “triantennular” form (Figure 4). The synthesis of such oligonucleotide conjugates is compatible with standard automated solid phase synthesis, where the GalNac modification is attached either at the 3′, 5′ extremities or within the oligonucleotide sequence.70 The non-nucleoside monomer is attached through a phosphodiester linkage during oligonucleotide synthesis as a synthetic phophoramidite. All of the different chemical modifications (2′-OMe, 2′-F, PTO), which can be inserted within the same sequence, are compatible with the GalNac conjugation approach. Manoharan et al. reported that these modifications improve the in vivo stability of siRNA–GalNAc conjugates.67 Interestingly, after subcutaneous injection, the siRNA–GalNAc conjugates distribute to the liver with subsequent decrease of gene expression of the targeted mRNA, thus enabling this approach for treating a wide range of liver diseases.71,72 The pharmacokinetics of GalNAc-siRNA conjugates were evaluated in monkeys73 and it was found that phosphorothioate linkages in the 5′-region of siRNA–GalNAc strands minimally affect plasma pharmacokinetics but result in a greater liver exposure. This provides a prolonged duration of gene silencing due to improved metabolic stability. The technology was also developed for ASOs targeting apolipoprotein with a successful improvement (10-fold) of inhibition effect in vivo.74,75 In this study, the active ASO is liberated only after internalization into cells and not in the plasma, demonstrating that ASO-GalNac conjugates are a functional prodrug. Many GalNAc conjugated oligonucleotides are currently in preclinical and clinical trials.76 For example, Fitusiran77,78 is a siRNA developed by Alnylam for the treatment of hemophilias. The collaboration between Ionis and Akcea yielded ASO IONIS-APO (a) LRX for the treatment of hyperlipoproteinemia and cardiovascular diseases. Additionally, two anti-miRNAs are being developed by Regulus to treat viral diseases, such as hepatitis C (RG-101), and a third is being developed in collaboration with AstraZeneca for the treatment of type 2 diabetes (RG-125).76
Peptide Sequences: Cell Penetrating Peptide (CPP) and RGD.
CPPs are short peptidic sequences, generally not exceeding 30 residues, which possess the ability to cross a cellular membrane and facilitate endosomal escape (Figure 6). CPP are classified into three categories: (1) protein-derived peptides; (2) chimeric peptides formed by the fusion of two natural sequences; and (3) synthetic peptides based on structure/function studies.79 These short peptides are ligands for specific receptors that facilitate cell internalization by endocytosis and destabilization of endosomes compartments. For example, Lönn et al. speculated that the PTD/CPP-EED domains enhance cellular delivery by insertion of a hydrophobic patch into the lipid bilayer at a critical distance (18 bonds) from the delivery domain. This resulting concentration in the endosome results in a strong localized membrane destabilization leading to enhanced escape into the cytoplasm.80 All CPPs do not exhibit the same penetration mechanism, membrane leakage rates, or toxicity. In 2007, a CPP ((RXR)4 (X = 6-aminohexanoic acid)) conjugated to a PMO oligomer was investigated.81 The CPP-PMO conjugates did not show toxicity below a dose of 15 mg/kg after intravenous injection (bolus). However, at a dose of 30 mg/kg, animal body weight decreased while at a dose of 150 mg/kg, the animals were lethargic with elevated levels of creatinine. Blood biochemisty analysis of albumin, electrolytes, and bilirubin showed constant levels at all doses.81
Figure 6.

CPP grafting strategy on siRNA. A: Noncleavable oligonucleotide CPP conjugate. B: Representation of the cleavable conjugate by reduction of the disulfide bridge: (1) Bioconjugate enters into the cell thanks to CPP. (2) Cleavage of the disulfide bridge allows detachment of the siRNA from the CPP moiety. (3) CPP enters into the nucleus. (4) siRNA elicits its inhibitory activity by an RNAi mechanism. Adapted with permission from Ye, J., Liu, E., Gong, J., Wang, J., Huang, Y., He, H., and Yang, V. C. (2017) High-Yield Synthesis of Monomeric LMWP(CPP)-SiRNA Covalent Conjugate for Effective Cytosolic Delivery of SiRNA. Theranostics 7, 2495–2508.83 Copyright 2017, Ivyspring.
A major design issue for these conjugates is the complexation between the anionic oligonucleotides and the cationic CPPs. In a previous review, Steven Dowdy focused on the potential use of CPP to deliver siRNA, and the reader is referred to this manuscript.82 The endocytosis-mediated uptake of such peptides depends on three important steps: cell association, internalization, and finally endosomal escape. Dowdy et al. also discuss the array of different cargos that have been delivered by cationic PTDs/CPPs as well as cellular processes and biological responses that have been modulated. CPPs share many common features responsible for efficient activity including positive amino acids (arginine and lysine) and hydrophobic residues (tryptophan and phenylalanine).83,84 Alternatively, nonionic oligonucleotides (PMO, PNA, phosphoryl guanidine) are good candidates to bypass this charge–charge complexation requirement. Such systems are used successfully for exon skipping.85–87
Conventional formulations that carry oligonucleotides are composed of CPP via physical mixing, but the conjugation of these molecules to the oligonucleotide by a covalent bond provides additional benefits. The chemical insertion of multiple peptides on the same oligonucleotide improves the sensitivity of the conjugate for its receptor. In the case of siRNA conjugated to cyclic RGD, the internalization mechanism is achieved by caveola-dependent endocytosis.88,89 In another study, the bombesin peptide, coupled to the 5′ end of a splice-shifting oligonucleotide (SSO), corrects splicing of an aberrant intron. This 14-amino-acid peptide, which is a neurotransmitter acting on the G protein receptor, facilitates oligonucleotide delivery to the nucleus of transfected prostate cancer PC3 cells.90–92 Ye et al.83 demonstrated that a 3′ CPP covalently conjugated to siRNA increased their intracellular delivery (Figure 6). The report investigated CPPs with different conjugation chemistries with a PEG space, one cleavable disulfide linkage, and another through a thiolmaleimide coupling. The incorporation of a stable versus cleavable linkage led to differences in knockdown and efficacy.
The linear tripeptide RGD and its cyclic-RGD analogs are well-known peptides for cell targeting. These peptide ligands bind αvβ3 integrins, which are overexpressed in several cancers, including melanomas,93 ovarian cancer,94 and breast cancer, as well as in metastasis.95 Grafting such ligands to therapeutic oligonucleotides provides a means for targeting and efficient delivery. However, a drawback of using the RGD as a targeting moiety is that the αvβ3 integrin is expressed on many cell types as well as platelets, and, as such, off-target effects and toxicities are of concern. Although this class of peptide-modified bioconjugates is being actively investigated for the delivery of nucleic acid in vitro, positive results still remain to be confirmed in vivo. Additionally, biodistribution and resistance to proteolytic degradation are not reported.
α-Tocopherol.
α-Tocopherol, commonly known as vitamin E, is used for the treatment of hypercholesterolemia. This liposoluble moiety is often coupled to oligonucleotides to facilitate cell membrane interactions.96 For example, Yutaro Asami et al. coupled tocopherol to a siRNA at the 5′ end of the 29-nucleotide antisense strand for reducing apolipoprotein B (ApoB) levels. Improved inhibitory activity is observed relative to the parent cholesterol derivative. In fact, only 2 mg/kg is needed to significantly reduce ApoB mRNA in mouse liver after intravenous injection. In similar conditions, the amount of cholesterol conjugate required to observe the same activity was 50 to 100 mg/kg.97 Additionally, α-tocopherol, coupled to an ASO, increases endogenous gene inhibition in a mouse liver compared to an unconjugated ASO.98 Simply conjugating tocopherol to the oligonucleotide structure did not induce an inhibitory effect, and a spacer of several (4–7) unlocked nucleic acids (UNA) was required between the oligonucleotide 5′ extremity and tocopherol to maintain biological activity. In this study, the oligonucleotides within the ASO selected were Gapmers, oligonucleotides containing a mixture of chemically modified nucleotides (LNA, 2′-O-MOE, for example) at each extremity and a central sequence of DNA (the “gap”), allowing the RNase H cleavage after forming the mRNA/DNA complementary heteroduplex. The UNA residues, on the spacer, are cleaved in the cell affording the ASO using the RNase H mechanism for inhibition of mRNA. The inhibitory efficacy increased by 3.5-fold compared to the intravenous injection of nonconjugated oligonucleotides. Importantly, tocopherol improved the pharmacokinetics of the ASO affording accumulation in the liver (9–14 μg/g and 2–5 μg/g were found in the liver 6 h after injection at a dose of 3 mg/kg for the conjugated and unconjugated ASO, respectively), unlike the unconjugated ASO, which distributed primarily to the kidneys (1–2 μg/g and 12–16 μg/g were found in the kidneys 6 h after injection at a dose of 3 mg/kg for conjugated and unconjugated ASO, respectively).
Aptamers.
Aptamers are oligonucleotide sequences (DNA or RNA) possessing a three-dimensional structure for biological recognition. Using a method called SELEX (Systematic Evolution of Ligands by EXponential enrichment) specific oligonucleotides are identified that bind a target with high affinity (Kd in the range of nanomolar to picomolar). Sullenger et al.99 used this approach to select an aptamer capable of targeting the prostate-specific membrane antigen (PSMA) receptor at the surface of prostate cells. The designed RNA molecule contained both the aptamer and siRNA moieties. This chimeric aptamer-siRNA features a dsRNA (siRNA) motif that is a substrate for the intracellular Dicer enzyme, an endoribonuclease. As a proof of concept, LNCaP and PC-3 prostate cells were treated with the chimeric aptamer-siRNA containing a therapeutic siRNA sequence that targets genes overexpressed in human tumors (PLK1 and BCL2). Successful knockdown resulted in a decrease in cell proliferation and an increase in apoptosis. As a control, an aptamer chimera bearing two point mutations exhibited an absence of specificity for its target, indicating the specific binding between the aptamer and its receptor. In another study, a siRNA targeting the tat/rev viral genes was conjugated via a short polynucleotide link to an aptamer that recognizes the glycoprotein gp120 of the HIV viral envelope. This conjugate suppressed HIV infection in human cell lines and in humanized mouse models.100–102 Aptamer-siRNA chimeras are of significant interest and the reader is referred to a recent review summarizing the state-ofthe-art.103 The additional advantages of such chimeras include lower production costs, low batch-to-batch variation, longer shelf life and little-to-no immunogenicity and toxicity compared to antibody conjugates (discussed below).
Given that the three-dimensional structure is required for recognition and that aptamers are degraded by nucleases, significant research efforts are directed at modifying nucleotides to increase the stability without losing target recognition. There are several locations and types of modifications being investigated including the terminals of nucleic acids, the phosphodiester linkage, the sugar ring and modifications on the bases, as well as 3′ end-capping with inverted thymidine and PEG conjugation.104
Antibodies.
Monoclonal antibodies are known for their recognition properties of biological targets. In 2005 Lieberman et al.105 reported the use of a monoclonal antibody conjugate for the delivery of siRNA in order to inhibit the protein production of HIV. The antibody was conjugated to the ASO oligonucleotide via a click reaction involving an azide-modified antibody and a cyclooctyne ASO. This strategy was further developed by Satake et al.106 to conjugate the anti-CD22 antibody to the MXD3 oligonucleotide. This oligonucleotide targets a dimeric protein (MXD3) associated with MYC, a transcription factor responsible for the survival of B cell precursor cells in acute lymphoblastic leukemia. The conjugate induces inactivation of the MXD3 protein responsible for apoptosis in vitro in leukemic cells as well as affords cytotoxicity in normal B cells but not in hematopoietic cells, including stem cells. The in vivo results are promising as only a dose of 0.2 mg/kg twice a week for 3 weeks is required to double survival (median 42.5 vs 20.5 days). Based on these encouraging results, antibody-ASO conjugates are a new therapeutic option to decrease the doses and to favor the accumulation of ASOs at designated locations. However, the selection of antibody or antibody fragment for targeting a specific cell type is challenging. For example, Genentech published an article describing the investigation of antibodies conjugated to ASO by a maleimide linkage, for targeting prostate carcinoma cells after systemic administration.101 This challenge is further complicated by the fact that the route of antigen internalization also affects silencing. Although antibody conjugates reach the intended target site, the subsequent steps of cellular internalization and endosomal escape are also required for efficacy.107
Cholesterol.
Lipid moieties, and particularly cholesterol derivatives, are often conjugated to oligonucleotides for delivery purposes (Figure 7). Oligonucleotides modified with cholesterol are recognized by high- and low-density lipoproteins (HDL and LDL) in vivo and internalized via cholesterol binding receptors. After intravenous or intraperitoneal injection, these oligonucleotide conjugates escape renal clearance, thus greatly impacting their pharmacokinetics by extending the time that the nucleic acids remain in the plasma. A number of studies have demonstrated the advantage of coupling a cholesterol moiety to the oligonucleotide in order to reduce its renal clearance. Also, cholesterol conjugation increases cellular uptake of oligonucleotides. Godeau et al. reported improved in vitro cellular internalization of an oligonucleotide functionalized with cholesterol via a “click chemistry” approach. Additionally, these conjugates inhibited viral translation of hepatitis via an IRES blocking mechanism.108 Additional studies in the KB-8–5 cell line showed that the cholesterol moiety allows endosomal escape without the use of any additional transfecting reagents. After 4 h, the siRNA conjugates accumulate in the intercellular space. After 24 h, the distribution became homogeneous with an accumulation of the oligonucleotides in the cytoplasm of the cells.109,110
Figure 7.

Molecular structure of a photocleavable cholesterol-conjugated oligonucleotide. Adapted with permission from Yang, J., Chen, C., and Tang, X. (2018) Cholesterol-Modified Caged SiRNAs for Photoregulating Exogenous and Endogenous Gene Expression. Bioconjug. Chem. 29, 1010–1015.111 Copyright 2018, American Chemical Society.
From a design perspective, the spacer between the oligonucleotide and cholesterol is crucial to maintain the biological activity. Directly grafting the cholesterol to the 5′ oligonucleotide end reduced the inhibitory activity of the nucleic acids (siRNA or ASO).109,111 In another study, ApoB was targeted to the liver after a daily intravenous injection of siRNA-cholesterol conjugates at a dose of 50 mg/kg.112 Additionally, the Huntingtin protein was inhibited after a single intrastriatal injection of a cholesterol-coupled siRNA. These two examples demonstrate that siRNA-cholesterol conjugation improves the delivery into different cell types other than liver cells.113 A photoresponsive cholesterol-siRNA conjugate (on both sense or antisense strands) is recently reported containing a photocleavable linkage (Figure 7).114 The orthonitrobenzyl group photocleaves in response to 365 nm UV light to yield the native 5′ end. Three different cholesterol derivatives were prepared to inhibit three different genes (two exogenous, luciferase and GFP; and an endogenous, Eg5) in hepatocarcinoma HepG2 cells. In the absence of light, this conjugation allows the internalization of the amphiphilic cholesterol-siRNA without inhibiting the target. In the presence of a light stimulus, the link between the cholesterol and the siRNA is cleaved, and the inhibitory activity of siRNA is introduced.111
Cholesterol is conjugated to oligonucleotides at different locations including the 5′ or 3′ end, as well as within the sequence. A recent study reported six constructs conjugated with cholesterol covalently bound either at the ends or after thymidines within the sequence. The oligonucleotide selected was a Gapmer with 2 and 3 LNAs at both the 5′ and 3′ ends, respectively. Higher in vitro activities are obtained when the cholesterol is inserted either at the 5′ end or within the sequence. In vivo, the cholesterol conjugates accumulate in the liver independent of the lipid conjugation location (3′, 5′-ends or within the sequence). Furthermore, a regular phosphodiester linkage exhibited superior in vivo performance in terms of biological activity (by a factor of 5) compared to the analog phosphorothioate as a link between the lipid and the ASO. Nakajima et al. suggests that the unmodified phosphate is cleaved in the liver freeing the nonconjugated ASO.115 In 2007, Moschos et al. covalently attached the cholesterol moiety to the oligonucleotide sequence via a disulfide linker that is cleaved under intracellular reductive conditions.116 In this study, the authors suggest that conjugation to cholesterol extends but does not increase siRNA-mediated mRNA knockdown in the lung (MAP kinase mRNA in mouse lung).
Squalene.
Squalene is a triterpene molecule and bio-synthetic precursor of cholesterol found in plants and animals. Squalene is often conjugated to the 3′ end of the sense strand of siRNA via a thiol-maleimide coupling. Due to the resulting amphiphilic character of the squalene-oligonucloetide conjugate, these conjugates spontaneously formed spherical assemblies of 165 ± 10 nm diameter with a zeta potential of −26 mV in aqueous media. These nanoparticles show enhanced oligonucleotide stability in the presence of serum and higher catalytic potency in vitro and in vivo against the targeted RET/PTC1 mRNA compared to the unmodified siRNA. Up to 70% inhibition of the tumor growth is observed after 19 days following intravenous injections of 2.5 mg/kg modified siRNA at day 0, 2, 4, 7 and 10.117,118
Fatty Acids.
Currently in clinical trial (phase 2) for the treatment of myelofibrosis, GRN163L is a telomerase targeting, antisense oligonucleotide of thio-phosphoramidate with a covalently linked palmitic acid at the 5′end (Figure 8).119–121 The palmitic acid moiety is conjugated though a PTO linkage to the backbone of the ASO. Telomerase activity is up-regulated in numerous cancers and, thus, is a viable target for treatment. Telomere shortening and lower cell viability are observed after inhibition of telomerase activity in cancer cell lines. The IC50 values ranged from 50 to 200 nM for telomerase expression in 10 different pancreatic cell lines in the presence of GRN163. Telomerase reactivation and elongation are also observed when the antisense is no longer present, indicating that sequences targeting the RNA template region of telomerase are effective inhibitors of telomerase. GRN163L is a potential adjuvant for cancer treatment as it halts the progression of the tumor by restoring a normal, correct phenotype for the cancerous cells.
Figure 8.

Molecular structure of GRN163L. The lipid oligonucleotide GRN163L, 5′-Palm-TAGGGTTAGACAA-NH2-3′, with a thiophosphoramidate internucleosidic linkage (top) and its structural formula (down).
The in vitro inhibition of telomerase activity by ASO is improved with the palmitoyle modification. An inhibition factor ranging from 1.4 to 39 is observed depending on the cell type (cervix, glioblastoma, hepatocarcinoma, lungs, melanoma, myeloma, ovary, and prostate). In vivo efficacy (up to 56% inhibition of telomerase activity after 24 h compared to the uncoupled antisense or PBS) is also observed in a murine xenograft model following intravenous injection (50 mg/kg) in the absence of any additional transfecting reagent.119 Additionally, investigations reveal a synergistic effect when GRN163L is coadministered with trastuzumab, a recombinant monoclonal antibody targeting HER2+ receptors in breast cancers.121 The composition of the lipid (oleic vs palmitic acid in the lipid oligonucleotide conjugate) was also evaluated, but with no noticeable difference in efficacy.
Ketal Nucleolipid Conjugate.
Nucleolipids, first reported by our groups, are also being investigated.48,52 For example, the ketal uridine phosphoramidite with two alkyl chains at the 2′ and the 3′ positions, anchors oligonucleotides to liposomal membranes.122 These lipid-oligonucleotides are amphiphilic with micellar aggregation properties that are used as a functional reservoir for hydrophobic drugs. The drug payload of the micelles is released in the presence of the oligonucleotides complementary to the lipid-DNA conjugate.123 Similarly modified ASOs are investigated for the treatment of prostate cancer.124 An antisense strategy based on targeting the translational controlled tumor protein (TCTP) is described for the treatment of castration resistant prostate cancer (CRPC), a disease currently with a very low survival rate (median survival reported between 9 and 30 months).125–127 TCTP is minimally expressed in healthy prostatic tissue, moderately in prostatic cancer cells, and overexpressed in CRPC cells. Therefore, designing an inhibitory sequence is key for this promising strategy for the treatment of CRPC. An ASO sequence of 20 nucleotides with PTO backbone was selected to impede TCTP expression. Normally, this ASO sequence requires the assistance of a vectorization agent such as oligofectamine to be efficient in vitro. The conjugation of the ASO with the ketal nucleolipid enables the inhibition of TCTP in the absence of transfecting agent in vitro and in vivo.124
Bis-Conjugates.
Multifunctionalized, bis-conjugated oligonucleotides are also being investigated. Tajik-Ahmadabad et al.128 designed an amphiphilic CPP-modified ASO (in the PMO series) capable of self-assembly. The ASO is functionalized by coupling a 3-maleimidopropanoic acid moiety to the free secondary amine group at the 3′ end. This maleimido-PMO was purified by HPLC method. The fatty-acid modified peptide was prepared by coupling myristic anhydride at the N-terminus of the resin-bound ApoE. After deprotection and resin cleavage, the ApoE peptide with C-terminal cysteine was conjugated to the ASO by thiol-maleimide click reaction. The spontaneous self-assembly of this ASO occurs when myristic acid is covalently attached to the N-terminus of the peptide. The bis-conjugated ASO inhibits exon 7 splicing of the premRNA SMN2 gene, which is responsible for the pathology of spinal muscular atrophy. These amphiphilic species exhibit a 4-fold increase in potency relative to the CPP-ASO mono-conjugate.
A similar approach is described by Wada et al., who used cholesterol and GalNac as modifiers at the 5′-end of an ASO targeting ApoB (Figure 9).129 The conjugation was prepared using phosphoramidite chemistry, amenable to solid support synthesis of oligonucleotides. The monovalent GalNAc phosphoramidite used was described earlier by Yamamoto et al.130 Such a dual conjugation strategy reduces the renal biodistribution of the oligonucleotide. In that case, the cholesterol-conjugated ASO also possessed a three-nucleotide-long linker with phosphodiester linkages susceptible to endonuclease hydrolysis in the targeted tissues or cells. Accumulation of the GalNac-modified ASO is low in the kidney just like the double 5′-modified cholesterol-GalNAc ASO. Renal accumulation is five times lower when the ASO is modified at both extremities one with the lipid and the other with GalNAc. As expected, the GalNac modified ASOs preferentially accumulate in the liver. Thus, the cholesterol–GalNAc dual strategy represents an effective strategy for reducing the nephrotoxic potential of ASOs, while maintaining the gene silencing activity in the liver.
Figure 9.

Cholesterol and GalNAc coupled ASO. TEG: Triethylene glycol; GalNAc: N-acetylgalactosamine; ASO: AntiSense Oligonucleotide.
Spherical Nucleic Acids.
Spherical nucleic acids (SNAs) are a class of nucleic acid conjugates differentiated from other conjugates here through their three-dimensional nanostructure.131 First developed by Chad Mirkin, this structure is composed of oriented single- or double-stranded nucleic acids in a dense spherical array (Figure 10). First-generation SNAs possessed a gold nanoparticle core with Au–S linkages to the nucleic acids, and since then have been expanded to contain other cores for specific applications.131 SNA structures are amenable to further modification to contain targeting ligands such as antibodies,132 and immune agents such as antigens.133,134 SNAs can also be formed with RNA,135 modified backbones,133 and sugar modifications.136 SNAs exhibit increased nuclease resistance due to their structure131 and are readily endocytosed137 into cells without additional transfection making them an attractive bioconjugate class to enhance delivery.137 Liposomal SNAs138 are formulated from 1,2-dioleoyl-sn-glycero-3-phosphocholine and a tocopherol-oligonucleotide conjugate which contains a phosphodiester linkage and a PEG spacer (discussed in its own section above). Tocopherol-oligonucleotide conjugates form micellar structures, but liposomal SNAs provide additional benefit.133 Liposomal SNA formulations are being evaluated as a novel therapy for immune modulation.116,133,134 For example, SNAs are effective for both immune TLR agonism and inhibition through either target sequence dependent binding to TLR receptors or the receptor for mRNA translation.133
Figure 10.

Spherical Nucleic Acids.131 A. SNAs present nucleic acids in an outward facing spherical array. B. Oligonucleotide-tocopherol conjugates form SNAs with liposomal cores. C. Structure of linkage and tocopherol moiety. D. TLR agonizing oligonucleotides conjugated to tocopherol can be combined with antigens through a complementary oligonucleotide and chemical conjugation through a linker. E. Different conjugate linkage chemistries explored.
Receptor agonism strategies focus on agonism through TLRs,133,134 and the activity of the SNA conjugates is dependent on linker chemistry. SNA and tocopherol oligonucleotide conjugates show effective immune stimulation and tumor growth reduction in vivo,133 and SNA formulations demonstrate the highest efficacy. In a recent publication,134 a melanoma-specific peptide antigen was conjugated via three different linkers to a TLR9 agonizing oligonucleotide adjuvant. The complementary oligonucleotide was conjugated to an antigen. Three different antigen-oligonucleotide linkages were evaluated: one stable (N-(β-maleimidopropyloxy) succinimide ester), one cleavable (succinimidyl 3-(2-pyridyldithio)-propionate), and a traceless linkage (4-nitrophenyl 2-(2-pyridyldithio)ethyl carbonate). See Figure 10 for structural comparison. The traceless linkage undergoes an internal cyclization in addition to a disulfide cleavage to yield the antigen’s native N-terminus. The choice of linker conjugation significantly influenced activity. The SNA formulations possessing the cleavable, traceless delivery linkage yielded the greatest T cell activation and proliferation. Importantly, these findings highlight the critical role the linker plays. Moreover, they also demonstrate the potential for oligonucleotide conjugates to expand to new applications and a few SNA formulations are progressing through preclinical into early clinical development (e.g., , Table 1 and discussion below).139,140
Table 1.
Bioconjugate Oligonucleotides in the Clinic
| name | target | linker | moiety | stage | trial reference |
|---|---|---|---|---|---|
| APOC-III-L-Rx | apoC-III | GalNAc | ii recriuting | NCT03385239 | |
| ARC-520 | HBV | Cholesterol | multiple i/ii | NCT01872065 | |
| DCR-PHXC-101 | GO | GalNAc | i active | NCT03392896 | |
| FXI-LRx | Factor XI | 2′-O-MOE | GalNAc | i recruiting | NCT03582462 |
| Inclisiran (ALN-PCSSC) | PCSK9 | GalNAc | multiple i/ii/iii | NCT03397121 | |
| Macugen | VEGF | PEG | Approved | ||
| Zimura (Avacincaptad pegol) | C5 | PEG | iib withdrawn | NCT03374670 | |
| Fovista (E10030) | PGDF | PEG | terminated | NCT01944839 | |
| Imetelstat Sodium (GRN163L) | telomerase | thio-phosphoramidate | Palmitate | ii completed | NCT01731951 |
| Revusiran (ALN-TTRSC) | TTR-FAC | GalNAc | discontinued | ||
| Fitusiran (ANL-AT3sc) | antithrombin | GalNAc | iii recruiting | NCT03549871 | |
| Cemdisiran (ALN-CC5) | Complement C5 | GalNAc | i/ii completed | NCT02352493 | |
| Givosiran (ALN-AS1) | ALAS1 | GalNAc | iii active | NCT03338816 | |
| Lumasiran (ALN-G01) | GO | GalNAc | ii enrolling | NCT03350451 | |
| ALN-HBV | HBV | GalNAc | i terminated | NCT02826018 | |
| ALN-AAT | alpha-1antitrypsin PiZZ allele | GalNAC | i/ii terminated | NCT02503683 | |
| RG-101 | HCV | GalNAc | terminated | ||
| ALN-TTRSC02 | TTR | GalNAc | i completed | NCT02797847 | |
| Akcea-APO(a)-L_rx | apoC-III | 2′-O-(2-methoxyethyl) | GalNAc | ii completed | NCT03070782 |
| IONIS-AGT-LRx | AGT | 5–10-5 MOE gapmer | GalNAc | i completed | NCT03101878 |
| IONIS-ANGPTL3-LRx | ANGPTL3 | Phosphorothioate and Phosphodiester | GalNAc | ii recruiting | NCT03371355 |
| IONIS-TMPRSS6-LRx | TMPRSS6 | GalNAc | i completed | NCT03165864 | |
| ARO-HBV | HBV | i/ii recruiting | NCT03365947 | ||
| IONIS-PKK-LRx | PKK | i completed | NCT03263507 | ||
| IONIS-GHR-LRx | GHR | ii recruiting | NCT03548415 | ||
| IONIS-AZ4–2.5-LRx | undisclosed | i recruiting | NCT03593785 | ||
| AMG-890 | cardiovascular disease | i recruiting | NCT03626662 | ||
| ARC-AAT | AAT | Cholesterol | ii terminated | NCT02363946 | |
| AST 008 | TLR9 | Spherical Nucleic Acids | i completed | NCT03086278 | |
| AST 008 and Pembrolizumab i | TLR9 | Spherical Nucleic Acids | i recruiting | NCT03684785 | |
| XCUR17 | IL17RA and TNFα | Spherical Nucleic Acids | i completed |
OLIGONUCLEOTIDE THERAPEUTIC APPLICATION
As exemplified in the preceding sections, numerous oligonucleotides have been investigated for a range of diseases. The majority of these bioconjugated oligonucleotides are in preclinical studies (i.e., in vitro and small animal in vivo efficacy studies). Importantly, several formulations are regulatory approved and used in the clinic (Table 1). The data accumulated in Table 1 are from company Web sites and the clinicaltrials.gov web resource. The first regulatory approved oligonucleotide (in 1998 by the FDA and in 1999 by the EMA (European Medicines Agency)) was for the treatment of cytomegalovirus retina, a viral infection of the eye. If untreated, vision loss can occur in immune compromised patients. Specifically, Fomivirsen (Vitravene) was developed by IONIS Pharmaceutical (ISIS at the time) in collaboration with Novartis Ophthalmics.141 This biomacromolecule, composed of 21 PTO nucleotides, is complementary to one viral mRNA. The posology consists of a weekly intravitreal injection of 165 μg of a Fomivirsen solution at 6.6 mg/mL.142 The market authorization was withdrawn on July 30th, 2002, at the request of the manufacturer for business reasons. Vitravene is still authorized in Switzerland and can be administered in the European Union upon specific request. Importantly, this success laid the foundation for a number of oligonucleotides being ushered into commercial development and the reader is referred to several reviews on this topic.68,143–146
As discussed in the Introduction of this Review, the design of oligonucleotides for use in the clinic requires specific design considerations and optimizations other than improved delivery.147 For enhanced in vivo efficacy, oligonucleotides containing chemical modifications such as 2′-O-MOE, 2′-OMe, LNA, and PMO are used to protect against degradation and increase target binding affinity. To date, the most common oligonucleotide modifications are PTO DNA and 2′-O-MOE. Additionally, 2′-OMe and LNA are used, but most of these oligonucleotides are still in phase 1 or 2 clinical trials.143,148 As of 2010, all of the oligonucleotides approved by the FDA and in phase 3 clinical trials contain PTO linkages.
The selection of oligonucleotide modifications defines the mechanism(s) of inhibition (Figure 2). The majority of oligonucleotides in phase 1 or 2 trial participate in an RNase H based inhibition mechanism, but are of more diverse chemical composition and functionalization. That said, the inhibition mechanisms available include intron–exon splicing, RNase H activation, or blockage of translation through steric hindrance. The following subsections are organized according to targeted disease with a discussion of the bioconjugated oligonucleotides used as well as their mechanism of action.
Bone Marrow Disorders.
(Myelofibrosis and myelodysplastic disorders). Myeleofibrosis is caused by the replacement of bone marrow with scar tissue. The resulting scarring leads to a reduction of red blood cell formation. Myelodysplastic syndrome is a type of cancer in the bone marrow. Risk factors are genetic or caused by or arise during some cancer treatments. Imetelstat is a palmitoyl lipid oligonucleotide conjugate in clinic trials to treat these diseases. This 13-nucleotide-long ASO targets telomerase activity, using palmitate as the targeting ligand, with thio-phospharamidite linkages including the linkage to the palmitate group (Figure 11).
Figure 11.

Structure of the thio-phospharamidite based ASO with a telomerase activity. The targeting moiety (palmitate) has been inserted at the 3′ extremity.149
The drug has completed multiple phase II clinical trials for metastatic breast cancer and multiple myeloma cancer patients. The dose is limited by thrombocytopenia. Imetelstat (GRN163L) received a fast track status in October 2017 from the FDA for certain patients with a myelodysplastic syndrome.149
Hepatitis B.
Hepatitis B is a common liver infection. The genome of the hepatitis B virus (HBV) is an attractive target because there are multiple sites on the HBV genome that can be targeted simultaneously. There are several conjugates in the pipeline for the treatment of this disease. For example, ARC-520 is an equimolar combination of two cholesterol conjugated siRNAs, siHBV74 and siHBV77, targeting an 18-nucleotide-long sequence in the HBV genome. In addition to the siRNAs, a GalNAc conjugated peptide is included as an excipient to aid in delivery and endosomal escape. A Phase I trial was completed in 2018, but the results have not been publicized as of September 2018. Other clinical trials with ARC-520 have been withdrawn/terminated, likely due to the difficulty in knocking down expression due to HBV DNA integrated into the host genome. Arrowhead Pharmaceuticals has a combined phase I/II clinical trial currently recruiting for ARO-HBV. Alnylam terminated a phase I clinical trial using ALN-HBV01, a 23-nucleotide-long, GalNAc conjugated, siRNA. This GalNAc conjugated siRNA incorporated numerous modifications for solubility and stability, including 2′-F and 2-OMe, with PS linkages only near the siRNA termini. The discontinuation is part of a new agreement with Vir Biotechnology to begin development on ALN-HBV02, also a GalNAc conjugate, citing reduced off-target effects due to the incorporation of glycol nucleic acids (GNAs) in the backbone.
Hemophilia.
Hemophilia is a blood clotting disorder. Fitusiran (ALN-AT3sc) is a GalNAc conjugate targeting antithrombin currently entering phase III clinical trials. In a phase I study, the mean antithrombin (AT) knockdown was reported to be 59%. The study included 4 healthy volunteers (A) and 12 patients with severe hemophilia (B). In one hemophilia patient, a maximal knockdown of 86% was reported along with 114 days bleed free. This conjugate was administrated via subcutaneous injection. First phase results showed the reduction of levels of the antithrombin protein and the restoration of the balance for the production of clotting factors in adult A and hemophilia B patients, after a single monthly injection. The second phase of the trial was suspended because one hemophilia A patient died due to a cerebral edema. The suspension of the clinical trial was lifted after the trial’s protocol was amended to better mitigate risks. Alnylam and Sanofi are scheduled to conduct a phase 3 clinical trial in 2018/2019 with the ATLAS program,150 which will include patients with hemophilia A and B with or without inhibitors, and patients previously receiving on-demand or prophylactic therapy.
Psoriasis.
Psoriasis is a chronic skin disease where skin cells undergo a rapid cell cycle. SNAs are being developed to treat dermatologic indications such as mild to moderate psoriasis.139,140 Recently completed results from Phase I studies for IL17RA and TNFα protein targets are promising (Purdue Pharma). The SNAs are absorbed through the skin and deliver siRNAs into the epidermis139 providing effective knockdown of targets within keratinocytes with minimal immune response. Topical administration offers a local treatment option, which minimizes systemic off-target effects.
Cancer.
Cancer is the unwanted abnormal growth and invasion of cells in the body. Of the various treatment classes—surgery, radiation, chemotherapy, and immunotherapy—immunotherapy is a particularly exciting one as it provides a means to use the immune system to fight the disease. For example, activation of Toll-like receptor 9 (TLR9) initiates a cascade of innate and adaptive immune responses including increased interferon gene expression. Increased interferon gene expression correlates with improved responses to programmed death 1 (PD-1) inhibition. Thus, patients who did not respond or stopped responding to anti-PD-1 therapy are potential candidates for combined TLR9 agonists and anti-PD-1 therapy. Exicure has successfully completed a Phase I trial of TLR9 agonizing SNAs administered subcutaneously in healthy volunteers (). A Phase Ib/II study is planned to study intratumorally delivered SNAs both alone and in combination with intravenously administered pembrolizumab, an antibody targeting the PD-1 receptor. The target cohort is in patients that have not yet responded to anti-PD-1 or anti-PD-L1 therapies ().
Age-Related Macular Degeneration.
Age-related macular degeneration (ARMD) is the gradual worsening of vision and eye health over time due to retina damage. The blurring or loss of vision associated with ARMD is characterized by the proliferation of blood vessels behind the retina. In “wet” macular degeneration, neovascularization can worsen symptoms and is activated by vascular endothelial growth factor (VEGF) signaling. Macugen (Pegaptanib) is a 27-nucleobase aptamer targeting VEGF165 binding,151,152 and possesses a 3′−3′ deoxythymidine extremity and a 5′ end with two branched polyethylene glycol chains of 20 kDa. The 2′ positions are modified for nuclease resistance purposes by 2′-OMe for bases purines and fluorinated pyrimidines (Figure 12). Importantly, the aptamer binds the heparin site of VEGF with a picomolar affinity. The FDA approved this aptamer for the treatment of “wet” or age-related macular degeneration. Specifically, Macugen, injected intravitreally, locally reduces the proliferation/expansion of blood vessels, and a lack of adverse events is still observed at a dose ten times higher than clinically used. The product is available in the form of prefilled syringes of 90 μL solution containing 0.3 mg of active substance.
Figure 12.

Secondary structure of Pegaptanib, active substance of Macugen. Reproduced with permission from Ng, E. W. M., Shima, D.T., Calias, P., Cunningham, E. T., Guyer, D. R., and Adamis, A. P. (2006) Pegaptanib, a Targeted Anti-VEGF Aptamer for Ocular Vascular Disease. Nat. Rev. Drug Discovery 5, 123–132.152 Copyright 2006, Nature Publishing.
Several other drugs have been developed for the treatment of ARMD. In the EU, Lucentis (ranibizumab, Novartis), a monoclonal antibody fragment that targets VEGF, and Eylea (aflibercept, Bayer) a recombinant fusion protein that binds to VEGF-A, VEGF-B, and placental growth factor (PIGF) with higher affinity than their native receptors. Avastin (bevacizumab, Roche) is a humanized recombinant monoclonal antibody that binds VEGF and competitively inhibits the binding between VEGF and Flt-1 and KDR. It adds to the list of Macugen competitors in the United States. The other nucleic acid treatment for ARMD is a modified siRNA called bevasiranib. It is a duplex of a 21-nt-long RNA that targets VEGF.153 Bevasiranib has not been approved but shows efficacy when combined with Lucentis in clinical trials.148 The effects of bevasiranib appear only 6 weeks after the beginning of the treatment, but the combination of Lucentis/bevasiranib provides better visual acuity than Lucentis alone.
Hepatic Porphyrias.
These conditions are a combination of rare diseases that all overlap in that they all feature the accumulation of porphyrins in the blood. The build-up of these proteins causes nervous or dermatological symptoms. Currently in an ongoing phase III clinical trial, Givosiran (ALN-AS1), is a GalNAc conjugate siRNA targeting aminovulinate synthase 1 (ALAS1) present in the liver. In a Phase I trial with 23 patients, 11 patients reported mild or moderate adverse events (AE) with 1 reporting an unrelated, severe AE of abdominal pain and another reported unrelated AE of bursitis. Common mild/moderate AEs of abdominal pain, diarrhea, nasopharyngitis, and hypoesthesia, pruritus, and rash were noted, but were not serious enough to stop treatment. The product was injected by single or multiple subcutaneous injections and compared to an injection of sterile normal saline solution. The purpose of the next phase 1 trial was to evaluate the safety and tolerability of Givosiran (ALN-AS1) in acute intermittent porphyria (AIP) patients as well as to characterize pharmacokinetics (PK) and pharmacodynamics (PD) of ALN-AS1 in AIP patients. The safety and efficacy of Givosiran was evaluated in 17 patients with acute AIP. The patients were initially monitored for three months and those who experienced one porphyria attack were eligible to receive once monthly injections of 2.5 or 5.0 mg/kg Givosiran or placebo for six months. Durable inhibition of ALAS1 was observed after Givosiran treatment. The lower tested dose of 2.5 mg/kg reduced the annualized attack rate by 83% and afforded a reduction of 88% of hemin compared to the placebo. A larger dose did not produce any amelioration. In 2018, data showed that at 22 months of therapy, patients were still experiencing a robust beneficial effect from Givosiran.
Famyloid Polyneuropathy.
Familial amyloid polyneuropathy (FAP) is a genetic autosomal dominant disorder resulting from a mutation of the TTR (transthyretin) gene, leading to TTR protein aggregation, which negatively affects the nervous system. Oligonucleotide therapeutic agents designed for the treatment of FAP, also known as transthyretin-related hereditary amyloidosis (ATTR), target the mRNA sequence of transthyretin (TTR) for degradation. Three formulations are in clinical trials to decrease expression of TTR: Inotersen (developed by Ionis and GSK), Onpattro (Patisiran developed by Alnylam), and Revusiran (developed in collaboration between Alnylam and Sanofi Genzyme).154 These therapeutics (2 siRNAs—one being a GalNac conjugate—and an ASO) are in phase 3 clinical trials. One study compared the inhibitory activity of Inotersen (ASO) and Patisiran (siRNA). Both treatments led to a 80% decrease in TTR expression.155 Inotersen (IONIS-TTRRx),156 is a 20-nucleotide-long gapmer PTO ASO with 5 2′-O-MOE modifications at both extremities. The ASO targets pre-mRNA that is degraded following RNase H activity.157 Inotersen sodium is subcutaneously injected weekly at a dose of 300 mg (284 mg of inotersen). Of note, three cases (out of the 172 patients tested over 64 weeks) reported thrombocytopenia. Inotersen has received marketing authorization approval from the European Commission (EC) for the treatment of stage 1 or stage 2 polyneuropathy in adult patients with hereditary transthyretin amyloidosis (hATTR). Inotersen is commercialized under the name of Tegsedi.
Patisiran (ALN-TTR02, Onpattro) is an advanced siRNA under development and is administered intravenously as lipidic nanoparticles (LNP) at noticeably low doses of 0.3 mg/kg every 3 weeks over an 18-month period. The most common adverse events observed in patients were peripheral edema and infusion related reactions.148,158 In August 2018, Patisiran received U.S. Food and Drug (FDA) approval for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. Regulatory filings in other markets, including Japan, are planned. The European Commission also granted marketing authorization for Onpattro on August 30th, 2018.
Revusiran, a GalNac siRNA conjugate administered subcutaneously, was previously undergoing phase 3 clinical trials. This molecule has two PTO linkages, 22 2′-OMe nucleotides, and another 22 modified residues with 2′-F modifications. However, development was stopped due to numerous AEs leading to deaths in phase 3 clinical studies. Causes of theses side effects have not been published but the high weekly dose of 500 mg was suspected.
Historic Timeline of Developments.
There have been numerous developments over the past 50 years that have led to the clinical efficacy of oligonucleotides for treating diseases.159 The timeline featuring significant accomplishments is presented in Figure 13. ASOs were the first oligonucleotides to reach clinical trials and to receive regulatory approval. Notably, this conjugated (pegylated) aptamer has been in use for more than 14 years. The development of siRNA therapeutics advanced significantly in the early 2000s and their first use in humans occurred in 2005 (bevasiranib). siRNA therapeutics that functioned as splicing modulators followed next in development and commercialization. Antagomirs (miRNA inhibitors) were developed in 2009 and were followed by miRNA mimetics in 2013. These two former classes of oligonucleotides utilize miRNAs to modulate the activity of a given gene. For example, Miravirsen sequesters microRNA-122 and is used for the treatment of chronic hepatitis C,160,161 whereas mimics of microARN-34a exhibit potentiated antitumor activity. Over 30 oncogenes are known to be inhibited by this microRNA.162
Figure 13.

Timeline for Key Advances in Therapeutic Oligonucleotides. The various substitutions and the advances of chemical modifications are shown in yellow, main discoveries and technological achievements are noted in blue, and clinical studies are highlighted in green. Adapted from Lundin, K. E., Gissberg, O., and Smith, C. I. E. (2015) Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 26, 475–485.159 Copyright 2015, Mary Ann Liebert, Inc.
6. FUTURE OPPORTUNITIES AND CONCLUSION
Several bioconjugated oligonucleotides are regulatory approved and used in the clinic. These bioconjugates possess a carbohydrate (Patisiran), a polymer (Pegaptanib), or a lipid (Imetelstat – GRN163L, currently on fast track status) covalently linked to the oligonucleotides. A number of additional ones are in preclinical development as well as clinical studies (see Table 1.). The primary function of these added conjugates is to enhance targeting, facilitate vectorization, reduce oligonucleotide susceptibility to nuclease activity, eliminate or reduce need for toxic transfection agents, and generally improve pharmacological activity and efficacy. Increasing the therapeutic window and reducing the likelihood for adverse events is a top priority, and the use of conjugated oligonucleotides is an effective approach. As discussed above, these conjugates are lipophilic or a targeting moiety.
Most of the therapies currently in late-stage development target the liver and liver associated diseases. This is largely due to the success of the GalNAc modification, and the fact that the liver is an efficient filter for the blood. Other highly efficient targeting moieties need to be developed to target other organs or tissues—lung, kidney, heart, bone marrow, brain, muscle, and so forth. Poor delivery to target sites necessitates higher doses, decreasing safety with greater chances of AEs. One strategy to reduce the severity of AEs is to modulate or knockdown the effect after administration. For example, a single subcutaneous 3 mg/kg GalNAc-siRNA dose can be effectively reversed with a 0.1 mg/kg GalNAc oligonucleotide conjugate targeting the seed region of the incorporated siRNA strand.163 The result demonstrates both effective knockdown and return of TTR protein levels in preclinical in vivo studies. The evolution of safe antidotes for oligonucleotide therapies, such as the REVERSIR strategy reported by Zlatev et al., poses benefits for safety and adoption into the clinic.164 The possibility to finely control RNAi pharmacology is a desired feature for highly efficient nucleic based therapeutic treatments.
At the molecular level, advances in oligonucleotide chemistry are providing new methods and site-specific reactions to modify oligonucleotides at the base, sugar, phosphate, or end groups. Additionally, natural bases can be substituted with synthetic analogs. These developments afford a large diversity of new and unique biomacromolecules. Still, significant opportunities exist in this area to improve on reaction efficiency, to expand on the compositional diversity of modifications, and to realize structure–property relationships for nanoscale, self-assembling, and supramolecular systems. These improvements will allow for more modular, diverse, and effective formulations. Similarly, molecular biology studies are providing new therapeutic targets with an improved understanding of pathways and resulting pathologies. As such, the development of combination therapies is forthcoming. The possible combinations include targeting either one or multiple mRNAs or combining a bioconjugated oligonucleotide with a small molecule or protein in order to improve efficacy and safety profiles.
As discussed in this Review, modifications such as GalNAc, cell penetrating peptides, α-tocopherol, aptamers, antibodies, cholesterol, squalene, fatty acids, and nucleolipids are new, promising therapeutic candidates in the field. Despite the clinical progress to date, the design of modified oligonucleotides encompassing both the optimal delivery and efficient biological activity remains an important challenge. Diseases such as coronary heart disease, lower respiratory infections, cancer, diabetes, and Alzheimer’s are in need of new therapies, and, as oligonucleotides can target and bind nearly any expressed mRNA, the possibility to develop oligonucleotide therapeutics exist. Key to these future successes will be understanding the pharmacokinetic and biodistribution profiles of these new therapeutics. Despite the promising bioconjugated oligonucleotides reported so far, oligonucleotide delivery represents a major hurdle still to be addressed. As mentioned previously, most of the oligonucleotide conjugates used in the clinic bioaccumulate in the liver. Furthermore, a clear relationship between the composition and structure of the chemical modification and its resulting effect on pharmacokinetic/biodistribution must be both quantified and established with appropriate positive and negative controls in place. For example, membrane interacting bioconjugates often interact with many targets and are known for their promiscuity. Such a molecular rationale would allow scientists to design the chemical structure of the oligonucleotides depending on the specific disease, target tissue/organ, cell type, and so forth.
In summary, the use of oligonucleotides in the clinic is in its infancy and the potential for continued significant advances in patient care exist. Given the clinical success of monoclonical antibodies (mAb) as a biologic, we speculate that oligonucleotides will have a greater impact due to the specificity inherent to the target sequence, the ability to target nearly any protein, and their stability both preadministration and in vivo. The full realization and maturation of the bioconjugated oligonucleotide therapeutic field will require time—e.g., 19 mAb drugs were on the market only after 20 years of development—but this should not curtail enthusiasm. In fact, findings from these mAb studies as well as from polymer conjugated enzymes and small molecules used in the clinic will catalyze development and subsequent clinical applicability.165 Bioconjugation chemistry is at the centerpiece on this therapeutic oligonucleotide revolution. We encourage all to investigate this area where conceptualizing, designing, and engineering at the molecular level new functionalized oligonucleotides is key to success in the clinic.
ACKNOWLEDGMENTS
This work was supported in part by funding from the Inserm transfert (IT), the National Institute of Health T32 Grant entitled Translational Research in Biomaterials (NIH T32EB006359, AM),166 and the Distinguished Professor of Translational Research Chair at Boston University (MWG). The authors (P.B. and S.B.) thank Dr Lara Moumne (IT) for fruitful discussions.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, and Darnell J (2000) Molecular Cell Biology, 4th ed., Freeman WH. [Google Scholar]
- (2).Belikova AM, Zarytova VF, and Grineva NI (1967) Synthesis of Ribonucleosides and Diribonucleoside Phosphates Containing 2-Chloroethylamine and Nitrogen Mustard Residues. Tetrahedron Lett. 8, 3557–3562. [DOI] [PubMed] [Google Scholar]
- (3).Summerton J, and Bartlett P. a. (1978) Sequence-Specific Crosslinking Agents for Nucleic Acids. J. Mol. Biol 122 (2), 145–162. [DOI] [PubMed] [Google Scholar]
- (4).Zamecnik PC, and Stephenson ML (1978) Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc. Natl. Acad. Sci. U. S. A 75 (1), 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stephenson ML, and Zamecnik PC (1978) Inhibition of Rous Sarcoma Viral RNA Translation by a Specific Oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. U. S. A 75 (1), 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Weiss B, Ed. (1997) Antisense Oligodeoxynucleotides and Antisense RNA Novel Pharmacological and Therapeutic Agents, 1st ed., CRC-Press, Roca Raton, FL. [Google Scholar]
- (7).Napoli C, Lemieux C, and Jorgensen R (1990) Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in Trans. Plant Cell 2 (4), 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, and Mello CC (1998) Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 391 (6669), 806–811. [DOI] [PubMed] [Google Scholar]
- (9).Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, and Bennett CF (1997) 2′-O-(2-Methoxy)Ethyl-Modified Anti-Intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 MRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem 272 (18), 11994–12000. [DOI] [PubMed] [Google Scholar]
- (10).Lengyel P, Speyer JF, and Ochoa S (1961) Synthetic Polynucleotides and the Amino Acid Code. Proc. Natl. Acad. Sci. U. S. A 47, 1936–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cazenave C, Stein CA, Loreau N, Thuong NT, Neckers LM, Subasinghe C, Héléne C, Cohen JS, and Toulmé JJ (1989) Comparative Inhibition of Rabbit Globin MRNA Translation by Modified Antisense Oligodeoxynucleotides. Nucleic Acids Res 17 (11), 4255–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chan JHP, Lim S, and Wong WSF (2006) Antisense Oligonucleotides: From Design to Therapeutic Application. Clin. Exp. Pharmacol. Physiol 33 (5–6), 533–540. [DOI] [PubMed] [Google Scholar]
- (13).Vickers TA, Wyatt JR, Burckin T, Bennett CF, and Freier SM (2001) Fully Modified 2′ MOE Oligonucleotides Redirect Polyadenylation. Nucleic Acids Res 29 (6), 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. (2007) A MicroRNA Component of the P53 Tumour Suppressor Network. Nature 447 (7148), 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, and Jacks T (2008) Suppression of Non-Small Cell Lung Tumor Development by the Let-7 MicroRNA Family. Proc. Natl. Acad. Sci. U. S. A 105 (10), 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Davis S, Propp S, Freier SM, Jones LE, Serra MJ, Kinberger G, Bhat B, Swayze EE, Bennett CF, and Esau C (2009) Potent Inhibition of MicroRNA in Vivo without Degradation. Nucleic Acids Res 37 (1), 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, et al. (2004) MicroRNA-143 Regulates Adipocyte Differentiation. J. Biol. Chem 279 (50), 52361–52365. [DOI] [PubMed] [Google Scholar]
- (18).Norton JC, Piatyszek MA, Wright WE, Shay JW, and Corey DR (1996) Inhibition of Human Telomerase Activity by Peptide Nucleic Acids. Nat. Biotechnol 14 (5), 615–619. [DOI] [PubMed] [Google Scholar]
- (19).Agrawal S, Temsamani J, Galbraith W, and Tang J (1995) Pharmacokinetics of Antisense Oligonucleotides. Clin. Pharmacokinet 28 (1), 7–16. [DOI] [PubMed] [Google Scholar]
- (20).Seth PP, Allerson CR, Berdeja A, Siwkowski A, Pallan PS, Gaus H, Prakash TP, Watt AT, Egli M, and Swayze EE (2010) An Exocyclic Methylene Group Acts as a Bioisostere of the 2′-Oxygen Atom in LNA. J. Am. Chem. Soc 132 (42), 14942–14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Khvorova A, and Watts JK (2017) The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol 35 (3), 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Manoharan M (2004) RNA Interference and Chemically Modified Small Interfering RNAs. Curr. Opin. Chem. Biol 8 (6), 570–579. [DOI] [PubMed] [Google Scholar]
- (23).Zamaratski E, Pradeepkumar PI, and Chattopadhyaya J (2001) A Critical Survey of the Structure-Function of the Antisense Oligo/RNA Heteroduplex as Substrate for RNase H. J. Biochem. Biophys. Methods 48 (3), 189–208. [DOI] [PubMed] [Google Scholar]
- (24).Stein CA, Benimetskaya L, and Mani S (2005) Antisense Strategies for Oncogene Inactivation. Semin. Oncol 32 (6), 563–572. [DOI] [PubMed] [Google Scholar]
- (25).Kupryushkin MS, Pyshnyi DV, and Stetsenko DA (2014) Phosphoryl Guanidines: A New Type of Nucleic Acid Analogues. Acta Naturae 6 (4), 116–118. [PMC free article] [PubMed] [Google Scholar]
- (26).Stetsenko DA, Kupryushkin MS, and Pyshnyi DV Modified Oligonucleotides and Methods for Their Synthesis, International Patent WO/2016/028187, February 25, 2016. [Google Scholar]
- (27).Summerton J, and Weller D (1997) Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev 7 (3), 187–195. [DOI] [PubMed] [Google Scholar]
- (28).Tros de Ilarduya C, Sun Y, and Düzgünes N (2010) Gene Delivery by Lipoplexes and Polyplexes. Eur. J. Pharm. Sci 40 (3), 159–170. [DOI] [PubMed] [Google Scholar]
- (29).Mintzer MA, and Simanek EE (2009) Nonviral Vectors for Gene Delivery. Chem. Rev 109 (2), 259–302. [DOI] [PubMed] [Google Scholar]
- (30).Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, and Anderson DG (2014) Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet 15 (8), 541–555. [DOI] [PubMed] [Google Scholar]
- (31).Wasungu L, and Hoekstra D (2006) Cationic Lipids, Lipoplexes and Intracellular Delivery of Genes. J. Controlled Release 116 (2), 255–264. [DOI] [PubMed] [Google Scholar]
- (32).Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK, and Duvall CL (2013) Balancing Cationic and Hydrophobic Content of PEGylated SiRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity In Vivo. ACS Nano 7 (10), 8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Verbaan FJ, Oussoren C, van Dam IM, Takakura Y, Hashida M, Crommelin DJA, Hennink WE, and Storm G (2001) The Fate of Poly(2-Dimethyl Amino Ethyl)Methacrylate-Based Polyplexes after Intravenous Administration. Int. J. Pharm 214(1), 99–101. [DOI] [PubMed] [Google Scholar]
- (34).Sakurai F, Terada T, Yasuda K, Yamashita F, Takakura Y, and Hashida M (2002) The Role of Tissue Macrophages in the Induction of Proinflammatory Cytokine Production Following Intravenous Injection of Lipoplexes. Gene Ther 9 (16), 1120–1126. [DOI] [PubMed] [Google Scholar]
- (35).Neuberg P, and Kichler A (2014) Recent Developments in Nucleic Acid Delivery with Polyethylenimines. Adv. Genet 88, 263–288. [DOI] [PubMed] [Google Scholar]
- (36).Chiper M, Tounsi N, Kole R, Kichler A, and Zuber G (2017) Self-Aggregating 1.8 kDa Polyethylenimines with Dissolution Switch at Endosomal Acidic PH Are Delivery Carriers for Plasmid DNA, MRNA, SiRNA and Exon-Skipping Oligonucleotides. J. Controlled Release 246, 60–70. [DOI] [PubMed] [Google Scholar]
- (37).Zhu Y, Liang G, Sun B, Tian T, Hu F, and Xiao Z (2016) A Novel Type of Self-Assembled Nanoparticles as Targeted Gene Carriers: An Application for Plasmid DNA and AntimicroRNA Oligonucleotide Delivery. Int. J. Nanomed 11, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Agarwal S, Zhang Y, Maji S, and Greiner A (2012) PDMAEMA Based Gene Delivery Materials. Mater. Today 15 (9), 388–393. [Google Scholar]
- (39).Jiang Y, Lu H, Khine YY, Dag A, and Stenzel MH (2014) Polyion Complex Micelle Based on Albumin–Polymer Conjugates: Multifunctional Oligonucleotide Transfection Vectors for Anticancer Chemotherapeutics. Biomacromolecules 15 (11), 4195–4205. [DOI] [PubMed] [Google Scholar]
- (40).Svyatchenko VA, Tarasova MV, Netesov SV, and Chumakov PM (2012) Oncolytic Adenoviruses in Anticancer Therapy: Current Status and Prospects. Mol. Biol 46 (4), 496–507. [PubMed] [Google Scholar]
- (41).Lukashev AN, and Zamyatnin AA (2016) Viral Vectors for Gene Therapy: Current State and Clinical Perspectives. Biochemistry 81 (7), 700–708. [DOI] [PubMed] [Google Scholar]
- (42).Merten O-W, and Gaillet B (2016) Viral Vectors for Gene Therapy and Gene Modification Approaches. Biochem. Eng. J 108, 98–115. [Google Scholar]
- (43).Zhao Y, and Huang L (2014) Lipid Nanoparticles for Gene Delivery. Adv. Genet 88, 13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Okamoto A, Asai T, Hirai Y, Shimizu K, Koide H, Minamino T, and Oku N (2018) Systemic Administration of SiRNA with Anti-HB-EGF Antibody-Modified Lipid Nanoparticles for the Treatment of Triple-Negative Breast Cancer. Mol. Pharmaceutics 15, 1495. [DOI] [PubMed] [Google Scholar]
- (45).Han J, Zern BJ, Shuvaev VV, Davies PF, Muro S, and Muzykantov V (2012) Acute and Chronic Shear Stress Differently Regulate Endothelial Internalization of Nanocarriers Targeted to Platelet-Endothelial Cell Adhesion Molecule-1. ACS Nano 6 (10), 8824–8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Han J, Shuvaev VV, Davies PF, Eckmann DM, Muro S, and Muzykantov VR (2015) Flow Shear Stress Differentially Regulates Endothelial Uptake of Nanocarriers Targeted to Distinct Epitopes of PECAM-1. J. Controlled Release 210, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Parhiz H, Shuvaev VV, Pardi N, Khoshnejad M, Kiseleva RY, Brenner JS, Uhler T, Tuyishime S, Mui BL, Tam YK, et al. (2018) PECAM-1 Directed Re-Targeting of Exogenous MRNA Providing Two Orders of Magnitude Enhancement of Vascular Delivery and Expression in Lungs Independent of Apolipoprotein E-Mediated Uptake. J. Controlled Release 291, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Baillet J, Desvergnes V, Hamoud A, Latxague L, and Barthélémy P (2018) Lipid and Nucleic Acid Chemistries: Combining the Best of Both Worlds to Construct Advanced Biomaterials. Adv. Mater 30, 1705078. [DOI] [PubMed] [Google Scholar]
- (49).Allain V, Bourgaux C, and Couvreur P (2012) Self-Assembled Nucleolipids: From Supramolecular Structure to Soft Nucleic Acid and Drug Delivery Devices. Nucleic Acids Res 40 (5), 1891–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, and Barthélémy P (2011) Nucleoside–Lipid-Based Nanoparticles for Cisplatin Delivery. ACS Nano 5 (11), 8649–8655. [DOI] [PubMed] [Google Scholar]
- (51).Chabaud P, Camplo M, Payet D, Serin G, Moreau L, Barthélémy P, and Grinstaff MW (2006) Cationic Nucleoside Lipids for Gene Delivery. Bioconjugate Chem 17 (2), 466–472. [DOI] [PubMed] [Google Scholar]
- (52).Gissot A, Camplo M, Grinstaff MW, and Barthélémy P (2008) Nucleoside, Nucleotide and Oligonucleotide Based Amphiphiles: A Successful Marriage of Nucleic Acids with Lipids. Org. Biomol. Chem 6 (8), 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Luvino D, Khiati S, Oumzil K, Rocchi P, Camplo M, and Barthélémy P (2013) Efficient Delivery of Therapeutic Small Nucleic Acids to Prostate Cancer Cells Using Ketal Nucleoside Lipid Nanoparticles. J. Controlled Release 172 (3), 954–961. [DOI] [PubMed] [Google Scholar]
- (54).Ceballos C, Prata CAH, Giorgio S, Garzino F, Payet D, Barthélémy P, Grinstaff MW, and Camplo M (2009) Cationic Nucleoside Lipids Based on a 3-Nitropyrrole Universal Base for SiRNA Delivery. Bioconjugate Chem 20 (2), 193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ceballos C, Khiati S, Prata CAH, Zhang X-X, Giorgio S, Marsal P, Grinstaff MW, Barthélémy P, and Camplo M (2010) Cationic Nucleoside Lipids Derived from Universal Bases: A Rational Approach for SiRNA Transfection. Bioconjugate Chem 21(6), 1062–1069. [DOI] [PubMed] [Google Scholar]
- (56).Oumzil K, Benizri S, Tonelli G, Staedel C, Appavoo A, Chaffanet M, Navailles L, and Barthélémy P (2015) PH-Cleavable Nucleoside Lipids: A New Paradigm for Controlling the Stability of Lipid-Based Delivery Systems. ChemMedChem 10 (11), 1797–1801. [DOI] [PubMed] [Google Scholar]
- (57).Järver P, Coursindel T, Andaloussi SE, Godfrey C, Wood MJ, and Gait MJ (2012) Peptide-Mediated Cell and In Vivo Delivery of Antisense Oligonucleotides and SiRNA. Mol. Ther.–Nucleic Acids 1, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Aigner A, and Kögel D (2018) Nanoparticle/SiRNA-Based Therapy Strategies in Glioma: Which Nanoparticles, Which SiRNAs? Nanomedicine 13 (1), 89–103. [DOI] [PubMed] [Google Scholar]
- (59).Reynolds PN, Zinn KR, Gavrilyuk VD, Balyasnikova IV, Rogers BE, Buchsbaum DJ, Wang MH, Miletich DJ, Grizzle WE, Douglas JT, et al. (2000) A Targetable, Injectable Adenoviral Vector for Selective Gene Delivery to Pulmonary Endothelium in Vivo. Mol. Ther 2 (6), 562–578. [DOI] [PubMed] [Google Scholar]
- (60).Reynolds PN, Nicklin SA, Kaliberova L, Boatman BG, Grizzle WE, Balyasnikova IV, Baker AH, Danilov SM, and Curiel DT (2001) Combined Transductional and Transcriptional Targeting Improves the Specificity of Transgene Expression in Vivo. Nat. Biotechnol 19 (9), 838–842. [DOI] [PubMed] [Google Scholar]
- (61).Ho W, Zhang X-Q, and Xu X (2016) Biomaterials in SiRNA Delivery: A Comprehensive Review. Adv. Healthcare Mater 5 (21), 2715–2731. [DOI] [PubMed] [Google Scholar]
- (62).Chiu YL, and Rana TM (2002) RNAi in Human Cells: Basic Structural and Functional Features of Small Interfering RNA. Mol. Cell 10 (3), 549–561. [DOI] [PubMed] [Google Scholar]
- (63).Bonora GM, Ivanova E, Zarytova V, Burcovich B, and Veronese FM (1997) Synthesis and Characterization of High-Molecular Mass Polyethylene Glycol-Conjugated Oligonucleotides. Bioconjugate Chem 8 (6), 793–797. [DOI] [PubMed] [Google Scholar]
- (64).Burcovich B, Veronese FM, Zarytova V, and Bonora GM (1998) Branched Polyethylene Glycol (Bpeg) Conjugated Antisense Oligonucleotides. Nucleosides Nucleotides 17 (9–11), 1567–1570. [Google Scholar]
- (65).Bonora GM (2002) Polymer-Conjugated Bioactive Oligonucleotides. J. Bioact. Compat. Polym 17 (5), 375–389. [Google Scholar]
- (66).Jeong JH, Kim SW, and Park TG (2003) Novel Intracellular Delivery System of Antisense Oligonucleotide by Self-Assembled Hybrid Micelles Composed of DNA/PEG Conjugate and Cationic Fusogenic Peptide. Bioconjugate Chem 14 (2), 473–479. [DOI] [PubMed] [Google Scholar]
- (67).Nair JK, Willoughby JLS, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, et al. (2014) Multivalent N-Acetylgalactosamine-Conjugated SiRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc 136 (49), 16958–16961. [DOI] [PubMed] [Google Scholar]
- (68).Girelli D, Busti F, Marchi G, Martinelli N, and Olivieri O (2018) Therapeutic Oligonucleotides in Cardiovascular and Metabolic Diseases: Insights for the Internist. Int. Emerg. Med 1–6, 1 DOI: 10.1007/s11739-018-1810-5. [DOI] [PubMed] [Google Scholar]
- (69).Springer AD, and Dowdy SF (2018) GalNAc-SiRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther 28 (3), 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Cedillo I, Dana C, Engle E, Chen L, McPherson A, and Rodriguez A (2017) Synthesis of 5′-GalNAc-Conjugated Oligonucleotides: A Comparison of Solid and Solution-Phase Conjugation Strategies. Molecules 22 (12), 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Rajeev KG, Nair JK, Jayaraman M, Charisse K, Taneja N, O’Shea J, Willoughby JLS, Yucius K, Nguyen T, Shulga-Morskaya S, et al. (2015) Hepatocyte-Specific Delivery of SiRNAs Conjugated to Novel Non-Nucleosidic Trivalent N-Acetylgalactosamine Elicits Robust Gene Silencing in Vivo. ChemBioChem 16 (6), 903–908. [DOI] [PubMed] [Google Scholar]
- (72).Matsuda S, Keiser K, Nair JK, Charisse K, Manoharan RM, Kretschmer P, Peng CG, V Kel’in A, Kandasamy P, Willoughby JLS, et al. (2015) SiRNA Conjugates Carrying Sequentially Assembled Trivalent N-Acetylgalactosamine Linked through Nucleosides Elicit Robust Gene Silencing in Vivo in Hepatocytes. ACS Chem. Biol 10 (5), 1181–1187. [DOI] [PubMed] [Google Scholar]
- (73).Yu RZ, Gunawan R, Post N, Zanardi T, Hall S, Burkey J, Kim T-W, Graham MJ, Prakash TP, Seth PP, et al. (2016) Disposition and Pharmacokinetics of a GalNAc3-Conjugated Antisense Oligonucleotide Targeting Human Lipoprotein (a) in Monkeys. Nucleic Acid Ther 26 (6), 372–380. [DOI] [PubMed] [Google Scholar]
- (74).Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, et al. (2014) Targeted Delivery of Antisense Oligonucleotides to Hepatocytes Using Triantennary N-Acetyl Galactosamine Improves Potency 10-Fold in Mice. Nucleic Acids Res 42 (13), 8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Yu RZ, Graham MJ, Post N, Riney S, Zanardi T, Hall S, Burkey J, Shemesh CS, Prakash TP, Seth PP, et al. (2016) Disposition and Pharmacology of a GalNAc3-Conjugated ASO Targeting Human Lipoprotein (a) in Mice. Mol. Ther.–Nucleic Acids 5, No. e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Huang Y (2017) Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther.–Nucleic Acids 6, 116–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Franchini M, and Mannucci PM (2018) Non-Factor Replacement Therapy for Haemophilia: A Current Update. Blood Transfus. Trasfus. Sangue 1–5, 1 DOI: 10.2450/2018.0272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, Bevan D, Austin S, Hay CR, Hegemann I, et al. (2017) Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N. Engl. J. Med 377 (9), 819–828. [DOI] [PubMed] [Google Scholar]
- (79).Bechara C, and Sagan S (2013) Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand? FEBS Lett 587 (12), 1693–1702. [DOI] [PubMed] [Google Scholar]
- (80).Lönn P, Kacsinta AD, Cui X-S, Hamil AS, Kaulich M, Gogoi K, and Dowdy SF (2016) Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN, Youngblood DS, and Iversen PL (2007) Pharmacokinetics, Biodistribution, Stability and Toxicity of a Cell-Penetrating Peptide–Morpholino Oligomer Conjugate. Bioconjugate Chem 18 (4), 1325–1331. [DOI] [PubMed] [Google Scholar]
- (82).Lönn P, and Dowdy SF (2015) Cationic PTD/CPP-Mediated Macromolecular Delivery: Charging into the Cell. Expert Opin. Drug Delivery 12 (10), 1627–1636. [DOI] [PubMed] [Google Scholar]
- (83).Ye J, Liu E, Gong J, Wang J, Huang Y, He H, and Yang VC (2017) High-Yield Synthesis of Monomeric LMWP(CPP)-SiRNA Covalent Conjugate for Effective Cytosolic Delivery of SiRNA. Theranostics 7 (9), 2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Meade BR, and Dowdy SF (2007) Exogenous SiRNA Delivery Using Peptide Transduction Domains/Cell Penetrating Peptides. Adv. Drug Delivery Rev 59 (2–3), 134–140. [DOI] [PubMed] [Google Scholar]
- (85).Betts CA, McClorey G, Healicon R, Hammond SM, Manzano R, Muses S, Ball V, Godfrey C, Merritt TM, and Westering T (2018) Cmah-Dystrophin Deficient Mdx Mice Display an Accelerated Cardiac Phenotype That Is Improved Following Peptide-PMO Exon Skipping Treatment. Hum. Mol. Genet, 1 DOI: 10.1093/hmg/ddy346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Blain AM, Greally E, McClorey G, Manzano R, Betts CA, Godfrey C, O’Donovan L, Coursindel T, Gait MJ, Wood MJ, et al. (2018) Peptide-Conjugated Phosphodiamidate Oligomer-Mediated Exon Skipping Has Benefits for Cardiac Function in Mdx and Cmah−/−Mdx Mouse Models of Duchenne Muscular Dystrophy. PLoS One 13 (6), No. e0198897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Gait MJ, Arzumanov AA, McClorey G, Godfrey C, Betts C, Hammond S, and Wood MJA (2018) Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther, 1 DOI: 10.1089/nat.2018.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Alam MR, Ming X, Fisher M, Lackey JG, Rajeev KG, Manoharan M, and Juliano RL (2011) Multivalent Cyclic RGD Conjugates for Targeted Delivery of Small Interfering RNA. Bioconjugate Chem 22 (8), 1673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Juliano RL, Ming X, Nakagawa O, Xu R, and Yoo H (2011) Integrin Targeted Delivery of Gene Therapeutics. Theranostics 1, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Gooding M, Malhotra M, Evans JC, Darcy R, and O’Driscoll CM (2016) Oligonucleotide Conjugates - Candidates for Gene Silencing Therapeutics. Eur. J. Pharm. Biopharm 107, 321–340. [DOI] [PubMed] [Google Scholar]
- (91).Ming X, Alam MR, Fisher M, Yan Y, Chen X, and Juliano RL (2010) Intracellular Delivery of an Antisense Oligonucleotide via Endocytosis of a G Protein-Coupled Receptor. Nucleic Acids Res 38 (19), 6567–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Nakagawa O, Ming X, Carver K, and Juliano R (2014) Conjugation with Receptor-Targeted Histidine-Rich Peptides Enhances the Pharmacological Effectiveness of Antisense Oligonucleotides. Bioconjugate Chem 25 (1), 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lobeek D, Franssen GM, Ma MT, Wester H-J, Decristoforo C, Oyen WJG, Boerman OC, Terry SYA, and Rijpkema M (2018) In Vivo Characterization of Four 68Ga-Labeled Multimeric RGD Peptides to Image Avβ3 Integrin Expression in Two Human Tumor Xenograft Mouse Models. J. Nucl. Med 59, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Shaw SK, Schreiber CL, Roland FM, Battles PM, Brennan SP, Padanilam SJ, and Smith BD (2018) High Expression of Integrin Αvβ3 Enables Uptake of Targeted Fluorescent Probes into Ovarian Cancer Cells and Tumors. Bioorg. Med. Chem 26 (8), 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Zhang J, Mao F, Niu G, Peng L, Lang L, Li F, Ying H, Wu H, Pan B, Zhu Z, et al. (2018) 68Ga-BBN-RGD PET/CT for GRPR and Integrin Αvβ3 Imaging in Patients with Breast Cancer. Theranostics 8 (4), 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Asami Y, Yoshioka K, Nishina K, Nagata T, and Yokota T (2016) Drug Delivery System of Therapeutic Oligonucleotides. Drug Discoveries Ther 10 (5), 256–262. [DOI] [PubMed] [Google Scholar]
- (97).Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, and Yokota T (2008) Efficient in Vivo Delivery of SiRNA to the Liver by Conjugation of Alpha-Tocopherol. Mol. Ther 16 (4), 734–740. [DOI] [PubMed] [Google Scholar]
- (98).Nishina T, Numata J, Nishina K, Yoshida-Tanaka K, Nitta K, Piao W, Iwata R, Ito S, Kuwahara H, Wada T, et al. (2015) Chimeric Antisense Oligonucleotide Conjugated to α-Tocopherol. Mol. Ther.–Nucleic Acids 4 (1), No. e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, and Giangrande PH (2006) Cell Type-Specific Delivery of SiRNAs with Aptamer-SiRNA Chimeras. Nat. Biotechnol 24 (8), 1005–1015. [DOI] [PubMed] [Google Scholar]
- (100).Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, Smith DD, Swiderski P, Rossi JJ, and Akkina R (2011) An Aptamer-SiRNA Chimera Suppresses HIV-1 Viral Loads and Protects from Helper CD4+ T Cell Decline in Humanized Mice. Sci. Transl. Med 3 (66), 66ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Zhou J, and Rossi J (2014) Cell-Type Specific Aptamer and Aptamer-SiRNA Conjugates for Targeted HIV-1 Therapy. J. Invest. Med 62 (7), 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Zhou J, Li H, Li S, Zaia J, and Rossi JJ (2008) Novel Dual Inhibitory Function Aptamer-SiRNA Delivery System for HIV-1 Therapy. Mol. Ther 16 (8), 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Kruspe S, and Giangrande PH (2017) Aptamer-SiRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines 5(3), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Ni S, Yao H, Wang L, Lu J, Jiang F, Lu A, and Zhang G (2017) Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci 18 (8), 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Song E, Zhu P, Lee S-K, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, et al. (2005) Antibody Mediated in Vivo Delivery of Small Interfering RNAs via Cell-Surface Receptors. Nat. Biotechnol 23 (6), 709–717. [DOI] [PubMed] [Google Scholar]
- (106).Satake N, Duong C, Yoshida S, Oestergaard M, Chen C, Peralta R, Guo S, Seth PP, Li Y, Beckett L, et al. (2016) Novel Targeted Therapy for Precursor B-Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med 22, 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu S-F, Wen X, Scales SJ, Gesch J, Davis D, van Brabant Smith A, et al. (2015) Systematic Evaluation of Antibody-Mediated SiRNA Delivery Using an Industrial Platform of THIOMAB–SiRNA Conjugates. Nucleic Acids Res 43 (2), 1189–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Godeau G, Staedel C, and Barthélémy P (2008) Lipid-Conjugated Oligonucleotides via “Click Chemistry” Efficiently Inhibit Hepatitis C Virus Translation. J. Med. Chem 51 (15), 4374–4376. [DOI] [PubMed] [Google Scholar]
- (109).Petrova NS, Chernikov IV, Meschaninova MI, Dovydenko Ii. S., Venyaminova AG, Zenkova MA, Vlassov VV, and Chernolovskaya EL (2012) Carrier-Free Cellular Uptake and the Gene-Silencing Activity of the Lipophilic SiRNAs Is Strongly Affected by the Length of the Linker between SiRNA and Lipophilic Group. Nucleic Acids Res 40 (5), 2330–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Chernikov IV, Gladkikh DV, Meschaninova MI, Ven’yaminova AG, Zenkova MA, Vlassov VV, and Chernolovskaya EL (2017) Cholesterol-Containing Nuclease-Resistant SiRNA Accumulates in Tumors in a Carrier-Free Mode and Silences MDR1 Gene. Mol. Ther.–Nucleic Acids 6, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Yang J, Chen C, and Tang X (2018) Cholesterol-Modified Caged SiRNAs for Photoregulating Exogenous and Endogenous Gene Expression. Bioconjugate Chem 29, 1010. [DOI] [PubMed] [Google Scholar]
- (112).Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, et al. (2004) Therapeutic Silencing of an Endogenous Gene by Systemic Administration of Modified SiRNAs. Nature 432 (7014), 173–178. [DOI] [PubMed] [Google Scholar]
- (113).DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, et al. (2007) Therapeutic Silencing of Mutant Huntingtin with SiRNA Attenuates Striatal and Cortical Neuropathology and Behavioral Deficits. Proc. Natl. Acad. Sci. U. S. A 104 (43), 17204–17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Yang J, Chen C, and Tang X (2018) Cholesterol-Modified Caged SiRNAs for Photoregulating Exogenous and Endogenous Gene Expression. Bioconjugate Chem 29, 1010. [DOI] [PubMed] [Google Scholar]
- (115).Nakajima M, Kasuya T, Yokota S, Onishi R, Ikehara T, Kugimiya A, and Watanabe A (2017) Gene Silencing Activity and Hepatic Accumulation of Antisense Oligonucleotides Bearing Cholesterol-Conjugated Thiono Triester at the Gap Region. Nucleic Acid Ther 27 (4), 232–237. [DOI] [PubMed] [Google Scholar]
- (116).Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, Barnes PJ, Sproat BS, Gait MJ, and Lindsay MA (2007) Lung Delivery Studies Using SiRNA Conjugated to TAT(48–60) and Penetratin Reveal Peptide Induced Reduction in Gene Expression and Induction of Innate Immunity. Bioconjugate Chem 18 (5), 1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Raouane M, Desmaële D, Urbinati G, Massaad-Massade L, and Couvreur P (2012) Lipid Conjugated Oligonucleotides: A Useful Strategy for Delivery. Bioconjugate Chem 23 (6), 1091–1104. [DOI] [PubMed] [Google Scholar]
- (118).Raouane M, Desmaele D, Gilbert-Sirieix M, Gueutin C, Zouhiri F, Bourgaux C, Lepeltier E, Gref R, Ben Salah R, Clayman G, et al. (2011) Synthesis, Characterization, and in Vivo Delivery of SiRNA-Squalene Nanoparticles Targeting Fusion Oncogene in Papillary Thyroid Carcinoma. J. Med. Chem 54 (12), 4067–4076. [DOI] [PubMed] [Google Scholar]
- (119).Herbert B-S, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, and Gryaznov SM (2005) Lipid Modification of GRN163, an N3′ → P5′ Thio-Phosphoramidate Oligonucleotide, Enhances the Potency of Telomerase Inhibition. Oncogene 24 (33), 5262–5268. [DOI] [PubMed] [Google Scholar]
- (120).Burchett KM, Yan Y, and Ouellette MM (2014) Telomerase Inhibitor Imetelstat (GRN163L) Limits the Lifespan of Human Pancreatic Cancer Cells. PLoS One 9 (1), e85155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Goldblatt EM, Erickson PA, Gentry ER, Gryaznov SM, and Herbert B-S (2009) Lipid-Conjugated Telomerase Template Antagonists Sensitize Resistant HER2-Positive Breast Cancer Cells to Trastuzumab. Breast Cancer Res. Treat 118 (1), 21–32. [DOI] [PubMed] [Google Scholar]
- (122).Gissot A, Di Primo C, Bestel I, Giannone G, Chapuis H, and Barthélémy P (2008) Sensitive Liposomes Encoded with Oligonucleotide Amphiphiles: A Biocompatible Switch. Chem. Commun 43, 5550–5552. [DOI] [PubMed] [Google Scholar]
- (123).Pokholenko O, Gissot A, Vialet B, Bathany K, Thiéry A, and Barthélémy P (2013) Lipid Oligonucleotide Conjugates as Responsive Nanomaterials for Drug Delivery. J. Mater. Chem. B 1 (39), 5329. [DOI] [PubMed] [Google Scholar]
- (124).Karaki S, Benizri S, Mejías R, Baylot V, Branger N, Nguyen T, Vialet B, Oumzil K, Barthélémy P, and Rocchi P (2017) Lipid-Oligonucleotide Conjugates Improve Cellular Uptake and Efficiency of TCTP-Antisense in Castration-Resistant Prostate Cancer. J. Controlled Release 258, 1–9. [DOI] [PubMed] [Google Scholar]
- (125).Acunzo J, Baylot V, So A, and Rocchi P (2014) TCTP as Therapeutic Target in Cancers. Cancer Treat. Rev 40 (6), 760–769. [DOI] [PubMed] [Google Scholar]
- (126).Baylot V, Katsogiannou M, Andrieu C, Taieb D, Acunzo J, Giusiano S, Fazli L, Gleave M, Garrido C, and Rocchi P (2012) Targeting TCTP as a New Therapeutic Strategy in Castration-Resistant Prostate Cancer. Mol. Ther 20 (12), 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Kirby M, Hirst C, and Crawford ED (2011) Characterising the Castration-Resistant Prostate Cancer Population: A Systematic Review. Int. J. Clin. Pract 65 (11), 1180–1192. [DOI] [PubMed] [Google Scholar]
- (128).Tajik-Ahmadabad B, Polyzos A, Separovic F, and Shabanpoor F (2017) Amphiphilic Lipopeptide Significantly Enhances Uptake of Charge-Neutral Splice Switching Morpholino Oligonucleotide in Spinal Muscular Atrophy Patient-Derived Fibroblasts. Int. J. Pharm 532 (1), 21–28. [DOI] [PubMed] [Google Scholar]
- (129).Wada F, Yamamoto T, Ueda T, Sawamura M, Wada S, Harada-Shiba M, and Obika S (2018) Cholesterol-GalNAc Dual Conjugation Strategy for Reducing Renal Distribution of Antisense Oligonucleotides. Nucleic Acid Ther 28 (1), 50–57. [DOI] [PubMed] [Google Scholar]
- (130).Yamamoto T, Sawamura M, Wada F, Harada-Shiba M, and Obika S (2016) Serial Incorporation of a Monovalent GalNAc Phosphoramidite Unit into Hepatocyte-Targeting Antisense Oligonucleotides. Bioorg. Med. Chem 24 (1), 26–32. [DOI] [PubMed] [Google Scholar]
- (131).Cutler JI, Auyeung E, and Mirkin CA (2012) Spherical Nucleic Acids. J. Am. Chem. Soc 134 (3), 1376–1391. [DOI] [PubMed] [Google Scholar]
- (132).Zhang K, Hao L, Hurst SJ, and Mirkin CA (2012) Antibody-Linked Spherical Nucleic Acids for Cellular Targeting. J. Am. Chem. Soc 134 (40), 16488–16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH, et al. (2015) Immunomodulatory Spherical Nucleic Acids. Proc. Natl. Acad. Sci. U. S. A 112 (13), 3892–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Skakuj K, Wang S, Qin L, Lee A, Zhang B, and Mirkin CA (2018) Conjugation Chemistry-Dependent T-Cell Activation with Spherical Nucleic Acids. J. Am. Chem. Soc 140 (4), 1227–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Barnaby SN, Perelman GA, Kohlstedt KL, Chinen AB, Schatz GC, and Mirkin CA (2016) Design Considerations for RNA Spherical Nucleic Acids (SNAs). Bioconjugate Chem 27 (9), 2124–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Seferos DS, Giljohann DA, Rosi NL, and Mirkin CA (2007) Locked Nucleic Acid–Nanoparticle Conjugates. ChemBioChem 8 (11), 1230–1232. [DOI] [PubMed] [Google Scholar]
- (137).Choi CHJ, Hao L, Narayan SP, Auyeung E, and Mirkin CA (2013) Mechanism for the Endocytosis of Spherical Nucleic Acid Nanoparticle Conjugates. Proc. Natl. Acad. Sci. U. S. A 110 (19), 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Banga RJ, Chernyak N, Narayan SP, Nguyen ST, and Mirkin CA (2014) Liposomal Spherical Nucleic Acids. J. Am. Chem. Soc 136 (28), 9866–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Zheng D, Giljohann DA, Chen DL, Massich MD, Wang X-Q, Iordanov H, Mirkin CA, and Paller AS (2012) Topical Delivery of SiRNA-Based Spherical Nucleic Acid Nano-particle Conjugates for Gene Regulation. Proc. Natl. Acad. Sci. U. S. A 109 (30), 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Lewandowski KT, Thiede R, Guido N, Daniel WL, Kang R, Guerrero-Zayas M-I, Seeger MA, Wang X-Q, Giljohann DA, and Paller AS (2017) Topically Delivered Tumor Necrosis Factor-α–Targeted Gene Regulation for Psoriasis. J. Invest. Dermatol 137 (9), 2027–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Piascik P (1999) Fomiversen Sodium Approved to Treat CMV Retinitis. J. Am. Pharm. Assoc 39 (1), 84–85. [DOI] [PubMed] [Google Scholar]
- (142).Kozak I, McCutchan JA, and Freeman WR (2013) Chapter 81 - HIV-Associated Infections In Retina, 5th ed. (Ryan SJ, Sadda SR, Hinton DR, Schachat AP, Sadda, Wilkinson CP, Wiedemann P, and Schachat AP, Eds.) pp 1441–1472, W.B. Saunders, London, DOI: 10.1016/B978-1-4557-0737-9.00081-3. [DOI] [Google Scholar]
- (143).Bennett CF, and Swayze EE (2010) RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol 50 (1), 259–293. [DOI] [PubMed] [Google Scholar]
- (144).Schoch KM, and Miller TM (2017) Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 94 (6), 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Evers MM, Toonen LJA, and van Roon-Mom WMC (2015) Antisense Oligonucleotides in Therapy for Neurodegenerative Disorders. Adv. Drug Delivery Rev 87, 90–103. [DOI] [PubMed] [Google Scholar]
- (146).Herrera VL, Colby AH, Ruiz-Opazo N, Coleman DG, and Grinstaff MW (2018) Nucleic Acid Nanomedicines in Phase II/III Clinical Trials: Translation of Nucleic Acid Therapies for Reprogramming Cells. Nanomedicine 13, 2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Juliano RL (2016) The Delivery of Therapeutic Oligonucleotides. Nucleic Acids Res 44 (14), 6518–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Crooke ST, Witztum JL, Bennett CF, and Baker BF (2018) RNA-Targeted Therapeutics. Cell Metab 27 (4), 714–739. [DOI] [PubMed] [Google Scholar]
- (149).Corporation Geron, Geron Announces Fast Track Designation Granted to Imetelstat for Lower Risk Myelodysplastic Syndromes, http://globenewswire.com/news-release/2017/10/31/1169498/0/en/Geron-Announces-Fast-Track-Designation-Granted-to-Imetelstat-for-Lower-Risk-Myelodysplastic-Syndromes.html (accessed Mar 8, 2018).
- (150).Machin N, and Ragni MV (2018) An Investigational RNAi Therapeutic Targeting Antithrombin for the Treatment of Hemophilia A and B. J. Blood Med 9, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Fine SL, Martin DF, and Kirkpatrick P (2005) Pegaptanib Sodium. Nat. Rev. Drug Discovery 4 (3), 187–188. [DOI] [PubMed] [Google Scholar]
- (152).Ng EWM, Shima DT, Calias P, Cunningham ET, Guyer DR, and Adamis AP (2006) Pegaptanib, a Targeted Anti-VEGF Aptamer for Ocular Vascular Disease. Nat. Rev. Drug Discovery 5 (2), 123–132. [DOI] [PubMed] [Google Scholar]
- (153).Garba AO, and Mousa SA (2010) Bevasiranib for the Treatment of Wet, Age-Related Macular Degeneration. Ophthalmol. Eye Dis 2, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Shen X, and Corey DR (2018) Chemistry, Mechanism and Clinical Status of Antisense Oligonucleotides and Duplex RNAs. Nucleic Acids Res 46 (4), 1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Niemietz CJ, Sauer V, Stella J, Fleischhauer L, Chandhok G, Guttmann S, Avsar Y, Guo S, Ackermann EJ, and Gollob J (2016) Evaluation of Therapeutic Oligonucleotides for Familial Amyloid Polyneuropathy in Patient-Derived Hepatocyte-Like Cells. PLoS One 11 (9), e0161455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, Kim T-W, Jesse Kwoh T, Matson J, Norris D, et al. (2016) Suppressing Transthyretin Production in Mice, Monkeys and Humans Using 2nd-Generation Antisense Oligonucleotides. Amyloid 23 (3), 148–157. [DOI] [PubMed] [Google Scholar]
- (157).Benson MD, Dasgupta NR, Rissing SM, Smith J, and Feigenbaum H (2017) Safety and Efficacy of a TTR Specific Antisense Oligonucleotide in Patients with Transthyretin Amyloid Cardiomyopathy. Amyloid 24 (4), 217–223. [DOI] [PubMed] [Google Scholar]
- (158).Rizk M, and Tüzmen Ş. (2017) Update on the Clinical Utility of an RNA Interference-Based Treatment: Focus on Patisiran. Pharmacogenomics Pers. Med 10, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Lundin KE, Gissberg O, and Smith CIE (2015) Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther 26 (8), 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, et al. (2013) Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med 368 (18), 1685–1694. [DOI] [PubMed] [Google Scholar]
- (161).van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, et al. (2014) Long-Term Safety and Efficacy of MicroRNA-Targeted Therapy in Chronic Hepatitis C Patients. Antiviral Res 111, 53–59. [DOI] [PubMed] [Google Scholar]
- (162).Beg MS, Brenner AJ, Sachdev J, Borad M, Kang Y-K, Stoudemire J, Smith S, Bader AG, Kim S, and Hong DS (2017) Phase I Study of MRX34, a Liposomal MiR-34a Mimic, Administered Twice Weekly in Patients with Advanced Solid Tumors. Invest. New Drugs 35 (2), 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Zlatev I, Castoreno A, Brown CR, Qin J, Waldron S, Schlegel MK, Degaonkar R, Shulga-Morskaya S, Xu H, Gupta S, et al. (2018) Reversal of SiRNA-Mediated Gene Silencing in Vivo. Nat. Biotechnol 36 (6), 509–511. [DOI] [PubMed] [Google Scholar]
- (164).Zlatev I, Castoreno A, Brown CR, Qin J, Waldron S, Schlegel MK, Degaonkar R, Shulga-Morskaya S, Xu H, Gupta S, et al. (2018) Reversal of SiRNA-Mediated Gene Silencing in Vivo. Nat. Biotechnol 36 (6), 509–511. [DOI] [PubMed] [Google Scholar]
- (165).Ekladious I, Colson YL, and Grinstaff MW (2018) Polymer-drug Conjugates for Cancer Treatment: Realities, Insights, and Prospects. Nat. Rev. Drug Discovery, 1 DOI: 10.1038/s41573-018-0005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Grinstaff MW, Kaplan HM, and Kohn J (2018) Predoctoral and Postdoctoral Training Pipeline in Translational Biomaterials Research and Regenerative Medicine. ACS Biomater. Sci. Eng 4, 3919. [DOI] [PMC free article] [PubMed] [Google Scholar]