

Abstract
BACKGROUND:
Research in transformed, immortalized cell lines indicates the cadherin-related family member 3 (CDHR3) protein serves as a receptor for human rhinovirus C (HRV-C). Similar experiments indicate the CDHR3 coding variant, rs6967330, increases CDHR3 protein surface expression.
OBJECTIVE:
To determine if CDHR3 is necessary for HRV-C infection of primary airway epithelial cells (AECs), and to identify molecular mechanisms by which CDHR3 variants confer risk for asthma exacerbations.
METHODS:
CDHR3 function and influence on HRV-C infection was investigated by single cell transcriptomics, CRISPR-Cas9 gene knockout, and genotype-specific donor experiments performed in primary AECs. Nasal airway epithelium cis-eQTL analysis of CDHR3 was performed followed by association testing for asthma hospitalization in minority children.
RESULTS:
CDHR3 lung expression is exclusive to ciliated AECs and is associated with basal bodies during and after motile ciliogenesis. Knockout of CDHR3 in human AECs did not prevent ciliated cell differentiation but was associated with a decrease in transepithelial resistance, and an 80% decrease in HRV-C infection of the mucociliary epithelium. AECs from subjects homozygous for the risk-associated rs6967330 SNP exhibited greater HRV-C infection compared to cells homozygous for the non-risk allele. AEC cis-eQTL analysis indicated rs6967330 and other SNPs are eQTLs for CDHR3. Only the eQTL block containing the rs6967330 SNP exhibited a significant association with childhood asthma hospitalization.
CONCLUSIONS:
Genetic deletion and genotype-specific studies in primary AECs indicate CDHR3 is critical to HRV-C infection of ciliated cells. The rs6967330 SNP confers risk of severe childhood asthma exacerbations likely through increasing HRV-C infection levels and protein surface localization.
Keywords: airway epithelium, CRISPR, rhinovirus, eQTL
Graphical Abstract
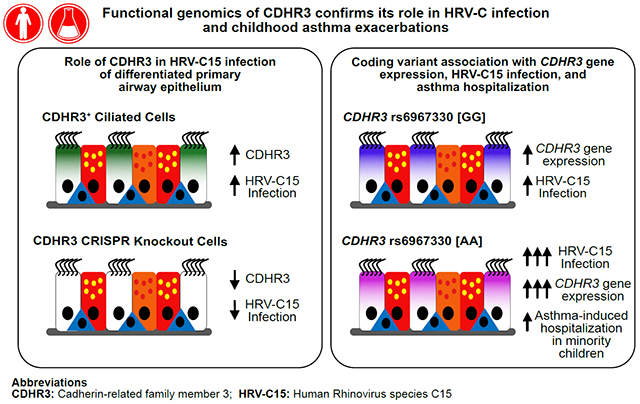
Capsule Summary
CRISPR-Cas9 studies of airway epithelial cells (AECs) confirm a role for CDHR3 in HRV-C infection and transepithelial resistance. A CDHR3 coding variant increases HRV-C infection and risk for asthma hospitalization in minority children.
INTRODUCTION
Asthma is the most common chronic disease of childhood, with severe exacerbations representing the cause of most patient burden and associated economic costs (1, 2). Asthma exacerbations are the result of complex interactions between environmental exposures and genetic predisposition, which are poorly understood. The environmental factor most commonly associated with asthma exacerbations is viral upper respiratory tract infections, occurring in 80% of exacerbations (3). Specifically, human rhinovirus species are found in >60% of these virus-associated exacerbations (4). Multiple studies have established infection with HRV-A is much more strongly associated with asthma exacerbations than infection with HRV-B (5–7). More recently, a third species, HRV-C, was discovered which several studies have found to be even more strongly associated with severe asthma exacerbations than HRV-A infections (8, 9).
The viral entry receptor for the majority of HRV-A and all HRV-B serotypes is the intercellular adhesion molecule 1 (ICAM-1), and the low density lipoprotein receptor (LDLR) acts as the receptor for some HRV-A serotypes (10, 11). Structure binding studies revealed HRV-C does not use either ICAM-1 or LDLR as its entry receptor (12). Rather, a recent genomic study identified a set of genes that are differentially expressed between HRV-C susceptible versus non-susceptible cell/tissue types. Heterologous expression of these genes in HeLa cells (a non-susceptible cell type) revealed that the expression of the cadherin-related family member 3 (CDHR3) allowed viral entry and replication in these cells, providing strong evidence that CDHR3 serves as the receptor for HRV-C (13). Moreover, although CDHR3 expression has been confirmed in the human airway epithelium, specifically in ciliated cells cultured in vitro (14), it is unclear if other cells in the lung express CDHR3. Supporting the possible expression in other lung cell types, a recent murine study described the mouse CDHR3 ortholog as a marker of an alveolar progenitor cell type (15).
A genome-wide scan for severe childhood asthma exacerbations identified CDHR3 among four genome-wide significant loci (16). The associated SNP, rs6967330, is a CDHR3 coding variant that results in a cysteine to tyrosine amino acid substitution at position 529 in the amino acid sequence. Heterologous expression of these allelic forms of CDHR3 in HeLa and HEK293T cells indicated that the asthma risk associated allelic form exhibited higher surface expression than the non-risk allelic form (13, 16). Taken together these data support a model where the rs6967330 variant increases asthma exacerbation risk by increasing surface expression of the CDHR3 protein and thus risk and possibly level of an HRV-C respiratory infection and illness. Several genetic studies have now associated the rs6967330 SNP with risk of asthma-related illnesses, including in Danish and Japanese patient cohorts (16–19).
Despite these findings many questions remain including: (1) whether lung cells other than ciliated cells express CDHR3, (2) if, as a cadherin-like protein, CDHR3 plays a role in cell adhesion or other ciliated cell functions, (3) if perturbation of CDHR3 expression in human AECs modulates HRV-C infection levels, (4) whether the rs6967330 variant of CDHR3 modifies HRV-C infection in AECs, and (5) whether rs6967330 or other cis-variants function as CDHR3 expression quantitative trait loci (eQTL) and modify risk for childhood asthma exacerbations. Here, we use CDHR3 CRISPR-Cas9 edited and CDHR3 risk genotype-specific primary AECs for functional experiments, as well as a comprehensive CDHR3 nasal airway epithelial eQTL analysis, and genetic association analysis for CDHR3 functional variants with childhood asthma exacerbations in order to answer these questions.
METHODS
Human Subject Information
Human lung cells for single cell RNA-sequencing and tracheal airway epithelial were isolated from de-identified lung donors whose lungs were not suitable for transplantation were obtained from International Institute for the Advancement of Medicine (Edison, NJ), and Donor Alliance of Colorado. The National Jewish Health Institutional Review Board (IRB) approved the research on lung cells under IRB protocol HS-3209. The tracheal airway epithelial cells were obtained in a de-identified fashion from the National Jewish Health (NJH) live cell core. The NJH Live Cell Core is an institutional review board-approved study (HS-2240) for the collection of tissues from consented patients for researchers at NJH. Nasal airway epithelial cells for culture and the eQTL study came from subjects recruited as part of the Genes-environments and Admixture in Latino Americans II (GALA II) childhood asthma cohort, which was approved by local institutional review boards (UCSF, IRB number 10–00889, Reference number 153543, NJH HS-2627). All subjects and their parents provided written informed assent and written informed consent, respectively (20, 21). Demographic and clinical variables for tissue donors used in this study are listed for all lung, tracheal, and nasal samples (Table E1) and for subjects in the genetic eQTL analysis (Table E2).
Single-Cell RNA Sequencing and Analysis
Single cell suspensions of elastase digested lung tissue was obtained as previously described (22). Cells were dispensed and imaged using the ICELL8™ Single-Cell System and samples sequenced with the Illumina HiSeq® 2500 System.
Culture of Primary Tracheal and Nasal Airway Epithelial Cells
Primary human tracheal and nasal AECs were expanded and differentiated at air-liquid interface (ALI) in vitro (23–26), and intact and dissociated cultures were harvested for Western blot, flow cytometry, immunofluorescence, and gene expression analyses (26).
Lentiviral CRISPR-Cas9 Gene Editing of Airway Basal Cells
The design of the CRISPR targeting guide sequences, addition of adaptors and cloning into the lentiCRISPR plasmid backbone, propagation and titration of lentivirus, and AEC transduction and selection were performed as previously described (23, 25).
Human Rhinovirus Infection
HRV-C15 virus was propagated using HeLa-E8 cells as previously described (12, 13). Cultures were infected with virus at an MOI of 0.2 for 4 hours at 34°C, washed, and incubated for a total of 48 hours at 34°C prior to harvest.
Whole Transcriptome Analysis, Genotyping, and eQTL Analysis
Whole transcriptome libraries of 695 nasal brushes from the GALA II childhood asthma cohort were constructed using the Beckman Coulter FXP automation system and samples sequenced using the Illumina HiSeq® 2500 system. DNA was genotyped using Affymetrix Axiom LAT 1 (World Array 4) and LAT plus HLA genome-wide arrays. Expression quantitative trait loci (eQTL) analysis followed the Genotype-Tissue Expression (GTEx) project protocol V7 for filtering and analysis (27).
Immunofluorescence Labeling
Intact ALI AEC culture inserts were fixed for 15 minutes at room temperature in 3.2% paraformaldehyde or with ice cold methanol at −20°C. Human lung tissue samples were fixed in 10% formalin prior to paraffin embedding and antibody labeling.
RESULTS
CDHR3 is exclusively expressed by ciliated airway epithelial cells
To identify CDHR3 expressing cells within the human lung, we performed single cell RNA sequencing (RNA-seq) of whole lung cell suspensions. By t-SNE clustering of cells’ gene expression we identified 13 distinct cell populations (Figure 1A). Differentially expressed genes between clusters were intersected with known cell type markers to determine the likely cell type identity of each cluster (Table E3). Cell types identified include airway epithelial cell (AEC) types (ciliated and secretory), alveolar type 1 and type 2 cells, lung immune cells, and endothelial cells. CDHR3 was highly and exclusively expressed in ciliated cells (Figure 1B), which were clearly identified as such by high expression of genes contained in cilia gene ontology (GO) categories (Figure E1). Immunofluorescence labeling of lung airway sections confirmed this result at the protein level as CDHR3 was only observed in ciliated AECs identified by acetylated alpha-Tubulin (ACT) and not secretory cells that are positive for MUC5AC (Figure 1C–D).
Figure 1: Single cell RNA-seq of the human lung reveals CDHR3 is exclusively expressed in ciliated airway epithelial cells.

A and B, RNA sequencing of single cells digested from a human lung identifies 13 distinct cell types using t-SNE analysis based on cell-specific gene markers (A) and demonstrates that CDHR3 gene expression is highly specific to ciliated cells in the lung (B) (n=1,142 individual cells analyzed from 1 digested lung and representative of single cell data from 6 total human lungs). C and D, Immunofluorescence labeling of whole lung tissue histology sections illustrates that CDHR3 localizes to the ciliated cells of the airway epithelium (C) but not to the MUC5AC+ secretory cells in the in vivo airway epithelium (D). Left panels – isotype control; Right panels – CDHR3 antibody. Bars - 50μm; white dotted line designates the basement membrane of the airway epithelium; image is representative of experiments completed using histology sections collected from 3 lung donors.
CDHR3 protein is apically distributed and is most highly expressed in immature ciliated cells
To study CDHR3 function we differentiated basal AECs into a mucociliary epithelium using the air liquid interface (ALI) cell culture model. Using time course analysis of CDHR3 gene and protein expression across mucociliary culture differentiation (Figure 2A), we found CDHR3 gene expression was evident by ALI day 5 and protein expression by day 6, prior to the appearance of motile cilia at day 11. Gene and protein expression increased until day 18 and were maintained at this level through ALI day 30.
Figure 2: CDHR3 exhibits an apical, cytoplasmic expression pattern highest in immature ciliating epithelial cells.
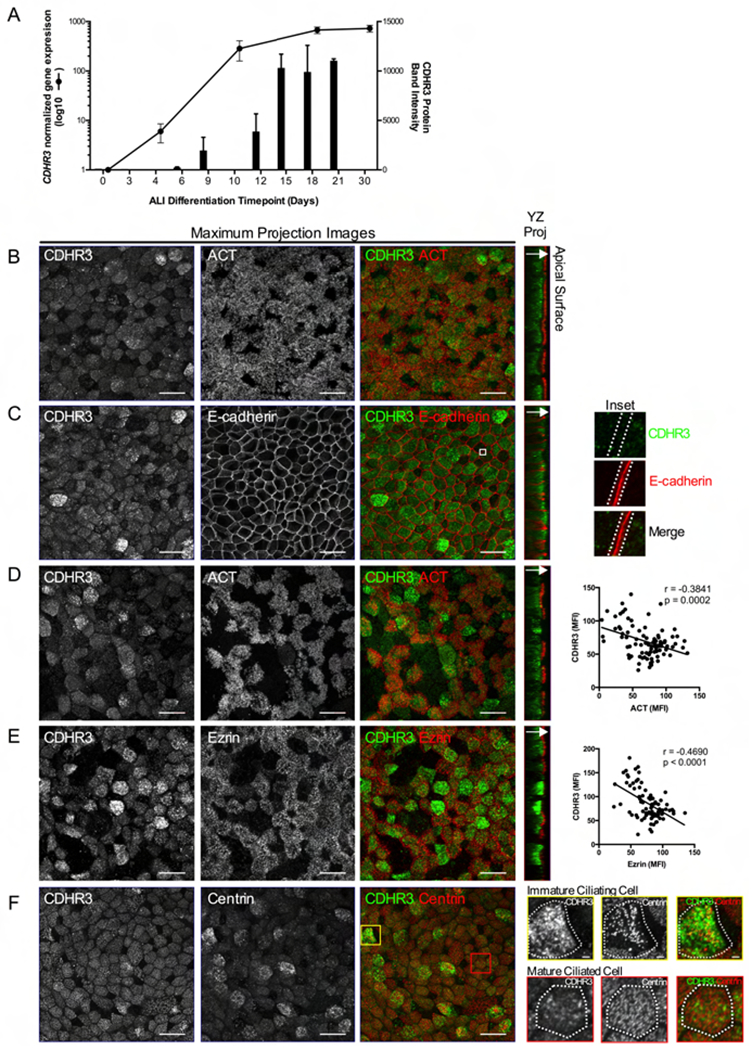
Tracheal basal AECs were differentiated in vitro at ALI for immunofluorescence labeling. A, qPCR and Western blot analyses were completed on cultures harvested over 30 days of differentiation for CDHR3 gene (n=3 donors) and protein expression (n=2 donors). B and C, ALI day 21 cultures were co-labeled for CDHR3 (green) and either ACT (red) (B) or E-cadherin (red) (C). Magnified inset panels indicate no co-localization of CDHR3 and E-cadherin at the cell-cell junction (C – far right panel). D and E, ALI day 14 cultures were co-labeled for CDHR3 (green) and either ACT (red) (D) or Ezrin (red) (E). MFI of each protein was quantified in individual cells in the culture for correlation analysis of intensities of CDHR3 and ACT or Ezrin (n=90 cells total) (D and E – far right panels). F, ALI day 21 cultures were co-labeled for CDHR3 (green) and centrin (red). Single cell panels illustrate the CDHR3 pattern in immature (top) and mature (bottom) cells as indicated by centrin pattern; single cell inset bars – 2μm. YZ projection images illustrate localization of proteins within the apical region of the epithelium (arrow indicates direction toward apical surface). Maximum projection bars - 20μm; images are representative of labeling performed in ALI cultures from 3 donors.
Co-labeling of CDHR3 and the cilia marker ACT in mature cultures (day 21) revealed the expected co-expression of these proteins (Figure 2B). Despite CDHR3 being a cadherin-related protein, it exhibited a punctate cytoplasmic rather than membrane localized staining pattern. Confirming this we co-labeled CDHR3 with E-cadherin, revealing no co-localization of these proteins. YZ-projection of confocal z-stacks revealed that CDHR3 puncta were localized in the cytoplasm, at the apical side of cells (Figure 2C). Given that CDHR3 is expressed early in ciliated cell development we also performed immunofluorescence labeling of day 14 cultures, which contain a mix of ciliating and mature ciliated cells. At ALI day 14, we identified many CDHR3+/ACT− cells (32.9% of total cell count; Figure 2D). These cells were rare at day 21 (3.5% of total cell count; Figure 2B), which led us to hypothesize that these were ciliating cells expressing CDHR3 prior to the development of cilia. Moreover, there was an inverse relationship between the mean fluorescent intensity (MFI) of CDHR3 and ACT within double positive cells at day 14 (r = −0.38, p = 0.0002, Figure 2D). To support the higher expression of CDHR3 in ciliating rather than mature ciliated cells, we co-labeled day 14 cultures for CDHR3 and ezrin, a marker of the apical membrane in mature ciliated cells. The brightest CDHR3 labeled cells were among the cells weakest for ezrin labeling, as reflected by the strong inverse correlation between CDHR3 and ezrin MFI values (r = −0.47, p< 0.0001, Figure 2E). The YZ-projection images also indicate that CDHR3 does not co-localize with ezrin to the apical membrane, but is localized to the apical sub-membrane compartment of ciliated cells (Figure 2E). To further validate that higher CDHR3 expression is found in ciliating cells in the differentiating AECs, we co-labeled cultures for centrin, a component of the basal body, which forms the intracellular base that anchors the cilium to the apical cell surface. During ciliogenesis, basal bodies are generated in the cytoplasm, traffic to and dock with the membrane, then elongate the cilium. Centrin has a well-defined pattern of basal body localization within ciliated cells during the course of ciliogenesis, which allows the identification of ciliating vs. mature ciliated cells (28). Image analysis revealed that cells with the strongest CDHR3 pattern co-expressed with centrin puncta in the cytoplasm indicative of ciliating cells with replicating or trafficking basal bodies. Alternatively, cells exhibiting centrin puncta evenly distributed at the apical surface to indicate the docked basal bodies of mature ciliated cells, exhibited dimmer expression of CDHR3 (Figure 2F).
CDHR3 gene knockout does not affect ciliated cell differentiation but results in reduced transepithelial membrane resistance
To investigate CDHR3 function in the airway epithelium we generated tracheal basal AECs knocked out for the CDHR3 gene using our lentiviral mediated CRISPR-Cas9 gene editing system (n=3 donors) (23, 25). The CDHR3 edited cells (CDHR3 KO) appeared normal and proliferated in culture similarly to the control edited basal cells (control). To assess whether the edited cells exhibited a loss of CDHR3 protein we differentiated the CDHR3 KO and control basal cells at ALI for 21+ days and performed quantitative Western blot analysis. We found the ALI cultures generated from CDHR3 KO cells exhibited 94.57% lower CDHR3 protein expression (Figure 3A, B; SD +/− 4.18%; p<0.0001) compared to cultures generated from control cells. Immunofluorescent labeling of cytospins prepared from differentiated ALI cultures also showed a marked decrease in CDHR3 protein within ACT+ ciliated cells (Figure 3C), indicating a substantial knockdown of CDHR3.
Figure 3: CDHR3 gene knockout decreases epithelium TEER and HRV-C infection.

A - E, CDHR3 KO or control (scrb) tracheal basal AECs were differentiated at ALI for 21 + days. Dissociated cultures were harvested for CDHR3 protein expression by Western blot (A and B), immunofluorescence labeling for CDHR3 (green) and ACT (red), bars – 20μm (C), and flow cytometry analysis (D and E). F, Transepithelial electrical resistance (TEER) measurements were taken at day 14 and day 21+ of ALI differentiation. G, Cultures were infected with HRV-C15 or HRV-A16 virus at an MOI of 0.2 for a total of 48 hours at 34°C prior to harvest and quantification of viral load by qPCR. Data are representative of CDHR3 KO or control (scrb) cultures grown and analyzed in parallel (n=3 independent donors).
Given the robust early ciliogenesis expression of CDHR3 we first wanted to determine if loss of CDHR3 expression affects the differentiation of ciliated cells. To address this, we performed flow cytometry analysis for CDHR3 and ACT in cells isolated from mature ALI cultures differentiated from CDHR3 KO and control basal cells (Figure 3D, E). We found 12.9% ± 1.43 of the control cells were ACT+/CDHR3+ versus only 2.34% ± 0.40 of the CDHR3 KO cells, an 81.67% ± 2.81 decrease with CDHR3 knockout (Figure 3E; p = 0.0140). In the CDHR3 KO cells we observed a corresponding 85.22% ± 1.33 increase in ACT+/CDHR3- cells, which represented 16.43 ± 5.95% of all cells versus only 2.65 ± 1.17 of all cells in the control population (Figure 3E; p = 0.0032). Importantly, we found the total percentage of ciliated cells (ACT+) was unchanged by CDHR3 knockout (KO = 18.8+/−%, Control = 15.6+/−%, p = 0.7531; Figure 3E). Additionally, we performed RNA-seq analysis of ALI differentiated control and KO edited cells from these 3 donors to determine whether expression of gene markers of ciliated cells and transcription factor drivers of ciliogenesis were affected by CDHR3 KO (Table E4). Supporting our flow cytometry data expression of FOXJ1, RFX2, RFX3, MYB, E2F4, and TP73 were not differentially expressed in the KO cells, despite significant downregulation of CDHR3 expression (padj = 4.6×10−18, Table E4). Further quantification of immunofluorescence labeling of FOXJ1+ nuclei in ALI cultures demonstrated that CDHR3 KO does not affect the expression of FOXJ1 or the ciliated cell fate of the airway epithelial cultures during differentiation (Figure E2). Moreover, club secretory and mucus secretory marker genes were unaffected by CDHR3 KO, suggesting loss of CDHR3 function has no effect on secretory cell formation. We did however find that among the 27 differentially expressed genes in CDHR3 KO cells (5% FDR and log2 fold change of >0.5 or <−0.5) that the basal cell marker genes KRT5, KRT14, NGFR were all upregulated, supporting that loss of CDHR3 either increases basal frequency or basal cell marker gene expression (Table E4).
Although we did not detect junctional localization, as a cadherin-related protein we speculated that CDHR3 KO may impact apical junctional integrity. We evaluated transepithelial electrical resistance (TEER) in AECs and observed that it was reduced in the CDHR3 KO cultures. We found that immature CDHR3 KO cultures (day 14), exhibited a 12.56% decrease in TEER compared to control cultures (Figure 3F; p<0.0001). Likewise, we found that mature differentiated ALI cultures from CDHR3 KO cells exhibited a 12.11% decrease in transepithelial resistance as compared to control cultures (Figure 3F; p<0.0001). These data indicate that CRISPR-Cas9 knockout of CDHR3 in primary AECs does not affect the development or differentiation of ciliated cells in the airway epithelium. However, the effect of CDHR3 knockout on epithelial TEER indicates a potential effect on barrier function, ion transport, or other epithelial organization despite a lack of exclusive CDHR3 junctional localization.
Deficiency in CDHR3 protein downregulates HRV-C15 infection of AECs
Our CDHR3 KO AECs allowed us to directly test whether HRV-C infection is dependent on CDHR3 protein expression in well-differentiated ALI-cultured human AECs. We examined both viral binding at four hours post-apical infection and infection at 48 hours by qPCR. We found no difference in HRV-C15 viral binding between control and KO cells (p = 0.1836; data not shown). However, at 48 hours post-infection, the CDHR3 KO epithelium exhibited 80.42% lower HRV-C15 RNA levels compared to control infected cultures (Figure 3G; SD ± 2.30%; p < 0.0001). To test if this effect on HRV-C15 infection was specific to the Rhinovirus C species, we performed the same experiment on CDHR3 KO cells with the HRV-A16 virus. We found no difference in the HRV-A16 RNA levels between control and CDHR3 KO infected cultures (Figure 3G; p = 0.9505).
CDHR3 rs6967330 genotype is associated with increased HRV-C infection of AECs
Our CDHR3 knockout experiments are supportive of CDHR3 levels influencing HRV-C infection. The CDHR3 coding risk variant rs6967330 has been shown to increase CDHR3 protein surface localization when allelic forms of the protein are expressed heterologously in HeLa and 293T cells (13, 16). Therefore, we hypothesized that donor AECs natively expressing the CDHR3 variant (A) allele would exhibit higher HRV-C infection levels than donor AECs expressing the alternate CDHR3 (G) allele. To test this hypothesis, we compared HRV-C15 infection levels between nasal AEC ALI cultures generated from six pediatric donors homozygous for the G allele (poorer surface localization) versus six donors homozygous for the A allele (better surface localization). Following HRV-C15 infection, we found a 4.3-fold increase in viral RNA isolated from the AA donor cells as compared to the GG donors at four hours post-infection (Figure 4; p = 0.0021). Likewise, at 48 hours we observed a 7.5-fold higher infection level in the AA versus GG subjects (Figure 4; p = 0.0390). These results show for the first time in primary human AECs differentiated from donors with different alleles for the CDHR3 SNP rs6967330, that carriage of the homozygous [A] genotype increases the risk of HRV-C15 infection at both the early and late stages of rhinovirus infection of the nasal airway epithelium.
Figure 4: HRV-C15 infection level is higher in AECs expressing the risk-associated CDHR3 rs6967330 genotype.
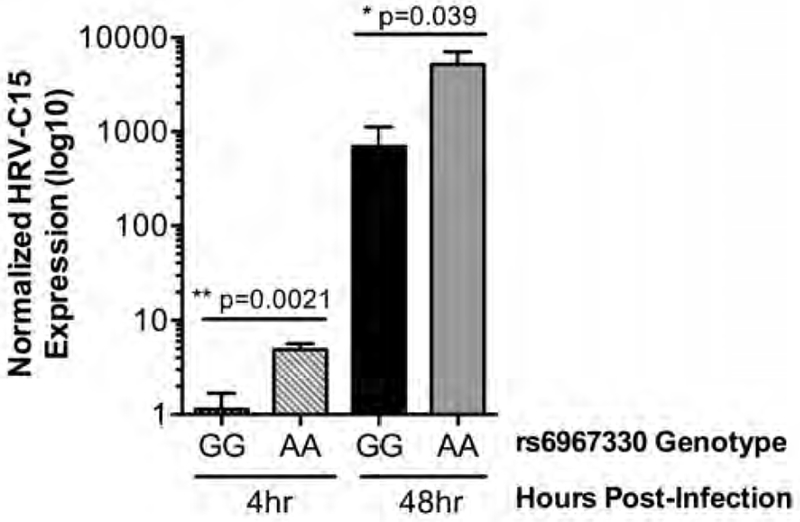
Nasal AECs homozygous for the rs6967330 G allele (n=6) or homozygous for the A allele (n=6) were differentiated at ALI. Cultures were infected with HRV-C15 at a MOI of 0.2 for 4 hours or a total of 48 hours at 34°C prior to quantitation of HRV-C15 by qPCR.
The rs6967330 variant is an eQTL associated with asthma hospitalization in minority children
We also considered whether the rs6967330 SNP might function as an expression quantitative trait loci (eQTL) in the airway epithelium to increase CDHR3 expression levels and thus susceptibility to HRV-C infection. To investigate this, we extracted CDHR3 eQTL data from the ongoing, genome-wide nasal airway epithelial eQTL study of the Genes-environments and Admixture in Latino Americans (GALA II) cohort (unpublished data). In this study we are using whole transcriptome sequencing expression data on nasal brushings from 431 asthmatic and 242 healthy control children, paired with genome-wide genotype data to perform a cis-eQTL analysis. We tested all genic SNPs and those 1Mb up- and down-stream of the gene for association with CDHR3 expression. We identified 11 independent eQTL loci for the CDHR3 gene (Table E5). A locus zoom plot, reveals the gene localization and linkage disequilibrium (LD) structure for the top four loci, all with association p values of <1×10−10 (Figure 5A). The CDHR3 eQTL SNP with the fourth strongest effect was part of an LD block which also included the rs6967330 coding risk SNP. We found the rs6967330 A allele, which we found was associated with increased HRV-C infection in primary AECs, was also associated with higher CDHR3 expression levels (Figure 5B; p = 2.16×10−9). In fact, we found there was a linear relationship between the number of variant alleles (higher expressed allele) for these top four eQTL SNPs a subject carried and CDHR3 gene expression levels (Figure 5C; p = 2.86×10−81). We tested the top four eQTL SNPs, as well as a variable for the total number of risk alleles carried, for association with asthma hospitalization risk in 670 Mexicans, 1153 Puerto Ricans, and 420 other Latino children with asthma by meta-analysis. We found only the rs73195680 SNP, located in the LD block with the rs6967330 loci, and rs6967330 itself were associated with asthma hospitalization risk (p = 5.0×10−3, 7.0×10−3 respectively, Table E6). These results reveal strong genetic regulation of the CDHR3 gene expression, but that only the rs6967330 coding variant LD block confers greater risk of asthma hospitalization in minority children.
Figure 5: The rs6967330 risk variant and other cis-variants are eQTLs for the CDHR3 gene in the nasal airway epithelium of children.
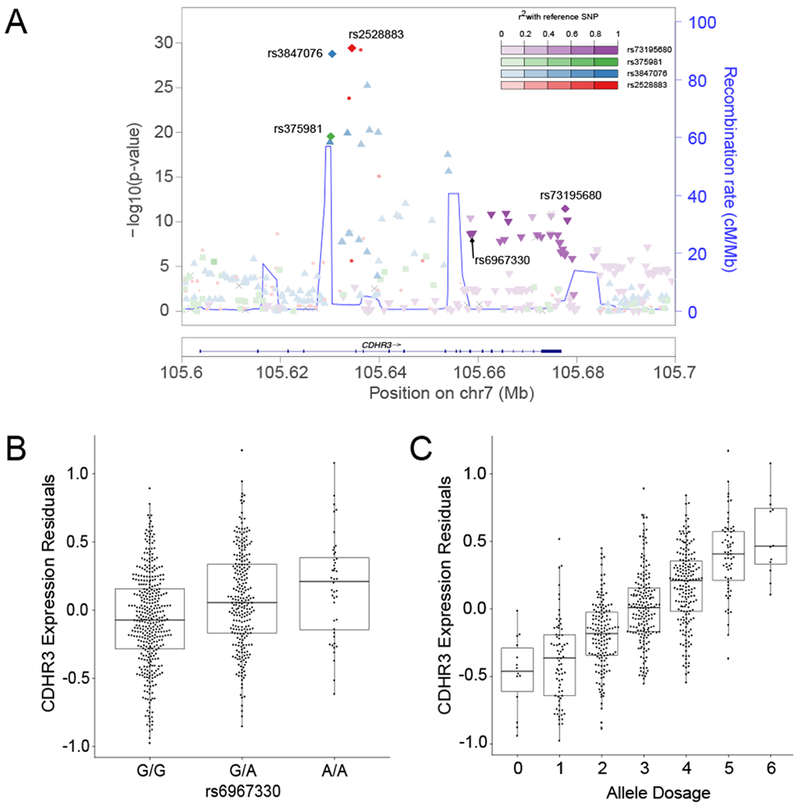
A, The top four independent cis-eQTLs for CDHR3 are depicted in the CDHR3 regional plot. B, The CDHR3 risk variant rs6967330 acts as an eQTL for CDHR3 gene expression. C, Additive effect of CDHR3 cis-eQTL variants on CDHR3 expression. Allele dosage was calculated as the sum of all eQTL associated (higher expression) effect alleles across the top four independent CDHR3 cis-eQTL variants.
DISCUSSION
The translation of novel asthma genetics findings into better understanding of molecular disease mechanisms has been impeded by a lack of suitable research methodologies in human airway tissue and primary cells. We applied a series of novel approaches to human airway epithelial cells including single cell transcriptomics, CRISPR gene editing, genotype-specific primary tissue culture, and eQTL studies to better understand the role of CDHR3 in viral induced asthma exacerbations.
Our single cell transcriptome data identifying most cell types in the human lung confirm a prior report of CDHR3 expression being restricted to the ciliated cells within airway epithelium (14). A recent report demonstrated that the mouse CDHR3 ortholog is expressed in a subset of alveolar cells, however, we did not detect CDHR3 in alveolar cells, thus this is likely not a conserved pattern of expression (15). As CDHR3 has been shown to be necessary for HRV-C infection, our data firmly establishes the tropism of HRV-C exclusively to ciliated cells in the human lung.
We leveraged the power of CRISPR-Cas9 technology to generate CDHR3 KO AECs. While KO did not abolish HRV-C infection, it was reduced by 80%, strongly supporting a significant role for CDHR3 in HRV-C infection. Surprisingly, we did not observe an effect of the KO on viral binding (although we did observe an effect of the CDHR3 variant risk on HRV-C binding). This result could be related to the timing or technical limitations of the binding assay, as well as higher sensitivity of the binding assay to incomplete KO. However, the reduced but measurable level of HRV-C infection we observed may also suggest the existence of co-receptors or weak alternative receptors among the mature mucociliary epithelium and/or other roles for CDHR3 in HRV-C infection including viral replication. In the future, development of an antibody against the extracellular domain of CDHR3 will allow a more complete investigation of the status of CDHR3 as the exclusive receptor for HRV-C in human AECs. Nonetheless, our CRISPR KO of CDHR3 allowed us to directly determine in primary well-differentiated AECs that robust HRV-C infection is dependent on expression of CDHR3.
As our data strongly support a role for CDHR3 in HRV-C infection of the human mucociliary airway epithelium, we addressed whether the rs6967330 asthma exacerbation risk variant could modulate HRV-C infection in well-differentiated AECs from genotyped donors. Our finding of a 7.5-fold increase in HRV-C infection among AECs derived from donors carrying the AA risk genotype strongly supports increased HRV-C infection as a mechanism for exacerbation risk associated with this variant. A prior study reported the CDHR3 protein carrying the rs6967330 risk allele (AA) results in greater protein surface localization than the non-risk allele (13, 16), providing a possible mechanism for our finding of increased HRV-C infection of AECs differentiated from donors with the AA genotype and consequently exacerbation risk. Unfortunately, the lack of an extracellular CDHR3 antibody has prevented formal confirmation of this increased surface localization of the endogenously expressed risk-associated rs6967330 allele in differentiated AECs.
Given that a large number of complex disease risk variants are regulatory in nature we decided to explore whether rs6967330 or other cis variants function as eQTLs for the CDHR3 gene. Our comprehensive cis-eQTL analysis of the nasal airway epithelium from >600 children established that CDHR3 gene expression modulated be genetic variation, with 11 independent eQTLs identified. This analysis allowed us to establish for the first time that the rs6967330 SNP is part of a CDHR3 eQTL linkage disequilibrium block. Since the risk A allele of the rs6967330 SNP was associated with higher CDHR3 gene expression, this presented the possibility that eQTL function rather than the previously suggested increase in CDHR3 protein localization at the cell surface, could explain the increased HRV-C infection and exacerbation risk associated with the rs6967330 variant. If this hypothesis was correct we would have expected the rs6967330 LD block, which was the 4th strongest eQTL loci, and the 3 stronger CDHR3 cis-eQTLs, to be associated with asthma exacerbation risk. Instead our finding that only the rs6967330 eQTL was associated with asthma hospitalization risk strongly supports an alternative mechanism for the variant risk, likely the reported change in surface localization. Moreover, our association between the rs6967330 variant and asthma-related hospitalization presents the first evidence that this variant confers risk in Hispanic populations. This extends a growing body of data implicating the rs6967330 variant in the risk of asthma exacerbations/hospitalization and related traits including early-life bronchiolitis, early-onset asthma, and chronic rhinosinusitis (16–19, 29).
Monitoring of gene and protein expression throughout AEC differentiation indicated robust CDHR3 expression prior to the appearance of cilia. Our finding of CDHR3 localization near the basal bodies in ciliating and mature ciliated cells, raised the possibility of its involvement in ciliogenesis. CDHR3 KO AECs differentiated at ALI allowed us to conclusively determine that CDHR3 loss does not block ciliated cell formation. We also localized CDHR3 to the apical surface in the vicinity of ezrin, a protein that links the apical cytoskeleton to the apical membrane. We speculate that CDHR3 may also be a component and possibly regulator of this network of actin and microtubules that is in constant contact with cilia, the cell surface, and the apical cell junctions. Through these interactions, CDHR3 may impact many vital ciliated cell processes, including barrier function, which is supported by the consistent decrease in transmembrane resistance upon CDHR3 knockout. If the reduced transmembrane resistance is mediated by a change in barrier function, this could allow greater penetration of allergen, toxins, and microorganisms in the airway, providing a potential mechanism for CDHR3 function influencing asthma exacerbation risk. Further work will be needed to confirm an effect of CDHR3 on barrier function.
In summary, our work here has provided some of the first data on CDHR3 expression, biology, and molecular genetics in the human airway epithelium. We have revealed the likely mechanism underlying the risk of asthma exacerbations conferred by the CDHR3 rs6967330 variant, while extending this risk to minority children with asthma. These data form the basis for both further human genetics and molecular mechanistic studies of CDHR3 involvement in childhood asthma and asthmatic airway epithelium.
Supplementary Material
Key Message.
Genetic deletion of CDHR3 in human mucociliary epithelial cultures strongly support involvement of CDHR3 in HRV-C infection of human airway epithelial cells .
A CDHR3 coding SNP (rs6967330) increases HRV-C infection of airway epithelial cells and is associated with asthma hospitalization in minority children.
ACKNOWLEDGEMENTS
We would like to thank Shirley Sobus and Josh Loomis at the National Jewish Flow and Microscopy core for guidance with instrument usage and analysis.
Funding Sources:
This work was supported by the R01 HL135156, R01 MD010443, R01 HL128439, P01 HL132821, P01 HL107202 and U01 HL138626.
Conflict of Interest
MAS reports grants from the NIH during the conduct of the study, and grants from Medimmune, Department of Defense, Pfizer, Genentech outside the submitted work, Patent invention “Transcriptomic response of airway epithelial cells to IL-13”, in process: File No. 2879–190-PROV-1 pending, Patent invention “Methods of diagnosing and treating subjects at risk of inflammation and/or exacerbation of a respiratory disease or condition”, in process: File No. 2879–191-PROV-1 pending, and invited lecture at Genentech. MAS and JLE report Patent invention “Methods of Identifying and treating subjects having inflammatory subphenotypes on asthma”, in process: File No. 2879–178-PROV pending. MN and DRV report grants from NIH and FAMRI during the conduct of this study focused upon mechanisms of anti-viral and anti-inflammatory actions of phospholipids, and are co-inventors of the Patent invention “Surfactant lipids, compositions thereof, and uses thereof” owned by National Jewish Health, 1400 Jackson St., Denver, CO 80206. The remaining authors report no relevant conflicts of interest for this study.
Abbreviations
- ALI
Air-Liquid Interface
- KO
knockout
- AEC
airway epithelial cell
- GALA II
Genes-environments and Admixture in Latino Americans II
- HRV
human rhinovirus
- CDHR3
cadherin-like family member 3
- eQTL
expression Quantitative Trait Loci
- ACT
acetylated alpha-Tubulin
- TEER
transepithelial electrical resistance
- GWAS
Genome Wide Association Study
- SNP
Single Nucleotide Polymorphism
- LD
Linkage Disequilibrium
- RNA-seq
RNA sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ivanova JI, Bergman R, Birnbaum HG, Colice GL, Silverman RA, McLaurin K. Effect of asthma exacerbations on health care costs among asthmatic patients with moderate and severe persistent asthma. J Allergy Clin Immunol. 2012;129(5):1229–35. [DOI] [PubMed] [Google Scholar]
- 2.Nurmagambetov T, Kuwahara R, Garbe P. The Economic Burden of Asthma in the United States, 2008–2013. Ann Am Thorac Soc. 2018;15(3):348–56. [DOI] [PubMed] [Google Scholar]
- 3.Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310(6989):1225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston SL, Sanderson G, Pattemore PK, Smith S, Bardin PG, Bruce CB, et al. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J Clin Microbiol. 1993;31 (1):111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khetsuriani N, Lu X, Teague WG, Kazerouni N, Anderson LJ, Erdman DD. Novel human rhinoviruses and exacerbation of asthma in children. Emerg Infect Dis. 2008;14(11):1793–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao M, Zhu WJ, Qian Y, Sun Y, Zhu RN, Deng J, et al. Association of Different Human Rhinovirus Species with Asthma in Children: A Preliminary Study. Chin Med J (Engl). 2016;129(13):1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen WJ, Arnold JC, Fairchok MP, Danaher PJ, McDonough EA, Blair PJ, et al. Epidemiologic, clinical, and virologic characteristics of human rhinovirus infection among otherwise healthy children and adults: rhinovirus among adults and children. J Clin Virol. 2015;64:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J. 2011. ;37(5):1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller EK, Khuri-Bulos N, Williams JV, Shehabi AA, Faouri S, Al Jundi I, et al. Human rhinovirus C associated with wheezing in hospitalised children in the Middle East. J Clin Virol. 2009;46(1):85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56(5):839–47. [DOI] [PubMed] [Google Scholar]
- 11.Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci U S A. 1994;91(5):1839–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochkov YA, Palmenberg AC, Lee WM, Rathe JA, Amineva SP, Sun X, et al. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat Med. 2011;17(5):627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A. 2015;112(17):5485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griggs TF, Bochkov YA, Basnet S, Pasic TR, Brockman-Schneider RA, Palmenberg AC, et al. Rhinovirus C targets ciliated airway epithelial cells. Respir Res. 2017;18(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555(7695):251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(1):51–5. [DOI] [PubMed] [Google Scholar]
- 17.Stenberg Hammar K, Niespodziana K, van Hage M, Kere J, Valenta R, Hedlin G, et al. Reduced CDHR3 expression in children wheezing with rhinovirus. Pediatr Allergy Immunol. 2018;29(2):200–6. [DOI] [PubMed] [Google Scholar]
- 18.Kanazawa J, Masuko H, Yatagai Y, Sakamoto T, Yamada H, Kaneko Y, et al. Genetic association of the functional CDHR3 genotype with early-onset adult asthma in Japanese populations. Allergol Int. 2017;66(4):563–7. [DOI] [PubMed] [Google Scholar]
- 19.Husby A, Pasanen A, Waage J, Sevelsted A, Hodemaekers H, Janssen R, et al. CDHR3 gene variation and childhood bronchiolitis. J Allergy Clin Immunol. 2017;140(5):1469–71 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neophytou AM, White MJ, Oh SS, Thakur N, Galanter JM, Nishimura KK, et al. Air Pollution and Lung Function in Minority Youth with Asthma in the GALA II (Genes-Environments and Admixture in Latino Americans) and SAGE II (Study of African Americans, Asthma, Genes, and Environments) Studies. Am J Respir Crit Care Med. 2016; 193(11):1271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, et al. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188(3):309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosmider B, Mason RJ, Bahmed K. Isolation and Characterization of Human Alveolar Type II Cells. Methods Mol Biol. 2018;1809:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu HW, Rios C, Huang C, Wesolowska-Andersen A, Burchard EG, O’Connor BP, et al. CRISPR-Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015;22(10):822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds SD, Rios C, Wesolowska-Andersen A, Zhuang Y, Pinter M, Happoldt C, et al. Airway Progenitor Clone Formation Is Enhanced by Y-27632-Dependent Changes in the Transcriptome. Am J Respir Cell Mol Biol. 2016;55(3):323–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everman JL, Rios C, Seibold MA. Primary Airway Epithelial Cell Gene Editing Using CRISPR-Cas9. Methods Mol Biol. 2018;1706:267–92. [DOI] [PubMed] [Google Scholar]
- 26.Everman JL, Rios C, Seibold MA. Utilization of Air-Liquid Interface Cultures as an In Vitro Model to Assess Primary Airway Epithelial Cell Responses to the Type 2 Cytokine Interleukin-13. Methods Mol Biol. 2018;1799:419–32. [DOI] [PubMed] [Google Scholar]
- 27.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vladar EK, Stearns T. Molecular characterization of centriole assembly in ciliated epithelial cells. J Cell Biol. 2007;178(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang EH, Willis AL, McCrary HC, Noutsios GT, Le CH, Chiu AG, et al. Association between the CDHR3 rs6967330 risk allele and chronic rhinosinusitis. J Allergy Clin Immunol. 2017;139(6):1990–2 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.