

Abstract
Dysferlinopathy is a progressive muscle disorder that includes limb‐girdle muscular dystrophy type 2B and Miyoshi myopathy (MM). It is caused by mutations in the dysferlin (DYSF) gene, whose function is to reseal the muscular membrane. Treatment with proteasome inhibitor MG‐132 has been shown to increase misfolded dysferlin in fibroblasts, allowing them to recover their membrane resealing function. Here, we developed a screening system based on myocytes from MM patient‐derived induced pluripotent stem cells. According to the screening, nocodazole was found to effectively increase the level of dysferlin in cells, which, in turn, enhanced membrane resealing following injury by laser irradiation. Moreover, the increase was due to microtubule disorganization and involved autophagy rather than the proteasome degradation pathway. These findings suggest that increasing the amount of misfolded dysferlin using small molecules could represent an effective future clinical treatment for dysferlinopathy. stem cells translational medicine 2019;8:1017–1029
Keywords: Induced pluripotent stem cells, Skeletal muscle, Drug target, Muscular dystrophy, Differentiation, Myosin heavy chain
Significance Statement.
Dysferlinopathy is one of the rare intractable diseases. There are no drugs to treat this disease yet. In this study, using myocytes differentiated from patient‐derived induced pluripotent stem cells, nocodazole was identified as a drug candidate. Microtubule disorganization and autophagy dysfunction was revealed as the mechanism of nocodazole. This finding should be useful information for future clinical treatment for dysferlinopathy.
Introduction
Dysferlin is a membrane protein composed of seven β‐sheet‐enriched C2 domains and is involved in Ca2+‐dependent membrane repair after wounding 1. Mutation of the dysferlin (DYSF) gene causes progressive muscle disorders, such as limb‐girdle muscular dystrophy type 2B and Miyoshi myopathy (MM) 2. Both disorders are autosomal recessive muscular dystrophies and are characterized by onset in young adults, a slow course of the disease, a massive increase in serum creatine kinase levels 3, and delayed muscular membrane repair after injury 4, 5.
Although several mutations in the DYSF gene have been reported, with clinical results showing the W999C missense mutation being associated with late onset of the disease 6, misfolded dysferlin has been found to possess residual membrane resealing function. Accordingly, W999C missense mutated dysferlin is still functional; however, it is structurally unstable due to misfolding and, therefore, its reduced levels contribute to the onset of dysferlinopathy. As a result, it is believed that stabilization of dysferlin through the blocking of protein degradation could offer an effective treatment for dysferlinopathy. In fact, treatment with proteasome inhibitor MG‐132 has been reported to increase the level of wild‐type, truncated, and misfolded dysferlin in human fibroblasts. Importantly, membrane resealing could be restored following treatment with MG‐132 in fibroblasts bearing a missense mutated DYSF 7. This finding suggested that misfolded dysferlin was degraded by the proteasome system. Additionally, MG‐132 has been reported to rescue the expression level and membrane localization of the SERCA1 missense mutant in a heterologous cell model of Chianina cattle congenital pseudomyotonia 8. Based on those results, we hypothesized that inhibiting the degradation of misfolded dysferlin would lead to its intracellular accumulation and, ultimately, an improvement in the dysferlinopathy phenotype. Based on that hypothesis, here, we established a system for the screening of small molecules that could increase the level of dysferlin in myocytes of patients with the W999C DYSF missense mutation.
Human induced pluripotent stem (iPS) cell‐based disease models represent a useful and powerful tool for drug discovery, especially for intractable and rare diseases, for which it is difficult to obtain cell samples from patients. Drug discovery using patient‐derived iPS cells with a certain genetic background has been established only over the past decade, yet several drug candidates against diseases such as amyotrophic lateral sclerosis 9 and fibrodysplasia ossificans progressiva 10 have already been reported.
Here, we established human iPS cells derived from a MM patient and induced skeletal myocytes by the forced expression of the skeletal muscle‐specific myogenic differentiation 1 (MyoD1) 11 transcription factor 12. To discover drugs against dysferlinopathy, we developed a screening system capable of identifying small molecules that increased the intracellular level of mutated dysferlin. We screened a limited number of off‐patent, Food and Drug Administration (FDA)‐approved drugs whose function were already elucidated, and we report that one of the identified molecules, nocodazole, effectively increased the level of misfolded dysferlin, and enhanced membrane resealing following injury by laser irradiation. Although nocodazole itself may not be the best choice for treating dysferlinopathy patients in the clinic due to its highly inhibitory effect on the cell cycle, the chemically induced increment of misfolded dysferlin could be a paradigm for future therapies against this disease.
Materials and Methods
Ethics
All experimental protocols in the study were approved by the Ethics Committee of the Graduate School and Faculty of Medicine, Kyoto University (approval numbers #R0091 and #G259), and Takeda Pharmaceutical Company Ltd. (GEN‐00000040‐008). The study was performed in accordance with the guidelines of the Declaration of Helsinki and conducted after obtaining written informed consent.
Establishment of Human iPS Cells
All human iPS cell lines used in this study were generated from fibroblasts. iPS cell lines were established using an episomal vector system as previously described 13. Fibroblasts used in these experiments were from a MM patient (46‐year‐old Japanese female), who has compound hetero mutations with c.2997 G>T (p. W999C) as missense mutation and c.1958delG as nonsense mutation. Control iPS cells were obtained from the younger sister of the MM patient (36‐year‐old Japanese female), who also has a c.2997 G>T (p. W999C) missense mutation on one allele but does not have the same mutation on the other allele. Because this genetic form of dysferlinopathy is autosomal recessive, the younger sister does not present any disease symptoms, and her iPS cells can be used as a control cell line. All iPS cell lines were cultured on mouse feeder cells in Primate Embryonic Stem (ES) Cell Medium (RCHEMD001; ReproCELL, Yokohama, Japan) containing 10 ng/ml of recombinant human basic fibroblast growth factor (Oriental Yeast, Tokyo, Japan).
Cell Culture
Screening and mechanistic studies were performed on myocytes differentiated from the iPS cells generated as mentioned above. iPS cells were cultured and maintained as a feeder‐free culture on iMatrix‐511 (892012; Nippi, Tokyo, Japan) in StemFit medium (AK02N; Ajinomoto, Tokyo, Japan) as previously described 14. Differentiation of MM iPS cells into myocytes was performed by the forced expression of MyoD1 under the control of doxycycline (D5897; Wako, Osaka, Japan) as previously described 12, 15. Briefly, on day 0, cells were plated on Matrigel‐coated CellCarrier‐96 or CellCarrier‐384 Ultra microplates (6057300; PerkinElmer, Waltham, MA) with Rock inhibitor (251‐00514; Wako); the next day, medium was switched to Primate ES Cell Medium. On day 2, doxycycline was added to the same medium, and the latter was switched to Minimum Essential Medium Eagle, Alpha Modification (α‐MEM) (21444‐05; Nakalai Tesque, Kyoto, Japan) containing 5% knockout serum replacement (10828028; Thermo Fisher Scientific, Waltham, MA) on day 3. The medium was changed every other day until day 8.
Screening Assay by Immunofluorescent Staining
Drug screening was performed on myocytes on a CellCarrier‐384 Ultra microplate. Small molecules in dimethyl sulfoxide (DMSO) were added at a concentration of 3 μM on day 7 and, 24 hours later, cells were fixed with 2% (wt/vol) paraformaldehyde (163‐20145; Wako) in phosphate‐buffered saline (PBS, 045‐29795; Wako) for 30 minutes. Cells were then blocked with 5% normal goat serum (16210064; Thermo Fisher Scientific) with 0.4% (vol/vol) Triton X‐100 in PBS for 1 hour before immunostaining. The following primary antibodies were used: anti‐dysferlin (1:800, ab124684; Abcam, Cambridge, U.K.) and anti‐myosin heavy chain (MHC; 1:800, MAB4470; R&D Systems, Minneapolis, MN). Secondary antibodies included Alexa Fluor 647 goat anti‐mouse (1:000, A21236) for MHC and Alexa Fluor 488 goat anti‐rabbit (1:1,000, A11034, both Thermo Fisher Scientific) for dysferlin. Nuclei were stained by Hoechst 33342 solution (1:2,000, 346‐07951; Wako). Stained cells were imaged using the Opera Phenix High Content Screening System (PerkinElmer) and the effect of small molecules was evaluated by the relative staining intensity of dysferlin to 0.1% DMSO control in each plate, as calculated by Harmony High Content Imaging and Analysis Software (PerkinElmer). All 622 small molecules tested in this study were from an external repositioning library of Takeda Pharmaceutical Company Ltd.
RNA Isolation and Quantitative Polymerase Chain Reaction
Total RNA was isolated and reverse‐transcribed using the TaqMan Gene Expression Cells‐to‐CT Kit (4399002; Thermo Fisher Scientific) according to the manufacturer's instructions. Quantitative polymerase chain reaction (PCR) for dysferlin was performed using TaqMan Fast Advanced Master Mix (4444557; Thermo Fisher Scientific). Assays were performed in multiple samples with the QuantiStudio7 Flex system (Thermo Fisher Scientific). Relative gene expression was calculated by the comparative Ct method using 36B4 as an endogenous control. PCR primers and TaqMan probe for human dysferlin (5′‐AGACGGATTACACCTTCCCACTTGCTGA‐3′) were purchased from Thermo Fisher Scientific (Hs01002513_m1). Human 36B4 forward (5′‐AAACGAGTCCTGGCCTTGTCT‐3′) and reverse primers (5′‐GCAGATGGATCAGCCAAGAAG‐3′) were purchased from Sigma–Aldrich, St. Louis, MO.
Western Blotting
After collection in ice‐cold PBS, cells were lysed in RIPA buffer containing 1 mM DTT, and protease and phosphatase inhibitors (89900; Thermo Fisher Scientific). The soluble fraction was subjected to SDS‐PAGE (456‐1086; Bio‐Rad, Hercules, CA) and protein transfer was performed using a Trans‐Blot Turbo Transfer System (1704156; Bio‐Rad) or Blocking One (03953‐95; Nakarai, Kyoto, Japan). The following primary antibodies were used: anti‐dysferlin (1:1,000, ab124684), anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; 14C10; 1:1,000–3,000, 2118; Cell Signaling Technology, Danvers, MA), anti‐p62 (1:1,000, 66184‐1‐JG; Proteintech, Rosemont, IL), and anti‐LC3 (1:1,000, M186‐3; MBL, Nagoya, Japan). Secondary antibodies included donkey Horse Radish Peroxidase (HRP)‐linked anti‐rabbit IgG (NA934‐1ML) and sheep HRP‐linked anti‐mouse IgG (NA931‐1ML), both from GE Healthcare, Little Chalfont, U.K. Antibodies were diluted in Canget Signal solution (NKB‐101; TOYOBO, Osaka, Japan) and blotting was performed using the iBind Western System (Thermo Fisher Scientific) at room temperature. Immunoblot detection was performed using ChemiDoc Touch Imaging System (Bio‐Rad) and band area was quantified by Image Lab (Bio‐Rad).
Membrane Resealing Assay
The membrane resealing assay was performed on day 8 using differentiated myocytes as previously described 12. Briefly, membrane damage was induced in the presence of FM 1‐43 dye (T35356; Thermo Fisher Scientific) with a confocal laser‐scanning microscope (LSM880; Zeiss, Oberkochen, Germany). Images were captured every 5 seconds for 165 seconds or 3 seconds for 99 seconds after irradiation. For every image taken, the fluorescence intensity at the site of damage was measured with Zeiss LSM 880 imaging software.
Chemicals
Nocodazole was synthesized by Takeda Pharmaceutical Company Limited. Colchicine (039‐03851) and Paclitaxel (163‐28163) were purchased from Fujifilm Wako Chemicals, Japan.
Statistical Analysis
Statistical analysis was performed in GraphPad Prism (GraphPad Software, San Diego, CA) by applying t test, Dunnett's, and Tukey's multiple comparison tests after analysis of variance. William's multiple comparison test was performed in EXSUS software (CAC Croit, Tokyo, Japan). n refers to the number of independent experiments or cultures of iPS cell‐derived myocytes. Data are reported as sample mean + SD. Significance levels of statistical tests are indicated as *, p < .025; **, p < .005; or ***, p < .0005 for William's test, and *, p < .05; **, p < .01; or ***, p < .001 for other tests.
Results
Dysferlin Expression in MM and Control Myocytes
First, we assessed the expression of dysferlin in control myocytes differentiated from iPS cells. To effectively induce myocytes from human iPS cells, the latter were transfected with MyoD1 using a tetracycline inducible piggyBac system 12. Control iPS cells were established from the sister of the MM patient, who did not present any diseased muscle phenotype. Control iPS cells (clone: Control#17) were differentiated in a 96‐multiwell plate as outlined in Figure 1A. Cell samples were fixed by paraformaldehyde on day 5, 6, 7, 8, 9, and 10 for immunostaining. Fixed cells were probed with antibodies against dysferlin and the muscle‐specific marker MHC. As shown in Figure 1B, expression of dysferlin and MHC increased gradually during the culture period. The intensity of dysferlin staining was highest on day 8 (Fig. 1C), and decreased on day10, whereas no difference in MHC expression could be observed between day 7 and day 9. To assess differentiation efficiency, the percentage of MHC‐positive myocytes among all cells (identified as Hoechst‐positive) was calculated. Accordingly, myocyte differentiation was highest on day 8 and day 98 (Fig. 1D).
Figure 1.
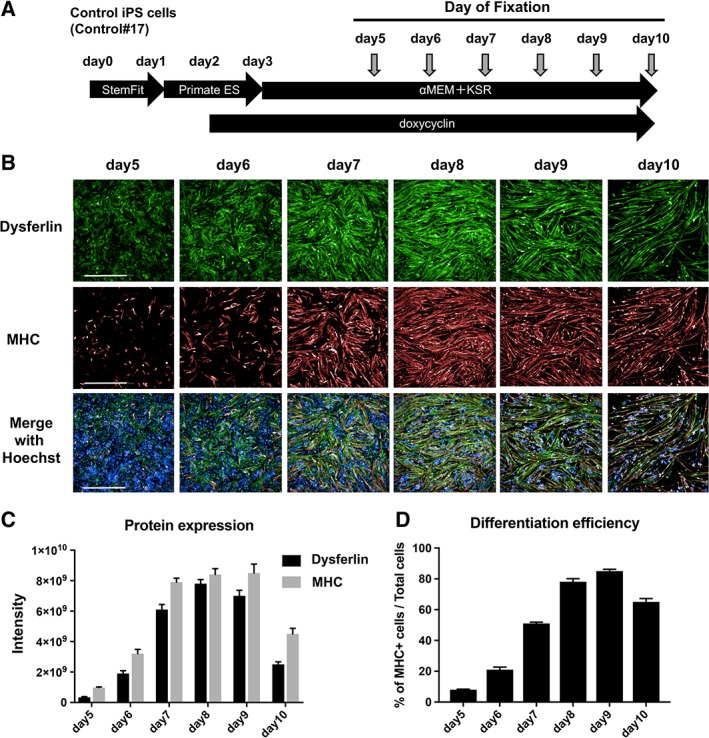
Protein expression in myocytes derived from control induced pluripotent stem (iPS) cells. (A): Schematic representation of the muscle differentiation protocol. Control iPS cells (Control#17) were used to establish the screening system. iPS cells were plated on a 96‐well plate on day 0 and cultured for 10 days. To determine protein expression patterns, cells were fixed on days 5, 6, 7, 8, 9, and 10. (B): Immunostaining of dysferlin and myosin heavy chain (MHC). Nuclei were stained by Hoechst 33342. Scale bar: 500 μm. (C): Staining intensity of dysferlin and MHC from panel B (n = 7). (D): Differentiation efficiency of myocytes with respect to total cells. The numbers of MHC‐positive cells and Hoechst‐positive cells were counted, and the percentage of the former relative to the latter was calculated (n = 7).
Next, dysferlin expression in myocytes of the MM patient (clone: MM#7) was evaluated using the same method. As shown in Figure 2A, immunostaining of dysferlin in MM cells was very faint, in contrast with MHC, which was clearly visible. Quantification of the staining revealed a slight increase in dysferlin on day 6 (Fig. 2B), which was more or less constant until day 8, after which it dropped. In contrast, MHC expression did not change during the course of the culture period. Again, differentiation efficiency was highest on day 8 (Fig. 2C). For the subsequent drug screening, we introduced the “dysferlin intensity” parameter, defined as the staining intensity of “dysferlin/MHC.” Accordingly, dysferlin intensity of MM myocytes (mean 0.25, MM#7) was three times lower than that of control myocytes (mean 0.93, Control#17). Based on the above results, it was determined that the effect of drug screening should be evaluated on day 8.
Figure 2.
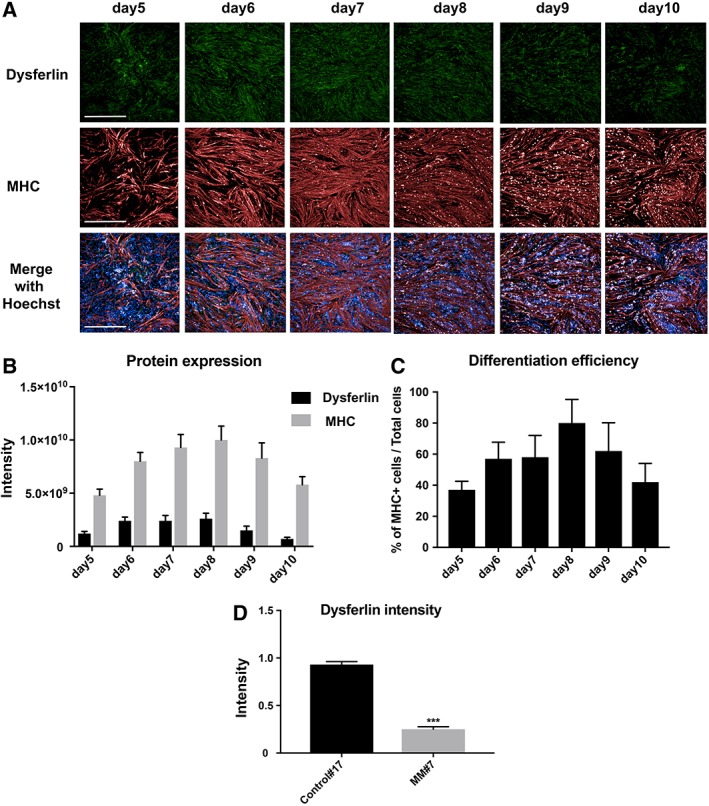
Protein expression in myocytes differentiated from Miyoshi myopathy (MM) patient‐derived induced pluripotent stem (iPS) cells (MM#7). (A): Immunostaining of dysferlin and myosin heavy chain (MHC) on days 5, 6, 7, 8, 9, and 10. Nuclei were stained by Hoechst. Scale bar: 500 μm. (B): Staining intensity of dysferlin and MHC from panel A (n = 7). (C): Differentiation efficiency of myocytes with respect to total cells. The numbers of MHC‐positive cells and Hoechst‐positive cells were counted, and the percentage of the former relative to the latter was calculated (n = 7). (D): Dysferlin intensity in control (mean: 0.93) and MM (mean: 0.25) myocytes on day 8. Statistical analysis was performed using t test. n = 7; ***, p < .001.
Construction of a Screening System Using Myocytes
A 384‐multiwell assay for the efficient drug screening of potential hit molecules was developed. For its validation, MM (MM#7) and control (Control#17) iPS cells were plated and differentiation efficiency on day 8 was measured by MHC immunostaining in each well (Fig. 3A, 3C). Efficiency of MM iPS cells' differentiation into myocytes was nearly the same in each well (Fig. 3A) and occurred in around 80% of cells (Fig. 3B), with coefficient of variation (cv) of 5.2% (MM#7, 320 wells). Differentiation efficiency in the 128 wells containing control myocytes was around 90% (data not shown) and cv was 4.2%. Representative images of 32 wells are shown in Figure 3C. These results confirmed the establishment of a very stable screening system for drug discovery using MM myocytes or, potentially, any type of myocytes differentiated from human iPS cells.
Figure 3.
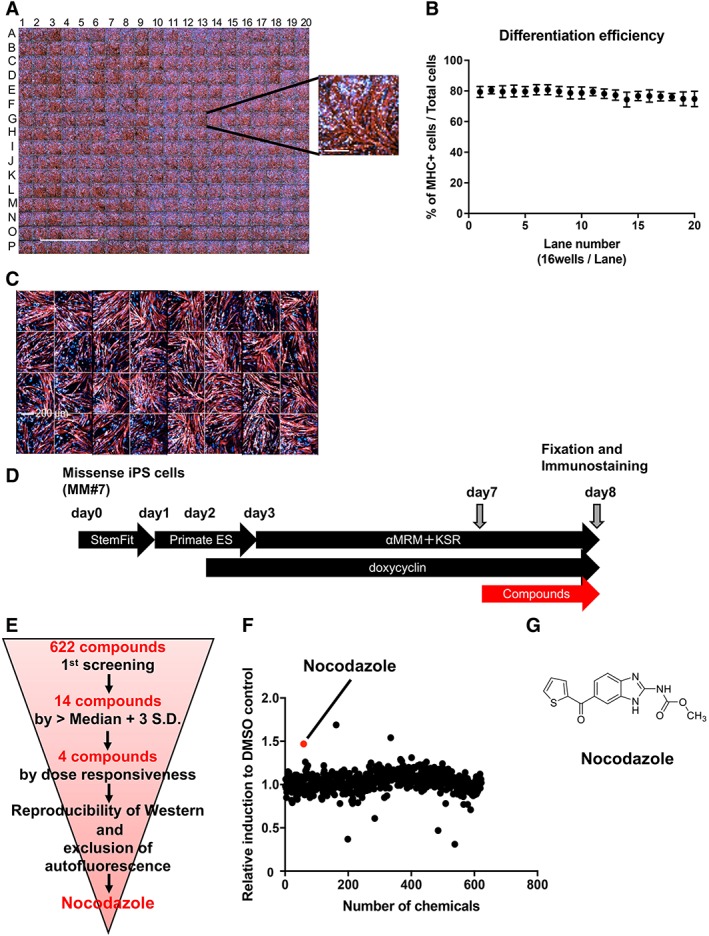
Construction of the Miyoshi myopathy (MM) myocytes (MM#7)‐based screening system and corresponding results. (A): Immunostaining of myosin heavy chain (MHC) in MM myocytes (MM#7) in a 384 multiwell plate on day 8. Scale bar: 5 mm. The inset on the right shows a high‐magnification image of a representative well (scale bar: 200 μm). (B): Mean differentiation efficiency of MM myocytes per lane (16 wells) in the same 384‐well plate described in panel (A). (C): Immunostaining of MHC in control myocytes (Control#17) plated in 32 wells of the 384‐well plate on day 8. Scale bar: 200 μm. (D): Schematic representation of the protocol for myocyte differentiation and small molecule screening. MM missense induced pluripotent stem cells were plated on a 384‐well plate on day 0 and cultured for 8 days. Small molecules were added on day 7. Cells were fixed on day 8, 24 hours after small molecule addition. (E): Schematic representation of the screening steps. Nocodazole was identified as the most effective molecule from the 622 compounds tested. (F): Result of the primary screen assay. Dot plot representation of the effects of 622 small molecules on dysferlin immunostaining relative to the dimethyl sulfoxide control; 3 μM and 0.3 μM of compounds (n = 1 each) were also evaluated (data not shown). (G): Structural formula of nocodazole.
Screening of a Small Molecule Library
Based on the dysferlin expression results, a phenotypic drug screening was performed on day 8. Briefly, 622 commercially available and functionally known small molecules from a Takeda external repositioning library were added to the medium (3 μM) of MM myocytes in the 384‐well plate on day 7. Cells were cultured until the next day (day 8) and fixed exactly 24 hours after small molecule addition (Fig. 3D). Cells were probed with anti‐dysferlin and anti‐MHC antibodies. Dysferlin intensity as defined before was used to evaluate the small molecules' impact, and the final readout was calculated as the fold increase in MM myocytes over 0.1% DMSO controls. For the selection of primary hit compounds, the hit value was set as fold increase >median + 3 SD relative to the DMSO control.
Schematic results of the screening are shown in Figure 3E. Accordingly, 14 out of 622 compounds were selected during the first screening. Following several rounds of validation, four compounds were selected according to dose‐responsiveness by immunostaining. Finally, based on the reproducibility of Western blotting results (data not shown) and the exclusion of small molecules with auto fluorescence, nocodazole was selected as the primary candidate (Fig. 3F, 3G). Nocodazole binds reversibly to tubulin 16 and alters tubulin‐associated GTP hydrolysis as well as microtubule dynamics in cells 17. Two additional hit molecules that induced dysferlin protein more potently than nocodazole during the first screening are shown in Figure 3F. They were identified as parbendazole and pyrvinium pamoate, two antiparasitic agents against pinworms. On the one hand, the effect of parbendazole could not be reproduced by the second screening. On the other hand, the effect of pyrvinium pamoate could be reproduced, but the molecule has poor solubility in water and is hardly absorbed by the digestive tract. From the perspective of therapeutic drug development, we judged these two small molecules were not suitable for our study. Thus, we excluded these two small molecules from further experiments.
Nocodazole Increases Dysferlin in Skeletal Myocytes
In our experiments, nocodazole was found to increase dysferlin intensity in MM myocytes. To clarify the result, dose‐responsiveness to nocodazole was checked in detail by immunostaining (Fig. 4A), and the corresponding dysferlin intensity was calculated (Fig. 4B). Thus, dysferlin was seen to increase in a dose‐responsive way upon addition of 1, 3, and 10 μM nocodazole (Fig. 4B) and was confirmed also in other clones (MM#1 and MM#3, shown in Supporting Information Figs. S1A, S1B, S2A, S2B, respectively). It should be noted that nocodazole treatment resulted also in a morphological change to myocytes, possibly due to its inhibitory effect on microtubules 16.
Figure 4.
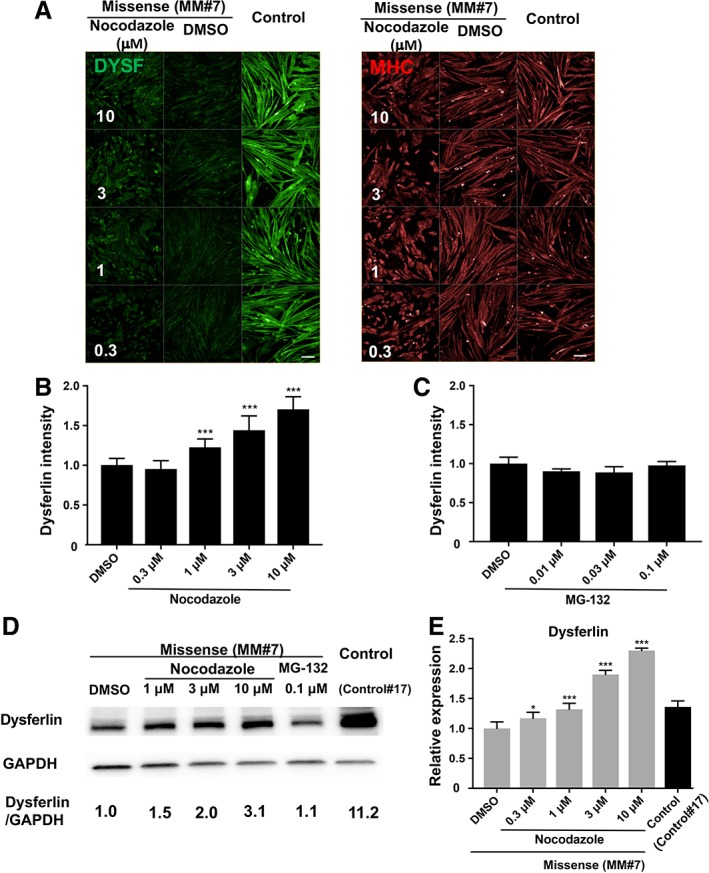
Effect of nocodazole and MG‐132 on Miyoshi myopathy (MM) myocytes (MM#7). (A): Immunostaining of myosin heavy chain and dysferlin, as well as Hoechst staining in MM myocytes (MM#7) on day 8 after a 24‐hours treatment with 0.3, 1, 3, and 10 μM nocodazole. Control myocytes (Control#17) were also treated with dimethyl sulfoxide (DMSO). n = 32 for control samples, and n = 4 for MM samples. Scale bar: 100 μm. (B): Dysferlin intensity calculated from data in panel (A). Statistical analysis was performed by William's multiple test using EXSUS software. ***, p < .0005. (C): Dysferlin intensity calculated from the staining of MG‐132‐treated MM myocytes (data not shown) on day 8. n = 32 for control samples and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. No significance was observed between any samples. (D): Western blotting of dysferlin and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) on day 8. MM myocytes were treated for 24 hours with DMSO, nocodazole, and MG‐132. Nontreated Control#17 cells were added as a control. Each band area was calculated and normalized to GAPDH. (E): qPCR analysis of dysferlin expression. MM myocytes were treated with DMSO or nocodazole and mRNA was isolated on day 8. Control#17 sample was added as a control. n = 12 for DMSO control samples and n = 4 for other samples. Statistical analysis was performed by William's multiple test using EXSUS software. *, p < .025 and ***, p < .0005.
To assess the effect of proteasome inhibitor treatment on the accumulation of misfolded dysferlin as reported previously 7, myocytes were treated with MG‐132 (Fig. 4C, Supporting Information Figs. S1C, S2C). Contrary to nocodazole addition, MG‐132 treatment did not result in any clear increment of dysferlin intensity based on either immunostaining (Fig. 4C, Supporting Information Figs. S1C, S2C) or blotting (Fig. 4D). These results suggest that the proteasome system may not be the dominant pathway for the degradation of misfolded dysferlin in human iPSC‐derived myocytes. Considering that MM iPS cells used in this experiment have both a c.2997 G>T (W999C) missense and a c.1958delG nonsense mutation, and control iPS cells have the same W999C mutation in one allele, expression of dysferlin in MM cells was expected to be half of that in control cells if the missense mutation had no bearing on protein degradation due to structural instability. However, judging from the result of Figure 4D, the overall level of dysferlin in MM myocytes was 13‐fold lower than that of control cells. This result suggested that the remaining misfolded dysferlin produced by MM myocytes was removed by post‐translational machinery. Furthermore, the effect of MG‐132 on dysferlin was not clear from our experiments (Fig. 4D; MM#7). In addition, dysferlin mRNA was increased in a dose‐dependent manner following treatment with nocodazole (Fig. 4E). Taken together, these data indicated that nocodazole increased both protein and mRNA expression of dysferlin.
Membrane Resealing Function Is Lower in Cells Bearing the Missense Mutation Than in Control Cells
To determine whether the decreased expression of dysferlin observed in MM patient‐derived myocytes led to a phenotypic defect such as dysfunction of membrane resealing, which is the basis of the pathological condition, we first compared the resealing disability of MM skeletal myocytes (MM#1, MM#3) to that of control myocytes (Control#17, Control#10; Fig. 5A). Myocytes were irradiated by laser light from a confocal microscope on day 8. The extent of cell membrane disruption was evaluated by the intensity of FM 1‐43 staining within cells. As shown in Figure 5A–5C, MM myocytes displayed uptake of FM 1‐43 dye in all cytoplasmic lesions, indicating defective membrane repair following laser‐induced injury. In contrast, control myocytes displayed only focal uptake of FM 1‐43 at the damaged area (see also Supporting Information Videos S1, S2 for MM#1 and MM#3, and S3‐4 for Control#10 and Control#17). These findings confirmed the expected pathological condition caused by mutated and misfolded dysferlin in MM myocytes.
Figure 5.
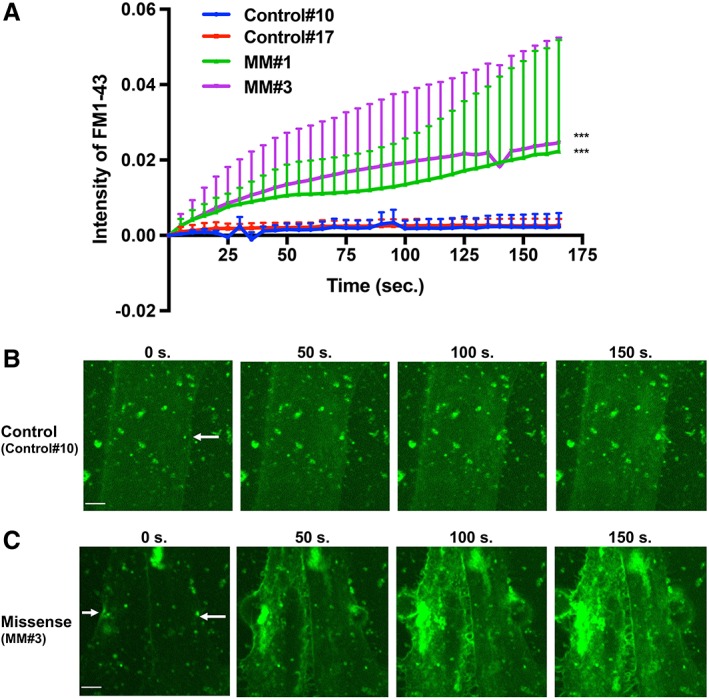
Membrane resealing assay using Miyoshi myopathy (MM) myocytes treated with nocodazole. (A): Graph of FM 1‐43 intensity in MM myocytes (MM#1 [n = 4], MM#3 [n = 9]) and control myocytes (Control#10 [n = 6], Control#17 [n = 5]) after laser irradiation. Statistical analysis was performed by Tukey's test for multiple comparison using GraphPad prism software. The intensity of FM 1‐43 on both of MM myocytes were significantly high compared with both of control myocytes. ***, p < .001. (B, C): Photographs taken at 0, 50, 100, and 150 seconds during resealing. Images were captured every 5 seconds for 165 seconds in control myocytes (Control#10; B) and missense myocytes (MM#3; C). Scale bar: 5 μm. White arrows indicate injury site.
Increased Dysferlin Following Nocodazole Treatment Effectively Improves Membrane Resealing
Finally, we investigated whether an increase in dysferlin following nocodazole treatment could ameliorate membrane resealing in MM myocytes. As shown in Figure 6A, MM#3 myocytes bearing the missense mutation were treated on day 7 with 3 μM nocodazole, 0.1 μM MG‐132, or 0.1% DMSO, and resealing function was evaluated on day 8 following laser irradiation and FM 1‐43 uptake. Compared with the DMSO control, uptake of FM 1‐43 was significantly lower in myocytes treated with nocodazole (Fig. 6A–6C; see also Supporting Information Videos S5, S6 for MM#3, Supporting Information Videos S8, S9 and Supporting Information Fig. S3 for MM#1). Thus, nocodazole effectively improved membrane resealing in MM myocytes bearing the DYSF missense mutation and characterized by high dysferlin levels. In contrast, the effect of MG‐132 could not be confirmed in this experiment (Fig. 6A, 6D; see also Supporting Information Video S7 for MM#3 and S10 for MM#1).
Figure 6.
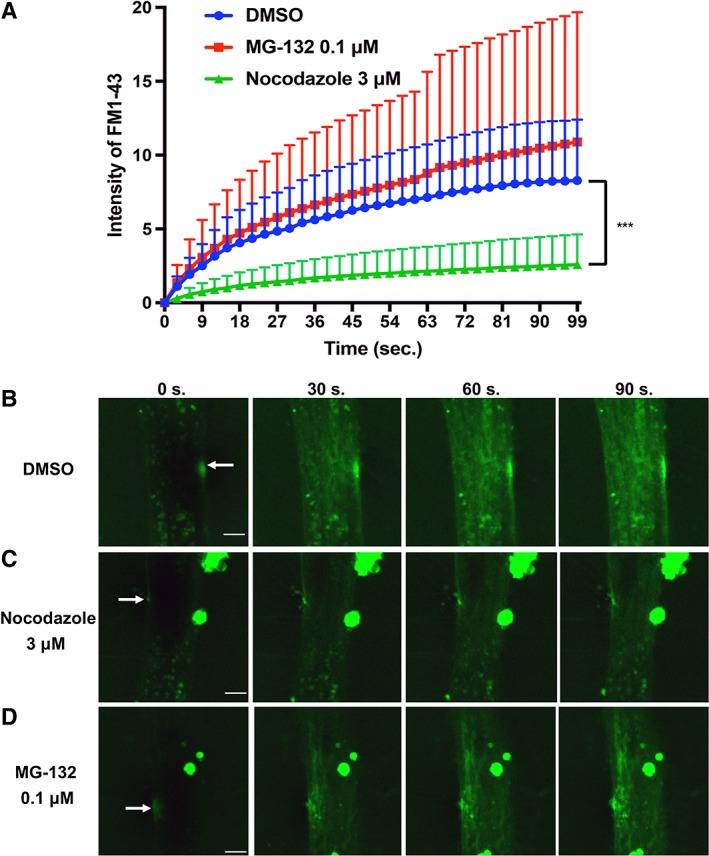
Membrane resealing assay using Miyoshi myopathy (MM) myocytes (MM#3) treated with nocodazole and MG‐132. (A): Graph of FM 1‐43 intensity in MM myocytes (MM#3) with dimethly sulfoxide (DMSO) (n = 9), treated with MG‐132 (n = 11), and treated with nocodazole (n = 11) after laser irradiation. Statistical analysis was performed by Dunnett's test for multiple comparison using the GraphPad prism software. ***, p < .001. (B–D): Photographs taken at 0, 30, 60, and 90 seconds during resealing. Images were captured every 3 seconds for 99 seconds in cells treated with DMSO (B), nocodazole (C), or MG‐132 (D). Scale bar: 5 μm. White arrows indicate injury site.
Mechanistic Analysis of Dysferlin Upregulation by Nocodazole
To better understand the mechanism by which nocodazole upregulated dysferlin in cells bearing the missense mutation, we assessed the impact of adding small molecules with the same effect, for example, colchicine 18, or opposite effect, for example, paclitaxel 19, as nocodazole. Immunostaining for dysferlin was performed in control (Control#10) myocytes treated with 0.1% DMSO and missense (MM#3) myocytes treated with 0.1% DMSO, nocodazole (3 μM), colchicine (10 μM), and paclitaxel (10 nM; Fig. 7A, 7B). At 24 hours after small molecules addition, dysferlin was observed to be upregulated by nocodazole and colchicine, but not by paclitaxel or 0.1% DMSO. Both nocodazole and colchicine cause microtubule disorganization, whereas paclitaxel causes microtubule organization. This finding suggests that upregulation of dysferlin was the result of microtubule disorganization by nocodazole or colchicine. Considering that one of the roles of microtubules is to transport proteins through the cytoplasm 20, inhibition of such transport route may induce cytoplasmic accumulation of misfolded dysferlin and prevent protein degradation by the microtubule‐dependent autophagy system. We hypothesized that disruption of autophagy system following nocodazole treatment would cause an increase of dysferlin in cells bearing the missense mutation. To confirm this hypothesis, we assessed the expression of dysferlin, autophagy markers (p62 and LC3), and GAPDH in missense (MM#3) myocytes following treatment with nocodazole (3 μM), colchicine (10 μM), paclitaxel (10 nM), and 0.1% DMSO (Fig. 7C, 7D). An increase in dysferlin, p62, and LC3 II/I was observed in myocytes treated with both nocodazole and colchicine, but not in those treated with either paclitaxel or 0.1% DMSO (see also Supporting Information Fig. S4 for MM#1). Accumulation of p62 was caused by microtubule disorganization, indicating blockage of the degradation pathway. In addition, accumulation of LC3 II following nocodazole or colchicine treatment suggested disruption of autophagy.
Figure 7.
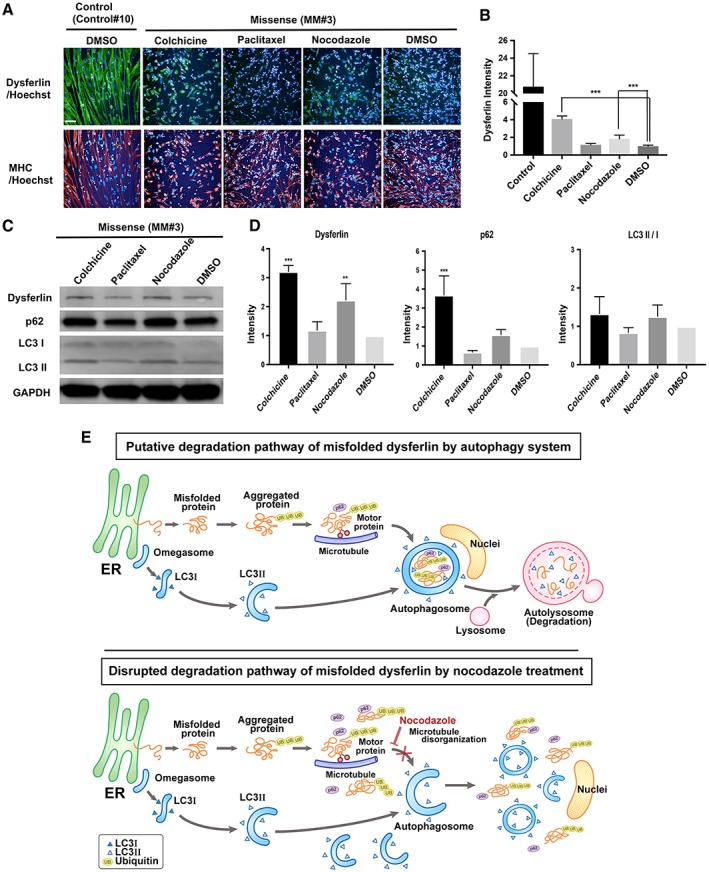
Mechanistic analysis of dysferlin upregulation by nocodazole. (A): Immunostaining of control (Control#10) and Miyoshi myopathy (MM; MM#3) myocytes treated with dimethyl sulfoxide (DMSO), nocodazole (3 μM), colchicine (10 μM), and paclitaxel (10 nM). Scale bar: 100 μm. (B): Graph of (A). Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad prism software. n = 8 for all samples. ***, p < .001. (C): Western blotting analysis of dysferlin, p62, glyceraldehyde 3‐phosphate dehydrogenase, LC3 I, and LC3 II in missense myocytes treated with DMSO, nocodazole (3 μM), colchicine (10 μM), and paclitaxel (10 nM). (D): Graph of (C). Data were presented as relative fold expression normalized to dimethyl sulfoxide control. Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad prism software. n = 3 for all samples. **, p < .01 and ***, p < .001. (E): Model of the effect of nocodazole. Upper panel: degradation pathway of misfolded proteins including dysferlin by the autophagy system. Lower panel: misfolded dysferlin is not degraded by autophagy after treatment with nocodazole.
In summary, these results indicate that the mechanism of dysferlin upregulation following nocodazole treatment is caused by disruption of autophagy and is a consequence of microtubule disorganization, as indicated in Figure 7E.
Discussion
In the present study, we established an immunostaining‐based 384‐multiwell drug screening system for dysferlinopathy using skeletal myocytes differentiated from a MM patient bearing the W999C DYSF missense mutation and control iPS cells. Cv of differentiation efficiency for all clones on day 8 was below 10%, indicating that the established screening system was suitable for all types of clones used in these experiments. Using this system, 622 small molecules were screened and nocodazole was identified as the most effective candidate for increasing dysferlin levels in myocytes bearing the MM W999C missense mutation and restoring membrane resealing function. This result indicates that pathways responsible for increasing the level of misfolded dysferlin could be targeted during clinical treatment of dysferlinopathy patients with the W999C missense mutation. The effect could be attained by blocking protein degradation, upregulating protein expression, and enhancing protein or mRNA stabilization.
As expected, an increased level of dysferlin in the cell effectively improved membrane resealing following injury. It was previously reported that misfolded dysferlin was degraded by the 26S proteasome system 7 and/or autophagy system 21. Based on those findings, we believe that the increment in dysferlin observed in our experiments was the result of blocking protein degradation by those systems following nocodazole treatment. However, in our screening assay, addition of the proteasome inhibitor MG‐132 alone was insufficient to cause an increment of dysferlin (Fig. 4C). Accordingly, the proteasome system might not constitute the main pathway responsible for the degradation of misfolded dysferlin in iPS cell‐derived myocytes.
Our data indicate that the main pathway for the degradation of dysferlin in iPS‐derived myocytes is provided by the autophagy system rather than the proteasome. As shown in Figure 7E (upper panel), during autophagy, misfolded proteins form small aggregates, which become ubiquitinated during the first step of degradation. Then, aggregated misfolded proteins are tagged by p62, to form large aggregations named “aggresomes” to be transported along microtubules. Aggregated proteins are stored in autophagosomes, and following fusion with lysosomes, are degraded by autolysosomes 22. Intracellular deposition of misfolded protein aggregates within ubiquitin‐rich cytoplasmic inclusions has been linked to the pathogenesis of many diseases 23. Recently, the degradation of aggresomes was reported to be induced by the autophagy system 24, and disruption of microtubules by nocodazole was described to block the formation of aggresomes 23. Based on our results, misfolded dysferlin might persist in the cells following nocodazole treatment due to inhibition of aggresome formation. As a result, misfolded dysferlin could not be detected in large aggregates, and its increased levels in the cytoplasm might contribute to improved membrane resealing. Additionally, as suggested by the increment in dysferlin mRNA (Fig. 4E), the observed effect of nocodazole treatment might be caused by a change in transcriptional regulation following blocking of the cell cycle 25. Elucidation of the exact sequence of events and mechanism of action will require further investigation.
We found nocodazole as one of the candidates for treatment of dysferlinopathy; however, there are also some limitations to the present study. For example, even though the membrane resealing assay is an effective method to assess the function of dysferlin in vitro, future in vivo assays should be performed to confirm present findings. Furthermore, overexpression of myoferlin in mice not expressing dysferlin could rescue the laser wounding phenotype in vitro, but not any of the dystrophic features 26. These results indicate that the role of dysferlin in skeletal muscle in vivo might not be adequately assayed in vitro. In addition, a cohort study revealed that exercise during adolescence was associated with earlier onset of dysferlinopathy 27. Macrophage infiltration was also observed in muscle tissue from the early phase, and myofiber damage was seen to accumulate over time in both dysferlinopathy patients and dysferlin‐deficient mice 28. Hence, to assess the effect of candidate drugs on these phenotypes, investigation of the enhanced resealing ability of candidate molecules in vitro should be accompanied by preclinical examinations using W999C knockin mice.
In the present study, we identified nocodazole as one of the small molecules responsible for increasing the intracellular level of dysferlin. Nocodazole has been often used as an anticancer drug due to its negative effect on microtubule formation and cell cycle arrest. In terms of clinical treatment of dysferlinopathy, nocodazole itself would likely be toxic to cells and inhibit the differentiation of satellite cells to mature myotubes. Such toxicity would have severe adverse effects in young dysferlinopathy patients. Other therapeutic agents with lower toxicity to the cells of dysferlinopathy patients should be developed in the future.
Although nocodazole itself is difficult to use for clinical purposes, a detailed analysis of the pathways involved in dysferlin degradation identified in this study, provides new insights on possible targets for drugs against dysferlinopathy. Ideally, such drugs would be highly effective at increasing misfolded dysferlin, while having low toxicity toward target cells. Furthermore, the screening system we pioneered in this study could represent a powerful tool for the future identification of new small molecule candidates for dysferlinopathy using large scale small molecule library. Indeed, in Japan, the W999C missense mutation is estimated to be present in over 25% of all dysferinopathy patients. Moreover, other intractable diseases caused by the specific type of missense mutated protein could also be the target of these new protein stabilizing drugs.
Conclusion
In this study, we performed phenotypic drug screening of a limited number of drugs, including off‐patent, FDA‐approved, and functionally known drugs, using skeletal myocytes differentiated from iPS cells derived from a dysferlinopathy patient. Nocodazole was identified as the most effective candidate for increasing dysferlin protein levels in myocytes bearing the MM W999C missense mutation and restoring membrane resealing function. The effect of nocodazole was the disruption of the autophagy system following microtubule disorganization. Nocodazole itself would be too toxic for long‐term administration for this disease; however, further research regarding the target of nocodazole responsible for upregulation of dysferlin would lead to new treatments in the future.
Author Contributions
Y.K.: conception and design, data analysis and interpretation, collection and assembly of data, discussed data, manuscript writing, final approval of manuscript; H.S.: conception and design, discussed data, manuscript writing, financial support, provision of study material, final approval of manuscript; T.N., K.S., T.O., K.M., A.K., M.F.: data analysis and interpretation, collection and assembly of data, discussed data; H.F., R.T.: discussed data.
Disclosure of Potential Conflicts of Interest
T.N., K.S., T.O., A.K., H.F., and R.T. are employees of Takeda Pharmaceutical Company Limited. Y.K., H.S. and M.F. declared research funding from Takeda Pharmaceutical Company Limited. The other author indicated no potential conflicts of interest.
Supporting information
Figure S1 Effect of nocodazole and MG‐132 on MM myocytes (MM#3). (A) Immunostaining of MHC and dysferlin, as well as Hoechst staining in MM myocytes on day 8 after a 24‐hours treatment with 0.3, 1, 3, and 10 μM nocodazole. Control myocytes (Control#17) were also treated with DMSO. N = 32 for control samples, and n = 4 for MM samples. Scale bar: 100 μm. (B) Dysferlin intensity calculated from data in panel (A). N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. ***P < .0005. (C) Dysferlin intensity calculated from the staining of MG‐132‐treated MM myocytes (data not shown) on day 8. N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. No significance was observed between any samples.
Figure S2 Effect of nocodazole and MG‐132 on MM myocytes (MM#1). (A) Immunostaining of MHC and dysferlin, as well as Hoechst staining in MM myocytes on day 8 after a 24‐hours treatment with 0.3, 1, 3, and 10 μM nocodazole. Control myocytes (Control#17) were also treated with DMSO. N = 32 for control samples, and n = 4 for MM samples. Scale bar: 100 μm. (B) Dysferlin intensity calculated from data in panel (A). N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. ***P < .0005. (C) Dysferlin intensity calculated from the staining of MG‐132‐treated MM myocytes (data not shown) on day 8. N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. No significance was observed between any samples.
Figure S3 Membrane resealing assay using MM myocytes (MM#1) treated with nocodazole. Graph of FM 1–43 intensity in MM myocytes (MM#1) without treatment (n = 14), treated with MG‐132 (n = 10), and treated with nocodazole (n = 12) after laser irradiation. Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad Prism software. ***P < .001.
Figure S4 Mechanistic analysis of dysferlin upregulation by nocodazole. (A) Western blotting analysis of dysferlin, p62, GAPDH, LC3 I, and LC3 II in missense myocytes (MM#1) treated with nocodazole (3 μM), colchicine (10 μM), and paclitaxel (10 nM). (B) Graph of Figure S4 (A). Data were presented as relative fold expression normalized to DMSO control. Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad Prism software. N = 3 for all samples. *P < .05 and **P < .01.
Video S1 Membrane resealing assay using MM myocytes (MM#1). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S2 Membrane resealing assay using MM myocytes (MM#3). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S3 Membrane resealing assay using control myocytes (Control#10). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S4 Membrane resealing assay using control myocytes (Control#17). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S5 Membrane resealing assay using MM myocytes (MM#3) treated with DMSO. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S6 Membrane resealing assay using MM myocytes (MM#3) treated with nocodazole. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S7 Membrane resealing assay using MM myocytes (MM#3) treated with MG‐132. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S8 Membrane resealing assay using MM myocytes (MM#1) treated with DMSO. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S9 Membrane resealing assay using MM myocytes (MM#1) treated with nocodazole. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S10 Membrane resealing assay using MM myocytes (MM#1) treated with MG‐132. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Acknowledgments
We thank Miki Nagai and Yukiyo Mikami for technical assistance. We also thank Misaki Ouchida for making illustrations. This work was partially supported by a grant from The Program for Intractable Diseases Research utilizing disease‐specific iPS cells and a grant from The Acceleration Program for Intractable Diseases Research utilizing disease‐specific iPS cells, both of which were provided by the Japan Agency for Medical Research and Development, AMED (to H.S.). This work was also supported by a grant from Takeda Pharmaceutical Company Limited.
Contributor Information
Yuko Kokubu, Email: yuko.kokubu@cira.kyoto-u.ac.jp.
Hidetoshi Sakurai, Email: hsakurai@cira.kyoto-u.ac.jp.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Bansal D, Miyake K, Vogel SS et al. Defective membrane repair in dysferlin‐deficient muscular dystrophy. Nature 2003;423:168–172. [DOI] [PubMed] [Google Scholar]
- 2. Aoki M. Dysferlinopathy In: Adam MP, Ardinger HH, Pagon RA. et al., eds. GeneReviews(R). University of Washington, Seattle, WA, 1993. [PubMed] [Google Scholar]
- 3. Nguyen K, Bassez G, Krahn M et al. Phenotypic study in 40 patients with dysferlin gene mutations: High frequency of atypical phenotypes. Arch Neurol 2007;64:1176–1182. [DOI] [PubMed] [Google Scholar]
- 4. Han R, Campbell KP. Dysferlin and muscle membrane repair. Curr Opin Cell Biol 2007;19:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Defour A, Van der Meulen JH, Bhat R et al. Dysferlin regulates cell membrane repair by facilitating injury‐triggered acid sphingomyelinase secretion. Cell Death Dis 2014;5:e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi T, Aoki M, Suzuki N et al. Clinical features and a mutation with late onset of limb girdle muscular dystrophy 2B. J Neurol Neurosurg Psychiatry 2013;84:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsuda C, Kiyosue K, Nishino I et al. Dysferlinopathy fibroblasts are defective in plasma membrane repair. PLoS Curr 2015;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bianchini E, Testoni S, Gentile A et al. Inhibition of ubiquitin proteasome system rescues the defective sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA1) protein causing Chianina cattle pseudomyotonia. J Biol Chem 2014;289:33073–33082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imamura K, Izumi Y, Watanabe A et al. The Src/c‐Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci Transl Med 2017;9:eaaf3962. [DOI] [PubMed] [Google Scholar]
- 10. Hino K, Horigome K, Nishio M et al. Activin‐A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest 2017;127:3339–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987;51:987–1000. [DOI] [PubMed] [Google Scholar]
- 12. Tanaka A, Woltjen K, Miyake K et al. Efficient and reproducible myogenic differentiation from human iPS cells: Prospects for modeling Miyoshi myopathy in vitro. PLoS One 2013;8:e61540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okita K, Yamakawa T, Matsumura Y et al. An efficient nonviral method to generate integration‐free human‐induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 2013;31:458–466. [DOI] [PubMed] [Google Scholar]
- 14. Nakagawa M, Taniguchi Y, Senda S et al. A novel efficient feeder‐free culture system for the derivation of human induced pluripotent stem cells. Sci Rep 2014;4:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uchimura T, Otomo J, Sato M et al. A human iPS cell myogenic differentiation system permitting high‐throughput drug screening. Stem Cell Res 2017;25:98–106. [DOI] [PubMed] [Google Scholar]
- 16. Florian S, Mitchison TJ. Anti‐microtubule drugs. Methods Mol Biol 2016;1413:403–421. [DOI] [PubMed] [Google Scholar]
- 17. Mejillano MR, Shivanna BD, Himes RH. Studies on the nocodazole‐induced GTPase activity of tubulin. Arch Biochem Biophys 1996;336:130–138. [DOI] [PubMed] [Google Scholar]
- 18. Brossi A, Yeh HJ, Chrzanowska M et al. Colchicine and its analogues: Recent findings. Med Res Rev 1988;8:77–94. [DOI] [PubMed] [Google Scholar]
- 19. Manfredi JJ, Parness J, Horwitz SB. Taxol binds to cellular microtubules. J Cell Biol 1982;94:688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Welte MA. Bidirectional transport along microtubules. Curr Biol 2004;14:R525–R537. [DOI] [PubMed] [Google Scholar]
- 21. Fanin M, Nascimbeni AC, Angelini C. Muscle atrophy, ubiquitin‐proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve 2014;50:340–347. [DOI] [PubMed] [Google Scholar]
- 22. Hyttinen JM, Amadio M, Viiri J et al. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev 2014;18:16–28. [DOI] [PubMed] [Google Scholar]
- 23. Johnston JA, Ward CL, Kopito RR. Aggresomes: A cellular response to misfolded proteins. J Cell Biol 1998;143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mackeh R, Perdiz D, Lorin S et al. Autophagy and microtubules—New story, old players. J Cell Sci 2013;126:1071–1080. [DOI] [PubMed] [Google Scholar]
- 25. Cho SG, Sihn CR, Yoo SJ et al. Analysis of gene expression induced by microtubule‐disrupting agents in HeLa cells using microarray. Cancer Lett 2006;241:110–117. [DOI] [PubMed] [Google Scholar]
- 26. Lostal W, Bartoli M, Roudaut C et al. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS One 2012;7:e38036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moore UR, Jacobs M, Fernandez‐Torron R et al. Teenage exercise is associated with earlier symptom onset in dysferlinopathy: A retrospective cohort study. J Neurol Neurosurg Psychiatry 2018;89:1224–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roche JA, Tulapurkar ME, Mueller AL et al. Myofiber damage precedes macrophage infiltration after in vivo injury in dysferlin‐deficient A/J mouse skeletal muscle. Am J Pathol 2015;185:1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of nocodazole and MG‐132 on MM myocytes (MM#3). (A) Immunostaining of MHC and dysferlin, as well as Hoechst staining in MM myocytes on day 8 after a 24‐hours treatment with 0.3, 1, 3, and 10 μM nocodazole. Control myocytes (Control#17) were also treated with DMSO. N = 32 for control samples, and n = 4 for MM samples. Scale bar: 100 μm. (B) Dysferlin intensity calculated from data in panel (A). N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. ***P < .0005. (C) Dysferlin intensity calculated from the staining of MG‐132‐treated MM myocytes (data not shown) on day 8. N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. No significance was observed between any samples.
Figure S2 Effect of nocodazole and MG‐132 on MM myocytes (MM#1). (A) Immunostaining of MHC and dysferlin, as well as Hoechst staining in MM myocytes on day 8 after a 24‐hours treatment with 0.3, 1, 3, and 10 μM nocodazole. Control myocytes (Control#17) were also treated with DMSO. N = 32 for control samples, and n = 4 for MM samples. Scale bar: 100 μm. (B) Dysferlin intensity calculated from data in panel (A). N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. ***P < .0005. (C) Dysferlin intensity calculated from the staining of MG‐132‐treated MM myocytes (data not shown) on day 8. N = 32 for control samples, and n = 4 for MM samples. Statistical analysis was performed by William's multiple test using EXSUS software. No significance was observed between any samples.
Figure S3 Membrane resealing assay using MM myocytes (MM#1) treated with nocodazole. Graph of FM 1–43 intensity in MM myocytes (MM#1) without treatment (n = 14), treated with MG‐132 (n = 10), and treated with nocodazole (n = 12) after laser irradiation. Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad Prism software. ***P < .001.
Figure S4 Mechanistic analysis of dysferlin upregulation by nocodazole. (A) Western blotting analysis of dysferlin, p62, GAPDH, LC3 I, and LC3 II in missense myocytes (MM#1) treated with nocodazole (3 μM), colchicine (10 μM), and paclitaxel (10 nM). (B) Graph of Figure S4 (A). Data were presented as relative fold expression normalized to DMSO control. Statistical analysis was performed by Dunnett's test for multiple comparison using GraphPad Prism software. N = 3 for all samples. *P < .05 and **P < .01.
Video S1 Membrane resealing assay using MM myocytes (MM#1). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S2 Membrane resealing assay using MM myocytes (MM#3). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S3 Membrane resealing assay using control myocytes (Control#10). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S4 Membrane resealing assay using control myocytes (Control#17). Images were captured every 5 seconds for 165 seconds after laser irradiation.
Video S5 Membrane resealing assay using MM myocytes (MM#3) treated with DMSO. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S6 Membrane resealing assay using MM myocytes (MM#3) treated with nocodazole. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S7 Membrane resealing assay using MM myocytes (MM#3) treated with MG‐132. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S8 Membrane resealing assay using MM myocytes (MM#1) treated with DMSO. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S9 Membrane resealing assay using MM myocytes (MM#1) treated with nocodazole. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Video S10 Membrane resealing assay using MM myocytes (MM#1) treated with MG‐132. Images were captured every 3 seconds for 99 seconds after laser irradiation.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.