

Abstract
Acute kidney injury (AKI) due to renal ischemia-reperfusion (I/R) is a major clinical problem without effective therapy. Ginger is one of the most widely consumed spices in the world, and 6-shogaol, a major ginger metabolite, has anti-inflammatory effects in neuronal and epithelial cells. Here, we demonstrate our novel findings that 6-shogaol treatment protected against renal I/R injury with decreased plasma creatinine, blood urea nitrogen, and kidney neutrophil gelatinase-associated lipocalin mRNA synthesis compared with vehicle-treated mice subjected to renal I/R. Additionally, 6-shogaol treatment reduced kidney inflammation (decreased proinflammatory cytokine and chemokine synthesis as well as neutrophil infiltration) and apoptosis (decreased TUNEL-positive renal tubular cells) compared with vehicle-treated mice subjected to renal I/R. In cultured human and mouse kidney proximal tubule cells, 6-shogaol significantly attenuated TNF-α-induced inflammatory cytokine and chemokine mRNA synthesis. Mechanistically, 6-shogaol significantly attenuated TNF-α-induced NF-κB activation in human renal proximal tubule cells by reducing IKKαβ/IκBα phosphorylation. Furthermore, 6-shogaol induced a cytoprotective chaperone heme oxygenase (HO)-1 via p38 MAPK activation in vitro and in vivo. Consistent with these findings, pretreatment with the HO-1 inhibitor zinc protoporphyrin IX completely prevented 6-shogaol-mediated protection against ischemic AKI in mice. Taken together, our study showed that 6-shogaol protects against ischemic AKI by attenuating NF-κB activation and inducing HO-1 expression. 6-Shogaol may provide a potential therapy for ischemic AKI during the perioperative period.
Keywords: acute kidney injury, apoptosis, inflammation, necrosis, neutrophil, reperfusion
INTRODUCTION
Despite more than seven decades of intense basic and clinical research, acute kidney injury (AKI) remains a major perioperative problem without effective clinical therapy or preventive methods (5). Indeed, perioperative AKI is a leading cause of patient mortality and morbidity and costs >$10 billion per year in the United States (5). Renal ischemia and reperfusion (I/R) injury is a major cause of clinical AKI, and patients subjected to major surgical procedures (cardiac, vascular, or liver transplantation) have ~50–80% chance of developing ischemic AKI, with mortality > 75% in severe AKI (3, 21).
The pathogenesis of ischemic AKI is due to a combination of renal tubular necrosis because of massive renal tubular cell energy loss during the ischemic period and the apoptotic programmed cell death pathway initiated during and after ischemic insult (18, 28, 29). Furthermore, a profound inflammatory response occurs during the reperfusion period due to the upregulation of chemokines and cytokines in renal tubular cells and resident intrarenal leukocytes that further attract infiltration of systemic leukocytes to induce additional renal tubular damage (27). Therefore, targeting any of the components of renal tubular cell death (necrosis and apoptosis) and inflammation would ameliorate ischemic AKI.
Ginger (Zingiber officinale) is one of the most popular spices in the world, with >1.8 million tons consumed globally per year (57). Anti-inflammatory activities of ginger and its metabolites have been previously reported (13, 22, 45). For example, ginger extract inhibits the inflammatory response and maintains barrier function in human colonic epithelial cells stimulated by a mixture of inflammatory mediators (IL-1β, LPS, TNF-α, and interferon-γ) (13). In LPS-stimulated microglial cells, ginger extract inhibits the production of nitric oxide and proinflammatory cytokines (22). There are also epidemiological suggestions that ginger may have renal protective effects in humans. India (Southern Asia) is the world’s leading ginger-consuming country, with ~790 thousand tons of ginger consumption, and ginger consumption per person in India is two times higher than the global average. Globally, the pooled incidence rate of AKI according to the Kidney Disease Improving Global Outcomes (KDIGO)-equivalent definition was >20%, and the incidence rate of AKI in Southern Asia was 7.5%, suggesting that populations that have a ginger-rich diet may have less AKI (35).
It is known that 6-shogaol is a major biologically active compound in ginger (45) and is the most potent anti-inflammatory and antioxidant component in ginger (8). Indeed, 6-shogaol attenuates neuroinflammation and brain injury in animal models of cerebral ischemia and dementia (37, 38). A previous study (34) in a rat model of renal I/R has shown that ginger pretreatment protects against renal I/R injury. However, it is unclear which of the active ginger metabolite(s) contributes the protective action, and the underlying mechanisms of protection are unclear. Moreover, the effects of 6-shogaol on ischemic AKI have not yet been elucidated. In the present study, we tested the hypothesis that 6-shogaol protects against ischemic AKI by reducing inflammation and kidney tubular cell death and investigated the underlying mechanisms of protection.
MATERIALS AND METHODS
Renal I/R injury in mice.
After Columbia University Institutional Animal Care and Use Committee approval, C57BL/6 male mice (~20–25 g, Jackson Laboratories) were anesthetized intraperitoneally with pentobarbital sodium (50 mg/kg body wt or to effect, Sigma, St. Louis, MO) and subjected to right nephrectomy and 30-min left renal ischemia as previously described (26, 31). Sham-operated animals underwent the same surgical procedures (anesthesia, laparotomy, right nephrectomy, bowel manipulations, and wound closure) without renal ischemia. A separate cohort of mice was intraperitoneally injected with 6-shogaol (20 mg/kg, Cayman Chemical, Ann Arbor, MI) or vehicle (3.6% DMSO in saline) 24 h and 15 min before surgery. Some mice were injected with zinc protoporphyrin IX [ZnPP; a heme oxygenase (HO)-1 inhibitor, 25 mg/kg, R&D System, Minneapolis, MN] 30 min before 6-shogaol treatment. For pain control, all mice received 0.5–1 mg/kg sc buprenorphine SR 24 h before surgery. Body temperature was maintained at ~37°C using a surgical heating pad during surgery and recovery from anesthesia. Mice were euthanized 24 h after renal I/R injury with an overdose of pentobarbital sodium followed by exsanguination.
Measurement of renal injury after kidney I/R injury.
Twenty-four hours after renal I/R injury, we measured plasma blood urea nitrogen (BUN) and creatinine using enzymatic urea nitrogen and creatinine reagent kits (ThermoFisher Scientific, Waltham, MA). We also performed quantitative RT-PCR for kidney neutrophil gelatinase-associated lipocalin (NGAL) mRNA from mice subjected to sham surgery or renal I/R injury. NGAL is an early and sensitive marker of renal tubular injury (36).
Histological detection of kidney injury.
Kidney hematoxylin and eosin-stained sections after renal I/R or sham surgery were evaluated by a pathologist (V. D. D’Agati) who was unaware of the treatment that each animal had received. An established grading scale of kidney necrotic I/R injury to the proximal tubules (renal injury score of 0−4) was used for histopathological assessment as outlined by Jablonski et al. (17).
Histological detection of kidney apoptosis.
We detected renal tubular apoptosis with TUNEL staining using a commercially available in situ cell death detection kit (Roche, Indianapolis, IN) 24 h after renal I/R or sham surgery as previously described (42). TUNEL-positive cells in the kidney were quantified in five to seven randomly chosen ×200 microscope image fields in the corticomedullary junction, and results are expressed as apoptotic cells counted per ×200 field.
Detection of kidney neutrophil infiltration.
Immunohistochemistry for neutrophils infiltrating the kidney was performed using rat anti-mouse Ly6G monoclonal antibody (ThermoFisher Scientific) as previously described (43, 44). Primary IgG2a antibody (AbD Serotec, Raleigh, NC) was used as a negative isotype control (data not shown). Neutrophils were quantified in five to seven randomly chosen ×200 microscope image fields (corticomedullary junction for kidney neutrophils), and results are expressed as neutrophils counted per ×200 field.
Quantitative RT-PCR.
We measured mRNAs encoding markers of inflammation [IL-6, keratinocyte chemoattractant (KC), ICAM-1, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and TNF-α] and HO-1 in the kidney after surgeries and cultured proximal tubule cells with quantitative RT-PCR as previously described (31, 43) with the primers shown in Table 1. Primer design was based on published GenBank sequences. Quantitative RT-PCR was performed using the MyiQ Real-Time Detection System (Bio-Rad, Hercules, CA) using FastStart Universal SYBR Green Master Mix (Roche). To confirm equal RNA input, GAPDH mRNA expression and the relative expression of proinflammatory mRNA was calculated with the ΔΔCt method (where Ct is threshold cycle). Specificity of the amplification was checked by melting curve analysis.
Table 1.
Primers used in quantitative RT-PCRs to amplify mouse cDNA based on previously published GenBank sequences
| Primers | Sequence | Annealing Temperature, °C |
|---|---|---|
| Mouse TNF-α | Sense: 5′-TACTGAACTTCGGGGTGATTGGTCC-3′ | 65 |
| Antisense: 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′ | ||
| Mouse MCP-1 | Sense: 5′-ACCTGCTGCTACTCATTCAC-3′ | 60 |
| Antisense: 5′-TTGAGGTGGTTGTGGAAAAG-3′ | ||
| Mouse MIP-2 | Sense: 5′-CCAAGGGTTGACTTCAAGAAC-3′ | 60 |
| Antisense: 5′-AGCGAGGCACATCAGGTACG-3′ | ||
| Mouse KC | Sense: 5′-CAATGAGCTGCGCTGTCAGTG-3′ | 60 |
| Antisense: 5′-CTTGGGGACACCTTTTAGCATC-3′ | ||
| Mouse IL-6 | Sense: 5′-CCGGAGAGGAGACTTCACAG-3′ | 62 |
| Antisense: 5′-GGAAATTGGGGTAGGAAGGA-3′ | ||
| Mouse ICAM-1 | Sense: 5′-TGTTTCCTGCCTCTGAAGC-3′ | 60 |
| Antisense: 5′-CTTCGTTTGTGATCCTCCG-3′ | ||
| Mouse NGAL | Sense: 5′-CACCACGGACTACAACCAGTTCGC-3′ | 66 |
| Antisense: 5′-TCAGTTGTCAATGCATTGGTCGGTG-3′ | ||
| Human TNF-α | Sense: 5′-CGGGACGTGGAGCTGGCCGAGGAG-3′ | 68 |
| Antisense: 5′-CACCAGCTGGTTATCTCTCAGCTC-3′ | ||
| Human MCP-1 | Sense: 5′-AGCAAGTGTCCCAAAGAAGC-3′ | 64 |
| Antisense: 5′-CTCAAAACATCCCAGGGGTA-3′ | ||
| Human MIP-2 | Sense: 5′-CTTGCCAGCTCTCCTCCTC-3′ | 64 |
| Antisense: 5′-GCTTTCTGCCCATTCTTGAG-3′ | ||
| Human IL-8 | Sense: 5′-TCTGCAGCTCTGTGTGAAGG-3′ | 64 |
| Antisense: 5′-ATTGCATCTGGCAACCCTAC-3′ | ||
| Human IL-6 | Sense: 5′-AAAGAGGCACTGGCAGAAAA-3′ | 64 |
| Antisense: 5′-CATGCTACATTTGCCGAAGA-3′ | ||
| Human ICAM-I | Sense: 5′-GCAGACAGTGACCATCTACAGC-3′ | 60 |
| Antisense: 5′-GCCATCCTTTAGACACTTGAGC-3′ | ||
| Mouse and human HO-1 | Sense: 5′-TGAAGGAGGCCACCAAGGAG-3′ | 65 |
| Antisense: 5′-GTGGGCCACCAGCAGCTC-3′ | ||
| GAPDH | Sense: 5′-ACCACAGTCCATGCCATCAC-3′ | 65 |
| Antisense: 5′-CACCACCCTGTTGCTGTAGCC-3′ |
The annealing temperatures used for each primer are also shown. HO-1, heme oxygenase-1; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; KC, keratinocyte chemoattractant; MCP-1, monocyte chemoattractant protein-1; MIP-2, macrophage inflammatory protein-2; NGAL, neutrophil gelatinase-associated lipocalin; TNF-α, tumor necrosis factor-α.
Human and mouse proximal tubule cell culture.
An immortalized human proximal tubular cell line (HK-2 cells, American Type Culture Collection, Manassas, VA) was kept in low glucose DMEM-F-12 medium supplemented with 10% FBS at 37°C in a humidified 5% CO2 atmosphere as previously described (26, 51). Mouse kidney proximal tubules were isolated using Percoll density gradient separation as previously described (58). Mouse proximal tubules were maintained in high-glucose DMEM-Ham’s F-12 medium plus 10% FBS, 2 mM l-glutamine, and 10 mM HEPES. After 16 h of serum deprivation, confluent cultured HK-2 cells were treated with 10 ng/ml human recombinant TNF-α. Some cells were pretreated with 10–150 µM 6-shogaol 1 h before vehicle or recombinant TNF-α treatment. Some HK-2 cells were pretreated with 20 µM SB203580 (a selective inhibitor of p38 MAPK, R&D Systems, Minneapolis, MN) 30 min before 6-shogaol treatment. Confluent cultured moue proximal tubule cells were treated with 10 ng/ml mouse recombinant TNF-α or 10 µg/ml LPS. Some cells were pretreated with 100 µM 6-shogaol 1 h before recombinant TNF-α or LPS treatment.
We have previously demonstrated that prior oxidant stress protects against subsequent necrosis induced by H2O2 via the p38 MAPK → HO-1 induction pathway (32). To determine whether oxidant precondition attenuates renal proximal tubular cytokine induction in renal proximal tubular cells, HK-2 cells were preconditioned with 500 µM H2O2 or vehicle (PBS) for 15 min. To determine the potential additive effects of oxidant preconditioning with 6-shogaol, some cells were preconditioned with 500 µM H2O2 16 h before and treated with 50 µM 6-shogaol 1 h before TNF-α treatment. With quantitative RT-PCR, we measured the mRNA expression of proinflammatory TNF-α, IL-8, MIP-2, and ICAM-1 in HK-2 cells.
Proximal tubule cell nuclear fraction isolation for immunoblot analysis for NF-κB pathway proteins.
Confluent cultured HK-2 cells were scraped in 100 μl of buffer A [10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 20% glycerol, 0.1 mM PMSF, and 1 M DTT plus Protease Inhibitor Cocktail (Roche)] and homogenized for 5 s to release nuclei. After an incubation on ice for 15 min and centrifugation at 10,000 g for 1 min at 4°C, the pellet containing nuclear proteins was resuspended in 50 μl of buffer B [20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 0.5 M NaCl, 0.5 mM EDTA, 25% glycerol, 0.1% Triton X-100, 0.1 M PMSF, and 0.5 mM DTT plus Protease Inhibitor Cocktail]. Nuclear fractions were subjected to immunoblot analysis for rabbit anti-NF-κB p65 subunit (Cell Signaling Technology, Beverly, MA). Some HK-2 cells were lysed in buffer containing 20 mM HEPES (pH 7.4), 5 mM EDTA, 1 mM EGTA, 1% Triton X-100, and 10% glycerol plus Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail (Sigma) and subjected to immunoblot analysis for rabbit anti-phosphorylated IκBα, mouse anti-total IκBα, and rabbit anti-phosphorylated IKKα/β (Cell Signaling Technology).
HO-1 immunoblot analysis in cultured HK-2 cells and mouse kidneys after 6-shogaol treatment.
HK-2 cells or kidney cortical tissues were lysed in buffer containing 20 mM HEPES (pH 7.4), 5 mM EDTA, 1 mM EGTA, 1% Triton X-100, and 10% glycerol plus Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail and subjected to immunoblot analysis for rabbit anti-HO-1 antibody (Cell Signaling Technology).
Statistical analysis.
Data were analyzed with a Student’s t-test to compare means between two groups or one-way ANOVA plus Tukey’s post hoc multiple-comparison test to compare multiple groups. The ordinal values of the renal injury scores were analyzed by a Mann-Whitney nonparametric test. In all cases, a probability statistic of P < 0.05 was taken to indicate significance. All data are expressed throughout the text as means ± SE.
RESULTS
6-Shogaol treatment attenuates TNF-α or LPS-induced proinflammatory mRNA synthesis in cultured proximal tubules.
HK-2 cells were pretreated with vehicle (0.1% DMSO) or 50, 100, or 150 µM 6-shogaol for 1 h before vehicle (PBS, n = 4–8) or 10 ng/ml TNF-α (n = 7–13) treatment for 6 h. Quantitative RT-PCR revealed that 6-shogaol pretreatment significantly attenuated the expression of proinflammatory cytokine and chemokine mRNAs in HK-2 cells [TNF-α, MCP-1, and ICAM-1 (50 µM and overdose 6-shogaol) as well as IL-6, IL-8, and MIP-2 (100 µM and overdose 6-shogaol); Fig. 1A]. Furthermore, 100 µM 6-shogaol pretreatment attenuated TNF-α (10 ng/ml; Fig. 1B) or LPS (10 µg/ml; Fig. 1C)-induced increases in proinflammatory cytokine and chemokine mRNA (TNF-α, IL-6, KC, MCP-1, MIP-2, and ICAM-1) synthesis in primary cultures of mouse proximal tubule cells (n = 3–4). In contrast, H2O2 oxidant preconditioning (500 μM H2O2 treatment and 16 h of recovery) had no effect on proinflammatory cytokine induction in HK-2 cells after TNF-α treatment (Fig. 1D). Furthermore, combination of oxidant precondition and 6-shogaol had no further anti-inflammatory effects compared with 6-shogaol alone after TNF-α treatment.
Fig. 1.

6-Shogaol treatment inhibits proinflammatory mRNA induction in renal proximal tubule cells after TNF-α or LPS treatment. A: human renal proximal tubule (HK-2) cells were treated with vehicle (PBS; n = 4–8) or 10 ng/ml human recombinant TNF-α (n = 7–13) for 6 h. Some cells were pretreated with 50–150 µM 6-shogaol 1 h before vehicle (PBS) or TNF-α treatment. With quantitative RT-PCR, we measured the mRNA expression of proinflammatory TNF-α, IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and ICAM-1 in HK-2 cells. Fold increases in proinflammatory mRNA normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA are shown. B and C: freshly isolated mouse proximal tubular cells were treated with vehicle (PBS) or 10 ng/ml murine recombinant TNF-α (B) or 10 µg/ml LPS (C) for 6 h. Some cells were pretreated with 100 µM 6-shogaol 1 h before vehicle, TNF-α, or LPS treatment. With quantitative RT-PCR, we measured the mRNA expression of proinflammatory TNF-α, IL-6, keratinocyte chemoattractant (KC), MCP-1, MIP-2, and ICAM-1 in murine proximal tubular cells. Fold increases in proinflammatory mRNA normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA (n = 3–4) are shown. D: H2O2 preconditioning had no anti-inflammatory effect in cultured HK-2 cells. HK-2 cells were treated with vehicle (PBS; n = 3) or 10 ng/ml human recombinant TNF-α (n = 4) for 6 h. Sixteen hours before TNF-α treatment, some cells were pretreated with 500 µM H2O2 for 15 min and some cells were treated with 50 µM 6-shogaol 1 h before TNF-α treatment after oxidant preconditioning. With quantitative RT-PCR, we measured the mRNA expression of proinflammatory TNF-α, IL-8, MIP-2, and ICAM-1 in HK-2 cells. Fold increases in proinflammatory mRNA normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA are shown. *P < 0.05 vs. vehicle-treated cells; #P < 0.05 vs. TNF-α- or LPS-treated cells. Error bars represent 1 SE.
6-Shogaol protects against renal I/R injury in mice.
We next tested whether 6-shogaol treatment protects against ischemic AKI in mice. Plasma creatinine (PCr) and BUN values as well as NGAL mRNA levels were similar between vehicle- and 6-shogaol-treated mice subjected to sham operation (anesthesia, laparotomy, right nephrectomy, and recovery, n = 3–6; Fig. 2). As expected, PCr, BUN, and NGAL mRNA synthesis increased significantly 24 h after 30 min of renal I/R injury in vehicle-treated mice. However, mice pretreated with 6-shogaol were significantly protected against renal injury, as evidenced by significantly lower PCr, BUN, and NGAL mRNA compared with vehicle-treated mice subjected to renal IR (n = 4–9; Fig. 2).
Fig. 2.
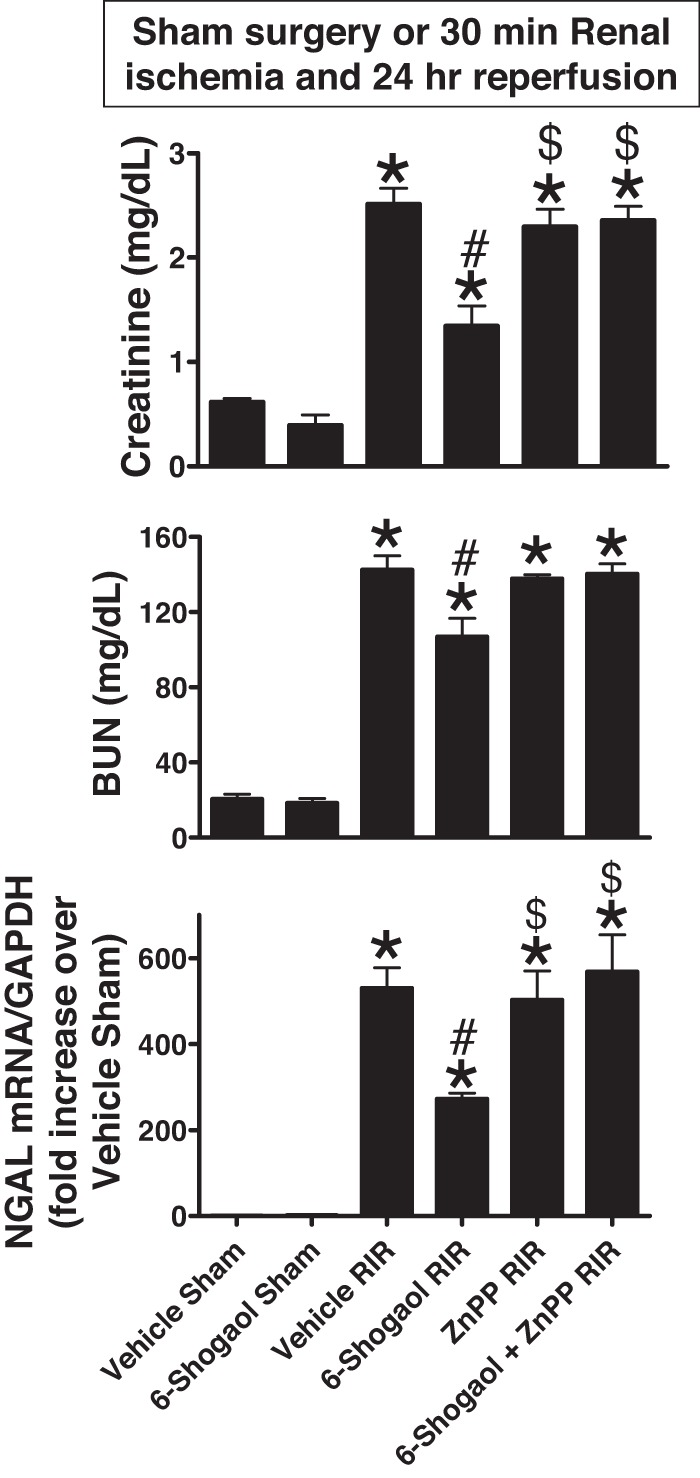
6-Shogaol protects against renal ischemia-reperfusion (RIR) injury via induction of the heme oxygenase (HO)-1 pathway. C57BL/6 male mice were subjected to sham operation or 30 min of RIR. Some mice were pretreated with 20 mg/kg 6-shogaol 24 h and 15 min before surgery. Plasma creatinine (PCr), blood urea nitrogen (BUN), and neutrophil gelatinase-associated lipocalin (NGAL) mRNA synthesis increased significantly 24 h after RIR injury. However, mice pretreated with 6-shogaol were significantly protected against renal injury, as evidenced by significantly lower PCr, BUN, and NGAL mRNA compared with vehicle-treated mice subjected to RIR (n = 4–9). To test whether HO-1 induction is critical for 6-shogaol-mediated renal protection, we injected HO-1 inhibitor [25 mg/kg zinc protoporphyrin IX (ZnPP)] before renal ischemia. Pretreatment with ZnPP significantly prevented 6-shogaol-mediated protection against ischemic acute kidney injuray (n = 4–9). *P < 0.05 vs. vehicle-treated mice subjected to sham surgery; #P < 0.05 vs. vehicle-treated mice subjected to RIR injury; $P < 0.05 vs. 6-shogaol-treated mice subjected to RIR injury. Error bars represent 1 SE.
6-Shogaol treatment protects against kidney necrosis and apoptosis after renal I/R.
Figure 3, A and B, shows representative hematoxylin and eosin-stained images of mice subjected to 30 min of renal ischemia and 24 h of reperfusion (A: magnification ×200 and B: magnification ×400, n = 4–6). As expected, kidneys of vehicle-treated mice subjected to 30 min of renal I/R showed severe tubular necrosis and proteinaceous casts as well as increased tubular dilatation and congestion. Consistent with the PCr data (Fig. 2), 6-shogaol-treated mice had decreased renal tubular necrosis, congestion, and cast formation compared with vehicle-treated mice subjected to renal I/R injury. The Jablonski scale renal injury score (scale: 0–4) for histology grading was used to grade renal tubular necrosis 24 h after renal I/R (Fig. 3C). Thirty minutes of renal I/R and 24 h of reperfusion resulted in severe renal tubular injury in vehicle-treated wild-type mice. In contrast, 6-shogaol-treated mice subjected to renal I/R had slightly but significantly lower (by ~21%) renal injury scores.
Fig. 3.

6-Shogaol attenuates renal tubular necrosis via the heme oxygenase (HO)-1 pathway after renal ischemia-reperfusion (RIR) injury. A and B: representative hematoxylin and eosin-stained images of mice subjected to 30 min of renal ischemia and 24 h of reperfusion (A: magnification ×200 and B: magnification ×400, n = 4–6). As expected, kidneys of vehicle-treated mice subjected to RIR showed severe tubular necrosis and proteinaceous casts as well as increased tubular dilatation and congestion. However, 6-shogaol-treated mice had decreased renal tubular necrosis, congestion, and cast formation compared with vehicle-treated mice subjected to RIR injury. Severe tubular necrosis and proteinaceous casts (*) are indicated. Arrowheads show increased tubular dilatation and congestion. C: Jablonski scale renal injury score (scale: 0–4) for histology grading was used to grade renal tubular necrosis 24 h after RIR. RIR for 30 min and 24 h of reperfusion resulted in severe renal tubular injury in vehicle-treated wild-type mice. In contrast, 6-shogaol-treated mice subjected to RIR had significantly lower (by ~21%) renal injury scores. HO-1 inhibition significantly prevented 6-shogaol-mediated protection against renal tubular necrosis. ZnPP, zinc protoporphyrin IX. *P < 0.05 vs. vehicle-treated mice subjected to RIR injury; #P < 0.05 vs. 6-shogaol-treated mice subjected to RIR injury. Error bars represent 1 SE.
TUNEL staining detected fragmented DNA suggestive of apoptosis in renal tubular cells in the kidneys of mice (×200, n = 4–6; Fig. 4A). Increased renal tubule cell apoptosis occurred in vehicle-treated mice subjected to 30 min of renal I/R. Again, 6-shogaol-treated mice subjected to 30 min of renal I/R had significantly decreased (by ~70%) TUNEL-positive renal tubule cells 24 h after reperfusion (Fig. 4B).
Fig. 4.

6-Shogaol decreases renal tubular apoptosis via the heme oxygenase (HO)-1 pathway after renal ischemia-reperfusion (RIR). A and B: representative images (from 4–6 experiments; A; magnification ×200) and quantifications of TUNEL staining to demonstrate renal tubular apoptosis per ×200 field (B; n = 4–6) in the kidneys of mice subjected to sham operation or 30 min of renal ischemia and 24 h of reperfusion. TUNEL staining shows increased renal tubule cell apoptosis in vehicle-treated mice subjected to RIR. In contrast, 6-shogaol-treated mice subjected to RIR had significantly decreased TUNEL-positive renal tubular cells 24 h after reperfusion. HO-1 inhibition significantly prevented the 6-shogaol-mediated decrease in TUNEL-positive renal tubular cells after RIR. ZnPP, zinc protoporphyrin IX. *P < 0.05 vs. vehicle-treated mice subjected to sham surgery; #P < 0.05 vs. vehicle-treated mice subjected to RIR injury; $P < 0.05 vs. 6-shogaol-treated mice subjected to RIR injury. Error bars represent 1 SE.
6-Shogaol treatment attenuates kidney neutrophil infiltration and induction of proinflammatory cytokines and chemokine synthesis after renal I/R injury.
Figure 5, A and B, shows representative images of immunohistochemistry for neutrophils (dark brown) in the kidneys of mice subjected to 30 min of renal ischemia and 24 h of reperfusion (A: magnification ×200 and B: magnification ×400, n = 4–6). Neutrophil infiltration markedly increased in vehicle-treated mice subjected to renal I/R concentrated near the corticomedullary junction. Again, 6-shogaol-treated mice subjected to renal I/R had decreased neutrophil infiltration (by ~60%) after 24 h of reperfusion (Fig. 5C).
Fig. 5.

6-Shogaol decreases neutrophil infiltration via the heme oxygenase (HO)-1 pathway after renal ischemia-reperfusion (RIR). A and B: representative images (from 4–6 experiments; A: magnification ×200 and B: magnification ×400) and quantification of infiltrated neutrophils per ×200 field (C; n = 4–6) of immunohistochemistry of neutrophil infiltration (dark brown) in the kidneys (corticomedullary junction) of mice subjected to sham operation or 30 min of renal ischemia and 24 h of reperfusion. Neutrophil infiltration markedly increased in mice subjected to RIR concentrated near the corticomedullary junction. In contrast, 6-shogaol-treated mice subjected to RIR had significantly decreased neutrophil infiltration after 24 h of reperfusion. HO-1 inhibition significantly prevented 6-shogaol-mediated protection against neutrophil infiltration after RIR. ZnPP, zinc protoporphyrin IX. *P < 0.05 vs. vehicle-treated mice subjected to sham surgery; #P < 0.05 vs. vehicle-treated mice subjected to RIR injury; $P < 0.05 vs. 6-shogaol-treated mice subjected to RIR injury. Error bars represent 1 SE.
Next, we measured the expression of proinflammatory cytokine and chemokine mRNAs in the kidneys (TNF-α, IL-6, KC, MCP-1, MIP-2, and ICAM-1) 24 h after renal I/R by quantitative RT-PCR (primer sequences shown in Table 1). All proinflammatory cytokine and chemokine mRNAs markedly increased in vehicle-treated mice subjected to renal I/R; inductions of IL-6, KC, MCP-1, and MIP-2 mRNA synthesis were attenuated by 6-shogaol pretreatment in mice subjected to renal I/R (n = 4–6; Fig. 6).
Fig. 6.

6-Shogaol attenuates the induction of proinflammatory cytokines via the heme oxygenase (HO)-1 pathway after renal ischemia-reperfusion (RIR) injury. With quantitative RT-PCR, we measured the expression of proinflammatory cytokine and chemokine mRNAs in the kidney [TNF-α, IL-6, keratinocyte chemoattractant (KC), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and ICAM-1] 24 h after sham operation or RIR. Fold increases in proinflammatory mRNA normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA (n = 4–6) are shown. All proinflammatory cytokine and chemokine mRNAs markedly increased in vehicle-treated mice subjected to RIR. However, induction of IL-6, KC, MCP-1, and MIP-2 was attenuated by 6-shogaol pretreatment in mice subjected to RIR. HO-1 inhibition significantly prevented the 6-shogaol-mediated decrease in IL-6, KC, MCP-1, and MIP-2 mRNA synthesis after RIR. ZnPP, zinc protoporphyrin IX. *P < 0.05 vs. vehicle-treated mice subjected to sham surgery; #P < 0.05 vs. vehicle-treated mice subjected to RIR injury; $P < 0.05 vs. 6-shogaol-treated mice subjected to RIR injury. Error bars represent 1 SE.
6-Shogaol attenuates TNF-α-induced nuclear translocation of the NF-κB transcription factor in cultured renal proximal tubules.
Because NF-κB is a well-known TNF-α-mediated transcription factor that promotes the synthesis of proinflammatory cytokines and chemokines and its pathway is a critical mediator of the inflammatory response after ischemia AKI (48), we tested whether 6-shogaol treatment attenuates NF-κB activation in cultured HK-2 cells (n = 3–4). Exogenous human recombinant TNF-α (10 ng/ml for 30 min) caused significant nuclear p65 NF-κB subunit translocation in cultured HK-2 cells (Fig. 7A). Additionally, we also discovered significantly increased IκBα degradation and phosphorylation of IκBα (Fig. 7B) and IKKα/β (Fig. 7C) after TNF-α treatment. These increases in nuclear p65 translocation, IκBα degradation, and phosphorylation of IκBα and IKKα/β were significantly attenuated when cells were pretreated with 6-shogaol, suggesting that 6-shogaol attenuates TNF-α induced NF-κB activity most likely via inhibiting IKKα/β and IκBα phosphorylation.
Fig. 7.

6-Shogaol attenuates TNF-α-induced activation of NF-κB in cultured renal proximal tubule cells by inhibiting IKKα/β and IκBα phosphorylation. Human renal proximal tubule (HK-2) cells were treated with vehicle (PBS) or 10 ng/ml human recombinant TNF-α for 6 h. Some cells were pretreated with 150 µM 6-shogaol 1 h before TNF-α treatment. A: representative p65 NF-κB subunit immunoblot analysis (top) and band intensity quantification (bottom) in nuclear fractions of cultured HK-2 cells 30 min after TNF-α treatment. B: representative phosphorylated IκBα and total IκBα immunoblot analysis (top) and band intensity quantification (bottom) in whole lysates of cultured HK-2 cells 30 min after TNF-α treatment. C: representative phosphorylated IKKα/β immunoblot analysis (top) and band intensity quantification (bottom) in whole lysates of cultured HK-2 cells 15 min after TNF-α treatment. Histone H3 (A) and β-actin (B and C) immunoblot analyses were performed to demonstrate equal nuclear and whole lysate protein loading, respectively. *P < 0.05 vs. vehicle-treated HK-2 cells; #P < 0.05 vs. TNF-α-treated HK-2 cells. Error bars represent 1 SE.
6-Shogaol induces cytoprotective chaperone HO-1 via p38 MAPK activation.
Because HO-1 plays a cytoprotective role in the modulation of tissue responses to injury in several pathophysiological states, including ischemic AKI, and previous studies have indicated that 6-shogaol induces HO-1 expression in some cell lines via Nuclear factor erythroid 2-related factor 2 (Nrf2) activation (4, 12, 25, 40), we tested whether 6-shogaol induces HO-1 expression in both human and mouse kidney cells. As shown in Fig. 8A, 6-shogaol treatment significantly induced HO-1 protein expression in HK-2 cells as well as in mouse kidneys (n = 3). Additionally, 6-shogaol treatment significantly induced HO-1 mRNA synthesis in HK-2 cells (Fig. 8B). This induction of HO-1 by 6-shogaol was inhibited by pretreatment with a p38 inhibitor (SB203580) in HK-2 cells, suggesting that 6-shogaol induces a HO-1 via p38 MAPK activation (n = 3–4; Fig. 8B).
Fig. 8.

6-Shogaol increases renal tubular heme oxygenase (HO)-1 via p38 MAPK activation. A: human renal proximal tubule (HK-2) cells were treated with 10 µM 6-shogaol (left) or mice were injected with 20 mg/kg 6-shogaol (right). Twenty-four hours after treatment, HK-2 cell lysates and kidney samples were subjected to immunoblot analysis using anti-HO-1 antibody. Representative HO-1 immunoblot analysis (top) and band intensity quantification (bottom) are shown (n = 3). B: HK-2 cells were treated with 100 µM 6-shogaol for 7 h. Some cells were pretreated with 20 µM SB203580 30 min before 6-shogaol treatment. With quantitative RT-PCR, we measured the mRNA expression of HO-1 in HK-2 cells. Fold increases in HO-1 mRNA normalized to GAPDH are shown (n = 3–4). *P < 0.05 vs. vehicle-treated HK-2 cells or mouse kidneys; #P < 0.05 vs. 6-shogaol-treated HK-2 cells. Error bars represent 1 SE.
Critical role for HO-1 in 6-shogaol-mediated protection against ischemic AKI in mice.
To test whether 6-shogaol protects the kidney against ischemic AKI via HO-1 induction, we injected mice with the HO-1 inhibitor ZnPP before renal ischemia. Pretreatment with ZnPP significantly attenuated 6-shogaol-mediated protection against ischemic AKI as evaluated by PCr, BUN, and NGAL mRNA (Fig. 2) as well as histological renal tubular cell necrosis (Fig. 3) and apoptosis (Fig. 4). Additionally, pretreatment with ZnPP significantly attenuated 6-shogaol-mediated protection against kidney neutrophil infiltration (Fig. 5) and proinflammatory cytokine mRNA induction (Fig. 6) after renal ischemia.
DISCUSSION
The major finding of the present study is that 6-shogaol treatment protects against ischemic AKI, resulting in decreased renal tubular necrosis, inflammation, and apoptosis. We deciphered the mechanisms of 6-shogaol-mediated renal protection by demonstrating that 6-shogaol attenuates proinflammatory NF-κB signaling and induces a cytoprotective heat shock protein HO-1 synthesis (Fig. 9).
Fig. 9.

Schematic of the proposed mechanisms for 6-shogaol-mediated protection against renal ischemia-reperfusion (IR) injury. Renal IR injury induces proinflammatory cytokines and chemokine synthesis via the NF-κB pathway and necrotic and apoptotic renal tubular cell death, leading to kidney tissue damage. 6-Shogaol protects against renal IR injury by inhibition of the canonical NF-κB pathway and activation of heme oxygenase (HO)-1 synthesis via p38 MAPK activation. Our previous study of oxidant preconditioning (500 µM H2O2 followed by 16 h of recovery) also protected against subsequent necrotic insult by lethal dose of H2O2 via the p38 MAPK → HO-1 pathway (32). However, unlike 6-shogaol-mediated attenuation in proinflammatory cytokine generation in renal proximal tubule cells, oxidant preconditioning → p38 MAPK activation failed to attenuate proinflammatory cytokine induction in human renal proximal tubule cells. This discrepancy in protection against proinflammatory cytokine generation further highlights gaps in our mechanistic understanding of stress-activated p38 MAPK signaling in renal protection.
Inflammation and recruitment of leukocytes play a major role in ischemic AKI. A number of potent mediators are generated by the injured kidney tubular epithelial cells, including proinflammatory cytokines and chemokines (TNF-α, IL-6, IL-8, MCP-1, and MIP-2), leading to rapid leukocyte activation and infiltration into the kidney after ischemic AKI. In particular, neutrophils are the earliest leukocytes to accumulate in the kidney after renal ischemia and play a critical role in renal tubular cell death. Indeed, blockade of or neutralization of neutrophils attenuates the severity and duration of ischemic AKI (6, 9, 15). In the present study, the decreased expression of neutrophil chemotactic cytokines KC and MIP-2 in 6-shogaol-treated mice is consistent with our finding that these mice had significantly attenuated neutrophil infiltration after renal I/R compared with vehicle-treated mice. Reduced kidney neutrophil infiltration most likely attenuated additional cytokine generation in 6-shogaol-treated mice that may have contributed to improved renal function after ischemic AKI. Furthermore, 6-shogaol-treated mice also had significantly reduced MCP-1 and IL-6 expression in the kidney after I/R. MCP-1 [chemokine (C-C motif) ligand 2 (CCL2)] is the major chemokine that promotes macrophage infiltration and migration after I/R (11, 20), and IL-6 is a major mediator of monocyte differentiation to macrophages as well as neutrophil activation (52). Moreover, we showed that 6-shogaol directly reduced proinflammatory cytokine and chemokine synthesis in cultured human and mouse renal proximal tubular cells (Fig. 1). We chose renal proximal tubule cells for our mechanistic in vitro experiments because renal proximal tubular cells are the most injury-susceptible cell type during and after renal I/R injury. In the present study, we found that 6-shogaol has anti-inflammatory effects on in vitro and in vivo models.
NF-κB is a family of inducible transcription factors critical for several biological processes, including inflammation, immunity, apoptosis, cell proliferation, and differentiation (14, 56). During and after ischemic AKI, NF-κB is rapidly activated and serves as a key mediator of inflammatory cytokine and chemokine generation (48). Indeed, NF-κB inhibitors attenuate the induction of renal tubular inflammation and chronic renal injury in the 5/6 renal ablation model (10). NF-κB inhibition by administration of IKKβ siRNA reduces ischemic kidney injury and inflammation in rats (59). Several studies have shown that 6-shogaol modulates the NF-κB pathway. For example, 6-shogaol inhibits NF-κB in an LPS-induced lung injury model in mice (60). 6-Shogaol also attenuates LPS-induced neuronal inflammation and microglial activation in the brain of mice via NF-κB modulation (14). However, the molecular mechanisms leading to inhibition of NF-κB activation by 6-shogaol remain unclear. Our present study shows that 6-shogaol exerts an anti-NF-κB effect through inhibition of phosphorylation of IKKα and IKKβ subunits of the IKK complex. This led to a blockade of IκBα phosphorylation and prevention of IκBα proteosomal degradation with a resulting decrease in p65 nuclear translocation and NF-κB transcriptional activation in TNF-α-treated kidney proximal tubule cells.
In the present study, we showed that 6-shogaol is a potent inducer of HO-1 [also known as heat shock protein 32 (HSP32)] in renal proximal tubule cells. HO-1 is a microsomal membrane enzyme that catalyzes the oxidative cleavage of heme molecules to carbon monoxide, iron, and biliverdin, which is subsequently reduced to bilirubin by the biliverdin reductase (46). Because HO-1 products (carbon monoxide and biliverdin) have potent antioxidant and anti-inflammatory properties, HO-1 has attracted major attention for therapeutic target for several inflammatory human diseases including AKI (16, 46). Indeed, numerous studies have reported the cytoprotective properties of HO-1 against kidney injury induced by cisplatin (1, 50), LPS (55), ureteral obstruction (20), and renal ischemia (49, 54). In previous in vitro studies, it has been reported that 6-shogaol induced HO-1 expression in a hepatoma cell line, colonic epithelial cell line (HCT-116), and primary cultured microglia cells (4, 25, 40). However, the molecular mechanisms of 6-shogaol-mediated HO-1 induction remains unclear.
The hmox1 gene encoding HO-1 is regulated by several transcription factors, including heat shock factor, NF-κB, nuclear factor-erythroid 2 (NF-E2), and activator protein-1 (AP-1) families and MAPKs (p38, ERK, and JNK) (2). Of these HO-1-inducing factors, p38 MAPK activation induces HO-1 expression in pulmonary epithelial cell (39), macrophages (41), and hepatocytes (24). In kidney tubule cells, we have previously demonstrated that oxidant preconditioning induces HO-1 via p38 MAPK activation in cultured HK-2 cells and protects HK-2 cells against lethal oxidant injury (32). Our present study also revealed that 6-shogaol treatment increases HO-1 protein and mRNA expression via p38 MAPK activation in renal proximal tubule cells because pretreatment with a selective p38 MAPK inhibitor (SB203580) prevented 6-shogaol-mediated HO-1 induction.
6-Shogaol is also a potent activator of Nrf2, which is one of the major HO-1 modulating transcription factors (4, 25, 61). Several previous studies have linked p38 MAPK activation to Nrf2 transcription factor regulation. p38 MAPK phosphorylation facilitates Nrf2 dissociation from Kelch-like ECH-associated protein 1 (Keap1) and its nuclear translocation for HO-1 induction (19). Moreover, 6-shogaol derivatives may directly interact with the reactive sulfhydryl groups of cysteine residues of Keap1, leading to Keap1-Nrf2 complex disruption and Nrf2 nuclear translocation (61). Although the detailed signal pathway of 6-shogaol inducing HO-1 remains to be defined, we can conclude that the renal protective effects of 6-shogaol are in part mediated by HO-1 upregulation because direct inhibition of HO-1 by ZnPP treatment completely prevented 6-shogaol-mediated protection against ischemic AKI in mice.
Although 6-shogaol completely abolished TNF-α-induced IKKα/β and IκB activation, it had only a partial effect on TNF-α-induced NF-κB activation (Fig. 7). It is possible that 6-shogaol may have an independent stimulatory effect on NF-κB translocation. It is also possible that the canonical NF-κB signal pathway mediated by phosphorylation of IKKα/β and phosphorylation and degradation of IκBα is modulated by 6-shogaol. However, the NF-κB pathway can also be activated by the noncanonical NF-κB signaling pathway mediated by B cell activation factor CD40, receptor activator for NF-κB, or lymphtoxin β-receptor. This leads to activation of IKKα by NF-κB-inducing kinase or atypical NF-κB mediated by ataxia telangiectasia-mutated checkpoint kinase and NF-κB essential modulator (NEMO) translocation signaling pathway (30, 53). The role of 6-shogaol on noncanonical or atypical NF-κB signal pathway activation is unknown. In the present study, we suggest that 6-shogaol inhibits NF-κB activation, at least in part, by suppressing the canonical NF-κB pathway. Furthermore, stress-activated p38 MAPK signaling can act both upstream and downstream of TNF-α in some cell types (33, 47). One of the limitations of this study is that our findings do not clearly delineate the extent or level of the p38 MAPK-TNF-α interaction. 6-Shogaol clearly promotes HO-1 induction via a mechanism involving p38 MAPK (similar to oxidant preconditioning), but the TNF-α dependence of these mechanisms remains to be elucidated. Similarly, 6-shogaol inhibits IKKα/β-IκB-NF-κB signaling and proinflammatory cytokine induction, but the p38 MAPK dependence of these effects also remains to be elucidated in future studies.
Although very powerful as translational studies, there are limitations with in vivo studies. With in vivo approaches, it is nearly impossible to dissect the cause-and-effect relationship. Moreover, there are multiple cell types and organ systems being affected by kidney I/R injury including renal tubular cells, endothelial cells, and leukocytes as well as the effects on the kidney produced by remote organ injury after renal I/R. Here, we complemented our in vivo experiments with mechanistic in vitro experiments. We show here that 6-shogaol failed to attenuate TNF-α mRNA induction after renal I/R in vivo. However, in cultured human and mouse proximal tubule cells, we showed that 6-shogaol inhibited TNF-α induction (Fig. 1) and modulated NF-κB pathway activation by exogenous TNF-α treatment (Fig. 7). We speculate that these discrepancies may be due to the inherent differences between in vivo and in vitro models. Moreover, the discrepancy between in vivo and in vitro TNF-α data may be due to the kinetics of TNF-α induction. TNF-α is one of the earliest cytokines induced after renal I/R injury. The tissue TNF-α mRNA level peaks at 30 min after reperfusion and then rapidly declines. The tissue TNF-α protein level peaks at 1 h after reperfusion, and this increase is sustained at 2 h of reperfusion and declines after 4 h of reperfusion (7). In addition, the plasma TNF-α level peaks at 2 h after reperfusion and declines to a near-normal level at 48 h after reperfusion (23). As 6-shogaol directly attenuates proinflammatory cytokines, attenuates NF-κB activation, and induces HO-1 in cultured cells, we can conclude that the anti-inflammatory effects of 6-shogaol are the one of the major mechanisms of protection against ischemic AKI in mice.
Our previous study (32) of oxidant preconditioning (500 µM H2O2 followed by 16 h of recovery) also protected against subsequent necrotic insult by a lethal dose of H2O2 via the p38 MAPK → HO-1 pathway. However, unlike the 6-shogaol-mediated attenuation in proinflammatory cytokine generation in renal proximal tubule cells, oxidant preconditioning → p38 MAPK activation failed to attenuate proinflammatory cytokine induction in HK-2 cells. This discrepancy in protection against proinflammatory cytokine generation further highlights gaps in our mechanistic understanding of stress-activated p38 MAPK signaling in renal protection. Future followup studies examining the potential additive renal protective effects of oxidant preconditioning and 6-shogaol would further elucidate the similarities and differences between oxidant preconditioning and 6-shogaol-mediated renal protection.
In summary, we demonstrated, in the present study, that 6-shogaol protects against murine ischemic AKI by preventing necrosis, apoptosis, and inflammation after I/R. In particular, 6-shogaol has powerful anti-inflammatory effects in renal proximal tubule cells by attenuating NF-κB activation and inducing HO-1 expression in renal proximal tubule cells. Our findings suggest that 6-shogaol may provide a potential therapy for ischemic AKI during the perioperative period.
GRANTS
This work was supported by the Department of Anesthesiology, Columbia University, and in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-109544 and R01-DK-115694 (to H. T. Lee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.J.H. and H.T.L. conceived and designed research; S.J.H. and M.K. performed experiments; S.J.H., M.K., V.D.D., and H.T.L. analyzed data; S.J.H., M.K., V.D.D., and H.T.L. interpreted results of experiments; S.J.H., M.K., and H.T.L. prepared figures; S.J.H. and H.T.L. drafted manuscript; S.J.H. and H.T.L. edited and revised manuscript; S.J.H., M.K., V.D.D., and H.T.L. approved final version of manuscript.
REFERENCES
- 1.Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA. Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat. Kidney Int 48: 1298–1307, 1995. doi: 10.1038/ki.1995.414. [DOI] [PubMed] [Google Scholar]
- 2.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36: 166–174, 2007. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 3.Aronson S, Blumenthal R. Perioperative renal dysfunction and cardiovascular anesthesia: concerns and controversies. J Cardiothorac Vasc Anesth 12: 567–586, 1998. doi: 10.1016/S1053-0770(98)90106-9. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Fu J, Chen H, Hu Y, Soroka DN, Prigge JR, Schmidt EE, Yan F, Major MB, Chen X, Sang S. Ginger compound [6]-shogaol and its cysteine-conjugated metabolite (M2) activate Nrf2 in colon epithelial cells in vitro and in vivo. Chem Res Toxicol 27: 1575–1585, 2014. doi: 10.1021/tx500211x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 6.De Greef KE, Ysebaert DK, Ghielli M, Vercauteren S, Nouwen EJ, Eyskens EJ, De Broe ME. Neutrophils and acute ischemia-reperfusion injury. J Nephrol 11: 110–122, 1998. [PubMed] [Google Scholar]
- 7.Donnahoo KK, Meng X, Ayala A, Cain MP, Harken AH, Meldrum DR. Early kidney TNF-α expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am J Physiol Regul Integr Comp Physiol 277: R922–R929, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Dugasani S, Pichika MR, Nadarajah VD, Balijepalli MK, Tandra S, Korlakunta JN. Comparative antioxidant and anti-inflammatory effects of [6]-gingerol, [8]-gingerol, [10]-gingerol and [6]-shogaol. J Ethnopharmacol 127: 515–520, 2010. doi: 10.1016/j.jep.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost 97: 738–747, 2007. doi: 10.1160/TH07-01-0022. [DOI] [PubMed] [Google Scholar]
- 10.Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM Jr, Zatz R. Chronic inhibition of nuclear factor-κB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol 292: F92–F99, 2007. doi: 10.1152/ajprenal.00184.2006. [DOI] [PubMed] [Google Scholar]
- 11.Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, Ishiwata Y, Asano M, Wang H, Matsushima K, Takeya M, Kuziel WA, Mukaida N, Yokoyama H. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 14: 2503–2515, 2003. doi: 10.1097/01.ASN.0000089563.63641.A8. [DOI] [PubMed] [Google Scholar]
- 12.Ha SK, Moon E, Ju MS, Kim DH, Ryu JH, Oh MS, Kim SY. 6-Shogaol, a ginger product, modulates neuroinflammation: a new approach to neuroprotection. Neuropharmacology 63: 211–223, 2012. doi: 10.1016/j.neuropharm.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Han SJ, Jang HS, Noh MR, Kim J, Kong MJ, Kim JI, Park JW, Park KM. Mitochondrial NADP+-dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia-reperfusion injury. J Am Soc Nephrol 28: 1200–1215, 2017. doi: 10.1681/ASN.2016030349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26: 203–234, 2012. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis 34: 384–399, 1999. doi: 10.1016/S0272-6386(99)70375-6. [DOI] [PubMed] [Google Scholar]
- 16.Hull TD, Agarwal A, George JF. The mononuclear phagocyte system in homeostasis and disease: a role for heme oxygenase-1. Antioxid Redox Signal 20: 1770–1788, 2014. doi: 10.1089/ars.2013.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jablonski P, Howden BO, Rae DA, Birrell CS, Marshall VC, Tange J. An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35: 198–204, 1983. doi: 10.1097/00007890-198303000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11: 88–101, 2015. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 19.Jiang G, Hu Y, Liu L, Cai J, Peng C, Li Q. Gastrodin protects against MPP+-induced oxidative stress by up regulates heme oxygenase-1 expression through p38 MAPK/Nrf2 pathway in human dopaminergic cells. Neurochem Int 75: 79–88, 2014. doi: 10.1016/j.neuint.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant 21: 1231–1239, 2006. doi: 10.1093/ndt/gfk047. [DOI] [PubMed] [Google Scholar]
- 21.Jones DR, Lee HT. Perioperative renal protection. Best Pract Res Clin Anaesthesiol 22: 193–208, 2008. doi: 10.1016/j.bpa.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Jung HW, Yoon CH, Park KM, Han HS, Park YK. Hexane fraction of Zingiberis Rhizoma Crudus extract inhibits the production of nitric oxide and proinflammatory cytokines in LPS-stimulated BV2 microglial cells via the NF-kappaB pathway. Food Chem Toxicol 47: 1190–1197, 2009. doi: 10.1016/j.fct.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 23.Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol 14: 1549–1558, 2003. doi: 10.1097/01.ASN.0000064946.94590.46. [DOI] [PubMed] [Google Scholar]
- 24.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem 278: 17927–17936, 2003. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 25.Kim JK, Jang HD. 6-shogaol attenuates H2O2-induced oxidative stress via upregulation of Nrf2-mediated γ-glutamylcysteine synthetase and heme oxygenase expression in HepG2 cells. Food Sci Biotechnol 25: 319–327, 2016. doi: 10.1007/s10068-016-0045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D’Agati VD, Lee HT. Selective renal over-expression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 299: F347–F358, 2010. doi: 10.1152/ajprenal.00194.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron, Exp Nephrol 109: e102–e107, 2008. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinsey GR, Okusa MD. Expanding role of T cells in acute kidney injury. Curr Opin Nephrol Hypertens 23: 9–16, 2014. doi: 10.1097/01.mnh.0000436695.29173.de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusch A, Hoff U, Bubalo G, Zhu Y, Fechner M, Schmidt-Ullrich R, Marko L, Müller DN, Schmidt-Ott KM, Gürgen D, Blum M, Schunck WH, Dragun D. Novel signalling mechanisms and targets in renal ischaemia and reperfusion injury. Acta Physiol (Oxf) 208: 25–40, 2013. doi: 10.1111/apha.12089. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651, 2009. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D’Agati VD, Cox GN. Interleukin-11 protects against renal ischemia and reperfusion injury. Am J Physiol Renal Physiol 303: F1216–F1224, 2012. doi: 10.1152/ajprenal.00220.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HT, Xu H, Ota-Setlik A, Emala CW. Oxidant preconditioning protects human proximal tubular cells against lethal oxidant injury via p38 MAPK and heme oxygenase-1. Am J Nephrol 23: 324–333, 2003. doi: 10.1159/000072914. [DOI] [PubMed] [Google Scholar]
- 33.Liu RY, Fan C, Liu G, Olashaw NE, Zuckerman KS. Activation of p38 mitogen-activated protein kinase is required for tumor necrosis factor-α-supported proliferation of leukemia and lymphoma cell lines. J Biol Chem 275: 21086–21093, 2000. doi: 10.1074/jbc.M001281200. [DOI] [PubMed] [Google Scholar]
- 34.Maghsoudi S, Gol A, Dabiri S, Javadi A. Preventive effect of ginger (Zingiber officinale) pretreatment on renal ischemia-reperfusion in rats. Eur Surg Res 46: 45–51, 2011. doi: 10.1159/000321704. [DOI] [PubMed] [Google Scholar]
- 35.Mehta RL, Cerdá J, Burdmann EA, Tonelli M, García-García G, Jha V, Susantitaphong P, Rocco M, Vanholder R, Sever MS, Cruz D, Jaber B, Lameire NH, Lombardi R, Lewington A, Feehally J, Finkelstein F, Levin N, Pannu N, Thomas B, Aronoff-Spencer E, Remuzzi G. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385: 2616–2643, 2015. doi: 10.1016/S0140-6736(15)60126-X. [DOI] [PubMed] [Google Scholar]
- 36.Mishra J, Mori K, Ma Q, Kelly C, Barasch J, Devarajan P. Neutrophil gelatinase-associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. Am J Nephrol 24: 307–315, 2004. doi: 10.1159/000078452. [DOI] [PubMed] [Google Scholar]
- 37.Moon M, Kim HG, Choi JG, Oh H, Lee PK, Ha SK, Kim SY, Park Y, Huh Y, Oh MS. 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochem Biophys Res Commun 449: 8–13, 2014. doi: 10.1016/j.bbrc.2014.04.121. [DOI] [PubMed] [Google Scholar]
- 38.Na JY, Song K, Lee JW, Kim S, Kwon J. Pretreatment of 6-shogaol attenuates oxidative stress and inflammation in middle cerebral artery occlusion-induced mice. Eur J Pharmacol 788: 241–247, 2016. doi: 10.1016/j.ejphar.2016.06.044. [DOI] [PubMed] [Google Scholar]
- 39.Ning W, Song R, Li C, Park E, Mohsenin A, Choi AM, Choi ME. TGF-β1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 283: L1094–L1102, 2002. doi: 10.1152/ajplung.00151.2002. [DOI] [PubMed] [Google Scholar]
- 40.Ohnishi M, Ohshita M, Tamaki H, Marutani Y, Nakayama Y, Akagi M, Miyata M, Maehara S, Hata T, Inoue A. Shogaol but not gingerol has a neuroprotective effect on hemorrhagic brain injury: Contribution of the α, β-unsaturated carbonyl to heme oxygenase-1 expression. Eur J Pharmacol 842: 33–39, 2019. doi: 10.1016/j.ejphar.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 41.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 42.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Human heat shock protein 27-overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol 297: F885–F894, 2009. doi: 10.1152/ajprenal.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 266–280, 2012. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SW, Kim M, Kim M, D’Agati VD, Lee HT. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int 80: 1315–1327, 2011. doi: 10.1038/ki.2011.281. [DOI] [PubMed] [Google Scholar]
- 45.Rahmani AH, Shabrmi FM, Aly SM. Active ingredients of ginger as potential candidates in the prevention and treatment of diseases via modulation of biological activities. Int J Physiol Pathophysiol Pharmacol 6: 125–136, 2014. [PMC free article] [PubMed] [Google Scholar]
- 46.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650, 2006. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 47.Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol 26: 237–245, 2014. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. NF-κB in renal inflammation. J Am Soc Nephrol 21: 1254–1262, 2010. doi: 10.1681/ASN.2010020218. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu H, Takahashi T, Suzuki T, Yamasaki A, Fujiwara T, Odaka Y, Hirakawa M, Fujita H, Akagi R. Protective effect of heme oxygenase induction in ischemic acute renal failure. Crit Care Med 28: 809–817, 2000. doi: 10.1097/00003246-200003000-00033. [DOI] [PubMed] [Google Scholar]
- 50.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 51.Song JH, Kim M, Park SW, Chen SW, Pitson SM, Lee HT. Isoflurane via TGF-β1 release increases caveolae formation and organizes sphingosine kinase signaling in renal proximal tubules. Am J Physiol Renal Physiol 298: F1041–F1050, 2010. doi: 10.1152/ajprenal.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su H, Lei CT, Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol 8: 405, 2017. doi: 10.3389/fimmu.2017.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res 21: 71–85, 2011. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, Kane GC, Terzic A, Griffin MD, Nath KA. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int 72: 1073–1080, 2007. doi: 10.1038/sj.ki.5002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tracz MJ, Juncos JP, Grande JP, Croatt AJ, Ackerman AW, Rajagopalan G, Knutson KL, Badley AD, Griffin MD, Alam J, Nath KA. Renal hemodynamic, inflammatory, and apoptotic responses to lipopolysaccharide in HO-1−/− mice. Am J Pathol 170: 1820–1830, 2007. doi: 10.2353/ajpath.2007.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol 27: 693–733, 2009. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 57.Vasala PA. Ginger. In: Handbook of Herbs and Spices (2nd ed.), edited by Peter KV. Philadelphia, PA: Woodhead, 2012, p. 319–335. [Google Scholar]
- 58.Vinay P, Gougoux A, Lemieux G. Isolation of a pure suspension of rat proximal tubules. Am J Physiol Renal Physiol 241: F403–F411, 1981. doi: 10.1152/ajprenal.1981.241.4.F403. [DOI] [PubMed] [Google Scholar]
- 59.Wan X, Fan L, Hu B, Yang J, Li X, Chen X, Cao C. Small interfering RNA targeting IKKβ prevents renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol 300: F857–F863, 2011. doi: 10.1152/ajprenal.00547.2010. [DOI] [PubMed] [Google Scholar]
- 60.Wang JC, Zhou LH, Zhao HJ, Cai SX. Examination of the protective effect of 6-shogaol against LPS-induced acute lung injury in mice via NF-κB attenuation. Arch Biol Sci 68: 633–639, 2016. doi: 10.2298/ABS151012055W. [DOI] [Google Scholar]
- 61.Zhu Y, Wang P, Zhao Y, Yang C, Clark A, Leung T, Chen X, Sang S. Synthesis, evaluation, and metabolism of novel [6]-shogaol derivatives as potent Nrf2 activators. Free Radic Biol Med 95: 243–254, 2016. doi: 10.1016/j.freeradbiomed.2016.03.026. [DOI] [PubMed] [Google Scholar]