

Abstract
GPCR 37 (GPR37) is a GPCR expressed in the CNS; its physiological and pathophysiological functions are largely unknown. We tested the role of GPR37 in the ischemic brain of GPR37 knockout (KO) mice, exploring the idea that GPR37 might be protective against ischemic damage. In an ischemic stroke model, GPR37 KO mice exhibited increased infarction and cell death compared with wild-type (WT) mice, measured by 2,3,5-triphenyl-2H-tetrazolium chloride and TUNEL staining 24 h after stroke. Moreover, more severe functional deficits were detected in GPR37 KO mice in the adhesive-removal and corner tests. In the peri-infarct region of GPR37 KO mice, there was significantly more apoptotic and autophagic cell death accompanied by caspase-3 activation and attenuated mechanistic target of rapamycin signaling. GPR37 deletion attenuated astrocyte activation and astrogliosis compared with WT stroke controls 24–72 h after stroke. Immunohistochemical staining showed more ionized calcium-binding adapter molecule 1–positive cells in the ischemic cortex of GPR37 KO mice, and RT-PCR identified an enrichment of M1-type microglia or macrophage markers in the GPR37 KO ischemic cortex. Western blotting demonstrated higher levels of inflammatory factors IL-1β, IL-6, monocyte chemoattractant protein, and macrophage inflammatory protein-1α in GPR37-KO mice after ischemia. Thus, GPR37 plays a multifaceted role after stroke, suggesting a novel target for stroke therapy.—McCrary, M. R., Jiang, M. Q., Giddens, M. M., Zhang, J. Y., Owino, S., Wei, Z. Z., Zhong, W., Gu, X., Xin, H., Hall, R. A., Wei, L., Yu, S. P. Protective effects of GPR37 via regulation of inflammation and multiple cell death pathways after ischemic stroke in mice.
Keywords: apoptosis, autophagy, mTOR, microglial, astrogliosis
Stroke is a leading cause of mortality and disability in adult populations around the world, yet few effective treatments for patients with acute stroke exist (1). Following a cerebral ischemic insult, a cascade of events involving multiple molecular and cellular mechanisms leads to neuronal death, suggesting that an effective therapy for stroke will require modulation of different cell death pathways in a variety of cell types (2, 3). Following acute cell death resulting from excitotoxic conditions, secondary damage can occur to surrounding and connected brain regions. Meanwhile, astrocyte activation and gliosis occur after ischemic stroke (4). We have demonstrated that, in wild-type (WT) mice, ischemia provokes significant astrocyte accumulation in the peri-infarct region within 6 h following stroke (3). Glial fibrillary acidic protein (GFAP)–positive astrocytes exhibit pronounced hypertrophy, and astrocyte-mediated gliosis is known to be an acutely regulated process that initially aids in the formation of a barrier around damaged tissue to limit the spread of noxious chemokines as well as excitotoxic and inflammatory factors (4, 5). To this end, a better understanding of the regulation of astrocyte gliosis and other cellular responses to ischemic insults, such as inflammation, programmed cell death, and intracellular signaling cascades, is essential to the development of effective acute and subacute treatments after ischemic strokes.
GPCR, the largest family of membrane receptors, converts information from a wide variety of neurotransmitters, hormones, neuropeptides, and chemokines into cellular and subcellular activities (6–8). Nearly one-third of all marketed drugs act by binding to GPCRs, including treatments for cancer, asthma, schizophrenia, Parkinson’s disease, and heart disease (9–11). GPCR 37 (GPR37, also known as Pael-R) is a GPCR and known substrate of parkin (12). This receptor plays an important role in the pathogenesis of Parkinson’s disease and possibly autism spectrum disorder (10, 13). GPR37 is predominantly expressed in the brain, with reported expression in a variety of regions, including the hippocampus and striatum (14, 15), and particularly high levels in white matter due to its expression in mature oligodendrocytes (16). There is evidence from in vitro studies that GPR37 may exert cytoprotective effects on cultured neurons or glial cells (17, 18). However, potential protective actions and mechanisms of GPR37 in vivo on the outcomes of ischemic stroke have not yet been explored.
Several ligands have been reported for GPR37, although a consensus has not yet been achieved as to whether any or all of these ligands are authentic endogenous agonists. The reported ligands for GPR37 include prosaposin and its active fragment prosaptide (17–19), an invertebrate peptide related to prosaptide known as head activator peptide (20, 21), and the lipid metabolite neuroprotectin D1 (22). Intriguingly, both prosaptide (23–25) and neuroprotectin D1 (26–28) have been reported to exert protective actions in rodent models of nerve injury or stroke. Moreover, a recent study reported that deletion of the GPCR GPR37-like 1 (GPR37L1), a relative of GPR37 that is expressed mainly in astrocytes, results in increased brain damage and cell death likely because of increased extracellular glutamate concentration and NMDA receptor activation (29). However, the various cellular and molecular roles that GPR37 may play following brain injury are unknown. Clarification of these mechanisms may yield a greater understanding of stroke pathophysiology and lead to a treatment strategy for ischemic stroke. Thus, we examined whether GPR37 might exert protective effects in a focal ischemic stroke model of the mouse. As shown below, these studies revealed GPR37-mediated regulation of multiple signaling pathways related to cell survival, astrocyte activation, inflammation, infarct formation, and functional outcomes after ischemic stroke, thereby identifying GPR37 as a novel therapeutic target for ameliorating ischemic brain damage.
MATERIALS AND METHODS
Animal and genotyping of GPR37 expression
GPR37 knockout (KO) (GPR37−/− or GPR37 KO) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) (strain Gpr37tm1Dgen, 005806). The mouse line was backcrossed with WT C57BL/6J mice (The Jackson Laboratory) for 10 generations each to ensure uniformity of genetic background. Genetic deletion of GPR37 was confirmed by DNA sequencing (30). All mice were maintained on a C57BL/6J background and housed in a 12-h light-dark cycle, with food and water available ad libitum. All experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Emory University. The researchers performing experiments and data analysis were blinded to the genotypes of the animals.
Genotyping was performed as previously described by Mohamad et al. (31). DNA for genotyping was extracted from tail snips. The primers used are listed in Table 1. Briefly, DNA (2 µl) was amplified on a thermocycler (MJ Minim, Personal Thermal Cycler, Bio-Rad, Hercules, CA, USA) for 40 cycles (95°C for 60 s, 58°C for 30 s, and 72°C for 60 s). After additional incubation at 72°C for 10 min and being transferred to 4°C, PCR products were subjected to electrophoresis in 1.5% agarose gel with ethidium bromide. Relative intensity of PCR bands was analyzed using the InGenius3 Manual Gel System (Syngene, Bangalore, India).
TABLE 1.
Primers used in genotyping of the WT and GPR37 KO brain
| Primer | Sequence, 5′–3′ |
|---|---|
| GPR37 mutant, forward | GGGTGGGATTAGATAAATGCCTGCTCT |
| GPR37 WT, forward | AACGGGTCTGCAGATGACTGGGTTC |
| Common, reverse | GGCCAAGAGAGAATTGGAGATCGTC |
Focal ischemic stroke model of the mouse
GPR37 KO, WT littermate controls, and C57BL/6J mice (2–3 mo old, male, 25–30 g) were used in this investigation. The sensorimotor cortex ischemic stroke was induced based on previous reports of the barrel cortex stroke, with modified artery occlusion procedures (3). Briefly, animals were subjected to ketamine or xylazine (ketamine 80–100 mg/kg, i.p., xylazine 10–12.5 mg/kg, i.p.) anesthesia, and the right middle cerebral artery supplying the sensorimotor cortex was permanently ligated by a 10-0 suture (Surgical Specialties, Wyomissing, PA, USA). The creation of the right sensorimotor cortex ischemia was completed by bilateral occlusions of the common carotid arteries for 7 min followed by reperfusion. This modified ischemic procedure was suitable and sufficient for the induction of focal ischemia in the mouse brain, resulting in a specific infarct formation in the right sensorimotor cortex. Animal body temperature was monitored during the surgery and recovery periods using a rectal probe and maintained at ±37°C by a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). Animals were kept in a ventilated, humidity-controlled incubator (ThermoCare, Paso Robles, CA, USA). All animals were given 1 dose of meloxicam (oral, 1 mg/kg) prior to surgery and 1 daily dose of meloxicam (1 mg/kg) for 3 d postsurgery. Furthermore, animals were monitored for 60 min following the surgery to ensure recovery from anesthesia as well as daily surveillance poststroke for illnesses and locomotor activity. Before and after surgery, the animals were housed at 5 animals per cage, with ad libitum access to food and water. Among all mice in this study, there was ∼5% rate of mortality. Mice were euthanized with an overdose of isoflurane and decapitated at specified days after stroke. The brains were immediately removed and preserved in optimal cutting temperature compound at −80°C until further processing. The animal protocols were approved by the Institutional Animal Care and Use Committee of Emory University School of Medicine. Animal procedures followed institutional guidelines that meet National Institutes of Health [(NIH), Bethesda, MD, USA] standards.
Infarct volume measurement
Three days after the onset of middle cerebral artery occlusion, animals in different groups were euthanized for assessment of brain infarct formation. 2,3,5-triphenyltetrazolium chloride (TTC; MilliporeSigma, Burlington, MA, USA) staining was used to reveal damaged or dead brain tissue as previously described by Wang et al. (32). Brains were removed and placed in a brain matrix, then sliced into 1-mm coronal sections. Slices were incubated in 2% TTC solution at 37°C for 5 min, imaged, then stored in 10% buffered formalin for 24 h. Digital images of the caudal aspect of each slice were obtained by a flatbed scanner prior to fixation. Infarct, ipsilateral hemisphere, and contralateral hemisphere areas were measured using ImageJ software (NIH). The indirect method (subtraction of residual right hemisphere cortical volume from cortical volume of the intact left hemisphere) was used for infarct volume calculation. Infarct measurements were performed under double-blind conditions.
TUNEL staining and cell death measurements
A TUNEL assay kit was used to examine cell death by detecting fragmented DNA in 10-µm–thick coronal fresh frozen sections as previously described by Lee et al. (33). After fixation [10% buffered formalin for 10 min and then ethanol:acetic acid (2:1) solution for 5 min] and permeabilization (0.2% Triton X-100 solution), brain sections were incubated in equilibration buffer for 10 min. Recombinant terminal deoxynucleotidyl transferase and nucleotide mixture were then added on the slide at 37°C for 60 min in the dark. Reactions were terminated by 2-times saline-sodium citrate solution for 15 min. Nuclei were counterstained with Hoechst 33342 (1:20,000; Molecular Probes, Eugene, OR, USA) for 5 min. Cell counting was performed as previously described by Lee et al. (33). ImageJ was used to analyze each picture. All analyses were performed in a double-blinded fashion.
Western blot analysis
Western blot analysis was used to detect the expression of a variety of cell markers. Brain cortical tissue was lysed in a lysis buffer containing 0.02 M Na4P2O7, 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton, 1 mM EGTA, 2 mM Na3VO4, and a protease inhibitor cocktail (MilliporeSigma). The supernatant was collected after centrifugation at 15,000 g for 10 min at 4°C. Protein concentration was determined with a bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA). Equivalent amounts of total protein were separated by MW on an SDS-polyacrylamide gradient gel, then transferred to a PVDF membrane. The blot was incubated in 5% bovine serum albumin for at least 1 h and then reacted with primary antibodies at 4°C for overnight. The primary antibodies used in this investigation are summarized in the table below (Table 2).
TABLE 2.
Antibodies used in Western blot analysis
| Marker | Dilution | Company |
|---|---|---|
| BCL2 | 1:1000 | Santa Cruz Biotechnology |
| Beclin1 | 1:1000 | Abcam |
| LC3 | 1:500 | MBL International |
| mTOR | 1:1000 | Cell Signaling Technology |
| p-MTOR | 1:1000 | Cell Signaling Technology |
| ERK | 1:1000 | Cell Signaling Technology |
| p-ERK | 1:1000 | Cell Signaling Technology |
| AKT | 1:1000 | Cell Signaling Technology |
| p-AKT | 1:1000 | Cell Signaling Technology |
| ULK1 | 1:1000 | Cell Signaling Technology |
| p-ULK1 | 1:1000 | Cell Signaling Technology |
| REDD1 | 1:1000 | Proteintech |
| p-p70 S6 kinase (Thr389) | 1:1000 | Cell Signaling Technology |
| p-p70 S6 kinase (Ser371) | 1:1000 | Cell Signaling Technology |
| P-4E-BP-1 | 1:1000 | Cell Signaling Technology |
After washing with Tris-buffered saline with Tween, membranes were incubated with alkaline phosphatase–conjugated or horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Waukesha, WI, USA) for 1–2 h at room temperature. After final washing with Tris-buffered saline with Tween, the signals were detected with bromochloroidolylphosphate/nitroblue tetrazolium solution (MilliporeSigma) or film. Signal intensity was measured by ImageJ and normalized to the actin signal intensity.
As a control assessment of the basal expression of apoptotic, autophagic, and inflammatory markers, Western blotting was used to measure and compare these proteins in cortical tissues of normal WT and GPR37 KO mice. As shown in the Supplemental Fig. S1, the baseline expression of the following proteins was similar between these 2 strains: Bcl-2, Beclin1, LC3, mechanistic target of rapamycin (mTOR), phopho (p)-mTOR, protein kinase B (AKT), p-AKT, Unc-51 like autophagy activating kinase-1 (ULK1), and p-UKL1.
Immunohistochemical assessment in brain sections
Frozen brain tissues were sliced into 10-µm–thick coronal sections using a cryostat vibratome (Leica CM 1950; Leica Microsystems, Buffalo Grove, IL, USA). Sections were dehydrated on a slide warmer for 30 min, fixed with 10% formalin buffer, washed with −20°C precooled ethanol:acetic acid (2:1) solution for 10 min, and finally permeabilized with 0.2% Triton X-100 solution for 5 min. All slides were washed 3 times with PBS (5 min each) after each step. Then, tissue sections were blocked with 1% fish gelatin (MilliporeSigma) in PBS for 1 h at room temperature and subsequently incubated with primary antibodies listed in the table below. The next day, the slides were washed 3 times with PBS for 5 min, then reacted with the secondary antibodies Alexa Fluor 488 goat anti-mouse or rabbit (1:300; Thermo Fisher Scientific) and cyanine 3–conjugated donkey anti-rabbit (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or cyanine 3–conjugated donkey anti-mouse or rabbit (1:400; Jackson ImmunoResearch Laboratories) for 80 min at room temperature. After 3 washes with PBS, nuclei were stained with Hoechst 33342 (1:20,000; Molecular Probes) for 5 min as a counterstain. The brain sections were then mounted, cover slipped, and imaged under a fluorescent microscope (BX51; Olympus, Tokyo, Japan) or laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany). The table below lists the reagents and related information in these experiments (Table 3).
TABLE 3.
Cellular markers in immunohistochemical experiments
| Marker | Dilution | Company | Catalog no. |
|---|---|---|---|
| NeuN | 1:400 | Abcam | ab104224 |
| GFAP | 1:400 | Abcam | ab4674 |
| IBA1 | 1:400 | Abcam | ab15690 |
| TUNEL kit | Per kit instructions | Promega | G3250 |
Cell counting in brain sections
Systematic random sampling was used to ensure accurate and nonredundant cell counting. Eight brain sections per animal were collected at 90-µm distances between sections for nonoverlapping multistage random sampling. For each animal, 8 area of interest regions per slide were selected. Each field was scanned at ×200 magnifications for cell counting.
RT-PCR assay
Real-time RT-PCR was used to detect the expression of mRNA. Total RNA was extracted from samples using Trizol (Thermo Fisher Scientific) as previously described by Lee et al. (34). The RNA samples were reverse transcribed to cDNA in 20 μl of a reaction mixture containing 20-times RT buffer and 20-times RT enzyme mix according to the manufacturer’s instruction (Thermo Fisher Scientific) at 37°C for 60 min as previously described by Lee et al. (35). The samples were then incubated at 95°C for 5 min and transferred to 4°C. Next, RT-PCR was carried out using a StepOnePlus System (Thermo Fisher Scientific) and a Sybr Green Kit (Thermo Fisher Scientific) according to the manufacturer protocols. Primers (Integrated DNA Technologies, Coralville, IA, USA) included inducible nitric oxide synthase (iNOS), IL-1β, IL-6, IL-10, YM1, arginase, chemokine ligand 2 (CCL2), CCL3, and TNF-α. The ΔΔCt method was used to compare relative gene expression between samples.
The primers used in this study are listed in Table 4.
TABLE 4.
Primers used in RT-PCR experiments
| Primer, 5′–3′ |
||
|---|---|---|
| Gene | Forward | Reverse |
| iNOS | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
| IL-1 β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| YM1 | CTGGAATTGGTGCCCCTACA | CAAGCATGGTGGTTTTACAGGA |
| Arginase | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| CCL2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| CCL3 | TTCTCTGTACCATGACACTCTGC | CGTGGAATCTTCCGGCTGTAG |
Behavioral tests
Adhesive-removal test for sensorimotor functions
The adhesive-removal test is a sensitive assay for sensorimotor deficits. Prior to the injury, mice were trained to remove a quarter-circle adhesive tape (Genesee Scientific, San Diego, CA, USA) from forelimb paws. The training was repeated 3 times for each paw for 3 d. Two latencies were recorded: latency to contact, which is the time the mouse took to recognize and demonstrate the conviction to remove the adhesive, and latency to removal. The maximum recording period was 180 s. Mice that were unable to rapidly remove the adhesive (<10 s) after the training period were excluded from behavior testing. Following the injury, the adhesive-removal task was performed at specified days after stroke. The latencies were scored with the experimenter blinded to the treatment conditions.
Corner test for sensorimotor function
The corner test monitors unilateral whisker deficits. Mice were allowed to roam freely in a star-shaped arena with 30° angles. Upon entering a corner, the mouse would rear and turn toward the wall, which was sensed by intact whisker sensations. In our right sensorimotor cortex ischemic stroke model, mice lost sensation in their left whiskers. The percentage of right turns was measured before and after stroke.
Statistical analysis
Prism 6 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis and graphic presentation. A 2-tailed Student’s t test was used for comparison of 2 experimental groups. For multiple comparisons, 1-way ANOVA analysis was used, followed by Bonferroni’s correction. For repeated measurements, 2-way ANOVA followed by Bonferroni’s correction was used. Significance was defined at P ≤ 0.05. All data are presented as means ± sem.
RESULTS
GPR37 deficiency exaggerated brain injury, apoptotic cell death, and functional deficits after stroke
A focal ischemic stroke targeting the right sensorimotor cortex was induced in adult male WT and GPR37 KO mice. The ischemia-induced cortical infarct was measured 24 and 72 h after stroke in WT and GPR37 KO mice. The infarct volume measurement of TTC staining showed significantly larger infarction in GPR37 KO mice compared with WT stroke controls (Fig. 1A, B).
Figure 1.

GPR37 deficiency exacerbates brain injury, apoptotic cell death, and functional deficits after stroke. A) Infarct volume was measured 72 h after stroke using TTC staining on brain sections. Representative photographs of TTC staining of brain coronal sections from WT and GPR37 KO mice show a white-colored area corresponding to the ischemic core of the injured cortex. TTC staining at 24 h after stroke showed a similarly enlarged infarction in GPR37 KO mice (not shown). B) Quantification of infarct volume at 72 h based on TTC staining calculated using the indirect method. Damage to the brain was significantly larger in GPR37 KO mice than that in WT mice (unpaired Student’s t test; n = 17 and 18 in WT and GPR37 KO group, respectively). **P < 0.01. C) Cell death in the ischemic cortex was detected using TUNEL staining (green) in brain sections from WT and GPR37 KO mice. Red, NeuN staining of neuronal cells; blue, Hoechst 33342 staining of all cells. D) Quantified data of TUNEL staining 24 h after stroke. GPR37 deficiency significantly increased the number of dead cells after stroke (unpaired Student’s t test; n = 7 animals/group). *P < 0.05 vs. WT stroke controls. E) Representative Western blots with anti–caspase-3 and anti–Bcl-2 antibodies in the peri-infarct cortical tissue. F) Quantified results showing caspase-3 activation during subacute phases (6 and 12 h after stroke). This apoptotic event was significantly higher in GPR37 KO brains than in WT brains (2-way ANOVA; F = 97.97; n = 4/group). *P < 0.05. G) Expression of antiapoptotic Bcl-2 was significantly decreased in GPR37 KO mice after stroke compared with the WT control group (2-way ANOVA; F = 46.98; n = 4/group). *P < 0.05. H–J) Sensorimotor and locomotor functions were tested 3 and 7 d after stroke. The adhesive-removal test revealed that GPR37 KO mice needed prolonged time to contact (H) and remove (I) the sticky dot attached to their affected forepaws [2-way ANOVA; F (2, 53) = 10.34, F (2, 53) = 9.532, respectively] *P < 0.05. In the corner test (J), GPR37 deficiency significantly impaired whisker sensory function after stroke [2-way ANOVA; F (2, 44) = 5.316; n = 7 and 8/group].
To understand how GPR37 might be regulating cell death mechanisms at the cellular level, the number of dead or dying cells were measured using the DNA fragmentation marker TUNEL staining. Consistent with the increased infarct volume in GPR37 KO mice, there were many more TUNEL-positive cells in the ischemic cortex of GPR37 KO mice 24 h after stroke than in WT controls (Fig. 1C, D). Because apoptosis has been implicated in ischemia-induced neuronal cell death (36), we also investigated the role of GPR37 in apoptotic cascades. Western blotting at 6 and 12 h after ischemia revealed the expression of cleaved caspase-3 in the peri-infarct region to be significantly higher in GPR37 KO mice compared with WT controls (Fig. 1E, F). Meanwhile, the antiapoptotic protein Bcl-2 in the peri-infarct region was found to be significantly lower in the GPR37 KO brain (Fig. 1E, G).
We observed exacerbated functional deficits after stroke in both GPR37 KO and WT mice (Fig. 1H–J). In the absence of ischemic insults, GPR37 KO mice do not exhibit striking differences from WT mice in motor tasks (14, 15, 37, 38), but following ischemic insults we found the GPR37 KO mice to exhibit greater deficits in tasks assessing sensorimotor function. In the adhesive-removal test of sensorimotor performance tested at 3 d after stroke, GPR37 KO mice took longer than WT mice to contact and remove the sticky dots on their affected left forepaws (Fig. 1H, I). In the corner test at 7 d after stroke, GPR37 KO mice exhibited greater rightward turning deviation compared with WT controls after stroke (Fig. 1J). These pathologic and functional data demonstrate that deletion of GPR37 resulted in exacerbated brain damage, increased cell death, and more severe functional deficits after focal ischemic stroke.
GPR37 deficiency induced autophagy by inhibiting the mTOR signaling pathway after stroke
Autophagic cell death has been implicated in ischemic stroke (39, 40). In immunohistochemical staining assays, autophagy-associated neuronal cell death was detected in cells triple positive to TUNEL, neuronal nuclei (NeuN), and LC3. Three days after stroke, we observed a trend toward increased numbers of triple-positive cells in the GPR37 KO mice (Supplemental Fig. S2). Given prior reports that GPR37 may regulate autophagy (41), we analyzed autophagic signaling molecules in the peri-infarct region of WT and GPR37 KO mice following ischemia. In Western blot assays, the baseline expression of autophagy factors was not significantly different between sham-treated WT and GPR37 KO mice (Fig. 2A–G). However, the expression of Beclin1, a protein critical for autophagosome formation, increased in the peri-infarct region of GPR37 KO mice at 6 and 12 h after stroke compared with WT controls (Fig. 2A, B). The LC3II-LC3I ratio (or cytosolic LC3 ratio) is regarded as a quantitative index of macroautophagy (42); this ratio also significantly increased in GPR37 KO mice compared with WT mice at 6 and 12 h after stroke (Fig. 2A, C). mTOR plays an inhibitory role in regulating autophagy in response to nutritional status, growth factors, and stress signals (43), and the ratio of phosphorylated mTOR to total mTOR in the peri-infarct region measured at 6 and 12 h after stroke was found to be significantly lower in GPR37 KO mice than that in WT mice (Fig. 2A, D). GPR37 deficiency also led to significantly reduced phosphorylation of a direct substrate of mTOR, 4E-binding protein 1 (phospho-4E-BP1, one of several rapamycin-sensitive sites) measured at 12 h after stroke (Fig. 2A, J). This observation was consistent with other evidence of suppressed mTOR signaling, such as decreased levels of phosphorylated p70 S6 kinase (Thr389) and phosphorylated p70 S6 kinase (Ser371) in the GPR37 KO brain after stroke (Fig. 2H, I). To further assess how GPR37 modulated mTOR activity, we examined the serine or threonine kinase Akt, which is an upstream positive regulator of both mTOR and ERK and known to exhibit extensive crosstalk with these kinases (44). In the peri-infarct region of the GPR37 KO brain, the phosphorylation of Akt was significantly reduced compared with WT controls (Fig. 2A, E), as was the phosphorylation of ERK (Fig. 2A, F). The activity of mTOR is known to prevent autophagy by promoting phosphorylation of the autophagy-initiating kinase ULK1 (45). We observed that phosphorylated ULK1 was much lower in GPR37 mice compared with WT mice (Fig. 2G), which is consistent with reduced phosphorylated mTOR levels and increased autophagy in the absence of GPR37.
Figure 2.

Increased autophagy in GPR37 KO mice after stroke associated with inhibited mTOR signaling. Western blot analysis was performed to measure several autophagy-related genes in the cortices of sham-treated controls and the peri-infarct region at 6 and 12 h after ischemic stroke. A) Representative Western blots with the indicated antibodies. B) Quantified results showed significant increases in relative Beclin-1 expression in GPR37 KO brain vs. WT control at 6 and 12 h after stroke [2-way ANOVA; F (2,9) = 72.63 l; n = 3/group]. *P < 0.05, ***P < 0.001 vs. WT controls, #P < 0.05 vs. sham-treated control. C) Greater increase in the conversion of LC3-I to LC3-II (larger ratio of LC3-II to LC3-1) after stroke was detected in GPR37 KO mice than that in WT control group [2-way ANOVA; F (2,12) = 95.22; n = 3/group]. *P < 0.05, **P < 0.0001, #P < 0.05 vs. sham-treated control. D) Phosphorylation of the autophagy inhibitory mTOR was significantly suppressed in GPR37 KO mice after stroke [2-way ANOVA; F (2,18) = 47.95; n = 3]. *P < 0.05, #P < 0.05 vs. sham-treated control. E) Phosphorylated AKT showed a trend of reduction in GPR37 KO mice after stroke [2-way ANOVA; F (2,9) = 7.346; n = 3]. #P < 0.05 vs. sham-treated control. F) Phosphorylation of ERK1 and 2 was significantly decreased in both WT and GPR37 KO mice at 12 h after stroke [2-way ANOVA; F (2,9) = 34.73; n = 3]. #P < 0.05 vs. sham-treated control. G) Levels of phosphorylated ULK1 were significantly decreased after stroke in GPR37 KO mice compared with WT mice [2-way ANOVA; F (2,9) = 180.7; n = 3/group]. *P < 0.05 compared with WT stroke controls, #P < 0.05 vs. sham-treated control. H, I) Western blotting of REDD1, an inhibitory factor on the mTOR pathway in the sham-treated and ischemic cortex. There was a transient down-regulation of this factor at 24 h after stroke in WT mice, whereas REDD1 expression was significantly enhanced at 72 h after stroke [F (2,9) = 24.18; n = 3/group]. *P < 0.05 vs. WT, #P < 0.05 vs. sham-treated control.
Regulated in development and DNA damage responses 1 (REDD1) is a target gene of hypoxia-inducible factor 1 and plays a crucial role in inhibiting mTOR signaling during hypoxic stress. At 24 h after stroke, REDD1 expression in WT mice was down-regulated, whereas the level in the GPR37 KO brain remained unchanged. Because this down-regulation was unexpected, we continued to monitor the REDD1 expression at 72 h after stroke. At this later time point, REDD1 levels were significantly increased in both WT and GPR37 KO mice, but the increase in GPR37 KO mice was much greater (Fig. 2L). These data support a crucial role of GPR37 in regulating autophagy after ischemic insults through the Akt-mTOR signaling pathway.
GPR37 deficiency perturbs glial scar formation in the peri-infarct cortex
A pathophysiological hallmark of ischemic stroke is astrocyte activation and gliosis, which may help to restrain the ischemic core region during the early stages of an ischemic insult and can benefit outcomes after stroke (46, 47). Because a larger infarct was observed in GPR37 KO mice, we hypothesized that GPR37 may play a regulatory role in astrocyte activation and formation of the glial scar in the peri-infarct region. In WT mice 24–72 h after the ischemic insult, GFAP immunostaining showed an accumulation of reactive astrocytes in the peri-infarct area, with this effect being substantially reduced in the same region of GPR37 KO mice (Fig. 3A–C). Given the interesting difference between WT and KO mice in GFAP staining, we examined a longer time point and found that the difference in astrocyte gliosis between WT and GPR37 KO mice was even greater 7 d after stroke (Fig. 3D). In addition, the morphology of GFAP-positive cells showed marked differences between WT and GPR37 KO mice. In the WT poststroke brain, GFAP-positive cells displayed dense and intensified GFAP fluorescence and ramification of primary processes, whereas these cells in the GPR37 KO brain showed a lesser degree of hypertrophy and exhibited shorter and sparser processes (Fig. 3A). These data suggest that GPR37 plays a key role in astrocyte reactivation and gliosis, which is considered an early protective mechanism after ischemic stroke (46, 47).
Figure 3.
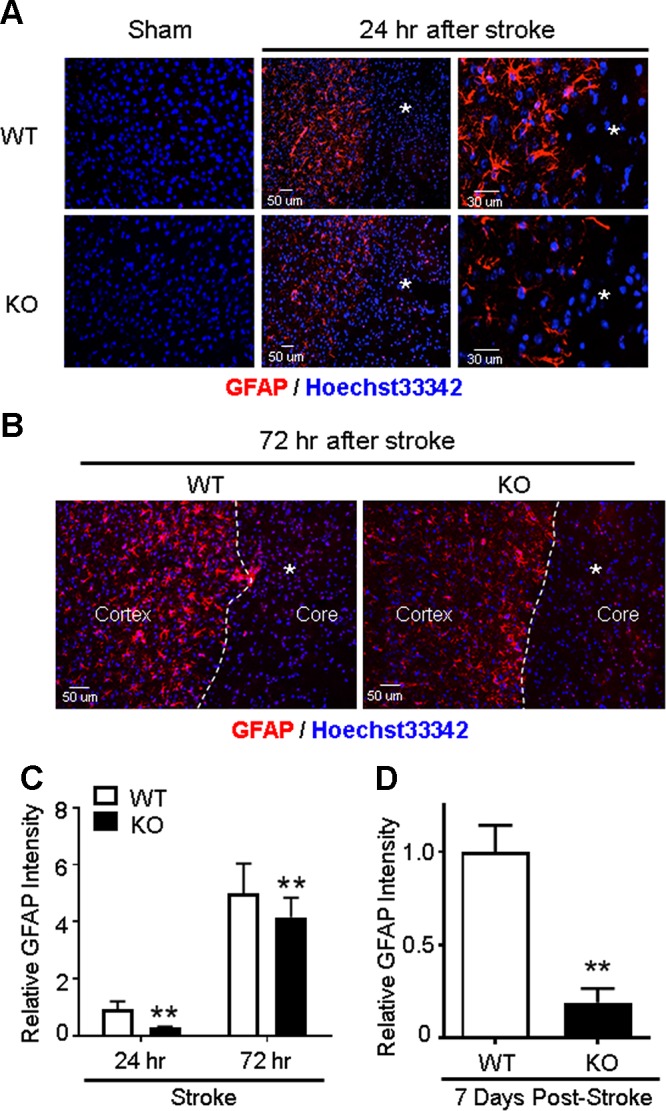
Impaired astrocyte activation and gliosis in GPR37 KO mice after stroke. Immunohistochemical staining measured reactive astrocytes in the ischemic cortex 24 and 72 h after stroke. A) Representative image of GFAP-positive reactive astrocytes in the sham-treated control cortex and the ischemic cortex after stroke. Reactive astrocytes accumulated in the peri-infarct region surrounding the ischemic core (asterisk). These astrocytes showed hypertrophic cell bodies and elongated processes in the WT brain, whereas these morphologic changes associated with astrogliosis were noticeably reduced in GPR37 KO mice. B) Immunostaining images of GFAP-positive astrocytes (red) at 72 h after stroke. There was higher density and greater number of GFAP-positive cells in WT mice next to the ischemic core (asterisk) compared with the GPR37 KO brain. C) Quantification of the GFAP fluorescence at 24 and 72 h after stroke. The increase of fluorescence intensity of reactive astrocytes was significantly less in GPR37 KO mice compared with WT mice (2-way ANOVA; F = 190.4; n = 5). **P < 0.01. D) At 7 d after stroke, we observed dramatically fewer GFAP-positive cells, consistent with the much-weakened glial scar formation at this delayed time point. Mann-Whitney U test nonparametric test was applied for the comparison; n = 5/group. **P = 0.0153 vs. WT stroke controls.
GPR37 deficiency increased microglial activation and M1 phenotype transformation after stroke
Microglial activation and the formation of M1-type microglia contribute to pathologic inflammation after stroke (48). To investigate the effects of GPR37 deficiency in these processes, the expression of microglia and macrophage activation marker ionized calcium-binding adapter molecule 1 (Iba-1) was examined in WT and GPR37 KO mice. Immunohistochemical staining showed that Iba-1–positive microglia in the cortex existed at comparably low levels in normal GPR37 KO and WT animals (Fig. 4A). At 24 h after stroke, although the total number of Iba-1–positive cells increased in both animals, the number of Iba-1–positive cells in the ischemic cortex of GPR37 KO mice was significantly higher than was observed for WT mice (Fig. 4A, B). To evaluate the M1-M2 phenotype polarization, we used RT-PCR to test the mRNA levels of M1- and M2-related genes in the ischemic cortex 24 and 72 h after stroke. The M1-type genes iNOS, IL-1β, and IL-6 were notably increased in GPR37 KO mice compared with WT mice (Fig. 4C–E). TNF-α, which is also used as an M1 marker, was not significantly different (Supplemental Fig. S2). The mRNA for M2-type genes IL-10 and β-N-acetylhexosaminidase Ym1 were not significantly different between the 2 groups (Fig. 4F, G), although there was a trend toward a smaller increase in arginase after stroke in GPR37 KO mice (Fig. 4H). Thus, deficiency of GPR37 may promote the formation of the proinjury M1-type microglia and macrophages after ischemia.
Figure 4.

GPR37 deficiency exacerbates microglial activation and inflammation in the poststroke brain. Immunohistochemical staining and RT-PCR were utilized to examine microglial activation and inflammatory factors in the poststroke brain. A, B) Immunostaining of Iba1-positive cells in brain tissue from WT and GPR37 KO mice. No differences were observed in Iba1-positive cells in uninjured cortex. Ischemic insults increased the numbers of microglia and macrophages (Iba1-positive cells) in the ischemic cortex 24 h after stroke, and even more Iba1-positive cells were seen in GPR37 KO mice. Quantified data are shown in the bar graph [2-way ANOVA; F (1,26) = 33.06; n = 8 independent assays]. **P < 0.001 vs. WT control, #P < 0.05 vs. sham-treated controls. C–E) M1 microglia markers were measured using RT-PCR. Ischemic insults increased the M1 markers iNOS, IL-1β, and IL-6 mRNA in both WT and GPR37 KO brains. Significantly greater increases in iNOS expression occurred in GPR37 KO mice [2-way ANOVA; F (1,23) = 199.3; n = 8 for sham-treated controls; n = 6 for stroke groups (C)] **P < 0.01 vs. WT stroke control, #P < 0.05 vs. sham controls. IL-1β mRNA increased at 24 and 72 h after stroke, with greater increases being seen in GPR37 KO mice at both time points [2-way ANOVA; F (2,8) = 48.89; n = 3 (D)]. *P < 0.05 vs. WT stroke control, #P < 0.05 vs. sham-treated control. Another M1 marker, IL-6, showed a similar expression pattern in the poststroke brain [F (2,6) = 45.51; n = 3 (E)]. **P < 0.01 vs. WT stroke control, #P < 0.05 vs. sham-treated control. F–H) Quantified analysis of mRNA levels showed similar reductions of the M2 cell marker IL-10 in both WT and GPR37 KO brains [2-way ANOVA; F (2,8) = 11.56; n = 3 (F)]. #P < 0.05 vs. sham-treated control. The M2 marker YM1 was not significantly changed after stroke in both strains [2-way ANOVA; n = 3 (G)]. Stroke increased the M2 marker arginase in WT mice but not in GPR37 KO mice [2-way ANOVA; F (1,23) = 4.431; n = 8 (H)]. #P < 0.05 vs. sham control. I, J) Ischemic insults showed time-dependent increases in expression of the inflammatory factors CCL2 and CCL3. At 24 h after stroke, larger increases in CCL2 expression were seen in GPR37 KO mice. These increases subsided at 72 h after stroke [2-way ANOVA; F (2,6) = 47.03 (I)]. ***P < 0.001 vs. WT, #P < 0.05 vs. sham-treateed controls. CCL3 mRNA was enhanced in the ischemic cortex in both WT and GPR37 KO mice, and there was a larger increase in the GPR37 KO cortex compared with WT stroke control at 24 h after stroke [2-way ANOVA; F (2,8) = 11.4; n = 3 (J)]. *P < 0.05 vs. WT stroke control, #P < 0.05 vs. sham-treated controls. Avg, average.
Inflammatory chemokines such as monocyte chemoattractant protein 1 (MCP-1 or CCL2) and macrophage inflammatory protein 1α (MIP-1α or CCL3) were also significantly higher in GPR37 KO mice, especially at the earlier time point of 24 h after stroke (Fig. 4I, J). CCL2 is normally expressed in microglial cells and acts as a chemokine that attracts cells involved in the immune and inflammatory response (49). Thus, the increased CCL2 in GPR37 KO mice may augment inflammation and immune-cell infiltration into the ischemic cortex.
DISCUSSION
The present investigation presents novel evidence for a neuroprotective role of GPR37 mediated by multiple cellular and molecular mechanisms in cerebral ischemia. We show that deletion of GPR37 increases apoptotic and autophagic cell death in vivo, involving caspase activation and reduced phosphorylation or activation of the mTOR pathway and downstream signaling during the early stage of stroke. GPR37 has previously been suggested to regulate autophagy (41), and the data shown here provide evidence that GPR37-mediated modulation of autophagy pathways plays a role in the protective actions of GPR37 following ischemia.
Another important observation made in the studies shown here is that GPR37 appears to be critical for astrocyte activation and glial scar formation in the peri-infarct region. This was evidenced by reduced accumulation of GFAP-positive astrocytes in the peri-infarct area and diminished astrogliosis in the GPR37 KO mice. Astrocyte regulation has been proposed to be a therapeutic target for stroke (50), and glial scar formation acutely after stroke is viewed mainly as a defensive response to cerebral ischemia that restrains expansion of the ischemic core (46, 47). The reaction of astrocytes in the subacute and acute phases of ischemic stroke is thought to be beneficial in some ways and pathogenic in others (51). This is further complicated by a range of factors including the severity of the injury with more severe insults resulting in disrupted homeostatic processes, such as inflammatory reactions of different cytokines and chemokines and microglia activation (47, 52).
The collective functional and morphologic transition of astrocytes, most markedly observed by the increased expression of GFAP in proximity to the site of infarction, demarcates reactive gliosis. Organized and complex interactions with the extracellular matrix form a glial scar that acts as a barrier to both the toxic ischemic environment as well as the invasion of immune cells into the periphery (53). It is likely that the reduced gliosis observed in GPR37 KO mice contributes to the enlarged infarct formation after stroke. GPR37 deficiency also resulted in up-regulation of chemoattractant chemokines that would be expected to attract more inflammatory and immune system cells into the ischemic region, with this augmented inflammatory microenvironment in the absence of GPR37 favoring M1 microglia transformation after stroke. These cellular and molecular events are likely the underlying mechanisms for the enlarged infarct volume that we observed in GPR37 KO mice after stroke. Consistent with the increased brain damage, GPR37 KO mice displayed more severe functional deficits correlating to the damage in the sensorimotor cortex after stroke. Taken together, these data implicate GPR37 as a potential therapeutic target for the treatment of ischemic stroke.
Overexpression of GPR37 has been shown to be cytotoxic, most likely due to protein misfolding (10). On the other hand, in vitro studies suggest that endogenously expressed GPR37 plays a cytoprotective role following cellular insults (17, 18). Moreover, deletion of GPR37 was found to result in greater demyelination in vivo following treatment of mice with cuprizone (54). The data shown in the present study are consistent with the idea that GPR37 plays a predominantly protective role against ischemic insults in vivo. Regarding the cell types in which GPR37 plays this protective role in the brain, GPR37 is known to be expressed in oligodendrocytes, astrocytes, and certain populations of neurons (10), so further work will be required to determine the cell types in which GPR37 must be expressed to mediate the protective effects against ischemia that were observed in the present work.
Several potential ligands for GPR37 have been reported, including prosaposin and its active fragment prosaptide (17–19), an invertebrate peptide called head activator that is structurally similar to prosaptide (20, 21), and the bioactive lipid neuroprotectin-1 (22). There is not yet a broad consensus as to whether any or all of these ligands represent authentic endogenous ligands for GPR37, and further studies in vivo will be necessary to address this important question. Nonetheless, it is intriguing to note that both prosaptide (23–25) and neuroprotectin D1 (26–28) have been shown to be protective against ischemia. Thus, the data shown here revealing that deletion of GPR37 results in greater damage in a mouse model of ischemia is consistent with a role for prosaptide or neuroprotectin D1 as modulators or agonists of GPR37.
Several groups have previously examined phenotypes of GPR37 KO or null mice (14–16, 30, 37, 38, 54–57). In the earliest studies on these mice, it was shown that GPR37 KO mice are viable, with normal brain development and anatomy. The mice were found to exhibit modestly reduced striatal dopamine content, reduced spontaneous locomotion in open field test, and impairment in motor coordination of rotarod test (37). The decreased locomotor activity with GPR37 KO, however, was not observed in a recent investigation (15). Our data demonstrate that, although their baseline performance was similar, GPR37 KO mice exhibited exacerbated functional deficits after stroke compared with WT mice. This is consistent with the observation that, without the stress from the ischemic insult, the levels of several survival and inflammatory factors were not significantly different between GPR37 KO and WT mice. The most striking differences between GPR37 KO and WT mice were observed in the postischemic brain, suggesting that GPR37 is an important mediator of protection against injurious insults.
A recent paper (29) described a neuroprotective effect of the GPR37 relative GPR37L1, as evidenced by a regulatory role on astrocyte glutamate transporters, reduced NMDA receptor current, and increased neuronal death after ischemic stroke in GPR37L1 KO mice (29). These receptors share 42% identity and are both widely expressed in the brain (58, 59). However, their patterns of distribution are distinct because the highest levels of GPR37L1 expression are in astrocytes and certain specialized cell types such as the Bergmann glia of the cerebellum (58). The high degree of homology between GPR37 and GPR37L1 suggests that they might share the same ligands, and moreover, KO of either receptor was found to result in increased seizure susceptibility, with genetic deletion of both receptors resulting in an even more dramatic increase in vulnerability to seizures (30). Given our findings that deletion of GPR37 increases damage induced by ischemia and the similar findings recently made for mice lacking GPR37L1 (29), it will be interesting in the future to explore whether loss of both these receptors together might result in even more profound susceptibility to ischemia-induced damage.
The mechanisms by which GPR37 might protect against apoptotic cell death have not previously been explored. In the current study, we found that the expression of proapoptotic protein Bax and caspase-3 cleavage were increased in GPR37 KO mice, whereas the expression of the antiapoptotic protein Bcl-2 significantly decreased. Both apoptosis and autophagy were increased in GPR37 KO mice. When autophagy occurs in excess, it can become cytotoxic and eventually lead to autophagic cell death (39, 60). Inhibition of autophagy has been shown to be beneficial for the outcome after CNS injury (61). In the present work, we observed an increased conversion of endogenous LC3-I to LC3-II and inhibition of the classic autophagic PI3K-AKT-mTOR signaling pathway in the poststroke GPR37 KO brain. LC3-II is implicated in autophagosome fusion with lysosomes during macroautophagy (43). The decrease in mTOR in GPR37 KO mice precedes or coincides with increases in markers of autophagy, such as pS317-ULK-1, Beclin-1, and the LC3-II:LC3-I ratio. This may indicate the induction of autophagy in neurons destined to die after infarction. The mTOR pathway is intimately involved in regulation of autophagy (62). In response to ischemia, phosphorylation of mTOR is significantly reduced in GPR37 KO mice, and the brake normally imposed on autophagy is released through phosphorylation of downstream substrates such as ULK1 or 2 and Atg13. Decreased phosphorylation of ULK-1 enables association with AMPK, which in turn activates Beclin-1 and promotes lipidation of LC3-I to generate LC3-II (63). It is possible that excessive autophagic flux contributed to increased neuronal damage, although we cannot rule out the reciprocal relationship (i.e., increased damage leading to a greater amount of autophagy). Furthermore, Parkin, for which GPR37 is a substrate, is known to regulate mitophagy (64). Mitophagy refers to a subset of autophagy implicated in recycling damaged mitochondria. Although not tested in the present study, an alternate hypothesis is that reduction in GPR37 may allow for aberrant Parkin-dependent increases in mitophagy. Excessive mitophagy following ischemia may also play a role in the increased autophagic cell death seen in GPR37-deficient mice. These data implicate GPR37 as a potential regulator of mTOR signaling and suggest the possibility that the lack of GPR37 activity may promote autophagic cell death after stroke.
Increasing evidence supports the idea that postischemic inflammatory responses play a detrimental role in the progression of stroke injury. Experimentally and clinically, stroke is followed by acute and prolonged inflammatory responses characterized by the production of inflammatory cytokines, leukocytes, and monocytes. Additionally, infiltration of inflammatory cells into the brain may contribute to ischemic brain injury. These cellular events collaboratively contribute to ischemic brain injury. In the present study, immunofluorescent staining for the microglia and macrophage marker Iba-1 and the astrocyte marker GFAP around the boundary of the infarction area provide several interesting insights about the potential role of GPR37. The activation of microglia cells in the absence of GPR37 leads to conditions favoring the M1 phenotype, which promotes inflammation in the poststroke brain. Concurrently, we noted significant increases in the levels of IL-1β, TNF-α, and IL-6, suggesting a coordinated proinflammatory response in the GPR37 KO brain.
In summary, our work illustrates novel and crucial roles for GPR37 in the regulation of mTOR signaling in neuronal cell death involving apoptosis and autophagy, the inflammatory responses to ischemia, and the glial scar formation in the poststroke brain. GPR37 may be a viable target to prevent expansion of infarct formation and inflammation during the early stages after ischemic stroke. Additional studies are needed to identify possible interactions between neurons and glial cells affected by GPR37 under pathologic conditions acutely as well as during chronic phases after stroke.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants NS085568 (to L.W. and S.P.Y.), NS091585 (to L.W.), NS088413 (to R.A.H.), and NS073378 (to S.P.Y.), a Veterans Affairs National Merit Grant RX000666 (to S.P.Y.), an American Heart Association Postdoctoral Fellowship POST25710112 (to Z.Z.W.), and a Predoctoral Fellowship PRE25710020 (to J.Z.). The authors declare no conflicts of interest.
Glossary
- AKT
protein kinase B
- CCL
chemokine ligand
- GFAP
glial fibrillary acidic protein
- GPR37
GPCR 37
- GPR37L1
GPR37-like 1
- Iba-1
ionized calcium-binding adapter molecule 1
- iNOS
inducible nitric oxide synthase
- KO
knockout
- mTOR
mechanistic target of rapamycin
- NeuN
neuronal nuclei
- REDD1
regulated in development and DNA damage responses 1
- TTC
2,3,5-triphenyl-2H-tetrazolium chloride
- ULK1
Unc-51 like autophagy activating kinase-1
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
M. R. McCrary, M. Q. Jiang, R. A. Hall, L. Wei, and S. P. Yu designed research; M. R. McCrary, M. Q. Jiang, M. M. Giddens, S. Owino, Z. Z. Wei, X. Gu, H. Xin, and L. Wei performed research; R. A. Hall, L. Wei, and S. P. Yu contributed new reagents or analytic tools; M. R. McCrary, M. Q. Jiang, M. M. Giddens, Z. Z. Wei, X. Gu, H. Xin, R. A. Hall, L. Wei, and S. P. Yu analyzed data; M. R. McCrary, M. Q. Jiang, and S. P. Yu wrote the manuscript; and M. M. Giddens, S. Owino, R. A. Hall, and S. P. Yu proof reading, editing, and corrections.
REFERENCES
- 1.Prabhakaran S., Ruff I., Bernstein R. A. (2015) Acute stroke intervention: a systematic review. JAMA 313, 1451–1462 [DOI] [PubMed] [Google Scholar]
- 2.Lee J. H., Zhang J., Yu S. P. (2017) Neuroprotective mechanisms and translational potential of therapeutic hypothermia in the treatment of ischemic stroke. Neural Regen. Res. 12, 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang M. Q., Zhao Y. Y., Cao W., Wei Z. Z., Gu X., Wei L., Yu S. P. (2017) Long-term survival and regeneration of neuronal and vasculature cells inside the core region after ischemic stroke in adult mice. Brain Pathol. 27, 480–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pekny M., Nilsson M. (2005) Astrocyte activation and reactive gliosis. Glia 50, 427–434 [DOI] [PubMed] [Google Scholar]
- 5.Sims N. R., Yew W. P. (2017) Reactive astrogliosis in stroke: contributions of astrocytes to recovery of neurological function. Neurochem. Int. 107, 88–103 [DOI] [PubMed] [Google Scholar]
- 6.Huang Y., Thathiah A. (2015) Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 589, 1607–1619 [DOI] [PubMed] [Google Scholar]
- 7.Sladek C. D., Song Z. (2012) Diverse roles of G-protein coupled receptors in the regulation of neurohypophyseal hormone secretion. J. Neuroendocrinol. 24, 554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.López de Maturana R., Sánchez-Pernaute R. (2010) Regulation of corticostriatal synaptic plasticity by G protein-coupled receptors. CNS Neurol. Disord. Drug Targets 9, 601–615 [DOI] [PubMed] [Google Scholar]
- 9.Eglen R. M., Bosse R., Reisine T. (2007) Emerging concepts of guanine nucleotide-binding protein-coupled receptor (GPCR) function and implications for high throughput screening. Assay Drug Dev. Technol. 5, 425–451 [DOI] [PubMed] [Google Scholar]
- 10.Smith N. J. (2015) Drug discovery opportunities at the endothelin B receptor-related orphan G protein-coupled receptors, GPR37 and GPR37L1. Front. Pharmacol. 6, 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alavi M. S., Shamsizadeh A., Azhdari-Zarmehri H., Roohbakhsh A. (2018) Orphan G protein-coupled receptors: the role in CNS disorders. Biomed. Pharmacother. 98, 222–232 [DOI] [PubMed] [Google Scholar]
- 12.Imai Y., Soda M., Inoue H., Hattori N., Mizuno Y., Takahashi R. (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902 [DOI] [PubMed] [Google Scholar]
- 13.Fujita-Jimbo E., Yu Z. L., Li H., Yamagata T., Mori M., Momoi T., Momoi M. Y. (2012) Mutation in Parkinson disease-associated, G-protein-coupled receptor 37 (GPR37/PaelR) is related to autism spectrum disorder. PLoS One 7, e51155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marazziti D., Mandillo S., Di Pietro C., Golini E., Matteoni R., Tocchini-Valentini G. P. (2007) GPR37 associates with the dopamine transporter to modulate dopamine uptake and behavioral responses to dopaminergic drugs. Proc. Natl. Acad. Sci. USA 104, 9846–9851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morató X., Luján R., López-Cano M., Gandía J., Stagljar I., Watanabe M., Cunha R. A., Fernández-Dueñas V., Ciruela F. (2017) The Parkinson’s disease-associated GPR37 receptor interacts with striatal adenosine A2A receptor controlling its cell surface expression and function in vivo. Sci. Rep. 7, 9452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H. J., Vainshtein A., Maik-Rachline G., Peles E. (2016) G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat. Commun. 7, 10884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer R. C., Giddens M. M., Schaefer S. A., Hall R. A. (2013) GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc. Natl. Acad. Sci. USA 110, 9529–9534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundius E. G., Vukojevic V., Hertz E., Stroth N., Cederlund A., Hiraiwa M., Terenius L., Svenningsson P. (2014) GPR37 protein trafficking to the plasma membrane regulated by prosaposin and GM1 gangliosides promotes cell viability. J. Biol. Chem. 289, 4660–4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu B., Mosienko V., Vaccari Cardoso B., Prokudina D., Huentelman M., Teschemacher A. G., Kasparov S. (2018) Glio- and neuro-protection by prosaposin is mediated by orphan G-protein coupled receptors GPR37L1 and GPR37. Glia 66, 2414–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rezgaoui M., Süsens U., Ignatov A., Gelderblom M., Glassmeier G., Franke I., Urny J., Imai Y., Takahashi R., Schaller H. C. (2006) The neuropeptide head activator is a high-affinity ligand for the orphan G-protein-coupled receptor GPR37. J. Cell Sci. 119, 542–549 [DOI] [PubMed] [Google Scholar]
- 21.Gandía J., Fernández-Dueñas V., Morató X., Caltabiano G., González-Muñiz R., Pardo L., Stagljar I., Ciruela F. (2013) The Parkinson’s disease-associated GPR37 receptor-mediated cytotoxicity is controlled by its intracellular cysteine-rich domain. J. Neurochem. 125, 362–372 [DOI] [PubMed] [Google Scholar]
- 22.Bang S., Xie Y. K., Zhang Z. J., Wang Z., Xu Z. Z., Ji R. R. (2018) GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Invest. 128, 3568–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotani Y., Matsuda S., Sakanaka M., Kondoh K., Ueno S., Sano A. (1996) Prosaposin facilitates sciatic nerve regeneration in vivo. J. Neurochem. 66, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 24.Igase K., Tanaka J., Kumon Y., Zhang B., Sadamoto Y., Maeda N., Sakaki S., Sakanaka M. (1999) An 18-mer peptide fragment of prosaposin ameliorates place navigation disability, cortical infarction, and retrograde thalamic degeneration in rats with focal cerebral ischemia. J. Cereb. Blood Flow Metab. 19, 298–306 [DOI] [PubMed] [Google Scholar]
- 25.Lu A. G., Otero D. A., Hiraiwa M., O’Brien J. S. (2000) Neuroprotective effect of retro-inverso Prosaptide D5 on focal cerebral ischemia in rat. Neuroreport 11, 1791–1794 [DOI] [PubMed] [Google Scholar]
- 26.Bazan N. G., Eady T. N., Khoutorova L., Atkins K. D., Hong S., Lu Y., Zhang C., Jun B., Obenaus A., Fredman G., Zhu M., Winkler J. W., Petasis N. A., Serhan C. N., Belayev L. (2012) Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp. Neurol. 236, 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao C., Zhang J., Chen F., Lin Y. (2013) Neuroprotectin D1 attenuates brain damage induced by transient middle cerebral artery occlusion in rats through TRPC6/CREB pathways. Mol. Med. Rep. 8, 543–550 [DOI] [PubMed] [Google Scholar]
- 28.Belayev L., Mukherjee P. K., Balaszczuk V., Calandria J. M., Obenaus A., Khoutorova L., Hong S. H., Bazan N. G. (2017) Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ. 24, 1091–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolly S., Bazargani N., Quiroga A. C., Pringle N. P., Attwell D., Richardson W. D., Li H. (2018) G protein-coupled receptor 37-like 1 modulates astrocyte glutamate transporters and neuronal NMDA receptors and is neuroprotective in ischemia. Glia 66, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giddens M. M., Wong J. C., Schroeder J. P., Farrow E. G., Smith B. M., Owino S., Soden S. E., Meyer R. C., Saunders C., LePichon J. B., Weinshenker D., Escayg A., Hall R. A. (2017) GPR37L1 modulates seizure susceptibility: evidence from mouse studies and analyses of a human GPR37L1 variant. Neurobiol. Dis. 106, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohamad O., Song M., Wei L., Yu S. P. (2013) Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J. Physiol. 591, 149–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L. L., Chen D., Lee J., Gu X., Alaaeddine G., Li J., Wei L., Yu S. P. (2014) Mobilization of endogenous bone marrow derived endothelial progenitor cells and therapeutic potential of parathyroid hormone after ischemic stroke in mice. PLoS One 9, e87284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J. H., Wei L., Gu X., Wei Z., Dix T. A., Yu S. P. (2014) Therapeutic effects of pharmacologically induced hypothermia against traumatic brain injury in mice. J. Neurotrauma 31, 1417–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J. H., Wei Z. Z., Cao W., Won S., Gu X., Winter M., Dix T. A., Wei L., Yu S. P. (2016) Regulation of therapeutic hypothermia on inflammatory cytokines, microglia polarization, migration and functional recovery after ischemic stroke in mice. Neurobiol. Dis. 96, 248–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J. H., Zhang J. Y., Wei Z. Z., Yu S. P. (2018) Impaired social behaviors and minimized oxytocin signaling of the adult mice deficient in the N-methyl-d-aspartate receptor GluN3A subunit. Exp. Neurol. 305, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marazziti D., Golini E., Mandillo S., Magrelli A., Witke W., Matteoni R., Tocchini-Valentini G. P. (2004) Altered dopamine signaling and MPTP resistance in mice lacking the Parkinson’s disease-associated GPR37/parkin-associated endothelin-like receptor. Proc. Natl. Acad. Sci. USA 101, 10189–10194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai Y., Inoue H., Kataoka A., Hua-Qin W., Masuda M., Ikeda T., Tsukita K., Soda M., Kodama T., Fuwa T., Honda Y., Kaneko S., Matsumoto S., Wakamatsu K., Ito S., Miura M., Aosaki T., Itohara S., Takahashi R. (2007) Pael receptor is involved in dopamine metabolism in the nigrostriatal system. Neurosci. Res. 59, 413–425 [DOI] [PubMed] [Google Scholar]
- 39.Tomita H., Ziegler M. E., Kim H. B., Evans S. J., Choudary P. V., Li J. Z., Meng F., Dai M., Myers R. M., Neal C. R., Speed T. P., Barchas J. D., Schatzberg A. F., Watson S. J., Akil H., Jones E. G., Bunney W. E., Vawter M. P. (2013) G protein-linked signaling pathways in bipolar and major depressive disorders. Front. Genet. 4, 297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang P., Shao B. Z., Deng Z., Chen S., Yue Z., Miao C. Y. (2018) Autophagy in ischemic stroke. Prog. Neurobiol. 163-164, 98–117 [DOI] [PubMed] [Google Scholar]
- 41.Marazziti D., Di Pietro C., Golini E., Mandillo S., Matteoni R., Tocchini-Valentini G. P. (2009) Induction of macroautophagy by overexpression of the Parkinson’s disease-associated GPR37 receptor. FASEB J. 23, 1978–1987 [DOI] [PubMed] [Google Scholar]
- 42.Kadowaki M., Karim M. R. (2009) Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 452, 199–213 [DOI] [PubMed] [Google Scholar]
- 43.Heras-Sandoval D., Pérez-Rojas J. M., Hernández-Damián J., Pedraza-Chaverri J. (2014) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 26, 2694–2701 [DOI] [PubMed] [Google Scholar]
- 44.Mendoza M. C., Er E. E., Blenis J. (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36, 320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J., Kundu M., Viollet B., Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayakawa K., Nakano T., Irie K., Higuchi S., Fujioka M., Orito K., Iwasaki K., Jin G., Lo E. H., Mishima K., Fujiwara M. (2010) Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 30, 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choudhury G. R., Ding S. (2016) Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 85, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girard S., Brough D., Lopez-Castejon G., Giles J., Rothwell N. J., Allan S. M. (2013) Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia 61, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selenica M. L., Alvarez J. A., Nash K. R., Lee D. C., Cao C., Lin X., Reid P., Mouton P. R., Morgan D., Gordon M. N. (2013) Diverse activation of microglia by chemokine (C-C motif) ligand 2 overexpression in brain. J. Neuroinflammation 10, 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niu B., Zhang T., Hu H., Cao B. (2017) Transcriptome sequencing reveals astrocytes as a therapeutic target in heat-stroke. Neurosci. Bull. 33, 627–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pekny M., Pekna M., Messing A., Steinhäuser C., Lee J. M., Parpura V., Hol E. M., Sofroniew M. V., Verkhratsky A. (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345 [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg P. A., Aizenman E. (1989) Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 103, 162–168 [DOI] [PubMed] [Google Scholar]
- 53.Barreto G. E., Sun X., Xu L., Giffard R. G. (2011) Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One 6, e27881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith B. M., Giddens M. M., Neil J., Owino S., Nguyen T. T., Duong D., Li F., Hall R. A. (2017) Mice lacking Gpr37 exhibit decreased expression of the myelin-associated glycoprotein MAG and increased susceptibility to demyelination. Neuroscience 358, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mandillo S., Golini E., Marazziti D., Di Pietro C., Matteoni R., Tocchini-Valentini G. P. (2013) Mice lacking the Parkinson’s related GPR37/PAEL receptor show non-motor behavioral phenotypes: age and gender effect. Genes Brain Behav. 12, 465–477 [DOI] [PubMed] [Google Scholar]
- 56.Lopes J. P., Morató X., Souza C., Pinhal C., Machado N. J., Canas P. M., Silva H. B., Stagljar I., Gandía J., Fernández-Dueñas V., Luján R., Cunha R. A., Ciruela F. (2015) The role of parkinson’s disease-associated receptor GPR37 in the hippocampus: functional interplay with the adenosinergic system. J. Neurochem. 134, 135–146 [DOI] [PubMed] [Google Scholar]
- 57.Rial D., Morató X., Real J. I., Gonçalves F. Q., Stagljar I., Pereira F. C., Fernández-Dueñas V., Cunha R. A., Ciruela F. (2017) Parkinson’s disease-associated GPR37 receptor regulates cocaine-mediated synaptic depression in corticostriatal synapses. Neurosci. Lett. 638, 162–166 [DOI] [PubMed] [Google Scholar]
- 58.Valdenaire O., Giller T., Breu V., Ardati A., Schweizer A., Richards J. G. (1998) A new family of orphan G protein-coupled receptors predominantly expressed in the brain. FEBS Lett. 424, 193–196 [DOI] [PubMed] [Google Scholar]
- 59.Donohue P. J., Shapira H., Mantey S. A., Hampton L. L., Jensen R. T., Battey J. F. (1998) A human gene encodes a putative G protein-coupled receptor highly expressed in the central nervous system. Brain Res. Mol. Brain Res. 54, 152–160 [DOI] [PubMed] [Google Scholar]
- 60.Choi K. E., Hall C. L., Sun J. M., Wei L., Mohamad O., Dix T. A., Yu S. P. (2012) A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J. 26, 2799–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin C. W., Chen B., Huang K. L., Dai Y. S., Teng H. L. (2016) Inhibition of autophagy by estradiol promotes locomotor recovery after spinal cord injury in rats. Neurosci. Bull. 32, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei H., Li Y., Han S., Liu S., Zhang N., Zhao L., Li S., Li J. (2016) cPKCγ-modulated autophagy in neurons alleviates ischemic injury in brain of mice with ischemic stroke through Akt-mTOR pathway. Transl. Stroke Res. 7, 497–511 [DOI] [PubMed] [Google Scholar]
- 63.Lee E. J., Tournier C. (2011) The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 7, 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.