

Abstract
Mesenchymal stromal cells (MSCs) have emerged as candidate cells with therapeutic potential to treat different pathologies. The underlying mechanism is paracrine signaling. The cells secrete proteins that can impact inflammation, apoptosis, angiogenesis, and cell proliferation. All are important in wound healing and tissue regeneration. Although the bone marrow has been the most widely used source of MSCs, umbilical cord tissue (CT) presents a source that is just starting to be used in the clinic, yet can be obtained with more ease and easily stored. Here, we characterize CT‐MSCs obtained from multiple donors by analyzing cell surface proteins, differentiation capacity, and proteome profile. Analysis of low, medium, and high passage cells indicates that the morphology and proliferation rate stay constant and with the exception of cluster of differentiation (CD) 105 at late passage, there are no changes in the cell surface protein characteristics, indicating the population does not change with passage. TNF‐stimulated gene 6 protein was measured in a subset of samples and variable expression was observed, but this did not impact the ability of the cells to enhance skin regeneration. In conclusion, CT‐MSC represents a consistent, easily accessible source of cells for cell therapy. stem cells translational medicine 2019;8:1041–1054
Keywords: Umbilical cord, Tissue regeneration, Stromal cells, Somatic cell therapy
Significance Statement.
Mesenchymal stromal cells (MSCs) from umbilical cords are becoming an important source of cells for cell therapy. Donor variation is a major concern for adult bone marrow donors of these cells. The variation has been attributed to the age of the donor, with older donors producing less‐effective cells. This study demonstrates that MSCs from full‐term umbilical cord demonstrate no variation between donors due to their being from newborns, thus mitigating any age‐related variation. In conclusion, MSCs from umbilical cords present a consistent and reliable source of cells for cell‐based therapies.
Introduction
In the 1960s and 1970s, Freidenstein et al. conducted a set of experiments that identified bone marrow (BM) stromal and osteogenic stem cells. These experiments established and solidified the existence of a nonhematopoietic cell population in the BM that supported the process of hematopoiesis, were able to differentiate to bone and also form colonies derived from single cells in tissue culture 1. In the 1990s, Arnold Caplan derived the term “mesenchymal stem cells,” and this nomenclature is frequently used in the scientific literature today 2. We will use the alternative terminology to describe mesenchymal stromal cells (MSCs), replacing “stem” with “stromal.” The reasoning being that no clonal studies have been done that demonstrated the presence of a multipotent stem cell. The population of cells depending on the method of isolation and culture can contain fibroblasts, muscle precursor cells, and endothelial precursor cells, thus giving a false indication of the presence of a stem cell. The population as a whole is a complex mix of cells that secrete a wide range of factors. Donor age seems to impact the secreted protein profile and proliferation ability of BM‐MSCs. Here, we investigate if cells from full‐term birth donors will reduce the variability observed with adult donors.
MSCs are of interest to the regenerative medicine field due to their ability to enhance tissue repair. They have been shown to act through paracrine signaling, whereby the cells respond to the injury and then secrete factors that enhance tissue repair, or through engraftment and differentiation into blood vessels, smooth muscle, fibroblasts, and osteogenic lineages. Due to these properties, MSCs have been used in a wide range of clinical trials to treat diabetic wounds, peripheral vascular disease, and osteoarthritis, to just name a few 3.
Due to of the wide range of tissue sources and the different methods used to isolate MSCs, the Mesenchymal Tissue and Stem Cell Committee of the International Society for Cellular Therapy (ISCT) has proposed a set of minimal criteria by which to phenotypically identify MSCs: the cells must adhere to plastic under standard culture conditions and must express CD105, CD73, CD44, and CD90, and lack expression of CD45, CD34, CD14 or CD11b, CD79a or CD19, and Human Leukocyte Antigen‐DR (HLA‐DR). Furthermore, the MSCs should be able to differentiate into osteocytes, adipocytes, and chondrocytes in vitro 4, 5.
Adult sources of MSCs are commonly isolated from the BM; however, this procedure is invasive and so alternative sources of MSCs have been investigated, such as the umbilical cord. MSCs isolated from the BM have a low cell yield, and the quality and quantity varies with the age of the donor 6. Studies have suggested that cell populations undergo changes when obtained from donors of advanced age, as the expression of surface makers CD44, CD90, CD105, and Stro‐1 changes with age 7. Furthermore, there is only a small frequency of MSCs present within the BM, with reports suggesting that ∼0.001%–0.01% of cells isolated are MSCs 8 and the frequency of MSCs declines with age, from 1/10,000 nucleated marrow cells in a newborn to approximately 1/1,000,000 nucleated marrow cells in an 80‐year‐old 9. This has also been shown for adipose tissue‐derived MSCs 10.
Due to the strong paracrine signaling effect of cord tissue (CT)‐MSCs, the secretome and surface protein profile is of interest 11, 12. Cytokines and chemokines have been shown to affect migration, proliferation, extracellular matrix (ECM) remodeling, and cell survival 13. Other proteins such as hemeoxygenase 1, cyclooxygenase 2, Interluekin (IL)1 receptor antagonist, and Transforming Growth Factor (TGF)‐β1 are correlative with the regenerative capacity of the cells 14, 15. Recent data have suggested that TNF‐stimulated gene 6 protein (TSG‐6) could be used as an informative biomarker to predict the in vivo efficacy of BM‐MSCs. TSG‐6 is a 35 kDa protein expressed in most BM‐MSCs. A study done by Lee et al. showed that there was a wide variation in the efficacy of human MSCs obtained from BM aspirates of healthy individuals, and the ability of MSCs to suppress inflammation in vivo was significantly positively correlated with TSG‐6 expression 16.
Here, we report that CT‐MSCs from multiple donors demonstrated little variation of their growth characteristics and phenotype (n = 110). Furthermore, analysis of 20 MSC lines over 10 passages revealed remarkable consistency. Analysis of TSG‐6 mRNA revealed differences in expression between different donor samples but unlike BM‐MSCs, this did not affect the regenerative ability of the CT‐MSCs as both low and high TSG‐6 expressing MSCs lines were able to accelerate healing in a diabetic wound model equally. Taken together, our results indicate that CT‐MSCs are a consistent, reliable, and cost‐effective source of MSCs for therapeutic use.
Materials and Methods
Umbilical Cord Collection and Preparation
Ethics approval was obtained from Mt. Sinai Hospital to obtain umbilical CT. Umbilical CT (n = 71) was obtained from full‐term, vaginal, and caesarean, deliveries from across Canada. Information pertinent to collection of the cord was recorded: the mother's age, the type of birth, baby gender, and weight was collected on 20 births. The majority of samples were vaginal birth and a maternal age range of 21–40 years old (Table 1). The data were used to determine if there were any confounding factors related to the establishment of an MSC line.
Table 1.
Personal data collected for each cord tissue sample, including maternal age, type of birth, gender of newborn, as well as weight of newborn (g).
| Cord tissue sample | Age of mother | Type of birth | Gender | Weight of baby (g) |
|---|---|---|---|---|
| CT 16 | 27 | Vaginal | Male | 3,164 |
| CT 17 | 35 | Vaginal | Female | 3,840 |
| CT 18 | 21 | Vaginal | Female | N/A |
| CT 19 | 39 | Vaginal | Female | N/A |
| CT 20 | 31 | Vaginal | Female | N/A |
| CT 21 | 34 | Vaginal | Male | 3,101 |
| CT 22 | 31 | Vaginal | Male | 2,795 |
| CT 23 | 33 | Vaginal | Female | 4,015 |
| CT 24 | 38 | Vaginal | Female | 2,380 |
| CT 25 | 41 | N/A | N/A | N/A |
| CT 26 | 34 | Vaginal | Male | 2,965 |
| CT 27 | 34 | Caesarian | Female | 3,528 |
| CT 28 | 30 | Caesarian | Female | 3,359 |
| CT 29 | 30 | Vaginal | Male | 3,294 |
| CT 30 | 31 | Vaginal | N/A | 3,355 |
| CT 31 | 34 | Vaginal | N/A | 2,930 |
| CT 32 | 29 | Vaginal | Female | 6,700 |
| CT 33 | 30 | Vaginal | Female | N/A |
| CT 34 | 35 | Caesarian | Female | 3,355 |
| CT 35 | 40 | Vaginal | Male | N/A |
TSG‐6 levels do not correlate with maternal age or type of birth.
Abbreviation: N/A, not available.
CT (approximately 4 cm long) was sprayed with 70% ethanol, and gauze was used to wipe down the entire length of the cord until the exterior surface was clean. The CT was transferred onto a petri dish and a scalpel was used to cutoff the end portions of the tissue (∼0.5 cm), which were discarded. The arteries and vessels were flushed with phosphate‐buffered saline (PBS) using an 18 gauge needle and 20 cc syringe to remove any blood clots. Five ringlets, each approximately 6 mm in width, were cut from each tissue, and washed twice with 10 ml of PBS then cut into four pieces, yielding 20 pieces per donor.
For freezing, all four pieces per ringlet were frozen together in one 2 ml cryovial and cryopreserved using a cryosolution (10% Dimethyl sulfoxide [Sigma, Mississauga, Ontario, Canada, Lot #RNBD5041], 90% fetal bovine serum [FBS]). Cryovials were placed in an isopropanyl buffered container for controlled freezing at a rate of −1°C/minute to −80°C. Frozen cords were then transferred to liquid nitrogen for long‐term storage.
MSCs Isolation
Pieces of frozen CT were thawed in a 37°C water bath for 2 minutes and then submerged in 10 ml of medium (complete α‐Minimal Essential Medium (MEM): α‐MEM with 10% fetal bovine serum, 1× antibiotic–antimycotic [Life Technologies, Burlington, Ontario, Canada, Lot #1631429]) to complete the thawing process and to wash away the cryopreservant. Tissue pieces were then evenly plated in a single well of a 6‐well polystyrene dish (Falcon, Ottawa, Ontario, Canada) and cultured in complete α‐MEM media. Medium was changed every 2 days. When cells reached 80% confluence (∼120,000 cells per cm2), the cells were passaged using 0.25% trypsin–EDTA solution and the whole contents of the plate were transferred on 10‐cm plates (Falcon, Ottawa, Ontario, Canada), labeled as P1 at 3,000 cells per cm2. At this point, tissue pieces were moved to a new well in a six‐well polystyrene dish, labeled as “P1ii.” The cells from P1ii were passaged when confluence was reached.
Cell Culture
For each subsequent passage, cells were plated at a density of ∼3,000 cells per cm2, in complete α‐MEM. Cultures of cells were maintained in an incubator at 37°C with 5% CO2. Complete medium change was done every 2 days. Once the cell cultures were 80%–90% confluent (∼120,000 cells per cm2 at day 4), cells were replated at ∼3,000 cells per cm2 and cultured in complete α‐MEM for expansion.
Flow Cytometry Analysis
Cell surface antigen expression was analyzed by flow cytometry for the following passages. p2 representing two population doublings (n = 40), p5 representing 10 population doublings (n = 20), and p10 representing 20 population doublings (n = 20) were harvested by treatment with 0.25% trypsin–EDTA, washed using 10 ml of PBS (Mg−/Ca−), and resuspended in antibody staining buffer (PBS Mg−/Ca− with 1% fetal bovine serum) at a concentration of 1 × 107 cells per milliliter. One hundred microliters of prepared cell suspension was aliquoted into a total of nine tubes. Cells were incubated with 2 μl of IgG from mouse serum (Sigma, Mississauga, Ontario, Canada) in the dark for 10 minutes. Cells were then stained using the human MSC analysis kit (BD Biosciences, Mississauga, Ontario, Canada) with the appropriate antibodies: expected positive: FITC CD90, PerCP‐Cy5.5 CD105, and Allophycocyanin CD73, Phycoerythrin (PE) CD44 and expected negative: PE CD45, PE CD34, PE CD11b, PE CD19, and PE HLA‐DR. After incubation for 30 minutes on ice in the dark, cells were washed twice with stain buffer and centrifuged at 400g for 2 minutes. Afterward, cells were resuspended in 300 μl of stain buffer and 0.5 μl of DAPI was added to each tube. Antibody binding was analyzed using a Beckman Coulter flow cytometer. Multicolour fluorescent beads (Flow‐Set Pro Fluorospheres, Beckman Coulter Ireland, Inc., Lot #3125121) were used for instrument standardization and reproducibility of each experiment, where the fluorescent of the beads in each channel was adjusted to match the fluorescence values from previous experiments. All plots were generated using Kaluza Flow Analysis Software (Beckman Coulter). Debris and auto‐fluorescence were removed by using forward scatter (FS) and side scatter (SS). Two light scatter parameters, FS and SS, were used to ensure a stringent gating of single cells. A dot plot depicting side scatter time of flight (SS‐TOF) versus SS peak was first used to gate single cells. Aggregates, which escaped the single cell gate could be seen as the few events which were high in FS‐TOF signal in the second dot plot. The 488 nm blue laser detected both scatter parameters. DAPI was then used to discriminate between live, apoptotic, and dead cell populations. A gate was used to select DAPI negative (live cells) on a FL9: DAPI versus FS peak dot plot. The maximum number of events that was used for each analysis was 15,000 cells. Isotype controls were used to identify positive and negative cell populations, ensuring that the observed staining is due to antibody binding to surface epitopes and to exclude nonspecific binding to Fc receptors. Compensation controls were done for each fluorochrome and this was used to subtract the spectral overlap of specific fluorochromes.
Adipocyte Differentiation
Cells were plated at 5,000 cells per cm2 in α‐MEM supplemented with 5% FBS and after 1 day the media was changed to the StemPro Adipogenic Media (ThermoFisher, Ottawa, Ontario, Canada, catalog no. A1007001). A total of 50% media change was done every other day for 14 days. Staining with Oil Red O stain: media was removed and the cells were washed 2× with PBS (Mg−/Ca−) and fixed in formalin at Reverse Transcriptase (RT) for 30 minutes then washed 2× with PBS (Mg−/Ca−). A total of 60% isopropanol was added to each well for 5 minutes and then removed. Five hundred microliters of adipocyte staining solution (StemPro Kit‐ThermoFisher, catalog no. A1007001) was added and placed on rocker for 30 minutes, followed by washing 4× with PBS (−/−). Stained cells were imaged by brightfield microscopy.
Osteocyte Differentiation
Cells were plated at 5,000 cells per cm2 and after 1 day, media was changed to the StemPro Osteogenic Media (ThermoFisher, catalog no. A1007201). Fifty percentage of media change was done every other day for 21 days.
Staining with 2% alizarin red solution: media was removed and the cells were washed 2× with PBS (Mg−/Ca−), and fixed in formalin at RT for 30 minutes, then washed 2× with PBS (Mg−/Ca−). Two percentage of alizarin red solution was added to cover the cells and incubated on a rocking platform for 30 minutes, washed 2× PBS (Mg−/Ca−), and imaged by brightfield microscopy.
RT‐q Polymerase Chain Reaction
Fifteen MSC samples (passage 4) were chosen from cryopreserved cells. RNA was extracted from each sample (Qiagen). RNA concentration was quantified using a Nanodrop Spectrophotometer and RNA integrity determined at using the Agilent RNA ScreenTape Assay (Agilent Technologies, Canada). cDNA synthesis was performed using reverse transcriptase (Superscript III Reverse Transcriptase; Invitrogen, Canada) and quantified using a Nanodrop Spectrophotometer. Real‐time polymerase chain reaction (PCR) analysis was performed in triplicate for human TSG‐6 and human glyceraldehyde‐3‐phosphate dehydrogenase for all samples (primer sequences in Supporting Information Table S1). SYBR Green PCR MasterMix (Invitrogen) was used for gene amplification and analyzed using the ABI Prism 7900HT sequence detection system (Invitrogen). Data were analyzed with the Sequence Detection Software 2.1 (Life Technologies, Burlington, Ontario, Canada) using the relative standard curve method. Normalized data are expressed as mean ± SEM.
Cytokine Analysis
The cytokine and growth factor expression profile of three umbilical cord samples were compared using a proteome assay (R&D Systems, Minneapolis, MN). Passage five cells were analyzed. Duplicate wells were produced for each cord sample. The supernatant was conditioned for 48 hours prior to the assay. All reagents and the protocol outline were conducted according to the manufacturer's instructions using the RayBiotech, Human Cytokine Array C1000: AAH‐CYT‐1000 (R&D Systems). This antibody array kit analyzed 56 different cytokines and chemokines in duplicate on each membrane. Briefly, the conditioned medium was collected and centrifuged at 290g for 5 minutes. The array membranes were incubated for 1 hour in blocking buffer. Membranes were then incubated with the prepared sample, containing cell supernatant with a detection antibody cocktail, overnight in a cold room (2°C–4°C) on a rocking platform. Membranes were then washed for 10 minutes on a rocking platform. This was repeated three times, after which diluted streptavidin‐Horse Radish Peroxidase was added. The membranes were incubated for 30 minutes at RT on a rocking platform. Afterward, membranes were washed three times for 10 minutes each followed by incubation for 1 minute with a Chemi Reagent Mix. Membranes were exposed to x‐ray film (Denville Scientific Inc., Metuchen, NJ), for 2, 5, and 10 minutes. Pixel density was measured using the (ImageJ, University of Wisconsin, Madison, WI) program. The concentrations of specific proteins were compared with the reference spots. Data analysis: pixel density was determined using (ImageJ, University of Wisconsin, Madison, WI). The pixel intensity of each cytokine value was determined by converting each image to grayscale. Each protein pixel density was calculated by subtracting the measured background pixel density from the measured protein pixel density, after which the net protein value was divided by the net loading control value. The four pixel density measurements (duplicate reads on two independent membranes) for each protein level from duplicate membranes were averaged. The standard error of the mean is shown in the graph. Protein networks were determined using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v.10.
Excisional Wound Mouse Model
Ten‐week‐old db/db C57BL/6 mice were purchased from The Jackson Laboratories (Bar Harbor, ME) (Stock #000642) and acclimatized in the facility for 2 weeks. db/db C57BL/6 mice were used because of their impaired wound healing. On the day of the surgeries, the back of each mouse was shaved with an electric clipper followed by the application of Nair, to ensure that all hair was removed. The surgery site was wiped clean with an alcohol swab. An isoflurane gas chamber was used to induce anesthesia in each mouse, at 1%–4% isoflurane. Each mouse was maintained at a 1%–2% isoflurane level during the surgery. Mice were also given a subcutaneous injection of buprenorphine Temgesic (0.15 mg/kg). A silicone splint was attached to the back of each mouse using an adhesive and four nylon sutures were used to ensure that the splint was fixed to the back of each mouse. A sterile 6‐mm biopsy punch was used to induce a full thickness wound extending through the panniculus carnosis, centered in the middle of the splint, on the back of each mouse. Three million CT‐MSCs, resuspended in 40 μl of PBS (Mg−/Ca−), were applied topically on each wound, followed by 20 μl of a fibrin sealant. For control mice, 20 μl of α‐MEM media was applied to the wound bed, followed by 20 μl of a fibrin sealant. A clear dressing (Tagaderm, St. Paul, MN) was placed over the entire wound area. Animals were transferred to a clean cage and placed under a warming lamp for 10 minutes to recover from the procedure. The mice were house in the animal facility at Lunenfeld Tanenbaum Research Institute.
Wound Analysis
Digital pictures of the wounds were taken with a Nikon camera at day 0, 3, 7, 10, and 14 after surgery. The wound area was analyzed by measuring the area of splint (the inner diameter only) and the area of the wound (by delineating the margins of the wound) on each respective day, using (ImageJ, University of Wisconsin, Madison, WI) software. The ratio of the percent of remaining wound relative to the splint was calculated. The final wound area was calculated as a percent of the original wound (denoted as 100%), created on day 0.
Tissue Collection and Fixation
An isoflurane gas chamber was used to induce anesthesia in each mouse, at a 1%–4% isoflurane concentration, for approximately 1 minute. Mice were euthanized by cervical dislocation. Tagaderm dressing was removed, and the wound area, including the splint was excised, cut in half, and fixed in 10% formalin for 120 minutes at 4°C. Tissues were washed in 1× PBS (−/−) and stored in 70% ethanol for 48 hours. The tissues were then dehydrated in a graded ethanol series (80% for 30 minutes, 95% for 45 minutes, and twice in 100% ethanol for 60 minutes). The tissues were then cleared in toluene two times for 60 minutes, immersed in paraffin at 65°C, and embedded in paraffin blocks. The embedded tissues were sectioned on a microtome and cut into 5 μm sections, placed on Fisherbrand Superfrost Plus (Fisher Scientific, Ottawa, Ontario, Canada) microscope slides, and allowed to dry overnight.
Tissue Staining
Tissue staining was done on paraformaldehyde fixed tissue sections. All samples were embedded in paraffin and underwent routine histological processing. Sections were deparaffinized in xylene and rehydrated using an ethanol gradient on a Leica staining machine. Sections were stained with hematoxylin and eosin (H&E). A wound was considered entirely healed if the wound bed was completely filled in with new tissue, spanning the area of the whole wound.
Masson's Trichrome Stain
Slides were stained as per standard protocols: nuclei—black, muscle, RBCs, fibrin—red, and collagen—green 17.
Sirius Red Staining
Slides were stained as per standard protocols. Collagen, reticulin, basement membrane—red, large fibers—yellow or orange birefringence, thin fibers—green birefringence, nuclei—black, and elastin, muscle, cytoplasm—yellow 18.
Statistical Analysis
Mouse sample size was chosen prior to the start of this study based on preliminary studies to determine adequate numbers required to reach statistical significance. Sample sizes are noted in the results section and figure legends. The data were subjected to a one‐way analysis of variance using the Prism Graph Pad program. This test compares paired groups with multiple members over time, and any mice that were not measured at every time point were excluded from the analysis. Total mice included per group per test are indicated in the figure legends. A probability (p) value <.05 was considered significant. Test used is indicated in the text. Data obtained from flow cytometry to determine the presence of surface proteins on the MSCs were analyzed using a Student's t test.
Results
MSCs Isolation Efficiency and MSCs Characteristics
Our original cohort of donor samples was n = 110, but 39 of these samples were used to establish a standard protocol for CT collection, transportation, and processing for freezing. We chose a nonenzymatic method in order to reduce the use collagenase therefore reducing the expense, time of preparation of the cord (stripping out the vessels and tying the ends together), and possible problems with clinical approval. With a focus on the nonenzymatic method, we determined that an ideal protocol with less manipulation of the CT would be advantageous and we wanted to determine if we could obtain cells from frozen tissues. Preparing cells from fresh tissue then freezing them for later use has a much higher upfront costs for the tissue banks compared with storing frozen tissue and only isolating cells when required. Although public banks are not currently storing CT or CT cells this should change as clinical trials using CT‐MSCs are increasing. Thus, keeping the cost down will make CT‐MSCs storage feasible for public and family banks. The method of finely chopping fresh CT and plating the cells for outgrowth presented us with two main questions. (a) Can this be successfully applied to frozen tissue? (b) How large can a tissue fragment be to be frozen successfully and still allow for the outgrowth of MSCs? Increasing the size of the tissue fragment would mean less chopping, which means less labor and therefore less cost. There was no precedent for the experiment. We ran experiments on 39 donor samples to establish the optimized protocol prior to the 71 reported in this article.
We expected that with thick pieces of tissue, only a portion of the tissue would freeze properly. We wanted to determine whether enough of the tissue would survive to yield cells in an explant culture. We tested CT sections of 4 mm, 6 mm, and 8 mm thickness. We froze the whole section as described in the article. After 7 days, the tissue was thawed, fixed, embedded, and sectioned for histology. We found that the 8 mm thick sections had necrotic centers but the 4 mm and 6 mm sections did not. We then thawed 4 mm and 6 mm sections in explant cultures. Keeping with the idea of minimal manipulation, we chose to cut each tissue section into just four pieces and plate each one into a separate well of a 6‐well plate.
The data for these initial 39 samples were similar to the data reported here for the 71 samples processed in our final protocol format. Cells were observed migrating from the piece of tissue at various days. Outgrowth was defined as five or more cells. The time to confluence in the well of the 6‐well plate for the first plating only also varied with each sample. However, after the first passage, the proliferation rate of each sample was surprisingly consistent. The piece of tissue would not be removed until confluence was reached. We found that we could move the piece of tissue to a fresh well and more cells would migrate out. We only tested this to three transfers. The finalized protocol consisted of cutting whole cords into 6 mm sections and freezing in cryopreservant for 7 days minimum before thawing, dividing the tissue section into four pieces and plating, as described in the Materials and Methods. These preliminary experiments (n = 39) provided the basis to run another 71 samples that are reported on here.
The next 71 samples were all collected and processed using our newly established Standard Operating Procedure (SOP). The protocol that resulted from this study included transporting the CT in a closed specimen container without liquid and processing the CT without the use of enzymatic digestion within 24 hours of collection. The tissues was then frozen and after a minimum of 7 days in liquid nitrogen, thawed, and the cells were isolated and characterized as described in the Materials and Methods section. All donor samples collected and processed this way resulted in viable and proliferating cells that were further characterized. Cell outgrowth appeared 5–38 days (average 14.5 days) after the initial plating for 100% of donors (n = 71; Table 2). Cells that grew out of the tissue were heterogeneous in size, had a spindle appearance, and had a typical MSC appearance. The cells migrated away from the tissue in a scattered formation. As the cells proliferated and became confluent, they aligned to form a parallel, organized sheet of cells (Fig. 1A). For all samples, the cells were not passaged until the 35 mm well was 80% confluent (∼120,000 cells per cm2; designated as passage 1). At this point, all samples exhibited similar proliferation characteristics, reaching confluence every 4 days after being plated 1:4 to keep an initial plating density of ∼3,000 cells per cm2 from passage 1 on. This rate of cell proliferation remained consistent for all donors followed through to P10 (n = 20).
Table 2.
Average time to cell outgrowth. Variation in first cell outgrowth ranges from 5 days to 38 days, with an average of 14.5 days.
| CT no. | Day first passage | CT no. | Day first passage | CT no. | Day first passage | CT no. | Day first passage |
|---|---|---|---|---|---|---|---|
| 1 | 8 | 21 | 22 | 41 | 5 | 61 | 38 |
| 2 | 14 | 22 | 22 | 42 | 17 | 62 | 17 |
| 3 | 8 | 23 | 16 | 43 | 38 | 63 | 17 |
| 4 | 10 | 24 | 15 | 44 | 17 | 64 | 7 |
| 5 | 14 | 25 | 14 | 45 | 20 | 65 | 7 |
| 6 | 10 | 26 | 27 | 46 | 5 | 66 | 7 |
| 7 | 14 | 27 | 16 | 47 | 5 | 67 | 7 |
| 8 | 10 | 28 | 16 | 48 | 20 | 68 | 7 |
| 9 | 8 | 29 | 14 | 49 | 8 | 69 | 7 |
| 10 | 10 | 30 | 16 | 50 | 20 | 70 | 7 |
| 11 | 35 | 31 | 14 | 51 | 5 | 71 | 7 |
| 12 | 10 | 32 | 16 | 52 | 5 | Average | 14.5 |
| 13 | 8 | 33 | 16 | 53 | 27 | ||
| 14 | 15 | 34 | 17 | 54 | 20 | ||
| 15 | 27 | 35 | 14 | 55 | 27 | ||
| 16 | 8 | 36 | 16 | 56 | 5 | ||
| 17 | 14 | 37 | 5 | 57 | 27 | ||
| 18 | 17 | 38 | 38 | 58 | 5 | ||
| 19 | 16 | 39 | 5 | 59 | 5 | ||
| 20 | 14 | 40 | 5 | 60 | 27 |
Bold entries highlight the sample number.
Figure 1.
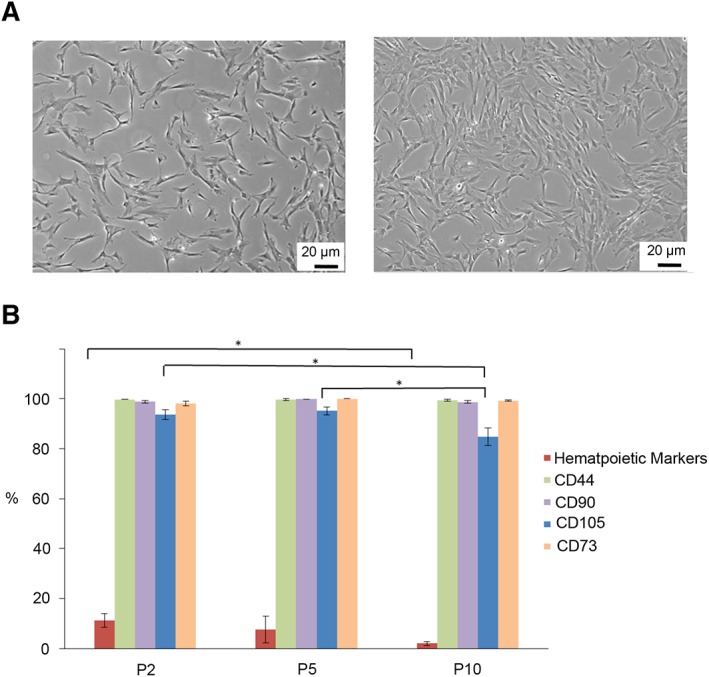
Immunophenotypic analysis of cord tissue (CT)‐mesenchymal stromal cells (MSCs). MSCs from multiple donors were analyzed at three different passages: passage 2 (n = 40), passage 5 (n = 20), and passage 10 (n = 20) to determine their MSC profile. (A): The cells migrate from the piece of CT and have the typical MSCs shape. As the cells reach confluence, they become aligned and elongated. (B): The percentage of cells expressing CD73, CD44, and CD90 remained consistent throughout the 10 passages, whereas the percentage of cells expressing CD105 declined after passage 5. Statistically significant differences are denoted by * (p < .05).
Immunophenotypic cell surface analysis of MSCs obtained from explant culture was done using flow cytometry at passage 2 (n = 40), passage 5 (n = 20), and passage 10 (n = 20; Supporting Information Fig. S1). All samples analyzed illustrated similar phenotypic cell surface expression profiles. We observed that all samples contained hematopoietic cells at the earliest passage analyzed (P2). The number of these cells decreased with passage indicating that the culture media selected for MSCs growth only (Fig. 1B).
Cell passaging did not affect MSCs marker expression, with the exception of CD105. There was no difference in the profiles of CD44 with 99.7% ± 0.059% positive cells at p2, 99.5% ± 0.374% at p5, and 99.2% ± 0.372% at p10. The same trend was observed for CD90+ cells; p2 = 99.7% ± 0.0787%, p5 = 99.8% ± 0.05029%, and p10 = 99.0% ± 0.568% and for CD73+ cells; p2 = 99.7% ± 0.0579%, p5 = 99.5% ± 0.0146%, and p10 = 99.2% ± 0.3415%. CD105 (Endoglin) expression decreased after passage 5. The intensity of CD105 expression of the positive cells remained high, but the frequency of cells expressing CD105 was reduced from p2 (95.4% ± 0.845%) to p5 (94.3% ± 0.435%) to p10 (84.7% ± 3.525%; p < .05).
A subset of CT‐MSCs was chosen randomly and tested for their ability to differentiate into adipose and osteoblasts. All samples tested (n = 7) differentiated into both cell types with equal efficiency (Fig. 2).
Figure 2.
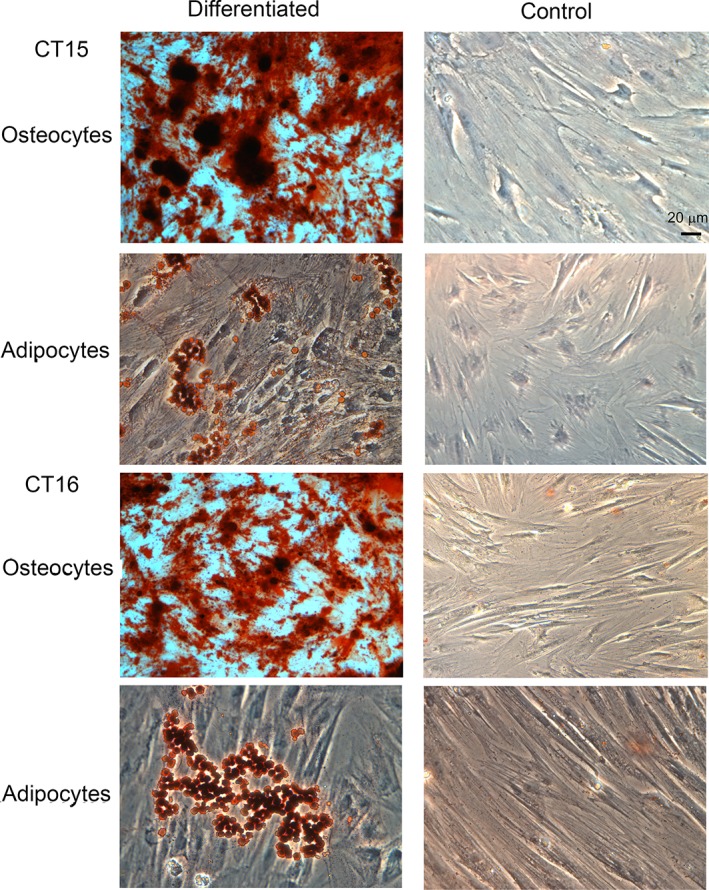
Cord tissue (CT)‐mesenchymal stromal cells (MSCs) have the capacity to differentiate to osteocytes and adipocytes. MSCs from seven donors were tested for their ability to differentiate into osteocytes and adipocytes. Two representative donors are displayed (CT15 and CT16). Both osteocytes and adipocytes could be obtained from all samples tested. Control cells were maintained in medium supplemented with serum for the same length of time. No spontaneous differentiation was detected (scale bar = 20 μm).
Proteome Profile
Several recent studies have suggested that a significant correlation was found between the gene expression of TSG‐6 and the therapeutic quality of BM‐MSCs. BM‐MSCs that express high levels of TSG‐6 mRNA show better efficacy 16. We examined the levels of TSG‐6 mRNA in a subset of CT‐MSCs samples (n = 15) all at the same passage, using RT‐qPCR. All of the samples analyzed had an RNA integrity numbers greater than 9.3. The expression levels of TSG‐6 mRNA varied widely among the samples indicating donor‐to‐donor variation (Fig. 3).
Figure 3.
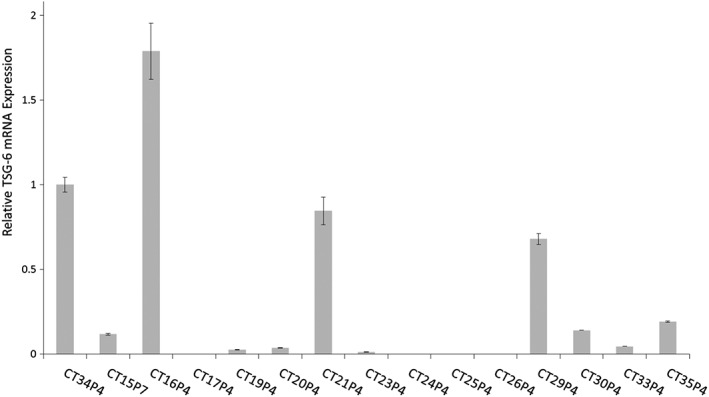
TNF‐stimulated gene 6 protein (TSG‐6) levels. TSG‐6 mRNA levels were tested in 15 cord tissue (CT)‐mesenchymal stromal cell samples and a wide range in expression levels were observed. CT34 was designated as 1 and all other samples are measured in relation to CT34. All samples were done in triplicate and CT15 was done twice in different experimental runs to check for consistency of the PCR assay.
We also examined whether there was a correlation between the TSG‐6 expression levels, the cytokine secretion profiles, and wound healing capabilities. A comparison was done between three cord samples with variations in TSG‐6 expression: CT15, CT16, and CT24. CT15 and CT24 both showed low TSG‐6 mRNA expression, and CT16 showed high TSG‐6 mRNA expression (Fig. 3). A cytokine antibody array kit was used to analyze the presence of 56 proteins in the conditioned medium of the three samples. Each array was done in duplicate and the results of those experiments were plotted (Fig. 4A–4C). The mean pixel density obtained for each cytokine from the control sample (media, no cells) was averaged and the value (0.022) was used to determine the lower threshold for cytokines expression (Fig. 4D). The results show that the cytokines profiles were very similar among the three different CT‐MSC donors with a few differences noted. ANG‐1, Hepatocyte Growth Factor (HGF), Granulocyte Macrophage Colony Stimulating factor (GM‐CSF), and Platelet Derived Growth Factor‐AA (PDGF‐AA) varied between the different donors. All four proteins have a role in wound healing. ANG‐1 was expressed at a higher level in only one of the low TSG‐6 expressing lines (CT15). HGF expression was high in the high TSG‐6 expressing line only. GM‐CSF was highly expressed in both the high TSG‐6 expressing line and one of the low TSG‐6 expressing lines. PDGF‐AA was highly expressed only in one of the low TSG‐6 lines. STRING analysis demonstrated that the 22 highest expressing proteins for all three CT‐MSC lines form a network that is enriched in pathways that have a role in tissue regeneration. Cell locomotion (IL8, Chemokine Ligand 2), migration (chemokine (C‐X‐C motif) ligand 16), angiogenesis (Vascular endothelial growth factor), and ECM protein remodeling and signaling (tissue inhibitor of metalloproteinases, Endoglin) were highly enriched functional pathways (Fig. 5).
Figure 4.
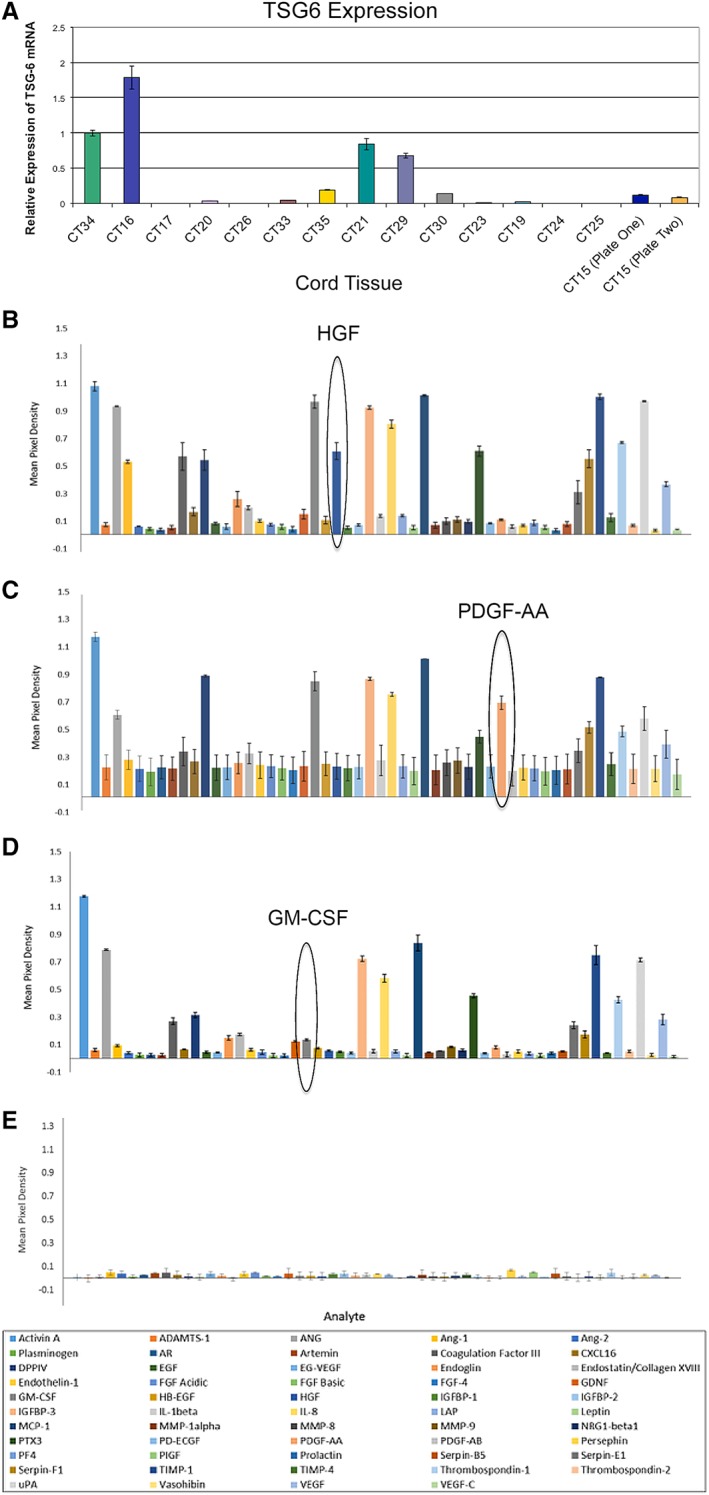
Cytokine profiles of low and high TNF‐stimulated gene 6 protein expressing lines. (A): CT15, (B) CT16, and (C) CT24 were analyzed for cytokine secretion. (D): Control profile consists of cell culture media not exposed to cells. No detectable cytokines were observed. CT15 and CT24 are low expressers of TSG6, whereas CT16 is a high expresser. Cytokine profiles were similar for most of the cytokines tested with the notable exception of ANG‐1, HGF, PDGF‐AA, and GM‐CSF.
Figure 5.

Proteins expressed by mesenchymal stromal cells (MSCs) are specific to tissue regeneration. Search Tool for the Retrieval of Interacting Genes/Proteins analysis was used to determine the biological pathways enhanced in MSCs. Twenty‐two highly expressed proteins from CT15, CT16, and CT24 proteome arrays revealed an enrichment for proteins involved in cell movement and migration, extracellular matrix protein signaling, and angiogenesis.
To further elucidate a basis for the range of TSG‐6 expression observed in the 15 samples, we compared TSG‐6 levels with maternal age and newborn weight to determine if these parameters were impacting TSG6 expression (Supporting Information Fig. S2). Maternal age and baby's weight had no impact on TSG‐6 levels.
Enhanced Diabetic Wound Healing with CT‐MSCs
We used a diabetic skin wound model to compare the wound healing efficiencies between a low TSG‐6 expresser (CT15) and a high TSG‐6 expresser (CT16), to determine if CT‐MSC TSG‐6 levels correlated with the therapeutic ability as observed with BM‐MSCs. Both lines were tested at P8. We used a murine excisional wound healing model with a splint, which closely mirrors human wound healing. Mouse skin heals mainly by contraction, due to a subcutaneous panniculosis carnosus muscle layer and human skin heals by granulation tissue formation and re‐epithelization 19. A full thickness wound, which effectively removes the panniculosis carnosis layer, is generated on the back of diabetic mice. Splints were then centered and fixed on the skin using an adhesive and four sutures. Five mice were used per group including a control group of mice administered only fibrin and media. Wound closure was measured on day 0, 3, 7, 10, and 14. Upon analysis, the low TSG‐6 expressing cells (CT‐15) and the high TSG‐6 expressing cells (CT‐16) gave similar results. CT‐MSCs‐treated mice showed a progressive closure of the wound lesion with both treatment groups demonstrating a faster rate of wound closure that was statistically significant compared with the nontreated controls (p < .05; Fig. 6).
Figure 6.
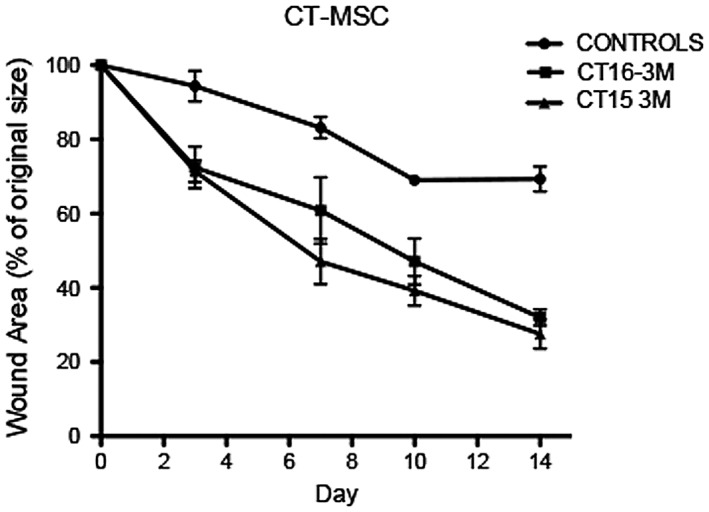
Rate and extent of wound healing was greater with cord tissue (CT)‐mesenchymal stromal cells (MSCs) compared with nontreated wounds. Cell‐treated wounds closed significantly faster than nontreated wounds for all time points (p < .05). There was no difference in the rate of wound closure between the low TNF‐stimulated gene 6 protein (TSG‐6) expresser line and the high TSG‐6 expresser CT‐MSCs line. Both lines were used at P8.
At day 14, the mice were euthanized and the wounds were excised for histological analysis. The tissue was stained with H&E stain that displays nuclei as blue and cytoplasm as pink. Masson's trichrome stain displays keratin (red), collagen (blue), erythrocytes (orange), and muscle (red). Sirius red staining displays keratin (yellow), collagen (red), erythrocytes (yellow), and muscle (yellow). H&E analysis of the wounds showed that the nontreated wounds were partially closed or fully closed but with only a thin layer of cells. Both the CT‐MSCs treated mice had full thickness wound coverage although the adipose layer had not been restored by day 14 (Fig. 7A).
Figure 7.
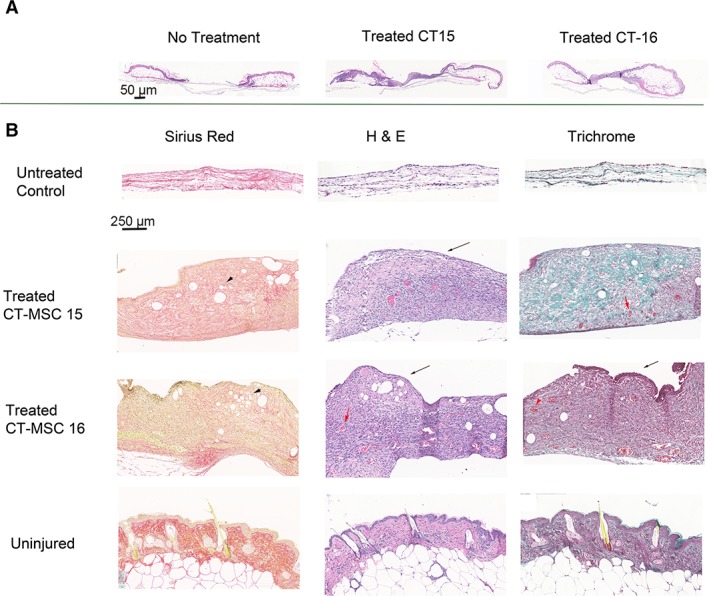
Histological analysis of the diabetic skin wounds from cord tissue (CT)‐mesenchymal stromal cell (MSC)‐treated and control mice. (A): Low magnification of the wound and surrounding tissue stained with hematoxylin and eosin (H&E) demonstrated that all wounds were covered but the nontreated wound only had a thin layer of cells covering the wound (magnification bar = 50 μm). (B): High magnification demonstrated that the control mice had a thin layer of collagen with some cells present. Blue nuclei from H&E and black nuclei from the Masson's trichrome stain can be seen in the untreated wound covering. Both CT‐MSCs treated groups have similar skin morphology and staining patterns. The wound was covered by a thick layer of cells and the extracellular matrix consisted mainly of collagen and laminin. Keratinocytes cover the top of the wound (black arrow). Neovascularization is observed in the new tissue (red arrows). Hair follicles are not present as more time is required for their formation (magnification bar = 250 μm).
High magnification of H&E, Masson's trichrome, and Sirius red stains illustrates the differences between CT‐MSCs treated and nontreated wounds (Fig. 7B). Within the thin layer covering the wounds of the untreated control mice, high levels of collagen were observed as seen by the uniform red staining (Sirius red) and uniform blue staining (Masson's trichrome). Nuclei were observed in the H&E stain. A layer of loosely packed cells was observed throughout the collagen layer. The untreated controls show no blood vessels, erythrocytes, muscle, or adipose tissue.
CT‐MSCs treated wounds look similar for both CT15 and CT‐16. These mice demonstrated thick granulation tissue formation. Keratinocytes can be noticed at the top of the wound (black arrows). The wound covering is thicker and contains a mix of collagen and keratin compared with control wounds. The muscle layer has not yet formed. Adipose cells (black arrowheads) were present. Furthermore, blood vessels with erythrocytes were present indicating vascularization has commenced (red arrows). At day 14, the sebaceous glands and hair follicles were not yet present (compared with uninjured skin).
Discussion
MSC's Isolation Efficiency and Characteristics
Donor age has a negative effect on the efficiency of isolation of MSCs from BM and the therapeutic quality of the cells 20. Because consistency is required for cells used in regenerative medicine, we sought to determine if these problems could be circumvented through the use of umbilical CT as the donor source of MSCs. Currently, most MSC‐based clinical trials use BM or adipose MSCs isolated from adult donors, but studies have demonstrated that there are differences in immunophenotype and wound healing capabilities between donors that maybe related to the age of the donor and may also be attributed to the different culture conditions being used. Donor age and/or culture conditions can have an effect on the secretion of cytokines and chemokines 16. These trophic factors have been shown to affect migration, proliferation, ECM remodeling, and survival of the cells surrounding the wound 13. As the number of clinical trials using CT‐MSCs is growing (n = 93)3, it was important to investigate if the same donor to donor variation and changes with passaging occurred with CT‐MSCs as observed with adult BM‐MSCs donors.
Our hypothesis is that term CT‐MSCs are all of a similar young age and the age related issues would not have an impact on the cells resulting in efficient isolation of a uniform MSC population of cells with little variation between donors, as well, less variation in proliferation rates and changes during passaging, compared with adult donor sources. In order to test our hypothesis, we collected and analyzed 71 donor samples using the SOP established during this study. Furthermore, the collection of umbilical cord does not impact the donor negatively since the collections were initiated after the baby was born and the cord had been clamped and cut. The CT samples were collected from across Canada, from both vaginal and caesarean delivery, from mothers age 21 to 40.
Viable CT‐MSCs have been obtained from both the enzymatic and explant method; however, some differences between the two methods have been noticed. The explant culture method has several advantages over enzymatic digestion. The explant method is more economical, there is less cellular damage, and cells isolated using this method release higher amounts of certain growth factors such as basic fibroblast growth factor (FGF) 21. Additionally, adherence and proliferation of CT‐MSCs after subculturing has also been shown to be more efficient with the explant method 22.
The ISCT has set guidelines that help to define MSCs by determining a set of positive and negative surface proteins and the ability of the MSCs to differentiate. All donor samples yielded cells with consistent surface protein profiles consistent with the ISCT definition for MSCs. Each donor sample analyzed was CD105+, CD73+, CD90+, and CD44+. The same samples were negative for CD45, CD34, CD11b, CD19, and HLA‐DR after passage 2 as expected for MSCs 23. This result establishes that CT is a reliable source of MSCs. The only variation we observed between donor samples was a range of time for the first cells to migrate from the CT and establish confluence. Despite this range, all samples yielded highly proliferative cells that would subsequently undergo two doublings every 4 days up to P10 (20 doublings ∼1 million‐fold increase). The variation in the migration of cells from the CT and proliferation rate had no correlation with the type of birth, age, or baby weight. This is similar to the result of Rezniczek et al. 24 as they also observed that maternal and gestational age did not influence MSC yield but birth weight did. Two studies observed maternal age influenced CD105+ cell yield but they did not have results on overall cell yield 25, 26.
Furthermore, immunophenotyping for MSC surface proteins during multiple passages demonstrated a consistent profile through 10 passages (20 doublings) with the only change being a decrease in CD105+ cells after passage 5. Others have observed stable CD105 in CT‐MSCs with extensive proliferation 27. The difference in results could be due to a difference in sample size, n = 71 compared with n = 3. The SEM observed in the DeWitte study indicates that variation occurs among individual samples. Despite the donor variation, they observe a stable or even increasing level of CD105 with passage. We can only speculate that the method of cell isolation, which is different between the two studies, has an effect on CD105 levels with passaging. We also demonstrated a strong consistency in the ability of the MSCs to differentiate into adipose cells and osetoblasts. All samples tested demonstrated robust differentiation, further identifying them as MSCs.
Proteome Profile and the Correlation to TSG‐6 Levels and Wound Healing
There are two main mechanisms, which convey therapeutic ability to MSCs. They can directly engraft and contribute to bone, cartilage, endothelial, muscle, and adipose tissue. MSCs are also capable of indirectly regenerating injured tissue through a paracrine secretion mechanism whereby the cells respond to the low oxygen, apoptotic cell environment, and stimulate local cells to commence tissue regeneration 28. The latter mechanism is the main mechanism observed in most models of tissue repair. The MSCs secrete a large number of cytokines and chemokines in response to tissue damage 29. Cytokines, chemokines, and cell surface proteins all have a role in the ability of MSCs to enhance tissue regeneration by regulating inflammation, enhancing cell proliferation, and cell survival and to remodel the ECM. Due to the variation in the therapeutic ability of BM‐MSCs to respond to and repair damaged tissue, studies have focused on correlating the expression of specific proteins to the therapeutic potential of individual donor MSCs.
Recently, TSG‐6 protein has been suggested as a biomarker that may be used to predict the in vivo efficacy of MSCs at reducing sterile inflammation in various animal models. TSG6 is activated by pro‐inflammatory cytokines that then leads to TSG6 downregulating inflammation and promoting tissue regeneration through the recruitment and retention of leukocytes at the wound site 16. A study done by Lee et al. demonstrated that there was a wide variation in the efficacy of human MSCs obtained from BM aspirates of healthy individuals at reducing inflammation in a chemical injury of the mouse cornea. They looked at a variety of biomarkers including TSG‐6, heme‐oxygenase, PGE‐2, IL‐1 receptor antagonist, TGF‐β1, and IDO1, however, only TSG‐6 was shown to have a significant positive correlation with the ability of the cells to suppress inflammation in vivo 16.
In our study, we investigated the TSG6 levels of 15 CT‐MSC donors. Expression levels were assessed on a relative basis to a randomly chosen donor that was set at a value of 1. There was a range of relative levels from 1.7‐fold higher (CT16) to 0.1 (CT24). This gives a relative expression range of 17‐fold. We also tested for possible correlatives such as maternal age or baby's weight but these showed no correlation with TSG6 levels. Low (CT24), moderate (CT15), and high expressing donor cells (CT16) were further analyzed for their cytokine secretion profile and wound healing ability. The cytokine profiles were strongly consistent but with some differences observed. HGF was produced by the high TSG6 expresser (CT16). HGF has a role in the wound healing process 30. PDGF‐AA was only expressed by the low TSG expresser (CT24). It has been postulated that low PDGF‐AA levels result in better skin architecture with less scarring during healing 31. Ang‐1 was upregulated in CT15 compared with the other two donors. Knockout of Ang‐1 increases fibrosis in damaged tissues 32. GM‐CSF expression is also important for wound healing and was present in both the high and low TSG6 expressing lines 33. In contrast, 18 other cytokines had similar profiles in all three donor groups, therefore, possibly overriding the differences observed with GM‐CSF, PDGF‐AA, ANG‐1, and HGF. Network analysis using STRING of the top 22 expressed cytokines, including GM‐CSF, PDGF‐AA, ANG‐1, and HGF were enriched for pathways involved in wound healing such as cell migration, ECM remodeling, and angiogenesis. Each of these pathways represent a distinct stage of wound healing 34.
To investigate if the three different donor cell lines had different therapeutic potential, we used a db/db mouse incisional wound model to explore the therapeutic potential of CT‐MSCs in a diabetic environment. We found that there was no significant difference in the rate of wound closure between different CT‐MSCs donors, and all animals treated with CT‐MSCs demonstrated a faster rate of healing compared with the untreated control. Histological analysis of the wounds illustrated that for the CT‐MSCs‐treated wounds, new tissue consisting of keratinocytes extended across the wound and the granulation tissue that formed was characterized by small capillaries and fibroblasts. There was no difference in the quality and the extent of healing that could be attributed to the CT donor age or sex, the maternal age, or baseline TSG‐6 levels. In contrast, the control animals had limited wound repair. Histology indicated that the cells were embedded mainly in collagen. Additionally, neovascularization was reduced in control mice. This may be due to the fact that diabetes impairs the growth and function of endogenous MSCs, which secrete many cytokines that play a role in angiogenesis, particularly VEGF and FGF2 35. Our study did not look at long‐term cell engraftment because we used a model that had similarities to a nonmatched allogeneic transplant model. We chose this model to specifically study the paracrine signaling effects without confounding factors related to the engraftment and differentiation of the MSCs.
Conclusion
From our study, we can conclude that cytokine expression was consistent between different donor samples and the different expression level observed for a few proteins; TSG6, PDGF‐A, HGF, and GM‐CSF does not seem to impact the ability of the cells to proliferate or help regenerate tissue. A recent study demonstrated that CT‐MSCs had a significant increase in TSG‐6 levels upon stimulation. This indicates that even units with low initial levels could be therapeutic due to stimulation of TSG‐6 expression by inflammatory factors found in the wound such as TNFα. This is a possible explanation of why our low TSG‐6 expressing cells had equivalent efficacy to the high expressing cells in skin regeneration. In conclusion, our results demonstrate that MSCs from different donors all have typical MSCs properties, similar cytokine profiles and show good therapeutic potential. CT‐MSCs maybe a better source of therapeutic cells compared with BM‐MSCs due to their ease of procurement, consistent proliferation and growth characteristics, and consistent therapeutic properties.
Disclosure of Potential Conflicts of Interest
I.M.R. declared honoraria and research funding from the Grant Agency Ontario Centers of Excellence (OCE). The other authors indicated no potential conflicts of interest.
Author Contributions
V.N.R., J.W., T.C., A.K., A.M.: designed experiments, performed research, analyzed data, wrote manuscript; A.K.: provided materials, interpreted results; I.M.R.: designed experiments, analyzed data, wrote manuscript, provided financial support.
Supporting information
Supplementary Figure 1 Flow cytometry of CT‐MSC
Supplementary Figure 2 TSG‐6 levels do not correlate with maternal age, baby weight and TSG‐6 levels.
Maternal age ranged from 18 to 41 years and baby weight at birth ranged from 2.4 kg to 4.0 kg. When compared to relative TSG‐6 mRNA levels no correlation was observed. Time in labor information was not available for analysis.
Supplementary Table 1 PCR primers.
Acknowledgments
This study was supported by CIHR #PPP110824, Center for the Commercialization of Regenerative Medicine‐001, Insception‐LifeBank‐003, and Stem Cell Network‐13/5226 (CT74). Umbilical cord blood and tissue collections: Research Centre for Women's and Infants' Health, Lunenfeld‐Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Canada, http://biobank.lunenfeld.ca. We acknowledge the histology assistance from the Lunenfeld‐Tanenbaum Research Institute's CMHD Pathology Core (www.cmhd.ca). V.N.R. is currently affiliated with the Institute for Molecular Bioscience, Queensland Bioscience Precinct, The University of Queensland, St Lucia, Queensland, Australia.
Data Availability Statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea‐pig bone marrow and spleen cells. Cell Tissue Kinet 1970;3:393–403. [DOI] [PubMed] [Google Scholar]
- 2. Caplan AI. Mesenchymal stem cells. J Orthop Res 1991;9:641–650. [DOI] [PubMed] [Google Scholar]
- 3. Can A, Celikkan FT, Cinar O. Umbilical cord mesenchymal stromal cell transplantations: A systemic analysis of clinical trials. Cytotherapy 2017;19:1351–1382. [DOI] [PubMed] [Google Scholar]
- 4. Karp JM, Leng Teo GS. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell 2009;4:206–216. [DOI] [PubMed] [Google Scholar]
- 5. Lv FJ, Tuan RS, Cheung KM et al. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014;32:1408–1419. [DOI] [PubMed] [Google Scholar]
- 6. Choudhery MS, Khan M, Mahmood R et al. Bone marrow derived mesenchymal stem cells from aged mice have reduced wound healing, angiogenesis, proliferation and anti‐apoptosis capabilities. Cell Biol Int 2012;36:747–753. [DOI] [PubMed] [Google Scholar]
- 7. Stolzing A, Jones E, McGonagle D et al. Age‐related changes in human bone marrow‐derived mesenchymal stem cells: Consequences for cell therapies. Mech Ageing Dev 2008;129:163–173. [DOI] [PubMed] [Google Scholar]
- 8. Pittenger MF, Mackay AM, Beck SC et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999;284:143–147. [DOI] [PubMed] [Google Scholar]
- 9. Bernardo ME, Locatelli F, Fibbe WE. Mesenchymal stromal cells. Ann N Y Acad Sci 2009;1176:101–117. [DOI] [PubMed] [Google Scholar]
- 10. Alt EU, Senst C, Murthy SN et al. Aging alters tissue resident mesenchymal stem cell properties. Stem Cell Res 2012;8:215–225. [DOI] [PubMed] [Google Scholar]
- 11. Lo Furno D, Mannino G, Giuffrida R. Functional role of mesenchymal stem cells in the treatment of chronic neurodegenerative diseases. J Cell Physiol 2018;233:3982–3999. [DOI] [PubMed] [Google Scholar]
- 12. Dabrowski FA, Burdzinska A, Kulesza A et al. Comparison of the paracrine activity of mesenchymal stem cells derived from human umbilical cord, amniotic membrane and adipose tissue. J Obstet Gynaecol Res 2017;43:1758–1768. [DOI] [PubMed] [Google Scholar]
- 13. Maxson S, Lopez EA, Yoo D et al. Concise review: Role of mesenchymal stem cells in wound repair. Stem Cells Translational Medicine 2012;1:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee HJ, Diaz MF, Ewere A et al. Focal adhesion kinase signaling regulates anti‐inflammatory function of bone marrow mesenchymal stromal cells induced by biomechanical force. Cell Signal 2017;38:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khan I, Zhang L, Mohammed M et al. Effects of Wharton's jelly‐derived mesenchymal stem cells on neonatal neutrophils. J Inflamm Res 2015;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee RH, Yu JM, Foskett AM et al. TSG‐6 as a biomarker to predict efficacy of human mesenchymal stem/progenitor cells (hMSCs) in modulating sterile inflammation in vivo. Proc Natl Acad Sci USA 2014;111:16766–16771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bancroft J. Theory and practice of histological techniques. Elsevier Health Sci 2008;1:1008. [Google Scholar]
- 18. Toki D, Zhang W, Hor KL et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant 2014;14:2126–2136. [DOI] [PubMed] [Google Scholar]
- 19. Wong VW, Sorkin M, Glotzbach JP et al. Surgical approaches to create murine models of human wound healing. J Biomed Biotechnol 2011;2011:969618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Puts R, Albers J, Kadow‐Romacker A et al. Influence of donor age and stimulation intensity on osteogenic differentiation of rat mesenchymal stromal cells in response to focused low‐intensity pulsed ultrasound. Ultrasound Med Biol 2016;42:2965–2974. [DOI] [PubMed] [Google Scholar]
- 21. Yoon JH, Roh EY, Shin S et al. Comparison of explant‐derived and enzymatic digestion‐derived MSCs and the growth factors from Wharton's jelly. Biomed Res Int 2013;2013:428726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Han YF, Tao R, Sun TJ et al. Optimization of human umbilical cord mesenchymal stem cell isolation and culture methods. Cytotechnology 2013;65:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–317. [DOI] [PubMed] [Google Scholar]
- 24. Rezniczek GA, Kumbruch S, Scheich J et al. Factors influencing yield and neuronal differentiation of mesenchymal stem cells from umbilical cord blood and matrix. Regen Med 2016;11:465–474. [DOI] [PubMed] [Google Scholar]
- 25. Alrefaei GI, Al‐Karim S, Ayuob NN et al. Does the maternal age affect the mesenchymal stem cell markers and gene expression in the human placenta? What is the evidence? Tissue Cell 2015;47:406–419. [DOI] [PubMed] [Google Scholar]
- 26. Alrefaei GI, Ayuob NN, Ali SS et al. Effects of maternal age on the expression of mesenchymal stem cell markers in the components of human umbilical cord. Folia Histochem Cytobiol 2015;53:259–271. [DOI] [PubMed] [Google Scholar]
- 27. de Witte SFH, Lambert EE, Merino A et al. Aging of bone marrow‐ and umbilical cord‐derived mesenchymal stromal cells during expansion. Cytotherapy 2017;19:798–807. [DOI] [PubMed] [Google Scholar]
- 28. Peng L, Jia Z, Yin X et al. Comparative analysis of mesenchymal stem cells from bone marrow, cartilage, and adipose tissue. Stem Cells Dev 2008;17:761–773. [DOI] [PubMed] [Google Scholar]
- 29. Ylostalo JH, Bartosh TJ, Tiblow A et al. Unique characteristics of human mesenchymal stromal/progenitor cells pre‐activated in 3‐dimensional cultures under different conditions. Cytotherapy 2014;16:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cowin AJ, Kallincos N, Hatzirodos N et al. Hepatocyte growth factor and macrophage‐stimulating protein are upregulated during excisional wound repair in rats. Cell Tissue Res 2001;306:239–250. [DOI] [PubMed] [Google Scholar]
- 31. Wagner W, Wehrmann M. Differential cytokine activity and morphology during wound healing in the neonatal and adult rat skin. J Cell Mol Med 2007;11:1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Humphreys BD. Chapter 28–Investigating the process of renal epithelial repair to develop new therapies In: Little M. Kidney Development, Disease, Repair and Regeneration. Amsterdam: Elsevier, 2016:381–393. [Google Scholar]
- 33. Lim JY, Choi BH, Lee S et al. Regulation of wound healing by granulocyte‐macrophage colony‐stimulating factor after vocal fold injury. PLoS One 2013;8:e54256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Franceschini A, Szklarczyk D, Frankild S et al. STRING v9.1: Protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013;41:D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin P. Wound healing—Aiming for perfect skin regeneration. Science 1997;276:75–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Flow cytometry of CT‐MSC
Supplementary Figure 2 TSG‐6 levels do not correlate with maternal age, baby weight and TSG‐6 levels.
Maternal age ranged from 18 to 41 years and baby weight at birth ranged from 2.4 kg to 4.0 kg. When compared to relative TSG‐6 mRNA levels no correlation was observed. Time in labor information was not available for analysis.
Supplementary Table 1 PCR primers.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.