

Abstract
The nearly axiomatic idea that de novo protein synthesis is necessary for long-term memory consolidation is based heavily on behavioral studies using translational inhibitors such as anisomycin. Although inhibiting protein synthesis has been shown to disrupt the expression of memory, translational inhibitors also have been found to profoundly disrupt basic neurobiological functions, including the suppression of ongoing neural activity in vivo. In the present study, using transverse hippocampal brain slices, we monitored the passive and active membrane properties of hippocampal CA1 pyramidal neurons using intracellular whole cell recordings during a brief ~30-min exposure to fast-bath-perfused anisomycin. Anisomycin suppressed protein synthesis to 46% of control levels as measured using incorporation of radiolabeled amino acids and autoradiography. During its application, anisomycin caused a significant depolarization of the membrane potential, without any changes in apparent input resistance or membrane time constant. Anisomycin-treated neurons also showed significant decreases in firing frequencies and spike amplitudes, and showed increases in spike width across spike trains, without changes in spike threshold. Because these changes indicated a loss of cellular energetics contributing to maintenance of ionic gradients across the membrane, we confirmed that anisomycin impaired mitochondrial function by reduced staining with 2,3,5-triphenyltetrazolium chloride and also impaired cytochrome c oxidase (complex IV) activity as indicated through high-resolution respirometry. These findings emphasize that anisomycin-induced alterations in neural activity and metabolism are a likely consequence of cell-wide translational inhibition. Critical reevaluation of studies using translational inhibitors to promote the protein synthesis dependent idea of long-term memory is absolutely necessary.
NEW & NOTEWORTHY Memory consolidation is thought to be dependent on the synthesis of new proteins because translational inhibitors produce amnesia when administered just after learning. However, these agents also disrupt basic neurobiological functions. We show that blocking protein synthesis disrupts basic membrane properties of hippocampal neurons that correspond to induced disruptions of mitochondrial function. It is likely that translational inhibitors cause amnesia through their disruption of neural activity as a result of dysfunction of intracellular energetics.
Keywords: CA1, cytochrome oxidase, mitochondria, neurosilencing, translational inhibition
INTRODUCTION
Despite many decades of research into the biological basis of learning and memory, we are still delving into the precise neurobiological mechanisms of memory consolidation. A dominant assumption in the field is that de novo protein synthesis (i.e., the translation of new proteins) in the period following a learned experience, is essential for the consolidation of long-term memory (for review see Davis and Squire 1984; Kandel 2004). The foundational support for this idea depends primarily on the amnestic effects of global translational inhibitors of protein synthesis (Hernandez and Abel 2008). Although initial studies using protein synthesis inhibitors suggested that they had additional disruptive effects on physiology and neurobiological function (Cohen et al. 1966; Cohen and Barondes 1967; Dahl 1969), the oft-cited review of this early literature (Davis and Squire 1984) argued that late-generation translational inhibitors such as anisomycin had fewer confounding effects, especially when administered directly into brain parenchyma. However, recent studies clearly indicate that translational inhibitors such as anisomycin and cycloheximide produce seriously confounding effects, likely as a consequence of global translational inhibition. For example, anisomycin interferes with neurobiological processes that would also be presumed to be important for memory consolidation and expression, including monoamine release and synthesis (Canal et al. 2007; Qi and Gold 2009), in addition to neural activity (Kleim et al. 2003; Schwartz et al. 1971; Sharma et al. 2012). Interestingly, if alterations in neurotransmitter release and neural activity are ameliorated pharmacologically and electrophysiologically, memory and plasticity impairments are no longer present despite the ongoing translational inhibition (Fonseca et al. 2006; Sadowski et al. 2011; for review see Gold 2008). Together these findings suggest that the neurobiological consequences of translational inhibition, if overridden, can rescue memory and plasticity, even during ongoing reductions of de novo protein synthesis.
In our own laboratory, both anisomycin and cycloheximide were found to catastrophically suppress ongoing hippocampal activity. This sustained “neurosilencing” occurred for several hours following local administration, with the level of neural inactivation correlated with reductions in ongoing protein translation as measured by amino acid incorporation (Sharma et al. 2012). Subsequently, we have found that anisomycin disrupts hippocampal involvement in the functional expression of both unconditioned fear and spatial navigation in freely behaving rats, similar to the actions of other compounds that also suppress neural activity, such as TTX (Dubue et al. 2015; Greenberg et al. 2014). Taken together, these findings suggests that anisomycin and other translational inhibitors disrupt behavior in general (and not just simply memory) by disrupting neural activity itself.
Given the confounding and detrimental effects of anisomycin demonstrated at both the behavioral and neuronal population levels, we aimed to assess how anisomycin might affect the membrane properties of individual hippocampal CA1 pyramidal cells. Using whole cell recordings in a brain slice preparation, we show in this report that even short-duration applications of anisomycin impair both the passive and active properties of CA1 pyramids. Because of the profile of neurophysiological alterations observed, we further studied the influences of anisomycin on mitochondrial function in hippocampal tissue and showed that anisomycin profoundly impairs cellular energetics and intracellular electron transfer. The goal of this study was to identify how inhibition of protein synthesis might contribute to impairments of neuronal activity at the membrane and cellular level to identify how translational inhibitors might impair neurobiological function and thus impact behavioral measures.
METHODS
Animals.
All procedures were performed on male Sprague-Dawley rats and were in accordance with Canadian Council of Animal Care guidelines. The ethics protocol (AUP#000092) was approved by the Biosciences Animal Care and Use Committee at the University of Alberta. The rats used in transverse hippocampal slice studies weighed 50–100 g (~3–4 wk old). On the day of experiment, rats were deeply anesthetized in a gas chamber containing 4% isoflurane in 100% O2 (Benson Medical Industry, Markham, ON, Canada) before rapid decapitation and brain slice preparation. The rats used for in vivo studies weighed 250–400 g (~2–3 mo old) and were anesthetized with urethane (0.67 g/mL to a final dose of 1.5–1.7 g/kg iv) before stereotaxic surgery for intracranial infusions.
Brain slice preparation.
Following loss of the righting reflex, rats were decapitated, and brains were placed into an ice-cold (0–4°C) slicing solution containing (in mM) 118 NaCl, 3 KCl, 1.4 NaH2PO4, 1.3 MgSO4, 26 NaHCO3, 1.5 CaCl2, and 10 glucose (Fisher Scientific, Toronto, ON, Canada) and saturated with carbogen (5% CO2-95% O2; Praxair Canada, Mississauga, ON, Canada), followed by dissection of both hippocampi on ice. Transverse hippocampal slices were cut at a thickness of 300 μm using a Leica VT1000S vibrating blade microtome (Leica, Bannockburn, IL) and placed in artificial cerebrospinal fluid (aCSF) solution containing (in mM) 124 NaCl, 3 KCl, 1.4 NaH2PO4, 1.3 MgSO4, 26 NaHCO3, 1.5 CaCl2, and 10 glucose, and saturated with carbogen. Slices were allowed to recover in carbogenated aCSF at room temperature for 1 h before any recordings were made. All extracellular solutions had an osmolarity of 303 ± 3 mosM.
In vivo preparation.
After jugular catheter implantation for intravenous urethane delivery under isoflurane anesthesia, each rat (n = 9) was placed in a stereotaxic apparatus (model 1900; David Kopf Instruments, Tujunga, CA) and had 30-gauge cannulas implanted bilaterally into the dorsal hippocampi (from bregma: anteroposterior −3.3 mm, mediolateral ±2.2 mm, and dorsoventral −3 mm from dura). For within-subject comparisons, one hippocampus was infused with 1 µL (at a rate of 0.5 µL/min; Harvard Apparatus infusion pump) of phosphate-buffered saline (PBS; 0.05 M), and the contralateral hippocampus was infused with anisomycin (100 µg/µL; 25 mg dissolved in 10 µL of 1 N HCl and brought to a final volume of 250 µL with PBS) To match the pH of the anisomycin and PBS solutions, the same proportion of HCl was added to the vehicle solution. Lateralization (left or right hemisphere) of each infusion was counterbalanced across all rats. Thirty minutes following the infusion, rats were euthanized by decapitation, and their hippocampi were extracted separately, weighed, and placed in ice-cold mitochondrial respiration medium 05 (MiR05; 0.5 mM EGTA, 3 mM MgCl2·6H2O, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/L BSA essentially fatty acid free, pH 7.1) before homogenization.
Membrane electrophysiology.
Following recovery, slices were transferred to a fixed stage of an upright microscope (Leica DM LFS) that was perfused with a continuous flow (2–4 mL/min) of aCSF maintained at 34.0 ± 1.0°C (Bipolar Temperature Controller, Warner Instruments, Hamden, CT) via a Minipuls 3 peristaltic pump (Mandel Scientific, Guelph, ON, Canada). For each slice (n = 45), we recorded and experimented on only one CA1 pyramidal cell (n = 45 cells: 16 control and 29 anisomycin treated). CA1 pyramidal neurons were visually identified using a ×40 water-immersion objective with infrared differential interference contrast imaging and a near-infrared charge-coupled device camera (Sony XC-75). Whole cell patch-clamp recordings were obtained using borosilicate glass pipettes (2–6 MΩ) pulled with a Flaming/Brown micropipette puller (model P97; Shutter Instrument, San Francisco, CA) and filled with intracellular solution containing (in mM) 135 K-gluconate, 10 HEPES, 10 EGTA, 7 KCl, 2 MgCl2, 0.3 Na2GTP, 3 MgATP, and 10 phosphocreatine (Sigma Aldrich, St. Louis, MO), pH 7.2–7.3, 290–295 mosM. Recordings were made using an Axon Multiclamp 700B amplifier (Axon Instruments/Molecular Devices, Sunnyvale, CA) and digitized at a sampling rate of 20 (continuous membrane voltage), 50 (current sweep and ramp protocols), or 250 kHz (single-spike protocols) using an Axon Digidata 1322A driven by pClamp software (Axon Instruments/Molecular Devices). The pipette offset was measured following the conclusion of the experiment by physical patch breakout and was corrected for individual experiments. The liquid-liquid junction potential was empirically determined to be −7 mV using the method of Neher (1992) but was not corrected. Whole cell patch-clamp recordings were made in visually identified CA1 pyramidal neurons and were allowed to stabilize for at least 5 min before the beginning of baseline recordings.
In every case, baseline measures of resting membrane potential (RMP) and single-spike thresholds were acquired. In some cases, a family of hyperpolarizing and depolarizing current pulses (in 50-pA steps) as well as current ramps were applied to also investigate baseline active membrane properties. The maximum deflection of the ramps was increased in 50-pA steps linearly from 0 pA (i.e., 0 to 50 pA, 0 to 100 pA, 0 to 150 pA, and so on), and were 1 s in duration. Following this baseline recording period, these measurements were repeated at 7-min intervals following perfusion with either the same control aCSF solution or one that contained 100 µM anisomycin. In the case of anisomycin perfusions, after 21 min, the perfusion medium was switched back to normal aCSF before the last acquisition period at 28 min. To assess input resistance and the membrane time constant, low-intensity (−10 pA) short (500 ms) hyperpolarizing pulses were applied every minute during continuous recordings. Input resistance (Rin) was calculated by measuring the peak deflection in voltage (ΔV) as a function of the amount of current (I) applied (Rin = V/I). The same hyperpolarizing pulses were used to calculate the membrane time constant (τ) by fitting a single-order exponential decay [f(t) = Aie−t/τ + C] to the membrane deflection. In every case in which membrane properties were assessed with current pulses or ramps, the resting membrane potential was equalized to baseline levels using current injections.
Single-spike characteristics were assessed by applying short (3.2 ms) depolarizing pulses to elicit a single action potential, whereas multiple spike characteristics were assessed using longer (1 s) depolarizing pulses. The threshold of the first action potential was calculated using MATLAB (The MathWorks, Natick, MA) and was defined as the point at which the differential of the voltage (Vm) as a function of time exceeded at least double the value of baseline fluctuations for seven consecutive data points, indicating the presence of an action potential. We confirmed visually that this method successfully demarcated spike threshold. Spike amplitude was measured as the difference in voltage between the threshold membrane potential and the peak amplitude of the spike. Spike duration at half-amplitude (half-width) was defined as the width of the spike at the half-point between threshold and the peak.
Multiple spike characteristics were assessed using long (1 s) current sweeps. The medium afterhyperpolarization was measured as the voltage deflection from baseline following the termination of depolarizing current sweeps for which multiple spikes were elicited. The frequency of firing in long depolarizing pulses was calculated by counting the number of action potentials elicited and dividing by the duration of the pulse. This was done at three intensity levels, which varied between cells. The incremental stimulus intensity was in 50-pA steps from the minimum stimulus intensity to evoke a spike.
Brain slice incubation procedures for autoradiography, histochemical staining, and high-resolution respirometry.
Slices were randomly divided into carbogenated incubation chambers containing either normal slice perfusate (aCSF) or 100 µM anisomycin in aCSF. The slices were incubated for 30 min before experimental assay.
Autoradiography.
The degree of protein synthesis inhibition was evaluated in vitro following bath incubation experiments using anisomycin. Slices (n = 7–9 slices for each individual experiment, N = 4 within-subject experiments in total) were prepared identically to those in the electrophysiological experiments and were incubated in carbogenated control aCSF or anisomycin solution (100 µM in aCSF). Slices were transferred to a solution with 10 mCi/mL [35S]-cysteine/methionine for 30 min and then dried overnight. Dried slices were placed under Kodak BioMax MR X-ray film (PerkinElmer, Waltham, MA) for 7–8 days. Control and anisomycin-treated slices from the same experiments were always developed on the same film to ensure identical exposure times. Once developed, grayscale digital images were scanned at a resolution of 2,400 dpi (HP ScanJet G3110; Hewlett-Packard, Palo Alto, CA) and analyzed using ImageJ (National Institutes of Health, Bethesda, MD). Light intensity values (from a standard scale ranging from 0 to 255, with 255 being the brightest and 0 being the darkest) were averaged across the entire slice following selection of the region of interest by hand, omitting any regions of non-tissue-related signal. In relation to this study, less amino acid incorporation, and therefore less protein synthesis, was indicated by a higher value on this scale. These raw values were then normalized to the background (lightest area surrounding each of the slices), resulting in a higher normalized value representing a darker image, and therefore interpreted as more protein synthesis.
Triphenyltetrazolium chloride staining.
Because of the pattern of changes observed in the electrophysiological properties of neurons with anisomycin treatment, we hypothesized that translational inhibition might interfere with cellular energy gradients. This was initially tested using a staining assay with 2,3,5-triphenyltetrazolium chloride (TTC; Sigma Aldrich) in slices following exposure to anisomycin or vehicle control solution for 30 min (detailed above). The conversion of white TTC to 1,3,5-triphenylformazan, which is red in color, has been previously used as a relatively nonspecific stain for mitochondrial health and integrity. Slices (n = 5 slices per treatment per experiment; N = 4 within-subject experiments) were incubated for 30 min in either carbogenated control aCSF or 100 µM anisomycin before being transferred to a 2% TTC solution in carbogenated aCSF for 30 min. Slices were then transferred to a 4% buffered formalin solution overnight before being mounted on hanging drop slides (Fisher Scientific). Slices were scanned in grayscale, analyzed using ImageJ, and quantified via light intensity values, similar to autoradiography experiments explained above, although images were scanned at 4,800 dpi.
High-resolution respirometry.
To further examine mitochondrial function, we used cellular respirometry. In slice experiments (N = 7 within-subject experiments), 5–10 transverse hippocampal slices from each rat were pooled in a bath treatment of control aCSF or 100 µM anisomycin. Following 30 min of bath incubation in either control or anisomycin solution, slices (28–56 mg) were weighed and transferred to 250 µL of ice-cold MiR05. The tissue was homogenized on ice for five passes at an intensity of 1.5 with a Potter-Elvehjem homogenizer (PRO Scientific, Oxford, CT) attached to an overhead stirrer (Wheaton Instruments, Millville, NJ). An aliquot (50 µL) of the homogenate (2.4–7.8 mg of tissue) was added to each oxygraph chamber (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria), previously calibrated according to the manufacturer’s instructions with 2 mL of MiR05. All experiments were run in simultaneous duplicate and averaged according to treatment. DatLab software (Oroboros Instruments) was used for data acquisition and analysis.
Because cellular energetics can be affected in slice preparations, we further assessed the effect of anisomycin on mitochondrial function using in vivo preparations (N = 9 within-subject experiments). Infusions of either anisomycin or vehicle were made alternately in the two hippocampi of each hemisphere (as described above), and after 30 min, each hippocampus (50–110 mg) was quickly dissected, weighed, and homogenized for five passes at an intensity of 1.5 in 2 mL of ice-cold MiR05. A volume of 25 µL of homogenate was added to each chamber (in 0.8–1.2 mg of brain tissue).
The protocol used for evaluating mitochondrial function included three different coupling states: 1) Leak represents the nonphosphorylating state in the absence of ADP, 2) OxPhos represents oxygen flux coupled to phosphorylation of ADP to ATP in the presence of saturating ADP, and 3) ET represents oxygen consumption after uncoupling, independent of the supply of ADP and the capacity of the phosphorylation system. The following substrates, uncouplers, and inhibitors were added (final concentration in the chamber): pyruvate (5 mM), malate (5 mM), ADP (2.5 mM), succinate (10 mM), rotenone (1 µM), antimycin A (5 µM), ascorbate (2 mM), tetramethyl phenylenediamine (TMPD; 0.5 mM), and azide (100 mM). This protocol allows evaluation of leak respiration in the presence of substrates feeding electrons into the NADH pathway (through complex I; pyruvate + malate; N pathway), OxPhos capacity after addition of saturating ADP and in the presence of substrates feeding electrons into the N pathway and the NADH and succinate pathway (feeding complexes I and II simultaneously; pyruvate + malate + succinate; NS pathway), maximal noncoupled ET capacity under NS pathways, ET capacity for the succinate pathway alone (after inhibition of complex I with rotenone; S pathway), and complex IV single-step activity (with ascorbate + TMPD). Mitochondrial respiration was corrected for instrumental background and for residual oxygen consumption after inhibition of complexes I and II with rotenone and antimycin A, respectively (Gnaiger et al. 1988). For complex IV activity, the chemical background measured in the presence of azide was subtracted.
In a separate set of experiments (N = 2 within-subject in slice; N = 2 within-subject in vivo), cytochrome c was added after ADP to test for the integrity of the mitochondrial outer membrane. The quality of the membrane was evaluated using the cytochrome c control factor, the fractional change of respiration from substrate state without cytochrome c to the substrate state with cytochrome c. The mitochondrial respiration rates are expressed in oxygen consumption per wet weight of tissue (pmol O2·s−1·mg−1). Alternatively, qualitative changes in the OxPhos system were expressed as flux control ratios (FCRs), normalized for maximal ET capacity in the presence of substrates feeding electrons into both the N and S pathways simultaneously (NS pathway). The FCR for Leak in the presence of pyruvate and malate gives an accurate estimation of mitochondrial coupling under conditions not limited by substrates and the phosphorylation system (Lemieux et al. 2017). In addition, the conventional respiratory control ratio (RCR; Chance and Williams 1956), calculated as OxPhos capacity/Leak respiration in the presence of substrates feeding electrons into the N pathway, was also used as an indicator of coupling.
Experimental design and statistical analysis.
To test for changes in protein translation in our preparation, autoradiography of amino acid incorporation was measured. Multiple consecutive tissue slices from n = 4 rats were treated with anisomycin or vehicle such that each rat contributed seven to nine slices to either control or anisomycin conditions (the assignment of slices to conditions was conducted using a predetermined sequence). For each rat, the average density of the slices in either condition was compared within animal. A paired Student’s t-test was used to assess differences between the average and normalized light intensity values of control and anisomycin-treated slices taken from the same animal on the same experimental day.
To test for basal cellular functional and activity changes due to anisomycin treatment over time, data were collected from cells that were bathed in anisomycin (n = 29) or control (n = 16) solution for 30 min and measurements of active and passive membrane properties were made across time and compared within a treatment group. Assignment to control or experimental groups occurred on an predetermined alternating basis, which allowed for a greater number of experimental observations compared with controls. Statistical analysis on electrophysiological data used individual experiment-based repeated-measures one-way analysis of variance (ANOVA) for both control and anisomycin data across all time points (0, 7, 14, 21, and 28 min after treatment onset), to assess for time-dependent changes in measures due to treatment. To control for multiple comparisons, Tukey’s honestly significant difference (HSD) post hoc tests were used to look for significant changes (α = 0.05) between baseline values and each time point (7, 14, 21, and 28 min) within a treatment when a main effect was observed.
To test for differences in cellular energetics using TTC staining, multiple consecutive tissue slices from n = 4 rats were treated with anisomycin or vehicle such that each rat contributed at least four slices to both control and anisomycin conditions. For each rat, the average density of the slices in either condition was compared within animal. A paired Student’s t-test was used to assess differences between normalized light intensity values of control and anisomycin-treated slices.
To test for differences in respirometry measurements, multiple consecutive tissue slices from n = 7 rats (for slice experiments) or unilateral hippocampus from n = 9 rats (for whole brain experiments) were treated with anisomycin or vehicle such that within-subject comparisons could be made. Paired Student’s t-tests compared the different coupling states (Leak, OxPhos, and ET) and steps or pathways of mitochondrial function in oxygen flux per mass in anisomycin-treated vs. control tissues; within-subject paired comparisons were made in both slice and in vivo experiments.
To compare mitochondrial function across experimental preparations (slice vs. whole brain), unpaired Student’s t-tests compared control-treated slices (in vitro; n = 7) with control-treated whole hippocampus (whole brain; n = 9) measures of FCR, RCR, and cytochrome c control ratio. All Student’s t-tests were two-tailed (α = 0.05).
RESULTS
Bath application of anisomycin inhibits protein synthesis in hippocampal slices.
To confirm that a 30-min application of a 100 µM solution of anisomycin inhibited translation in transverse hippocampal slices, we used autoradiography to measure the incorporation of [35S]-cysteine/methionine (n = 4 rats). Average light intensity values of developed autoradiographs from individual slices were quantified using ImageJ and were normalized (from 0 to 1) to the minimum (i.e., background) and maximum values for each experimental scan batch that included both anisomycin and control conditions. Following this normalization, a value of 1 represented the darkest intensity value (i.e., the maximal autoradiographic signal) whereas 0 represented the background level of intensity. As shown in Fig. 1, anisomycin-treated slices exhibited less incorporation of radiolabeled amino acids compared with control slices (average intensity of 0.48 ± 0.05 and 0.86 ± 0.02, respectively). This confirmed that our procedure and concentration of anisomycin significantly suppressed ongoing protein synthesis in hippocampal slices by 46% (t3 = 8.5; P = 0.006, paired t-test).
Fig. 1.
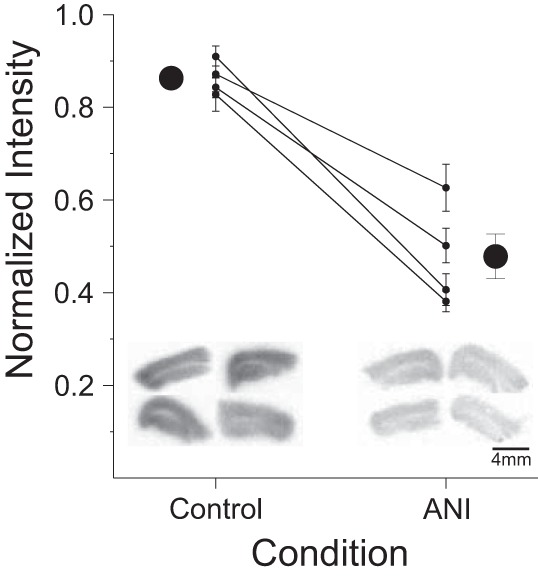
Anisomycin (ANI) inhibits protein synthesis in transverse hippocampal brain slice preparations. Data are normalized light intensity from autoradiographs of 35S-radiolabeled amino acid incorporation experiments across control and ANI incubation conditions. Large circles are scatter plots showing averages (±SE); smaller circles with adjoining lines across conditions show within-experiment averages (±SE). There was a significant decrease in the normalized amount of incorporated 35S-labeled cysteine and methionine in slices treated with 100 µM ANI for 30 min compared with controls. *P < 0.01, ANI vs. control. Insets show example background-normalized autoradiographs from 4 transverse hippocampal slices from each group. Darker staining and a higher normalized intensity equate to more amino acid incorporation and therefore a greater level of protein synthesis in control compared with ANI-treated slices.
Anisomycin depolarizes membrane potential without changing other passive membrane properties in CA1 neurons.
To investigate the effect of anisomycin on neuronal membrane properties, we targeted CA1 pyramidal cells for whole cell recordings in transverse hippocampal slices. The RMP was reported as the transmembrane voltage when no holding current was applied (control: n = 16 cells; anisomycin: n = 29 cells). The initial RMP was extracted from spontaneous recordings during long (minimum 5 min) stable baseline conditions and at three time points spaced at 7-min intervals following addition of anisomycin (7, 14, and 21 min), as well as a further 7 min following restoration of control aCSF perfusion (i.e., 28 min following the initiation of anisomycin application). In control cells, measurements were taken at the same time points in the absence of anisomycin. Figure 2, A and C, shows example traces over the 28-min recording period from a control and anisomycin-treated cell, respectively. The subthreshold transient fluctuations observed in these traces correspond to spontaneous synaptic activity.
Fig. 2.

Anisomycin caused a significant depolarization in CA1 pyramidal neurons over time but did not change input resistance (IR) or membrane time constant measures. A: sample trace of a continuous whole cell patch-clamp recording from a CA1 pyramidal neuron under control perfusion conditions. As demonstrated, there is a relatively steady resting membrane potential (RMP) throughout the duration of the recording period. Hyperpolarizing deflections at 1-min intervals reflect current pulses administered to track apparent IR. Transient depolarizations reflect ongoing synaptic activity. B: superimposed traces from the same cell showing membrane responses to hyperpolarizing current pulses of −10 pA administered at numbered time points (1–3) and asterisks in A. As demonstrated in control cells, peak deflections remained relatively constant across the 3 time points, as did the exponential decay of the voltage traces. C: sample trace of a similar recording from an ANI-treated neuron showing a marked depolarization of the RMP following 100 µM bath application of ANI (shaded area). As in A, hyperpolarizing deflections at 1-min intervals reflect time points where current pulses were injected to track apparent input resistance. At time points when the RMP was depolarized, steady holding current was injected to bring the membrane potential back to its baseline value (dotted line) before hyperpolarizing current pulses were administered. D: superimposed traces from the same cell showing membrane responses to hyperpolarizing current pulses of −10 pA (from holding current levels) administered at numbered time points (1–3) and asterisks in C. As with the control recordings in A and B, peak deflections remained relatively constant across the 3 time points [in this case corresponding to baseline (black), ~13 min after ANI application (red), and ~5 min after the washout period (gray)]. In addition, the exponential decay of the voltage traces also overlapped across time points. E: average RMP in control (black squares; left) and ANI-treated cells (red circles; right) as a function of time (B, baseline; 7, 7 min of ANI or vehicle perfusion; 14, 14 min of ANI or vehicle perfusion; 21, 21 min of ANI or vehicle perfusion; 28, 7 min after washout and 28 min postperfusion). Whereas there were no changes of RMP in control conditions, there was a significant depolarization of RMP in ANI-treated cells at all time points following bath application (shaded area; 7, 14, and 21) as well as into the washout period (28). F: there were no significant changes in the average apparent input resistance in either control (black squares; left) or ANI perfusion conditions (red circles; right) at any time point. G: there were also no significant changes in the average membrane time constant in either control (black squares; left) or ANI perfusion conditions (red circles; right) across the same time points. *P < 0.05 vs. baseline (in E–G). HC, holding current; I, hyperpolarizing current; Vm, membrane potential.
As expected, control cells showed stable RMP values across the entire 28-min period (Fig. 2, A and E). During applications of anisomycin, however, RMP values significantly depolarized by 5–10 mV during the 21 min of perfusion as demonstrated by a one-way repeated-measures ANOVA [F(4,112) = 23.02, P < 0.0001; Fig. 2, C and E]. Tukey’s HSD post hoc tests revealed that the average RMP significantly depolarized from −70 ± 1 mV at baseline to −68 ± 1 mV at 7 min post-anisomycin application (P < 0.05), to −66 ± 1 mV at 14 min post-anisomycin application (P < 0.01), and to 63 ± 1 mV at 21 min post-anisomycin application (P < 0.01) and was maintained at −63 ± 1 mV at 7 min following reperfusion with regular aCSF (28 min following the initiation of anisomycin perfusion; P < 0.01). In contrast, there were no significant changes over time in control cells at identical recording times [F(4,60) = 1.03, P = 0.39]. Average RMP responses for control and anisomycin data are summarized in Fig. 2E.
In a subset of these experiments (control: n = 7 cells; anisomycin: n = 12 cells), we monitored other passive membrane properties such as apparent input resistance and the membrane time constant. In these cases, we injected small (−10 pA) and brief (500 ms) hyperpolarizing pulses every minute in both control (Fig. 2, A and B) and anisomycin conditions (Fig. 2, C and D). In these protocols, holding current was applied when necessary, to bring the membrane potential back to its baseline RMP value before the application of the pulse to compensate for any changes in apparent input resistance caused by voltage-dependent channel activation or deactivation. Care was also taken to ensure the bridge was balanced during these manipulations using the built in bridge balance function and visual confirmation using pClamp software (Axon Instruments).
Input resistance was calculated by measuring the maximal voltage deflection (ΔV) as a function of the amount of current (I) applied (Rin = ΔV/I). There were no significant changes in input resistance over time as measured by a one-way repeated-measures ANOVA in either control [n = 7; F(4,24) = 0.19, P = 0.94] or anisomycin-treated cells [n = 12; F(4,40) = 0.43, P = 0.78; Fig. 2F]. Raw traces show examples of the responding voltage deflections before, during, and after control (Fig. 2B) or anisomycin bath application (Fig. 2D).
The membrane time constant (τ) was calculated by fitting a single-order exponential to the initial portion of the membrane deflection caused by the square-wave current pulse. As shown in Fig. 2, B and D, deflections across time did not show any major differences. Across all cells tested, there were no significant changes in τ in either control [n = 7; F(4,24) = 0.39, P = 0.81, one-way repeated-measures ANOVA] or anisomycin-treated cells [n = 12; F(4,40) = 1.24, P = 0.31, one-way repeated-measures ANOVA; Fig. 2G].
Anisomycin alters single action potential properties of CA1 neurons.
To assess for changes in active membrane properties, we injected just-threshold depolarizing current pulses to elicit a single spike in each neuron across specified time points (0, 7, 14, 21, and 28 min) in both control and anisomycin conditions (see Fig. 3, A and B, respectively, for representative traces). For each time point, we measured spike threshold, spike peak, spike height, spike half-width, and spike rise and decay slopes in both control and anisomycin conditions.
Fig. 3.

Anisomycin (ANI) diminished spike height and maximum rise slope of single spikes in CA1 pyramidal neurons over time. A: superimposed traces of single action potentials during the baseline (black), 21-min (red), and 28-min postperfusion periods (gray) for a CA1 pyramidal neuron in control conditions. Arrows and lines indicate how and where spike threshold, spike peak, spike amplitude, and half-amplitude duration were measured. Across all time points, the overlay of spike shape and measures appears highly consistent. Inset shows high temporal resolution of the depolarizing phase of the action potentials. B: superimposed traces of single action potentials during the same time periods for an ANI-treated neuron, and inset shows the high temporal resolution of the depolarizing phase of spikes. Although threshold and half-amplitude duration measures appear similar in this case, spike peak, spike height, and maximum rise slope appear to be decreased following ANI treatment. C: average values for spike threshold across time in both control (black squares; left) and ANI conditions (red circles; right) show no significant changes from baseline values (B, baseline; 7, 7 min of ANI or vehicle perfusion; 14, 14 min of ANI or vehicle perfusion; 21, 21 min of ANI or vehicle perfusion; 28, 7 min postwashout and 28 min postperfusion). D: average values for spike height across the same time periods show a lack of significant change in control conditions (black squares; left) with a marked decrease in ANI conditions (red circles; right) beginning at 14 min following ANI treatment. E: average values for spike half-amplitude duration across the same time points in both control (black squares; left) and ANI conditions (red circles; right) show no significant changes in either condition. F: average values for maximum rise slope across the same time periods in control conditions (black squares; left) show no significant differences, whereas in the ANI conditions (red circles; right) there is a significant and continuing decrease beginning at 14 min postinfusion. *P < 0.05 vs. baseline.
Spike thresholds were measured as the voltage at which the initial deflection point of the action potential occurred (see Fig. 3, A and B). In control conditions, the spike threshold decreased only slightly across time (n = 8 cells; Fig. 3C); conversely, in anisomycin conditions, a slight increase in spike threshold values occurred (n = 20 cells; Fig. 3C). However, one-way repeated-measures ANOVA within treatment over time found no significant changes in either group [control: F(4,28) = 1.39, P = 0.26; anisomycin: F(4,76) = 0.92, P = 0.45].
The spike peak amplitude was defined as the maximum voltage value of the spike, and spike height was measured as the voltage difference between the threshold and spike peak of the first action potential elicited by the minimal depolarizing current pulse for both control and experimental situations across time. Although there were no changes in spike height in control conditions across time [n = 8 cells; F(4,28) = 2.61, P = 0.23, one-way repeated-measures ANOVA; Fig. 3D], there was a significant decrease in spike height in anisomycin-treated neurons across time [n = 20 cells; F(4,76) = 16.48, P < 0.0001, one-way repeated-measures ANOVA; Fig. 3D]. Tukey’s HSD post hoc tests found that within 7 min of anisomycin incubation, spike height was not different from baseline (P > 0.05). However, within 14 (P < 0.01), 21 (P < 0.01), and 28 min (7 min after the return of regular aCSF; P < 0.01) following bath application of anisomycin, spike height was significantly decreased from baseline.
The spike half-width of the action potential was measured as the width of the first action potential evoked by minimal depolarizing current pulses at a point halfway between the threshold and spike peak. One-way repeated-measures ANOVA found no significant changes in spike half-width across time points measured in control [n = 8 cells; F(4,28) = 1.45, P = 0.24] or anisomycin conditions [n = 20 cells; F(4,76) = 1.42, P = 0.23; Fig. 3E].
The maximum rise slope was measured as the slope of the rising phase of the action potential at its highest value (see raw traces expanded from time bar in Fig. 3, A and B). Although the one-way repeated-measures ANOVA across time points showed a significant decrease of these values in control cells [n = 8 cells; F(4,28) = 3.09, P = 0.03; Fig. 3F], no post hoc tests were significant when values at later time points were compared with baseline measures. In contrast, rise slope in anisomycin-treated cells significantly decreased across time points [n = 16 cells; F(4,60) = 12.35, P < 0.0001; one-way repeated-measures ANOVA; Fig. 3F], and Tukey’s HSD post hoc tests found significant differences in the maximum rise slope when baseline values were compared with those at 14, 21, and 28 min (but not 7 min) following anisomycin (P < 0.05).
The maximum decay slope was measured as the maximum slope during the repolarization phase of the action potential. There were no significant changes in maximum decay slope in either control [n = 8 cells; F(4,28) = 2.23, P = 0.09; one-way repeated-measures ANOVA] or anisomycin conditions [n = 16 cells; F(4,60) = 2.21, P = 0.07; one-way repeated-measures ANOVA] across time points measured.
Anisomycin alters multiple-spike characteristics in CA1 neurons.
We also monitored the number of action potentials generated during longer (1 s) depolarizing pulses in which multiple spikes were consistently elicited during baseline conditions (see raw traces in Fig. 4A). Across time, although there was an apparent increase in spike numbers in control conditions, these changes were not significant [n = 7 cells; F(4,24) = 1.02, P = 0.41; one-way repeated-measures ANOVA; Fig. 4B]. Conversely, in anisomycin conditions, there was a significant net reduction in the number of spikes elicited across time points [n = 16 cells; F(4,60) = 3.36, P = 0.01; one-way repeated-measures ANOVA; Fig. 4B]. Tukey’s HSD post hoc tests revealed significant decreases from baseline measures at 21 and 28 min following anisomycin administration.
Fig. 4.

Anisomycin (ANI) detrimentally affected spike train features, including diminished spike frequency and increased spike duration measures, in CA1 pyramidal neurons. A: spike train and membrane responses for 2 different neurons in response to 1-s-duration suprathreshold current pulses for control (left) and ANI perfusion conditions (right) across time (B, baseline, black traces; 21, 21 min of ANI or vehicle perfusion, red traces; 28, 7 min postwashout and 28 min postperfusion, gray traces). Whereas spike frequency remains consistent in the control perfusion case, the spike frequency markedly declines with time following ANI perfusion. Inset shows an overlaid expansion of the period following the pulse and the medium afterhyperpolarizing potential, which appears to be similar across time in the control case but to increase in amplitude in the ANI perfusion case. B: average number of spikes elicited by the same suprathreshold depolarizing pulse in control (black squares; left) and ANI conditions (red circles; right) across time (B, baseline; 7, 7 min of ANI or vehicle perfusion; 14, 14 min of ANI or vehicle perfusion; 21, 21 min of ANI or vehicle perfusion; 28, 7 min postwashout and 28 min postperfusion). Whereas there was a nonsignificant trend toward increasing spike numbers across time points in the control conditions, there was an opposite and significant decrease in the number of spikes elicited in the ANI perfusion conditions that began at 21 min and remained following ANI washout at the 28-min time point. C: average differences in firing rates as a function of 3 incremental intensities of depolarizing current pulses for control (black squares; left) and ANI conditions (red circles; right) across the baseline and 21-min sample. As demonstrated, whereas neurons in control conditions had a nonsignificant tendency to increase firing frequency across this time period, ANI-perfused neurons showed a significant decrease at higher intensities. D: average values for the differences in half-amplitude duration between the first and last spikes in a train as a function of the same time periods as described in B for both control (black squares; left) and ANI conditions (red circles; right). As demonstrated, there is a nonsignificant tendency in control conditions for the last spike to show duration increases across time, whereas this same tendency is statistically significant in ANI-treated neurons. This difference becomes significant at 21 min and continues 7 min postwashout at 28 min following ANI exposure. *P < 0.05 vs. baseline.
To follow up on these spike frequency differences, we also characterized and compared the spike frequency by current intensity characteristics in neurons at baseline and 21 min for both control and anisomycin conditions (Fig. 4C) at three consistent increasing intensities. Similar to single-spike intensity results, a two-way repeated-measures ANOVA revealed a main effect for treatment with significant decreases in spike frequency for the anisomycin group [control (n = 7 cells) vs. anisomycin (n = 16 cells): F(1,21) = 5.99, P = 0.008].
Although we found no significant differences in the half-width duration of single spikes evoked by just-threshold depolarizing pulses, we did observe changes in spike widths for later occurring spikes in spike trains evoked by suprathreshold depolarizing pulses in the anisomycin treatment group. To track changes across time, we computed the difference in half-width duration between the last and first spikes in a train across time for both control and anisomycin conditions (Fig. 4D). Whereas there were no significant differences in control-treated cells across time points [n = 7 cells; F(4,24) = 0.86, P = 0.49; one-way repeated-measures ANOVA], there were marked significant differences across time in anisomycin conditions [n = 16 cells; F(4,60) = 7.42, P < 0.0001; one-way repeated-measures ANOVA; Fig. 4D]. Tukey’s HSD post hoc tests revealed significant increases in half-width differences between baseline and the time points at 21 (P < 0.01) and 28 min (P < 0.01) following anisomycin.
Anisomycin impairs mitochondrial activity.
The prominent membrane-depolarizing effects of anisomycin occurred in the absence of any changes in apparent input resistance, similar to what is seen following inhibition of the Na+-K+-ATPase exchanger (Glitsch, 2001; Van Der Heyden and Docampo 2002; Vassalle 1987). Thus we hypothesized the effects of anisomycin on membrane potential might have been mediated by a loss of cellular ATP, therefore impairing Na+-K+-ATPase function. Although mitochondrial protein synthesis is not known to be directly hindered by anisomycin, mitochondria are dependent on more than 2,000 nuclear-encoded proteins, which make up more than 98% of the total protein content of the organelle. Indeed, mitochondrial protein synthesis itself is reliant on intact and well-balanced cytosolic translation of nuclear-encoded proteins (for reviews, see Christian and Spremulli 2012; Ryan and Hoogenraad 2007). Given our results demonstrating that anisomycin significantly diminished cytosolic translation (e.g., Fig. 1), it appeared likely that mitochondrial function was impaired.
Mitochondrial function across control and anisomycin-treated hippocampal slices was first assessed by TTC staining. As shown in Fig. 5, there was a significant decrease in the degree of TTC staining in anisomycin-treated compared with control slices (n = 4 within subject; t3 = 5.12, P = 0.01, paired t-test). Representative TTC-stained slices from control and anisomycin conditions are also shown in Fig. 5 (inset).
Fig. 5.
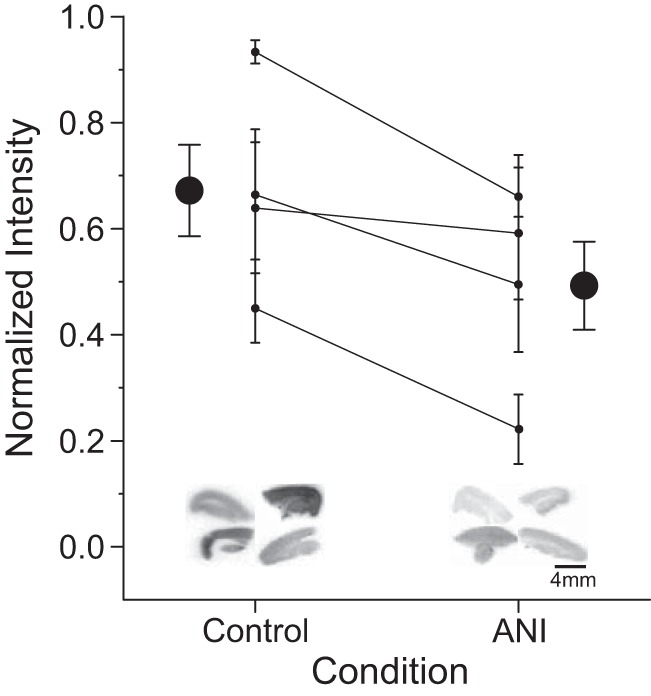
Anisomycin (ANI) decreases triphenyltetrazolium chloride (TTC) staining in transverse hippocampal brain slices. Data are normalized light intensity from TTC-stained slices across control and ANI conditions. Large circles are scatter plots showing averages (±SE); smaller circles with adjoining lines across conditions show within-experiment averages (±SE). There was a significant decrease in the normalized degree of TTC staining in slices treated with 100 µM ANI for 30 min compared with controls. Insets show example background-normalized staining scans from 4 transverse hippocampal slices in each condition from a representative within-subject experiment. A darker stain produces a higher normalized intensity. *P < 0.05, ANI vs. control.
Because of the nonspecificity of the TTC stain, we employed a targeted technique, high-resolution respirometry, to assess for specific changes in integrated mitochondrial function due to anisomycin. Two separate experimental preparations were used to make within experimental comparisons following administration of anisomycin or vehicle in 1) transverse hippocampal slices (in vitro; n = 5) and 2) whole brain intra hippocampal infusions (in vivo; n = 7).
Anisomycin-treated slices showed a general decrease in oxygen consumption across all states, pathways, and steps (see Fig. 6A). The trend reached significance only for complex IV activity, with an average decrease of 27 ± 5% in anisomycin-treated compared with control slices (t4 = 3.5, P = 0.02, paired t-test; Fig. 6A). All other pathways in the mitochondrial electron transfer system showed nonsignificant decreases in anisomycin-treated samples, including the OxPhos capacity of the N pathway (t4 = 1.67, P = 0.18, paired t-test) and the NS pathways (t4 = 2.13, P = 0.10, paired t-test) and the ET capacity of the NS pathways (t4 = 1.98, P = 0.12, paired t-test) and the S pathway (t4 = 2.11; P = 0.10, paired t-test; Fig. 6A). There was also no significant change across treatments in FCR for Leak (N pathway: t4 = 0.4, P = 0.64, paired t-test), indicating a similar mitochondrial coupling in control and anisomycin groups.
Fig. 6.

Anisomycin (ANI) impairs mitochondrial complex IV activity in vitro and in vivo. Data are high resolution respirometry results for in vitro (A) and in vivo experiments (B). For each paired experiment (connected by a single line), the measures of O2 flux for each component are shown (control, black; ANI treatment, red). Error bars on either side of the paired comparisons reflect the SE around group means, shown by the bisecting horizontal line. A: although there was a trend for overall decrease in O2 flux in ANI-treated slices, there was a significant decrease only in complex IV activity (CIV) in ANI-treated vs. control slices. B: in ANI-treated hippocampi in vivo, there was an overall decrease in O2 flux in all measures of oxidative phosphorylation (OxPhos), substrate oxidation (ET), and CIV, but not Leak respiration, compared with the contralateral control hippocampi. ECF, extracellular fluid; N pathway, NADH pathway (through complex I; pyruvate + malate); NS pathway, NADH and succinate pathways (feeding complexes I and II simultaneously; pyruvate + malate + succinate); S pathway, succinate pathway alone (after inhibition of complex I with rotenone). Refer to methods for detailed explanation of N and NS pathways, Leak, OxPhos, and ET. *P < 0.05, ANI vs. control.
A subset of experiments was performed to assess if treatment with anisomycin modified the integrity or the sensitivity of the mitochondrial outer membrane. Addition of exogenous cytochrome c caused an increase in oxygen consumption in both anisomycin-treated and control tissues, indicating possible damage to membrane integrity in the slice preparations. However, there was no effect of anisomycin treatment (t1 = 0.73, P = 0.59, paired t-test) on membrane integrity compared with control tissue, as defined by cytochrome c control ratio (control: 0.22 ± 0.08 vs. anisomycin: 0.14 ± 0.02).
In the in vitro slice preparation, due to the nature of the protocol, the extracted tissue is incubated at room temperature for at least 90 min. Short- or long-term exposure of tissue or mitochondria to room temperature is known to affect the coupling and integrity of the mitochondria (e.g., Hillered and Chan 1988, Lemieux et al. 2010, 2017). However, in a whole brain in vivo preparation, which more closely matches protocols used in behavioral paradigms, anisomycin or control treatment is delivered to a live animal 30 min before fresh tissue extraction. Following extraction, the tissue is immediately cooled before respirometry protocols. Because the results in the slice preparation may be exacerbated by changes in the integrity of mitochondria related to the conditions of the protocol itself, a second set of within-subject experiments was performed. Rat hippocampi were infused with both anisomycin and vehicle (each on opposite hemispheres) 30 min before tissue extraction. Immediately following tissue extraction, the tissue was cooled at 4°C, to more closely assess the effects of anisomycin on intact mitochondrial function.
Compared with contralateral vehicle-treated hippocampi, ipsilateral anisomycin-treated hippocampi significantly decreased oxygen consumption across all states, pathways, and steps (N pathway OxPhos: t6 = 3.09, P = 0.02, −9.4 ± 5%; NS pathways OxPhos: t6 = 2.68, P = 0.03, −9.6 ± 5%; NS pathways ET: t6 = 2.53, P = 0.04, −8 ± 5%; S pathway ET: t6 = 3.14, P = 0.02, −15 ± 5%; complex IV single step: t6 = 2.70, P = 0.03, −14 ± 5%, all paired t-test; Fig. 6B). The one exception to this significant decrease was the Leak respiration (t6 = 0.23, P = 0.78, paired t-test; Fig. 6B). These results, together with those for the slice preparation, indicate at least a defect in complex IV, which is needed in all others states and pathways as the final electron acceptor.
The FCRs showed no significant difference between control and anisomycin-treated tissues for Leak (t6 = 0.5, P = 0.63), OxPhos N pathway (t6 = 1.7, P = 0.13), OxPhos NS pathways (t6 = 1.02, P = 0.34), and complex IV single step (t6 = 1.7, P = 0.13). The cytochrome c control ratios were zero in all experiments, and therefore the mitochondrial outer membrane integrity was fully preserved in both anisomycin and control groups in the whole brain in vivo preparation.
Indeed, when tissues from the slices (n = 5) were compared with the in vivo preparations (n = 7), the slices showed overall decrements in mitochondrial integrity with significantly less coupled mitochondria as shown by a higher FCR for Leak (t10 = 4.29, P = 0.001, unpaired t-test; mean in vitro: 0.08 ± 0.006 vs. in vivo: 0.02 ± 0.004) and lower RCR values (t10 = 5.24, P = 0.0003, unpaired t-test; mean in vitro: 3 ± 0.5 vs. in vivo: 23 ± 3.9). Compared with the in vivo preparation, the slice preparation also showed a general decrease in FCR for the OxPhos state (via the N pathway; t10 = 16.45, P < 0.0001, unpaired t-test; mean in vitro: 0.22 ± 0.01 vs. in vivo: 0.59 ± 0.01; via the NS pathways: t10 = 7, P < 0.0001, unpaired t-test), whereas the FCR for complex IV was increased (t10 = 10.9, P < 0.0001) in slice vs. whole brain preparations. Previous work in the rat heart has shown that complex IV can better preserve its function during short incubation at high temperature compared with the NADH pathway or various complexes of the electron transfer system (Lemieux et al. 2010). This is clearly exacerbated during longer incubation, given that the complex IV capacity relative to the maximal OxPhos capacity with combined pathways is higher in the in vitro slice preparation compared with the in vivo preparation (1.62 ± 0.13 and 0.42 ± 0.01, respectively). The increase in cytochrome c leakage was not statistically significant between preparations (slice, n = 2; whole brain, n = 2; t1 = 3, P = 0.21, unpaired t-test). Compared with other results previously obtained in brain or other tissue homogenates (Dejos et al. 2018; Han et al. 2017; Larsen et al. 2014; Makrecka-Kuka et al. 2015; Woodman et al. 2018), the FCRs for Leak indicate similar or higher mitochondrial coupling, and the cytochrome c effect showed well-preserved integrity of the outer mitochondrial membrane.
DISCUSSION
Our results demonstrate that anisomycin has significant detrimental effects on basic membrane properties (both passive and active) in hippocampal CA1 pyramidal neurons as measured using whole cell techniques in brain slices. These changes were consistent with a breakdown of transmembrane ionic gradients and were accompanied by significant decreases in cellular energy production. Our findings suggest that the ramifications of translational blockade go well beyond the simple reduction of newly synthetized proteins and that agents such as anisomycin severely impact cellular health and function. Indeed, we propose that the use of translational inhibitors in behavioral and neurobiological studies should be completely eliminated and that prior data acquired using these substances be extensively reevaluated in the scope of our findings of impaired activity and metabolism.
We showed that the time course of our bath applications of 100 µM anisomycin significantly reduced protein synthesis by 46% in hippocampal brain slices as measured by a reduction of radiolabeled amino acid incorporation. Most in vitro studies of synaptic plasticity use lower concentrations of anisomycin (20–25 μM; Fonseca et al. 2006; Frey et al. 1988; Frey and Morris 1997; Sajikumar and Frey 2003), although the bath application durations tend to be far longer than reported here. Most in vivo behavioral studies, on the other hand, use concentrations of more than three to four orders of magnitude higher (75–235 mM; Wanisch and Wotjak 2008). Our own work has demonstrated that whereas concentration has a modest positive relationship with “neurosilencing,” it is actually the degree of translational inhibition that is critically related to neural activity impairments (Sharma et al. 2012). Thus the influence of anisomycin on neurophysiological measures is directly related to its potency at blocking de novo protein synthesis and is likely not a result of some off-target mechanism, but instead a direct consequence of translational inhibition itself. In the present experimental situation, we were successful in reducing protein synthesis by about half during our brief (30 min) profusion period. We would expect that reductions differing from this would also have varying levels of neurophysiological effects, with more translational inhibition producing more profound neurophysiological disruptions.
Our baseline values for CA1 cell membrane properties of control-treated slices were consistent with previous studies: RMP between –70 and −68 mV, thresholds for action potentials being ~20 mV above rest (between −50 and −40 mV; Spruston and Johnston 1992; Staff et al. 2000), spike peaks at 40–50 mV, and spike amplitudes of 80–120 mV (Scammell et al. 2003). The lack of change in membrane properties across time points measured in the control-treated cells indicates that changes identified in anisomycin-treated slices are due to the anisomycin treatment itself, and not to cell run-down from the in vitro preparation or whole cell technique. It was interesting to note that following a 7-min washout of anisomycin, we observed no recovery of our electrophysiological measures, suggesting that this period was not sufficient to completely rid the slice of anisomycin or, alternatively, that intracellular loading and its pharmacological effect were not sufficiently affected in this time period. Interestingly, the time course of anisomycin-induced inhibition of protein synthesis, even in the in vivo case in which local applications are presumably being actively washed by blood perfusion and fluid exchange, is on the order of 9 h (Wanisch and Wotjak 2008). In our own experiments, the detrimental effect of anisomycin on ongoing field activity was shown to last in the longest cases for 6 h and beyond (Sharma et al. 2012). Thus reversibility may not be achievable at all in the limited time frame of the ex vivo (brain slice) preparation.
Our electrophysiological findings were consistent with a loss of ionic concentration gradients, especially for Na+. Membrane depolarization was unaccompanied by any change in membrane resistance or changes in the membrane time constant, suggesting that they were unlikely to be due to changes in channel activity or to an increase in mixed ionic leak currents. Furthermore, having observed no change in the spike threshold despite the reduction in spike amplitude, together with a reduction of maximum slope on the depolarizing phase of the spike, is consistent with a reduced driving force on Na+ ions. Spike train features following anisomycin suggested more severe impairments in neuronal excitability, with a reduction in spike frequency and an increase in spike width across spike trains. This is consistent with an inability to effectively repolarize, which again could be due to reductions in driving forces that accumulate across the depolarizing pulse and spike train itself. Collectively, these changes would have broad ramifications within larger neuronal networks. Indeed, with the magnitude of protein synthesis inhibition as reported in in vivo experiments (Sharma et al. 2012; Wanisch and Wotjak 2008), the influence on collective neural activity in larger neuronal ensembles would be expected to be catastrophic, consistent with what we have previously shown (Sharma et al. 2012).
The observed influences of anisomycin on membrane properties of CA1 pyramids suggested deficiencies in cellular energetics, especially related to the operation of the Na+-K+-ATPase exchanger, which actively maintains resting ionic gradients and the high driving force on Na+ at rest. Similarly, specific blockade of Na+-K+-ATPase function using ouabain produces membrane depolarization without other passive changes in electrically excitable cells (Glitsch 2001; Van Der Heyden and Docampo 2002; Vassalle 1987). Although anisomycin does not directly impair mitochondrial protein synthesis, it is well known that mitochondrial function is directly dependent on the translation of nuclear-derived proteins (for reviews, see Christian and Spremulli 2012; Ryan and Hoogenraad 2007), many of which have an especially short half-lives (Cohen et al. 2013). Indeed, both synaptic function and plasticity may be impaired via anisomycin through a targeted influence on pre (and likely post)-synaptic mitochondria and their demonstrated dependence on local protein translation in these zones (Hillefors et al. 2007; reviewed in Cohen et al. 2013; Kaplan et al. 2009). We believe that the effect of translational inhibition on mitochondrial function, and more importantly, the inhibition of cellular energy production, is the cause of the neurophysiological effects noted in the present study. However, it would be of interest to compare the neurophysiological similarities of directed translational inhibition of mitochondrial vs. nuclear proteins.
Consistent with the idea of reduced mitochondrial function, we observed a significant decrease in cellular energetics as measured by both TTC staining and cellular respirometry. The decreased TTC staining following anisomycin treatment can be very loosely interpreted as decreased dehydrogenase activity (Nachlas et al. 1957; Riepe et al. 1996) and/or decreased oxygen consumption (Dettmers et al. 1994; Rich et al. 2001). Our respirometry experiments using various substrates and inhibitors localized the anisomycin-induced mitochondrial defect to the impairment of complex IV activity. In the slice preparation, anisomycin induced nearly a 25% decrease in complex IV activity. However, a potential problem for the respirometry assay in the slice experiments concerned the maintenance of the slices at room temperature, which likely reduced the capacities of the ET complexes upstream of complex IV, which could artificially increase complex IV excess capacity, thus reducing the impact of a defect in the final electron acceptor on the OxPhos and ET capacities. Paralleling and expanding on results from hippocampal slices, when infused into the in vivo hippocampus, anisomycin ubiquitously not only reduced complex IV capacity but also caused a general decrease in OxPhos and ET capacity within all pathways. The inhibition of complex IV disrupts the flow of ET and OxPhos pathways, thus reducing the proton motive force driving ATP-synthase, which slows overall ATP synthesis. Thus our collective findings from TTC staining and high-resolution respirometry experiments can be interpreted as decreased oxygen consumption due to complex IV inhibition. The resulting cellular energy deficit, due to inhibition of complex IV by anisomycin, would additionally disrupt ongoing resting and activity-related neuronal processes and thus explain the observed neurophysiological disruptions.
A major limitation of the current study, as with other studies, is the focus on neuronal properties while ignoring glial physiology. For example, astrocytes store and break down glycogen while serving vital oxidative fuel to neurons during periods of activation (Pellerin and Magistretti 2012). It previously has been shown that the transcriptional and translational inhibitors actinomycin D and cycloheximide inhibit activity-dependent glycogen replenishment (Bélanger et al. 2011; Sorg and Magistretti 1992). Understanding that glycogenolysis and its subsequent metabolic provision of lactate to neurons is vital for neural function as well as learning and memory (Gold et al. 2013; Pellerin and Magistretti 2012; Sorg and Magistretti 1992), we would presume that any impairment of astrocytic operations that are also dependent on de novo protein synthesis would likely have a further catastrophic effect on neural operations. It would be of interest to determine if these detrimental effects could be reversed by manipulations designed to rescue cellular and network metabolism. That being said, given our observation of mitochondrial disruption additionally induced by translational inhibition, it may be difficult to rescue anisomycin effects with metabolic substrate alone, unless they are processes dependent on glycolytic and not oxidative metabolism. Indeed, there is a critical need for further in-depth analysis of anisomycin effects on astrocytes and other glial cell types, in terms of brain physiology impacted by glial function, including cerebral blood flow, brain metabolism, and neural activity.
Future studies should additionally assess the impact of anisomycin on basal synaptic transmission. Lower concentrations of anisomycin have been shown to have little to modest effects on stimulus-evoked responses in vitro. In vivo studies using higher concentrations clearly demonstrate anisomycin-induced abolition of evoked potential responses (Kleim et al. 2003; Sharma et al. 2012), and local applications also induce gross aberrations in spontaneous monoamine release (Canal et al. 2007). However, no studies to our knowledge have assessed ongoing spontaneous synaptic activity in vitro as a function of differing degrees of translational inhibition. Given that synaptic proteins, many of them mitochondrial, have short half-lives (Cohen et al. 2013), we hypothesize that increased translational inhibition would significantly disrupt both evoked and spontaneous synaptic activity in these conditions.
Our findings suggest that the effects of anisomycin are manifold, parallel, and interacting. Given that spontaneous network activity and oxidative metabolism is important for memory encoding and consolidation (for reviews, see Dickson 2010; Hasselmo 1999; Kapogiannis and Mattson 2011; Marshall and Born 2007; Small et al. 2000), we propose that the compound suppression of normal activity, oxidative metabolism, and ongoing protein translation synergistically contribute to anisomycin’s amnestic effects. Similarly to anisomycin, manipulations or disease states that inhibit neural activity and/or metabolism impair learning and memory consolidation (e.g., Dubue et al. 2015; Gold et al. 2013; Kapogiannis and Mattson 2011; Newman et al. 2011; Parent and McGaugh 1994). Indeed, it is well known that manipulations that circumvent the neurobiological deficits induced by translational inhibitors can restore mnemonic function. Such demonstrations include the use of increasing intensities of footshock (Gold and Wrenn 2012), direct optogenetic driving of engram cells for contextual fear (Ryan et al. 2015), and limitations imposed on the aberrant release of monoamines (Qi and Gold 2009), all of which help ameliorate the activity and metabolic disturbances induced by translational inhibition. It also is the case that many manipulations known to modulate memory either directly or indirectly impact neural activity (e.g., Dickson 2010; Dubue et al. 2015; Marshall et al. 2006) or metabolism (Gold et al. 2013; Korol and Gold 1998). Given these findings, it is tempting to speculate that most to all the behavioral and memory deficits reported with the use of anisomycin or other translational inhibitors may be due to their detrimental effects on activity and neurobiology. Of course, this conjecture would severely question the validity of the serial de novo protein synthesis dependence of long-term memory.
Given our current findings showing disruption of both intrinsic neural properties and cellular oxidative metabolism, in addition to the known inhibition of ongoing protein translation, it should come as no surprise that anisomycin treatments impair both behavior and memory. The processes affected by translational inhibition reported in the present study—disrupted neuronal excitability and oxidative metabolism—are essential to brain function. We would hope that this, in addition to our repeated demonstrations of the activity-dependent and neurobehavioral consequences of translational inhibitors (Dubue et al. 2015; Greenberg et al. 2014; Sharma et al. 2012), would convince behavioral neuroscientists to abandon their use and to critically reevaluate prior data that has used them to advance the de novo protein synthesis hypothesis of long-term memory.
GRANTS
This work was supported by the Natural Science and Engineering Council of Canada Discovery Grants 249861 and 2016-06576 (to C. T. Dickson), 04843 (to T. J. Hamilton), and 402636 (to H. Lemieux), in addition to start-up funds from Faculty Saint-Jean and an equipment grant from the Canadian Foundation for Innovation (to H. Lemieux).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.J.L., T.J.H., and C.T.D. conceived and designed research; C.J.S., M.J.L., and T.J.H. performed experiments; C.J.S., M.J.L., F.N., H.L., T.J.H., and C.T.D. analyzed data; C.J.S., M.J.L., H.L., T.J.H., and C.T.D. interpreted results of experiments; C.J.S., M.J.L., H.L., and C.T.D. prepared figures; C.J.S., M.J.L., T.J.H., and C.T.D. drafted manuscript; C.J.S., F.N., H.L., T.J.H., and C.T.D. edited and revised manuscript; C.J.S., M.J.L., F.N., H.L., T.J.H., and C.T.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Lisa Rimstad for her contribution to drug preparations.
REFERENCES
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14: 724–738, 2011. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intraamygdala injections of a protein synthesis inhibitor. Proc Natl Acad Sci USA 104: 12500–12505, 2007. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem 17: 65–134, 1956. [DOI] [PubMed] [Google Scholar]
- Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 1819: 1035–1054, 2012. doi: 10.1016/j.bbagrm.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HD, Barondes SH. Puromycin effect on memory may be due to occult seizures. Science 157: 333–334, 1967. doi: 10.1126/science.157.3786.333. [DOI] [PubMed] [Google Scholar]
- Cohen HD, Ervin F, Barondes SH. Puromycin and cycloheximide: different effects on hippocampal electrical activity. Science 154: 1557–1558, 1966. doi: 10.1126/science.154.3756.1557. [DOI] [PubMed] [Google Scholar]
- Cohen LD, Zuchman R, Sorokina O, Müller A, Dieterich DC, Armstrong JD, Ziv T, Ziv NE. Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8: e63191, 2013. doi: 10.1371/journal.pone.0063191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl NA. Nerve electrical activity; depression by puromycin not related to inhibited protein synthesis. J Neurobiol 1: 169–180, 1969. doi: 10.1002/neu.480010205. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull 96: 518–559, 1984. doi: 10.1037/0033-2909.96.3.518. [DOI] [PubMed] [Google Scholar]
- Dejos C, Kuny S, Han WH, Capel H, Lemieux H, Sauvé Y. Photoreceptor-induced RPE phagolysosomal maturation defects in Stargardt-like maculopathy (STGD3). Sci Rep 8: 5944, 2018. doi: 10.1038/s41598-018-24357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmers C, Hartmann A, Rommel T, Krämer S, Pappata S, Young A, Hartmann S, Zierz S, MacKenzie ET, Baron JC. Immersion and perfusion staining with 2,3,5-triphenyltetrazolium chloride (TTC) compared to mitochondrial enzymes 6 hours after MCA-occlusion in primates. Neurol Res 16: 205–208, 1994. doi: 10.1080/01616412.1994.11740228. [DOI] [PubMed] [Google Scholar]
- Dickson CT. Ups and downs in the hippocampus: the influence of oscillatory sleep states on “neuroplasticity” at different time scales. Behav Brain Res 214: 35–41, 2010. doi: 10.1016/j.bbr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Dubue JD, McKinney TL, Treit D, Dickson CT. Intrahippocampal anisomycin impairs spatial performance on the Morris water maze. J Neurosci 35: 11118–11124, 2015. doi: 10.1523/JNEUROSCI.1857-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Bonhoeffer T. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci 9: 478–480, 2006. doi: 10.1038/nn1667. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57–65, 1988. doi: 10.1016/0006-8993(88)90008-X. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature 385: 533–536, 1997. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev 81: 1791–1826, 2001. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- Gnaiger E, Kuznetsov AV, Lassnig B, Fuchs A, Reck M, Renner K, Stadlmann S, Rieger G, Margreiter R. High-resolution respirometry. Optimum permeabilization of the cell membrane by digitonin. In: BioThermoKinetics in the Post Genomic Era, edited by Larsson C, Påhlman IL, Gustafsson L. Göteborg, Sweden: Chalmers Reproservice, 1988, p. 89–95. [Google Scholar]
- Gold PE. Protein synthesis inhibition and memory: formation vs amnesia. Neurobiol Learn Mem 89: 201–211, 2008. doi: 10.1016/j.nlm.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold PE, Newman LA, Scavuzzo CJ, Korol DL. Modulation of multiple memory systems: from neurotransmitters to metabolic substrates. Hippocampus 23: 1053–1065, 2013. doi: 10.1002/hipo.22182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold PE, Wrenn SM. Cycloheximide impairs and enhances memory depending on dose and footshock intensity. Behav Brain Res 233: 293–297, 2012. doi: 10.1016/j.bbr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg A, Ward-Flanagan R, Dickson CT, Treit D. ANI inactivation: unconditioned anxiolytic effects of anisomycin in the ventral hippocampus. Hippocampus 24: 1308–1316, 2014. doi: 10.1002/hipo.22312. [DOI] [PubMed] [Google Scholar]
- Han WH, Gotzmann J, Kuny S, Huang H, Chan CB, Lemieux H, Sauvé Y. Modifications in retinal mitochondrial respiration precede type 2 diabetes and protracted microvascular retinopathy. Invest Ophthalmol Vis Sci 58: 3826–3839, 2017. doi: 10.1167/iovs.17-21929. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci 3: 351–359, 1999. doi: 10.1016/S1364-6613(99)01365-0. [DOI] [PubMed] [Google Scholar]
- Hernandez PJ, Abel T. The role of protein synthesis in memory consolidation: progress amid decades of debate. Neurobiol Learn Mem 89: 293–311, 2008. doi: 10.1016/j.nlm.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillefors M, Gioio AE, Mameza MG, Kaplan BB. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell Mol Neurobiol 27: 701–716, 2007. doi: 10.1007/s10571-007-9148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillered L, Chan PH. Effects of arachidonic acid on respiratory activities in isolated brain mitochondria. J Neurosci Res 19: 94–100, 1988. doi: 10.1002/jnr.490190113. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialog between genes and synapses. Biosci Rep 24: 475–522, 2004. doi: 10.1007/s10540-005-2742-7. [DOI] [PubMed] [Google Scholar]
- Kaplan BB, Gioio AE, Hillefors M, Aschrafi A. Axonal protein synthesis and the regulation of local mitochondrial function. In: Cell Biology of the Axon, edited by Koenig E. Berlin: Springer, 2009, p. 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol 10: 187–198, 2011. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleim JA, Bruneau R, Calder K, Pocock D, VandenBerg PM, MacDonald E, Monfils MH, Sutherland RJ, Nader K. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron 40: 167–176, 2003. doi: 10.1016/S0896-6273(03)00592-0. [DOI] [PubMed] [Google Scholar]
- Korol DL, Gold PE. Glucose, memory, and aging. Am J Clin Nutr 67: 764S–771S, 1998. doi: 10.1093/ajcn/67.4.764S. [DOI] [PubMed] [Google Scholar]
- Larsen S, Kraunsøe R, Gram M, Gnaiger E, Helge JW, Dela F. The best approach: homogenization or manual permeabilization of human skeletal muscle fibers for respirometry? Anal Biochem 446: 64–68, 2014. doi: 10.1016/j.ab.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Lemieux H, Blier PU, Gnaiger E. Remodeling pathway control of mitochondrial respiratory capacity by temperature in mouse heart: electron flow through the Q-junction in permeabilized fibers. Sci Rep 7: 2840, 2017. doi: 10.1038/s41598-017-02789-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux H, Tardif JC, Blier PU. Thermal sensitivity of oxidative phosphorylation in rat heart mitochondria: Does pyruvate dehydrogenase dictate the response to temperature? J Therm Biol 35: 105–111, 2010. doi: 10.1016/j.jtherbio.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Makrecka-Kuka M, Krumschnabel G, Gnaiger E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules 5: 1319–1338, 2015. doi: 10.3390/biom5031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, Born J. The contribution of sleep to hippocampus-dependent memory consolidation. Trends Cogn Sci 11: 442–450, 2007. doi: 10.1016/j.tics.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature 444: 610–613, 2006. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Nachlas MM, Tsou KC, De Souza E, Cheng CS, Seligman AM. Cytochemical demonstration of succinic dehydrogenase by the use of a new p-nitrophenyl substituted ditetrazole. J Histochem Cytochem 5: 420–436, 1957. doi: 10.1177/5.4.420. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207: 123–131, 1992. doi: 10.1016/0076-6879(92)07008-C. [DOI] [PubMed] [Google Scholar]
- Newman LA, Korol DL, Gold PE. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6: e28427, 2011. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent MB, McGaugh JL. Posttraining infusion of lidocaine into the amygdala basolateral complex impairs retention of inhibitory avoidance training. Brain Res 661: 97–103, 1994. doi: 10.1016/0006-8993(94)91186-X. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32: 1152–1166, 2012. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Gold PE. Intrahippocampal infusions of anisomycin produce amnesia: contribution of increased release of norepinephrine, dopamine, and acetylcholine. Learn Mem 16: 308–314, 2009. doi: 10.1101/lm.1333409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR, Mischis LA, Purton S, Wiskich JT. The sites of interaction of triphenyltetrazolium chloride with mitochondrial respiratory chains. FEMS Microbiol Lett 202: 181–187, 2001. doi: 10.1111/j.1574-6968.2001.tb10801.x. [DOI] [PubMed] [Google Scholar]
- Riepe MW, Niemi WN, Megow D, Ludolph AC, Carpenter DO. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp Neurol 138: 15–21, 1996. doi: 10.1006/exnr.1996.0042. [DOI] [PubMed] [Google Scholar]
- Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem 76: 701–722, 2007. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Engram cells retain memory under retrograde amnesia. Science 348: 1007–1013, 2015. doi: 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski RN, Canal CE, Gold PE. Lidocaine attenuates anisomycin-induced amnesia and release of norepinephrine in the amygdala. Neurobiol Learn Mem 96: 136–142, 2011. doi: 10.1016/j.nlm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU. Anisomycin inhibits the late maintenance of long-term depression in rat hippocampal slices in vitro. Neurosci Lett 338: 147–150, 2003. doi: 10.1016/S0304-3940(02)01400-3. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW. Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci 23: 5762–5770, 2003. doi: 10.1523/JNEUROSCI.23-13-05762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JH, Castellucci VF, Kandel ER. Functioning of identified neurons and synapses in abdominal ganglion of Aplysia in absence of protein synthesis. J Neurophysiol 34: 939–953, 1971. doi: 10.1152/jn.1971.34.6.939. [DOI] [PubMed] [Google Scholar]
- Sharma AV, Nargang FE, Dickson CT. Neurosilence: profound suppression of neural activity following intracerebral administration of the protein synthesis inhibitor anisomycin. J Neurosci 32: 2377–2387, 2012. doi: 10.1523/JNEUROSCI.3543-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA 97: 6037–6042, 2000. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg O, Magistretti PJ. Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J Neurosci 12: 4923–4931, 1992. doi: 10.1523/JNEUROSCI.12-12-04923.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol 67: 508–529, 1992. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M, Spruston N. Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol 84: 2398–2408, 2000. doi: 10.1152/jn.2000.84.5.2398. [DOI] [PubMed] [Google Scholar]
- Van Der Heyden N, Docampo R. Proton and sodium pumps regulate the plasma membrane potential of different stages of Trypanosoma cruzi. Mol Biochem Parasitol 120: 127–139, 2002. doi: 10.1016/S0166-6851(01)00444-3. [DOI] [PubMed] [Google Scholar]
- Vassalle M. Contribution of the Na+/K+-pump to the membrane potential. Experientia 43: 1135–1140, 1987. doi: 10.1007/BF01945511. [DOI] [PubMed] [Google Scholar]
- Wanisch K, Wotjak CT. Time course and efficiency of protein synthesis inhibition following intracerebral and systemic anisomycin treatment. Neurobiol Learn Mem 90: 485–494, 2008. doi: 10.1016/j.nlm.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Woodman AG, Mah R, Keddie D, Noble RM, Panahi S, Gragasin FS, Lemieux H, Bourque SL. Prenatal iron deficiency causes sex-dependent mitochondrial dysfunction and oxidative stress in fetal rat kidneys and liver. FASEB J 32: 3254–3263, 2018. doi: 10.1096/fj.201701080R. [DOI] [PubMed] [Google Scholar]