

Abstract
Hypericum perforatum L. (St. John's wort) is used to treat mild‐to‐moderate depression. Its potential safety risks are pharmacokinetic drug interactions via cytochrome P450 (CYP) enzymes and P‐glycoprotein, presumably caused by hyperforin. In a phase I, open‐label, nonrandomized, single‐sequence study, the low‐hyperforin Hypericum extract Ze 117 was investigated using a drug cocktail in 20 healthy volunteers. No pharmacokinetic interactions of Ze 117 were observed for CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, and P‐glycoprotein. Area under the curve (AUC) and peak plasma concentration (Cmax) of the used probe drugs showed 90% confidence intervals (CIs) of the geometric mean ratios of the drugs taken together with Ze 117 vs. probe drug alone, well within the predefined bioequivalence range of 80–125%. Though Ze 117 did not induce dextromethorphan metabolism by CYP2D6, it weakly increased dextromethorphan AUC ratio (mean 147.99, 95% CI 126.32–173.39) but not the corresponding metabolic ratio. Ze 117 does not show clinically relevant pharmacokinetic interactions with important CYPs and P‐glycoprotein.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ St. John's wort preparations may induce cytochromes and transport proteins such as P‐glycoprotein (P‐gp), which could result in drug–drug interactions (DDIs). The constituent hyperforin has been demonstrated to be responsible for many of these DDIs.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Evidence suggests that low‐dose hyperforin extracts have a reduced risk of DDIs. Therefore, the low‐hyperforin extract Ze 117 might have a lower potential for DDIs than other high‐hyperforin St. John's wort preparations.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ No evidence for a pharmacokinetic interaction (neither induction nor inhibition) of the low‐hyperforin extract Ze 117 was observed for CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, and P‐gp. Ze 117 likewise showed no induction of CYP2D6 but rather a weak and not clinically relevant inhibition of this enzyme.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Low‐hyperforin St. John's wort extracts such as Ze 117 might have a major advantage in drug safety compared with high‐hyperforin St. John's wort preparations, avoiding unnecessary safety risks in co‐medication therapy.
St. John's wort dry extract Ze 117 is approved in several countries for the short‐term treatment of mild‐to‐moderate depressive disorders (ICD‐10 F32.0 and F32.1). Various clinical studies have shown that St. John's wort preparations are as efficacious as synthetic antidepressants but are usually better tolerated than their chemical counterparts1 and were granted with the well‐established use status by the European Medicines Agency (EMA).2, 3 Major constituents in St. John's wort extracts are hypericin, hyperforin, various flavonoids, and procyanidines.4 According to the monograph of the Committee on Herbal Medicinal Products (HMPC) of the EMA,2, 3 Hypericum extracts can contain variable amounts of constituents up to a maximum of 6.0%, 0.1–0.3% hypericin, and a minimum of 6.0% flavonoids. The commercial extract Ze 117 contains up to 0.3% hypericin and low amounts of hyperforin (≤ 0.2%).
The mechanism of action of St. John's wort is still not fully elucidated in detail, but the accepted hypothesis is that St. John's wort exerts its antidepressant effects by inhibiting a reuptake of norepinephrine, serotonin, and dopamine in the presynaptic cleft and through a modulating effect on neurotransmitters at the post‐synaptic membrane.5, 6
However, the contribution of individual Hypericum constituents to the overall efficacy is still a matter of debate. In vitro and in vivo data are either in favor or against hyperforin being the major active principle, but based on published data, a definite conclusion is still not possible.6 Considering the clinical data, extracts devoid of hyperforin have been proven to be efficacious, and thus it appears that hyperforin is not an essential constituent in Hypericum preparations. These studies demonstrated that Hypericum extract (Ze 117) is as efficacious as imipramine and fluoxetine and superior to placebo7, 8, 9 in the treatment of depressive disorders.
Nevertheless, the widely differing hyperforin amounts in commercial Hypericum extracts need to be considered when focusing on drug interactions with St. John's wort.
Several studies have shown that, in a dose‐proportional manner, hyperforin is responsible for many of the observed drug interactions.10, 11, 12 There is thus reasonable evidence to suggest that low‐dose hyperforin extracts exert no significant effects on cytochrome (CYP) enzymes, such as CYP3A4, or on transport proteins such as P‐glycoprotein (P‐gp).13, 14, 15, 16 Therefore, due to its low hyperforin content, Ze 117 might present with a lower potential for drug interactions than other St. John's wort preparations. Since in vitro data are not always predictive of in vivo behavior of the compound tested, a study in healthy human participants was performed. The aim of this study was to investigate the effect of the Hypericum extract Ze 117 (low in hyperforin) on the interaction potential with relevant CYP enzymes and P‐gp transporter.
The study design was standard for drug–drug interaction studies and was based on US Food and Drug Administration (FDA)17, 18 and EMA19 regulatory guidance (see Figure 3 for study design).
Figure 3.
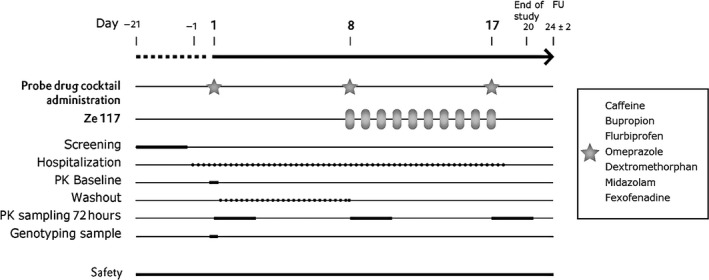
Study design. FU, follow‐up; PK, pharmacokinetic.
Cocktail approaches for phenotyping involving the administration of multiple CYP‐specific or P‐gp‐specific probe drugs were used to simultaneously assess the activities of these enzymes and the transporter P‐gp. Many phenotyping cocktails have been developed and used in recent years.20, 21, 22, 23, 24, 25 For this study, the compilation of the probe drugs was selected according to a validated phenotyping cocktail (Geneva cocktail26, 27), which also included a probe drug for P‐gp (fexofenadine). To our knowledge, this is the first seven‐probe drug cocktail interaction study investigating St. John's wort.
The primary objective of this study was to evaluate a possible interaction (induction or inhibition) of St. John's wort dry extract Ze 117 on several cytochrome P450 (CYP 450) enzymes and P‐gp. We therefore compared the pharmacokinetic (PK) end points (area under the curve from time 0 to the last sample drawn (AUC0–t) and metabolic ratios) of selective substrates for specific CYPs and P‐gp administered as a cocktail obtained on Day 8 (reflecting inhibition) and on Day 17 (reflecting induction) with Day 1.
Furthermore, the safety of Ze 117 alone and in combination with the probe drugs was also evaluated.
Results
Demographics
Twenty healthy participants of European ancestry (10 men, 10 women) entered the study. Participants were eligible if they were aged between 18 and 55 years, had a mean body weight of 72.3 kg (range 53.6–88.9 kg), and had a mean body mass index between 19.5 and 28.3 kg/m.2
Pharmacokinetics
Table 1 summarizes the PK results of probe drugs and the main metabolites: median plasma concentration/time curves are displayed in Figure 1 and Figures S1 and S2 .
Table 1.
Primary and secondary end points of probe drugs and metabolites
| End points | Ratio of geometric LS mean (%) | 90% CI of the ratio (%) | Intra‐subject CV (%) | Ratio of geometric LS mean (%) | 90% CI of the ratio (%) | Intra‐subject CV (%) |
|---|---|---|---|---|---|---|
| CYP1A2 | Caffeine | Paraxanthine | ||||
| AUC0–t Day 17/Day 1 | 99.10 | 89.35–109.90 | 18.56 | 86.48 | 76.86–97.31 | 21.21 |
| AUC0–t Day 8/Day 1 | 105.13 | 94.34–117.16 | 19.44 | 92.54 | 81.06–105.64 | 23.87 |
| Cmax Day 17/Day 1 | 99.08 | 85.83–114.37 | 25.94 | 84.93 | 75.36–95.72 | 21.50 |
| Cmax Day 8/Day 1 | 104.33 | 93.72–116.15 | 19.24 | 90.97 | 79.87–103.61 | 23.44 |
| CYP2B6 | Bupropion | 4‐Hydroxybupropion | ||||
| AUC0–t Day 17/Day 1 | 107.41 | 93.43–123.47 | 25.16 | 109.57 | 100.61–119.32 | 15.25 |
| AUC0–t Day 8/Day 1 | 107.02 | 96.03–119.27 | 19.43 | 105.54 | 95.88–116.18 | 17.19 |
| Cmax Day 17/Day 1 | 111.58 | 88.50–140.67 | 43.00 | 105.80 | 97.66–114.61 | 14.29 |
| Cmax Day 8/Day 1 | 100.81 | 84.37–120.47 | 32.47 | 95.48 | 87.78–103.85 | 15.02 |
| CYP2C9 | Flurbiprofen | 4‐Hydroxyflurbiprofen | ||||
| AUC0–t Day 17/Day 1 | 103.54 | 97.02–110.51 | 11.61 | 123.43 | 112.25–135.71 | 16.99 |
| AUC0–t Day 8/Day 1 | 99.07 | 93.83–104.61 | 9.69 | 117.89 | 109.88–126.49 | 12.56 |
| Cmax Day 17/Day 1 | 111.14 | 102.09–120.99 | 15.19 | 119.86 | 111.75–128.56 | 12.50 |
| Cmax Day 8/Day 1 | 109.67 | 100.68–119.46 | 15.29 | 117.36 | 111.35–123.69 | 9.36 |
| CYP2C19 | Omeprazole | 4‐Hydroxyomeprazole | ||||
| AUC0–t Day 17/Day 1 | 101.04 | 89.31–114.30 | 22.19 | 105.82 | 97.02–115.43 | 15.54 |
| AUC0–t Day 8/Day 1 | 118.65 | 102.64–137.15 | 26.19 | 111.33 | 102.48–120.94 | 14.79 |
| Cmax Day 17/Day 1 | 100.64 | 83.50–121.29 | 34.11 | 104.58 | 89.54–122.14 | 28.13 |
| Cmax Day 8/Day 1 | 109.70 | 91.51–131.51 | 33.08 | 104.99 | 92.33–119.38 | 23.14 |
| CYP2D6 | Dextromethorphan | Dextrorphan | ||||
| AUC0–t Day 17/Day 1 | 147.99 | 126.32–173.39 | 28.72 | 108.50 | 93.84–125.46 | 24.61 |
| AUC0–t Day 8/Day 1 | 162.23 | 141.02–186.64 | 25.30 | 108.07 | 92.48–126.29 | 26.47 |
| Cmax Day 17/Day 1 | 154.57 | 131.73–181.36 | 29.00 | 113.16 | 92.83–137.93 | 33.98 |
| Cmax Day 8/Day 1 | 163.29 | 143.98–185.19 | 22.65 | 111.61 | 94.44–131.91 | 28.46 |
| CYP3A4 | Midazolam | 1‐Hydroxymidazolam | ||||
| AUC0–t Day 17/Day 1 | 111.33 | 100.56–123.25 | 18.23 | 115.51 | 98.78–135.06 | 28.35 |
| AUC0–t Day 8/Day 1 | 120.53 | 111.10–130.77 | 14.57 | 117.82 | 103.65–133.92 | 23.07 |
| Cmax Day 17/Day 1 | 117.23 | 103.20–133.16 | 22.95 | 117.61 | 93.58–147.82 | 42.37 |
| Cmax Day 8/Day 1 | 123.50 | 108.61–140.43 | 23.14 | 117.04 | 97.21–140.91 | 33.92 |
| P‐gp | Fexofenadine | |||||
| AUC0–t Day 17/Day 1 | 101.33 | 89.00–115.81 | 23.73 | |||
| AUC0–t Day 8/Day 1 | 97.11 | 87.48–107.82 | 18.74 | |||
| Cmax Day 17/Day 1 | 116.76 | 99.41–137.13 | 29.18 | |||
| Cmax Day 8/Day 1 | 102.13 | 89.55–116.47 | 23.68 | |||
Primary end points (bold): geometric mean ratios of AUC0–t (Day 17 vs. Day 1) of probe drugs; secondary end points: all of the other end points.
AUC, area under the curve; CI, confidence interval; Cmax, peak plasma concentration; CV, coefficient of variance; LS, least squares; P‐gp, P‐glycoprotein.
Figure 1.
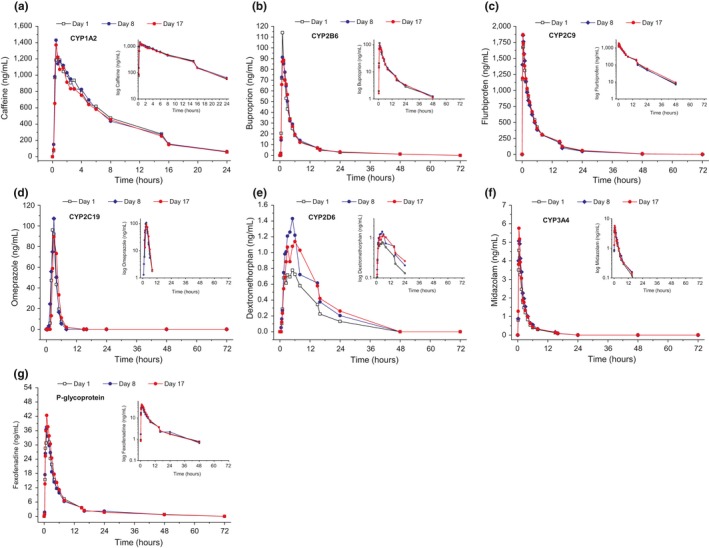
Median plasma concentrations of probe drugs (n = 19–20).
With regard to the ratio of geometric least square means (%) and their 90% confidence interval (CI), neither an inhibition (AUC0–t Day 8/Day 1) nor an induction (AUC0–t Day 17/Day 1) of metabolism for caffeine, bupropion, flurbiprofen, omeprazole, or midazolam was observed, nor was an effect on fexofenadine transport seen. When the drugs were given with and without Ze 117, the AUC ratios of the probe drugs and their respective metabolites were generally within the extended bioequivalence ranges. Dextromethorphan fulfilled the criteria for weak inhibition after acute administration (162.23% (141.02–186.64%)) and 10‐day treatment with Ze 117 (147.99% (126.32–173.39%)) (see Table 1).
Metabolic ratios (see Table 2) did not change among the different treatments. Although a larger decrease in the metabolic ratio was observed for dextromethorphan, thereby reflecting weak CYP2D6 inhibition, this was not statistically significant either for the comparison of Day 8 vs. Day 1 or when comparing Day 17 vs. Day 1.
Table 2.
Metabolic ratio of metabolite AUC0–t over probe drug AUC0–t
| Probe drug | Mean metabolic ratios ± SEMa | Comparison | Mean difference (−) a | 95% CI (−) | P‐value b | ||
|---|---|---|---|---|---|---|---|
| Day 1 | Day 8 | Day 17 | |||||
| Caffeine (CYP1A2) | 0.74 ± 0.04 | 0.66 ± 0.04 | 0.67 ± 0.05 | Day 8 vs. Day 1 | −0.08 | −0.21 to 0.05 | 0.21 |
| Day 17 vs. Day 1 | −0.07 | −0.20 to 0.05 | 0.25 | ||||
| Bupropion (CYP2B6) | 16.45 ± 1.83 | 16.80 ± 2.20 | 16.41 ± 1.75 | Day 8 vs. Day 1 | 0.35 | −5.10 to 5.81 | 0.90 |
| Day 17 vs. Day 1 | −0.03 | −5.49 to 5.42 | 0.99 | ||||
| Flurbiprofen (CYP2C9) | 0.05 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.01 | Day 8 vs. Day 1 | 0.01 | 0.01 to 0.04 | 0.27 |
| Day 17 vs. Day 1 | 0.01 | 0.02 to 0.02 | 0.35 | ||||
| Omeprazole (CYP2C19) | 0.81 ± 0.09 | 0.79 ± 0.09 | 0.84 ± 0.09 | Day 8 vs. Day 1 | −0.02 | −0.28 to 0.24 | 0.88 |
| Day 17 vs. Day 1 | 0.04 | −0.22 to 0.30 | 0.77 | ||||
| Dextromethorphan (CYP2D6) | 1.52 ± 0.38 | 0.91 ± 0.39 | 0.96 ± 0.39 | Day 8 vs. Day 1 | −0.61 | −1.70 to 0.47 | 0.26 |
| Day 17 vs. Day 1 | −0.56 | −1.65 to 0.53 | 0.30 | ||||
| Midazolam (CYP3A4) | 0.41 ± 0.05 | 0.40 ± 0.05 | 0.41 ± 0.05 | Day 8 vs. Day 1 | −0.02 | −0.15 to 0.11 | 0.78 |
| Day 17 vs. Day 1 | −0.01 | −0.14 to 0.12 | 0.89 | ||||
AUC, area under the curve; CI, confidence interval; SEM, standard error of the mean.
aCalculated as ratio of metabolite AUC0–t over probe drug AUC0–t. bLinear mixed model analysis.
Genotyping
Genotyping was performed for CYP2B6, CYP2C9, CYP2C19, CYP2D6, and ABCB1 (P‐gp) (for details see Table S1): All 20 participants were extensive metabolizers for CYP2B6 according to Turpeinen and Zanger.28 Two intermediate metabolizers (IMs) and one poor metabolizer (PM) were identified for CYP2C9 according to Deenen,29 and according to Hicks,30 two ultrarapid metabolizers, six rapid metabolizers, two IMs, and one PM were detected for CYP2C19,30 and one ultrarapid metabolizer, one IM, and one PM were determined for CYP2D6.
Three polymorphisms were investigated for P‐gp (rs2032583, rs2235015, and rs2235015). Based on Hicks,30 the rs2032583 haplotypes showed only normal activity, for rs2235015, five intermediate activity and 15 normal activity were observed, and for rs1045642, six intermediate, five reduced and nine normal activity were seen. The AUC0–t did not significantly change between Day 1, Day 8, and Day 17 for any of the probe drugs (see Figure 2). Despite this genetic variability, no alterations in the PK of the probe drugs or interactions with Ze 117 were observed for CYP2B6, CYP2C9, CYP2C19, or ABCB1 (P‐gp).
Figure 2.
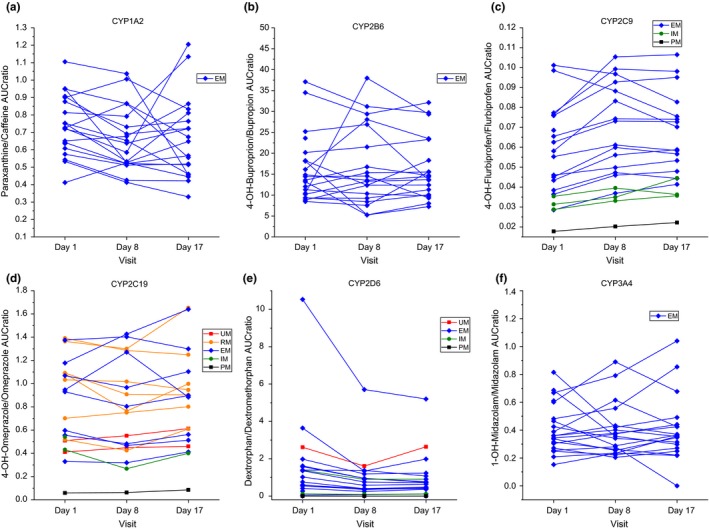
Metabolic ratios (AUC 0–t) of probe drugs for different visits and metabolic genotypes. Metabolizer status: UM = ultrarapid, RM = rapid, EM = extensive, IM = intermediate, and PM = poor metabolizers (n = 19–20). AUC, area under the curve.
Considering the genotype of the participants, it could be proven that metabolizer and transporter function status did not exert a significant effect on the PK of the probe drugs (see Figures 1 and 2) among the visits on Day 1, Day 8, and Day 17.
Tolerability
Sixteen participants reported 31 treatment‐emergent adverse events (TEAEs) of mild to moderated intensity (the most prevalent being headache and fatigue). Ze 117 given alone and in combination with the cocktail probe drugs was well tolerated. Four participants (21.1%) reported six Ze 117‐related TEAEs after dosing of the Ze 117 on Days 10 to 16, when Ze 117 was administered alone; and four participants (20.0%) reported four probe drug–related TEAEs on Day 1. One participant was withdrawn because of adverse effects on Day 5 (tonsillitis), and this was deemed unrelated to the study medication. No drug‐related TEAEs started after the last dose of Ze 117 administered together with the Geneva cocktail.
The most frequently reported TEAE was headache (overall four participants (20.0%) reported six TEAEs), followed by fatigue (overall three participants (15.0%) reported three TEAEs).
All TEAEs were of mild or moderate intensity and recovered/resolved by the end of the study.
Vital signs showed no clinically relevant changes after dosing, except for an increased body temperature in three participants (15.0%) during the period between the first cocktail administration and before the start of the Ze 117 dosing. These were considered unlikely to be related to probe drug administration.
All electrocardiograms were without any clinically significant findings.
Discussion
Since 1999, when the first clinical study focusing on PK interactions involving a commercial Hypericum extract and digoxin was reported,31 drug interactions with St. John's wort have received much attention. The first clinically relevant interaction of a Hypericum extract and the immunosuppressant cyclosporine was published in 2000 and detailed an acute rejection in two transplant patients.32 Interestingly, these initial reports of clinically relevant interactions with Hypericum preparations occurred after 1998, when many manufacturers changed the St. John's wort extraction procedure, thereby resulting in hyperforin‐rich extracts (for review, see 33).
In general, the majority of drug–drug interactions (and herb–drug interactions) are metabolism‐related interactions; indeed, most xenobiotics are capable of interacting with CYP enzymes in various ways, resulting in either enzyme induction or inhibition. When enzymes are induced by enhanced enzyme synthesis, decreased drug serum levels and thus a decreased drug response are achieved; enzyme inhibition (reversible or irreversible) results in higher serum concentrations of the drug. Depending on the magnitude of the effect, clinically relevant adverse drug reactions are possible. In addition to an induction/inhibition of CYP enzymes in the liver and intestines, the P‐gp efflux transporter interferes with drug absorption and controls tissue penetration (e.g., into the brain).
Published in vitro and in vivo data indicate that, in a manner dependent on the hyperforin concentration in the extract, St. John's wort preparations may induce CYP enzymes and P‐gp transport protein.10, 11, 12, 34
It has been shown that hyperforin activates the PXR nuclear receptor, which regulates the expression of various CYP enzymes and P‐gp.10
Since Ze 117 is a low‐hyperforin St. John's wort extract, it was this study's aim to determine its potential for interaction with relevant CYP enzymes and the P‐gp transporter in human healthy participants using a cocktail of probe drugs.
PK interaction was excluded when the 90% CIs for the geometric mean ratios were within the bioequivalence limits of 80–125% or the extended bioequivalence limits of 70–143%, also accepted for phenotyping metrics.19, 35
It was found that the 90% CIs for the geometric least square mean ratios for the probe drugs together with Ze 117 vs. probe drug alone of the PK end point AUC0–t for CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, and P‐gp were well within the bioequivalence acceptance range of 80–125%, thereby indicating no CYP or P‐gp induction or inhibition. In general, these results were confirmed by the secondary end points such as peak plasma concentration (Cmax) and metabolic ratios, which were all within the bioequivalence range of 80–125% or the predefined acceptance range of 70–143% or not significantly different between the study day (metabolic ratios). Thus, a PK interaction between Ze 117 and drugs that are substrates of these enzymes could be excluded.
Thus, in contrast to previous data with high‐hyperforin Hypericum perforatum preparations,31, 32, 36, 37, 38, 39, 40, 41, 42 no induction of CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4, or P‐gp by Ze 117 was observed in our study. Comparison of dextromethorphan AUC0–t on Day 8 and Day 17 with baseline suggested a weak inhibition of CYP2D6. In addition, the corresponding metabolic ratio showed a numerical decrease, but without reaching statistical significance (see Table 2).
An interaction of St. John's wort with CYP2D6 has been previously controversially described in the literature. Hyperforin, hypericin, and the flavonoid I3,II8‐biapigenin showed a clear inhibition of enzyme preparations in vitro;43 however, no effect of hyperforin and hypericin on CYP2D6 activity was seen in primary human hepatocytes.44 In a phenotyping experiment in elderly patients as well as in healthy volunteers, no significant effects of a St. John's wort extract on CYP2D6 activity after long‐term administration were observed.23, 36, 45, 46, 47 In our study, the extent of CYP2D6 inhibition could be classified as weak according to FDA guidelines.18 Although the mechanism of this effect is not yet understood, it appears unlikely to be related to PXR activation, which should have been associated with an induction of the CYPs assessed (except CYP1A2 and 2D6) and of P‐gp. To date, other clinical trials investigating the effect of St. John's wort preparations have shown no PK interaction with dextromethorphan as a CYP2D6.
In summary, given the well‐established use of Ze 117 in the treatment of mild‐to‐moderate depressive episodes,2, 3 these study results provide evidence of a major safety advantage of Ze 117 compared with high‐hyperforin St. John's wort extracts. This underscores the current expert opinions recommending low‐hyperforin extracts below a daily hyperforin dose of 1 mg to alleviate or mitigate potential unnecessary safety risks in co‐medication therapy.2, 3
Methods
This study (EudraCT number 2017‐003760‐12) was conducted at the Center for Human Pharmacology in Neu‐Ulm, Germany, from February 6, 2018 (first participant signed the informed consent), until March 23, 2018 (last participant last contact). All study procedures were approved by the Ethics Committee (EC) of the Bavarian State Medical Council, Munich, Germany (No. 17085, approval 18.01.2018), and by the Federal Institute for Drugs and Medical Devices (BfArM, 4042510, approval 24.01.2018). This trial was registered at ClinicalTrials.gov (NCT 03482817). All patients provided written informed consent before study entry. Additional informed consent was obtained for genotyping.
Participants
Healthy male and female volunteers were eligible to be enrolled in the study if they were physically and mentally healthy, of European ancestry, aged inclusive of and between 18 and 55 years, body mass index inclusive of and between 19 and 29 kg/m2, body weight less than or equal to 90 kg, non‐smoker, and not pregnant.
Participants of childbearing potential had to practice an acceptable method of contraception/birth control. Physical (including electrocardiogram) and laboratory examinations (including drug screening) had to be without any pathological findings.
Sample size
The intra‐subject coefficients of variation (CVs) for the AUCs of the administered cocktail probe drugs vary between 8% and 30% as reported by Bosilkovska.27 A sample size of 16 participants allowed a rejection of each null hypothesis “interaction present induction/inhibition” with α = 0.05 (one‐sided) and a power of at least 99% for a tolerance zone of 0.50–2.0 assuming as a conservative approach that the intra‐individual CVs exceed 30% and the true ratio of μtest/μreference = 1.0.19 A sample size of 16 participants allowed a rejection of each null hypothesis “interaction present induction/inhibition” with α = 0.05 (one‐sided) and a power of at least 90% for a tolerance zone of 0.70–1.43 assuming that the intra‐individual CVs exceed 30% and the true ratio of μtest/μreference = 1.0.35 As a safety margin, an additional four participants were included to account for dropouts, resulting in a total sample size of N = 20. Sample size calculations were performed using SAS 9.4 (SAS Institute, Cary, NC) for equivalence of a one‐sample mean in a multiplicative model.
Study design
The study was conducted as a phase I, open‐label, nonrandomized trial with single‐sequence design (Figure 3).
Participants were hospitalized from Day‐1 until discharge on the morning of Day 18. They received orally administration of 500 mg Ze 117 in the morning of Day 8 to Day 17. A cocktail consisting of seven probe drugs was given three times orally as a single dose each (Day 1, Day 8, Day 17). The compilation of the probe drugs was made according to a validated cocktail (Geneva cocktail27) and contained the following probes: caffeine (CYP1A2), bupropion (CYP2B6), flurbiprofen (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), midazolam (CYP3A4), and fexofenadine (P‐gp). The cocktail of seven probe drugs was given in the morning of Day 1, Day 8 and Day 17. All probe drugs were marketed drugs.
Plasma samples for determination of the probe drugs and their metabolites were taken pre‐dose and post‐dose until 72 hours after each administration of the cocktail. The primary objective was to evaluate a possible interaction (induction or inhibition) of St. John's wort dry extract Ze 117 on several CYP P450 enzymes and the transporter P‐gp by comparing the AUC0‐t of Days 1 and 17 of the cocktail substrates. Secondary objectives were to evaluate a possible interaction (induction or inhibition) of St. John's wort dry extract Ze 117 on several CYP P450 enzymes and the transporter P‐gp by comparing Cmax of the cocktail substrates and the metabolic ratios of the cocktail substrates and their relevant metabolites. Additionally, we sought to evaluate the safety of Ze 117 alone and in combination with the probe drugs.
Study drug and doses
St. John's wort dry extract Ze 117 (Rebalance 500, containing 0.96 mg hyperforin per film‐coated tablet) was taken as single tablet with 240 mL of water for 10 days (Day 8 to Day 17). The dose of 500 mg St. John's wort dry extract Ze 117 selected for this study is the registered recommended daily dose of this herbal medicinal product. This dose is acknowledged by the monograph of the Herbal Medicines Committee of the EMA.2, 3 Tablets were manufactured by Max Zeller Soehne AG, Romanshorn, Switzerland (the composition of the marketed product is given in Table S2).
The cocktail of seven marketed probe drugs was given in the morning of Day 1, Day 8, Day 17. Compilation of the probe drugs according to the validated Geneva cocktail: 50 mg caffeine tablet (CYP1A2), 75 mg bupropion HCl (CYP2B6), 10 mg flurbiprofen oral solution (CYP2C9), 10 mg omeprazole capsule (CYP2C19), 10 mg dextromethorphan oral solution (CYP2D6), 1 mg midazolam oral solution (CYP3A4), and 25 mg fexofenadine oral suspension (P‐gp). (Details are available in Table S2.)
The probe drugs were taken “at once” without test drug Ze 117 (Day 1) or together with the test drug Ze 117 (Day 8 and 17) together with 240 mL of water. Ze 117 was administered for 10 days (Days 8 to Day 17), and the cocktail was administered three times (Day 1, Day 8, Day 17). The participants received standardized meals and co‐medication was controlled. Dosing of the probe drug cocktail together with Ze 117 was performed under fasting conditions. Dosing of Ze 117 on the other study days was performed in the fasting state.
PK sampling schedule and measurement
Blood samples for determining the concentration of probe drugs and their metabolites were collected by venous puncture or indwelling venous catheter as closely as possible to the following time points: pre‐dose and 10, 20, 30, 45 minutes and 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 16, 24, 48, and 72 hours after each cocktail administration. Blood samples were analyzed by Nuvisan GmbH Bioanalytics, Neu‐Ulm, Germany, for the probe drug and metabolite concentrations using a validated liquid chromatography‐tandem mass spectrometry method. Analytes were caffeine and paraxanthine, bupropion and 4‐hydroxybupropion, flurbiprofen and 4‐hydroxyflurbiprofen, omeprazole and 5‐hydroxyomeprazole, dextromethorphan and dextrorphan, midazolam and 1‐hydroxymidazolam, and fexofenadine. The assay was carried out in accordance to Good Laboratory Practice regulations and the EMA reflection paper Good Clinical Practice.48
Bioanalytical measurement
For the quantitative determination of flurbiprofen, 4‐hydroxyflurbiprofen, bupropion, 4‐hydroxybupropion, dextromethorphan, dextrorphan, fexofenadine, omeprazole, 5‐hydroxyomeprazole, caffeine, paraxanthine, midazolam, and 1‐hydroxymidazolam in human K3‐EDTA plasma, an assay was developed and validated at Nuvisan GmbH, Neu‐Ulm, Germany, in accordance to the EMA Guideline on bioanalytical method validation.49 For quantification, isotopic labeled internal standards were used.
For flurbiprofen and 4‐hydroxyflurbiprofen, an assay based on supported liquid extraction (Isolute SLE 200 μL) of 100 μL plasma followed by a chromatographic separation on a C18 column (Waters Aquity UPLC BEH C18, 50 × 2.1 mm; 1.7 μm) under gradient conditions and liquid chromatography‐tandem mass spectrometry detection (AB SCIEX API5500) of negative ions was established. The remaining analytes were determined by protein precipitation of 50.0 μL plasma with 300 μL acetonitrile followed by a chromatographic separation on a C18 column (the same as for flurbiprofen) under gradient conditions and liquid chromatography‐tandem mass spectrometry detection of positive ions.
The following lower limits of quantification were obtained: 0.05 ng/mL for dextromethorphan, dextrorphan, midazolam, and 1‐hydroxymidazolam; 0.5 ng/mL for fexofenadine; 1.0 ng/mL for 4‐hydroxyflurbiprofen, bupropion, 4‐hydroxybupropion, omeprazole, and 5‐hydroxyomeprazole; 5.0 ng/mL for flurbiprofen; and 10 ng/mL for caffeine and paraxanthine (characteristics of the validated analytical methods are given in Table S3).
PK and statistical end points
Using Phoenix WinNonlin 7.0 (Certara, Princeton, NJ), the following non‐compartmental PK end points were calculated from the individual blood concentration–time data of the probe drugs and their metabolites on Days 1, 8, and 17: AUC0–t (AUC from time 0 to the last sample drawn), AUC0–inf (AUC extrapolated to infinite time), Cmax (peak plasma concentration), λz (terminal elimination rate constant), t 1/2 (terminal elimination half‐life) and tmax (time to reach Cmax). PK end points were calculated by noncompartmental or model‐free methods, e.g., linear trapezoidal rule for AUC, log‐linear regression for λz, etc. Missing data were not replaced or imputed in any way. Concentrations below the lower limit of quantification were treated as zero. In cases with multiple peaks, Cmax and tmax referred to the highest measured concentration even if earlier peaks were present. In cases with two or more samples having the same concentration, tmax referred to the earliest reading. The data points to be used for calculation of λz were determined by visual inspection of concentration‐time curves in log‐linear scaling. The calculation was considered sufficiently reliable in case where the coefficient of determination r 2 > 0.85 and unreliable in cases where r 2 < 0.8. Cases in between were considered on a case‐by‐case basis. The value of AUC0–inf was considered unreliable but was reported if the terminal area beyond the last quantified sample was >20% of the total AUC0–inf.
To estimate the changes in the activity of the investigated enzymes, the metabolic ratios based on AUC0–t (metabolite/parent compound) were calculated for each probe drug. These ratios were compared (Day 1 vs. Day 8 and Day 1 vs. Day 17) by linear mixed model analysis (IBM SPSS Software, version 25, IBM, Zürich, Switzerland).
Genotyping
Genotyping was performed by IMGM Laboratories GmbH, Martinsried, Germany. CYP2B6, CYP2C9, CYP2C19, and ABCB1 genotyping was performed by quantitative polymerase chain reaction (PCR) and CYP2D6 genotyping by dye terminator sequencing and agarose gel electrophoresis in 20 human blood samples using appropriate positive and negative controls.
DNA was extracted from PAXgene Blood DNA tubes using the QIAamp DNA Blood Midi Kit (Qiagen) according to manufacturer's instructions. Concentration (ng/μL) and purity (absorbance ratios 260 nm/280 nm and 260 nm/230 nm) were determined using a NanoDrop ND‐1000 UV‐VIS spectrophotometer.
Genotyping of CYP2B6, CYP2C9, CYP2C19, and ABCB1 was performed by quantitative PCR using the single nucleotide polymorphism genotyping assays listed in Table S1. From the genomic DNA 10 ng was amplified in duplicates using the ViiA7TM system (ThermoFisher Scientific) with the TaqMan Universal Master Mix II “No AmpErase UNG” and the following reaction conditions: temperature was 60°C for 30 seconds, 95°C for 10 minutes, 40 cycles of (95°C for 15 seconds followed by –60°C for 1 minutes), 60°C for 30 seconds. Data analysis and genotype calling were performed using the TaqMan Genotyper Software (ThermoFisher Scientific). Respective star alleles analyzed are listed in Table S1. Haplotypes were predicted according to Table S1.
Genotyping of CYP2D6 (alleles *2, *3, *4, *6, *7, *8, *9, *10, *14, *17, *19, *29, *35, *38, *41) was performed by dye terminator sequencing of selected PCR products using specific PCR primers followed by capillary electrophoresis. Sequencing data were analyzed using the SeqPilot Software (JSI Medical Systems). Alleles *5 (deletion) and *xN (multiplication) were analyzed by agarose gel electrophoresis using appropriate controls. Haplotypes (see Table S1), diplotypes, and phenotypes were predicted based on sequencing and agarose gel electrophoresis data.
Funding
The study costs were covered by Max Zeller Söhne AG: C.Z., E.K., J.U., M.L., M.H., S.N., J.D. S.K. did not receive any funding.
Conflict of Interest
C.Z., E.K., J.U., S.N., J.D. are employees of Max Zeller Söhne AG. M.L., and M.H. are employees of Nuvisan GmbH. S.K. declared no conflict of interest.
Author Contributions
E.K., S.N., S.K., and J.D. wrote the manuscript; C.Z., J.D., J.U., E.K., and S.N. designed the research; M.H. and M.L. performed the research; C.Z., E.K., J.U., S.N., S.K., and J.D. analyzed the data.
Supporting information
Figure S1. (a–g) Mean ± SEM plasma concentrations of probe drugs after acute administration of Ze 117 (Day 8) compared with baseline (Day 1) (n = 19–20).
Figure S2. (a–g) Mean ± SEM plasma concentrations of probe drugs after multiple administrations of Ze 117 (Day 17) compared with baseline (Day 1) (n = 19–20).
Table S1. Genotyping information.
Table S2. Applied drugs.
Table S3. Characteristics of the validated method.
Acknowledgments
The authors thank Veronika Butterweck for her support during the preparation of this article and Kathleen Bucher for careful reading of the manuscript. We thank Mikael Hansson for statistical analysis and Olivier Simonides and Carina Buehler for independent study monitoring.
Contributor Information
Simon Nicolussi, Email: simon.nicolussi@zellerag.ch.
Jürgen Drewe, Email: juergen.drewe@zellerag.ch.
References
- 1. Linde, K. , Berner, M.M. & Kriston, L. St John's wort for major depression. Cochrane Database Syst. Rev. 4, CD000448 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. EMA/HMPC . Assessment Report on Hypericum perforatum L., herba. EMA/HMPC/101303/2008. (European Medicines Agency, London, 2009) [Google Scholar]
- 3. EMA/HMPC . Community herbal monograph on Hypericum perforatum L., herba (traditional use). EMEA/HMPC/745582/2009 (European Medicines Agency, London, 2009). [Google Scholar]
- 4. Wichtl, M. Herbal Drugs and Phytopharmaceuticals, 3rd edn. (CRC Press, Boca Raton, FL, London, New York, NY, Washington, DC, 2004). [Google Scholar]
- 5. Russo, E. et al Hypericum perforatum: pharmacokinetic, mechanism of action, tolerability, and clinical drug‐drug interactions. Phytother. Res. 28, 643–655 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Schmidt, M. & Butterweck, V. The mechanisms of action of St. John's wort: an update. Wien. Med. Wochenschr. 165, 229–235 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Schrader, E. , Meier, B. & Brattström, A. Hypericum treatment of mild‐moderate depression in a placebo‐controlled study. A prospective, double‐blind, randomized, placebo‐controlled, multicentre study. Hum. Psychopharmacol. 13, 163–169 (1998). [Google Scholar]
- 8. Schrader, E. Equivalence of St John's wort extract (Ze 117) and fluoxetine: a randomized, controlled study in mild‐moderate depression. Int. Clin. Psychopharmacol. 15, 61–68 (2000). [DOI] [PubMed] [Google Scholar]
- 9. Woelk, H. Comparison of St John's wort and imipramine for treating depression: randomised controlled trial. Br. Med. J. 321, 536–539 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore, L.B. et al St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc. Natl Acad. Sci. USA 97, 7500–7502 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mannel, M. Drug interactions with St John's wort: mechanisms and clinical implications. Drug Saf. 27, 773–797 (2004). [DOI] [PubMed] [Google Scholar]
- 12. Rahimi, R. & Abdollahi, M. An update on the ability of St. John's wort to affect the metabolism of other drugs. Expert Opin. Drug Metab. Toxicol. 8, 691–708 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Arold, G. et al No relevant interaction with alprazolam, caffeine, tolbutamide, and digoxin by treatment with a low‐hyperforin St John's wort extract. Planta Med. 71, 331–337 (2005). [DOI] [PubMed] [Google Scholar]
- 14. Whitten, D.L. , Myers, S.P. , Hawrelak, J.A. & Wohlmuth, H. The effect of St John's wort extracts on CYP3A: a systematic review of prospective clinical trials. Br. J. Clin. Pharmacol. 62, 512–526 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Will‐Shahab, L. , Bauer, S. , Kunter, U. , Roots, I. & Brattström, A. St John's wort extract (Ze 117) does not alter the pharmacokinetics of a low‐dose oral contraceptive. Eur. J. Clin. Pharmacol. 65, 287–294 (2008). [DOI] [PubMed] [Google Scholar]
- 16. Mueller, S.C. et al No clinically relevant CYP3A induction after St. John's wort with low hyperforin content in healthy volunteers. Eur. J. Clin. Pharmacol. 65, 81–87 (2009). [DOI] [PubMed] [Google Scholar]
- 17. FDA . Guidance for industry. Drug interaction studies—Study design, data analysis, and implications for dosing and labeling. Draft guidance (Department of Health and Human Service. Food and Drug Administration, Rockville, MD, USA, 2012). [Google Scholar]
- 18. FDA . Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications Guidance for Industry (U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Rockville, MD, 2017). [Google Scholar]
- 19. EMA . Guideline on the investigation of drug interactions, Committee for Human Medicinal Products (CHMP), CPMP/EWP/560/95/Rev. 1 Corr. 2** (European Medicines Agency, London, 2012). [Google Scholar]
- 20. Goh, B.C. et al An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin. Pharmacol. Ther. 88, 652–659 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Magalhaes, P. , De Andres, F. , Falcao, A. , LLerena, A. & Alves, G. Can the CEIBA cocktail designed for human cytochrome P450 enzymes be used in the rat for drug interaction studies? J. Pharm. Pharm. Sci. 19, 520–529 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Lenuzza, N. et al Safety and pharmacokinetics of the CIME combination of drugs and their metabolites after a single oral dosing in healthy volunteers. Eur. J. Drug Metab. Pharmacokinet. 41, 125–138 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Wang, Z. , Gorski, J.C. , Hamman, M.A. , Huang, S.M. , Lesko, L.J. & Hall, S.D. The effects of St John's wort (Hypericum perforatum) on human cytochrome P450 activity. Clin. Pharmacol. Ther. 70, 317–326 (2001). [PubMed] [Google Scholar]
- 24. Berger, B. et al Comparison of liver cell models using the basel phenotyping cocktail. Front. Pharmacol. 7, 443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rowland, A. , van Dyk, M. , Warncken, D. , Mangoni, A.A. , Sorich, M.J. & Rowland, A. Evaluation of modafinil as a perpetrator of metabolic drug‐drug interactions using a model informed cocktail reaction phenotyping trial protocol. Br. J. Clin. Pharmacol. 84, 501–509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bosilkovska, M. et al Geneva cocktail for cytochrome p450 and P‐glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 96, 349–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bosilkovska, M. et al Evaluation of mutual drug‐drug interaction within geneva cocktail for cytochrome P450 phenotyping using innovative dried blood sampling method. Basic Clin. Pharmacol. Toxicol. 119, 284–290 (2016). [DOI] [PubMed] [Google Scholar]
- 28. Turpeinen, M. & Zanger, U.M. Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metabol. Drug Interact. 27, 185–197 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Deenen, M.J. , Cats, A. , Beijnen, J.H. & Schellens, J.H. Part 2: pharmacogenetic variability in drug transport and phase I anticancer drug metabolism. Oncologist 16, 820–834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hicks, J.K. et al Clinical Pharmacogenetics Implementation Consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 102, 37–44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johne, A. , Brockmöller, J. , Bauer, S. , Maurer, A. , Langheinrich, M. & Roots, I. Pharmacokinetic interaction of digoxin with an herbal extract from St John's wort (Hypericum perforatum). Clin. Pharmacol. Ther. 66, 338–345 (1999). [DOI] [PubMed] [Google Scholar]
- 32. Ruschitzka, F. , Meier, P.J. , Turina, M. , Lüscher, T.F. & Noll, G. Acute heart transplant rejection due to Saint John's wort. Lancet 355, 548–549 (2000). [DOI] [PubMed] [Google Scholar]
- 33. Madabushi, R. , Frank, B. , Drewelow, B. , Derendorf, H. & Butterweck, V. Hyperforin in St. John's wort drug interactions. Eur. J. Clin. Pharmacol. 62, 225–233 (2006). [DOI] [PubMed] [Google Scholar]
- 34. Gutmann, H. , Poller, B. , Berger Büter, K. , Pfrunder, A. , Schaffner, W. & Drewe, J. Hypericum perforatum: which constituents may induce intestinal MDR 1 and CYP3A4 mRNA expression? Planta Med. 72, 685–690 (2006). [DOI] [PubMed] [Google Scholar]
- 35. Doroshyenko, O. et al Drug cocktail interaction study on the effect of the orally administered lavender oil preparation silexan on cytochrome P450 enzymes in healthy volunteers. Drug Metab. Dispos. 41, 987–993 (2013). [DOI] [PubMed] [Google Scholar]
- 36. Markowitz, J.S. et al Effect of St John's wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA 290, 1500–1504 (2003). [DOI] [PubMed] [Google Scholar]
- 37. Mai, I. et al Hyperforin content determines the magnitude of the St John's wort‐cyclosporine drug interaction. Clin. Pharmacol. Ther. 76, 330–340 (2004). [DOI] [PubMed] [Google Scholar]
- 38. Mueller, S.C. et al Effect of St John's wort dose and preparations on the pharmacokinetics of digoxin. Clin. Pharmacol. Ther. 75, 546–557 (2004). [DOI] [PubMed] [Google Scholar]
- 39. Wang, L.S. et al St John's wort induces both cytochrome P450 3A4‐catalyzed sulfoxidation and 2C19‐dependent hydroxylation of omeprazole. Clin. Pharmacol. Ther. 75, 191–197 (2004). [DOI] [PubMed] [Google Scholar]
- 40. Wang, L.‐S. et al The influence of St. John's wort on CYP2C19 activity with respect to genotype. J. Clin. Pharmacol. 44, 577–581 (2004). [DOI] [PubMed] [Google Scholar]
- 41. Xu, H. , Williams, K.M. , Liauw, W.S. , Murray, M. , Day, R.O. & McLachlan, A.J. Effects of St John's wort and CYP2C9 genotype on the pharmacokinetics and pharmacodynamics of gliclazide. Br. J. Pharmacol. 153, 1579–1586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lundahl, A. , Hedeland, M. , Bondesson, U. , Knutson, L. & Lennernäs, H. The effect of St. John's wort on the pharmacokinetics, metabolism and biliary excretion of finasteride and its metabolites in healthy men. Eur. J. Pharm. Sci. 36, 433–43 (2009). [DOI] [PubMed] [Google Scholar]
- 43. Obach, R.S. Inhibition of human cytochrome P450 enzymes by constituents of St. John's wort, an herbal preparation used in the treatment of depression. J. Pharmacol. Exp. Ther. 294, 88–93 (2000). [PubMed] [Google Scholar]
- 44. Komoroski, B.J. et al Induction and inhibition of cytochromes P450 by the St. John's wort constituent hyperforin in human hepatocyte cultures. Drug Metab. Dispos. 32, 512–518 (2004). [DOI] [PubMed] [Google Scholar]
- 45. Wenk, M. , Todesco, L. & Krähenbühl, S. Effect of St John's wort on the activities of CYP1A2, CYP3A4, CYP2D6, N‐acetyltransferase 2, and xanthine oxidase in healthy males and females. Br. J. Clin. Pharmacol. 57, 495–499 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gurley, B.J. et al Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St John's wort, garlic oil, Panax ginseng and Ginkgo biloba . Drugs Aging 22, 525–539 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gurley, B.J. et al Clinical assessment of CYP2D6‐mediated herb‐drug interactions in humans: effects of milk thistle, black cohosh, goldenseal, kava kava, St. John's wort, and echinacea. Mol. Nutr. Food Res. 52, 755–763 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. EMA . GCP Inspection Working Group. Reflection paper for laboratories that perform the analysis or evaluation of clinical trial samples. EMA/INS/GCP/532137/2010, February 2012 (European Medicines Agency, London, 2012). [Google Scholar]
- 49. EMA . Guideline on bioanalytical method validation, EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2**, 21 July 2011, Committee for Medicinal Products for Human Use (CHMP) (European Medicines Agency, London, 2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a–g) Mean ± SEM plasma concentrations of probe drugs after acute administration of Ze 117 (Day 8) compared with baseline (Day 1) (n = 19–20).
Figure S2. (a–g) Mean ± SEM plasma concentrations of probe drugs after multiple administrations of Ze 117 (Day 17) compared with baseline (Day 1) (n = 19–20).
Table S1. Genotyping information.
Table S2. Applied drugs.
Table S3. Characteristics of the validated method.